 Francesca De Martini1†,§
Francesca De Martini1†,§ Nicole L. Coots1†
Nicole L. Coots1† Daniel E. Jasso-Selles1Jordyn Shevat1
Daniel E. Jasso-Selles1Jordyn Shevat1 Alison Ravenscraft2‡
Alison Ravenscraft2‡ Petr Stiblík3‡Jan Šobotník3,4‡
Petr Stiblík3‡Jan Šobotník3,4‡ David Sillam-Dussès5‡
David Sillam-Dussès5‡ Rudolf H. Scheffrahn6Tiago F. Carrijo7‡
Rudolf H. Scheffrahn6Tiago F. Carrijo7‡ Gillian H. Gile1‡*
Gillian H. Gile1‡*- 1School of Life Sciences, Arizona State University, Tempe, AZ, United States
- 2Department of Biology, The University of Texas at Arlington, Arlington, TX, United States
- 3Faculty of Forestry and Wood Sciences, Czech University of Life Sciences, Prague, Czechia
- 4Faculty of Tropical AgriSciences, Czech University of Life Sciences, Prague, Czechia
- 5Laboratory of Experimental and Comparative Ethology (LEEC), University of Sorbonne Paris Nord, Villetaneuse, France
- 6Fort Lauderdale Research and Education Center, University of Florida, Davie, FL, United States
- 7Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, São Bernardo do Campo, Brazil
The eukaryotic microbiome of “lower” termites is highly stable and host-specific. This is due to the mutually obligate nature of the symbiosis and the direct inheritance of protists by proctodeal trophallaxis. However, vertical transmission is occasionally imperfect, resulting in daughter colonies that lack one or more of the expected protist species. This phenomenon could conceivably lead to regional differences in protist community composition within a host species. Here, we have characterized the protist symbiont community of Heterotermes tenuis (Hagen) (Blattodea: Rhinotermitidae) from samples spanning South and Central America. Using light microscopy, single cell isolation, and amplicon sequencing, we report eight species-level protist phylotypes belonging to four genera in the phylum Parabasalia. The diversity and distribution of each phylotype’s 18S rRNA amplicon sequence variants (ASVs) mostly did not correlate with geographical or host genetic distances according to Mantel tests, consistent with the lack of correlation we observed between host genetic and geographical distances. However, the ASV distances of Holomastigotoides Ht3 were significantly correlated with geography while those of Holomastigotoides Ht1 were significantly correlated with host phylogeny. These results suggest mechanisms by which termite-associated protist species may diversify independently of each other and of their hosts, shedding light on the coevolutionary dynamics of this important symbiosis.
Introduction
The degree of intraspecific variation in the insect gut microbiota varies greatly among lineages. At one extreme, many insects, including butterflies and walking sticks, have such highly variable gut microbial communities that it has been argued they have no specific microbiome (Shelomi et al., 2013; Hammer et al., 2019; Ravenscraft et al., 2019). This high variability likely stems from molting, which disrupts the gut microbiota, and from the lack of a mechanism for vertical transmission of microbes. At the other extreme, in social insects, intimate interactions among nestmates create a route by which the microbiome can be transmitted and stably maintained (Engel and Moran, 2013; Sanders et al., 2014). As a result, many social insects host a stable and consistent core microbiome at both the intra-colony and intra-species levels (e.g., corbiculate bees, Kwong et al., 2017).
In termites, the social behavior of proctodeal trophallaxis (anal feeding) provides a mechanism for both vertical inheritance of gut microbes and for re-establishment of the microbiota after each molt (Brune and Dietrich, 2015). This behavior emerged in the ancestor of termites and their sister lineage Cryptocercus and likely enabled the transition to wood feeding by trapping cellulolytic protists and their nitrogen-fixing endosymbionts in an increasingly specialized digestive symbiosis (Nalepa, 2015). As a result, termites exhibit relatively stable and largely co-diversifying gut microbial communities (Bourguignon et al., 2018).
The eukaryotic component of “lower” termite microbiomes (all families except Termitidae) consists of highly specialized wood-feeding flagellates (protists) that cannot live outside their host. This means that unlike prokaryotic microbiota, the protist communities do not include a transient, environmentally acquired component. Because of this strict vertical inheritance, protist communities are strongly influenced by termite phylogeny and are not expected to have a biogeographical influence independent of their hosts (Tai et al., 2015; Taerum et al., 2018). However, there are two documented evolutionary mechanisms consistent with direct termite-to-termite transmission that could conceivably introduce a biogeographical component to protist distribution within a single host species. These are (1) transmission of protist species from one host species to another, and (2) lineage-specific losses of symbiont species.
Horizontal symbiont transfer (HST) from one termite lineage to another is theoretically possible, according to experiments on laboratory colonies (Light and Sanford, 1928; Dropkin, 1946). It may have taken place between Hodotermopsis (Archotermopsidae) and the ancestor of Reticulitermes (Rhinotermitidae) (Kitade, 2004; Tai et al., 2015). This would explain the distinct protist community found in Reticulitermes as compared to its rhinotermitid relatives (Kitade, 2004). Another ancient HST may have occurred in the ancestor of Serritermes and Glossotermes (Serritermitidae), again explaining why the symbionts of these host genera differ from those of their rhinotermitid relatives (Radek et al., 2018). To date these are the only two documented inferences of HST in termites. However, if HST is more common than currently appreciated, it could lead to regional differences in protist communities across a host species’ range, especially when different parts of that range overlap with different termite species.
Lineage-specific loss of protist symbionts is much more common than HST. The absence of one or two of the expected protist species from a termite colony has been documented by both morphological and molecular methods (Kitade and Matsumoto, 1993; Kitade et al., 2012, 2013; Taerum et al., 2018; Michaud et al., 2020). Such losses are more common for some host species than others, and for some protist species than others (Kitade and Matsumoto, 1993; Kitade et al., 2013; Michaud et al., 2020). Protists are inherited biparentally: both king and queen contribute their symbionts to the nascent colony’s microbial community (Shimada et al., 2013; Brossette et al., 2019). Host outcrossing can therefore allow complementation of these deficient communities, helping to maintain a consistent protist community across the host species’ range (Michaud et al., 2020). However, if gene flow between host populations becomes restricted, the opportunity to re-acquire a protist species by host inter-population mating would cease, and regional differences in protist communities could emerge.
If these mechanisms can, in fact, lead to a geographical influence on protist distribution, such an influence should be more readily detected in a broadly distributed host. Heterotermes tenuis (Hagen) (Blattodea: Rhinotermitidae) occupies a vast geographical range from as far north as southern Mexico and the West Indies down to northern Argentina, and from Ecuador to eastern Brazil (Constantino, 1998; Krishna et al., 2013; Carrijo et al., 2020). It is one of the most common termite species in both dry and forested areas of South America (Mathews, 1977; Davies et al., 2003; Ackerman et al., 2009). Like other Heterotermes species, it forages underground and consumes wood in contact with soil (Mathews, 1977). It has been reported to cause damage to buildings and crops, including sugarcane and eucalyptus plantations (Constantino, 2002; Batista-Pereira et al., 2004; Haifig et al., 2008). Heterotermes tenuis exhibits both morphological and molecular diversity, suggesting that it may be considered a species complex rather than a singular species (Constantino, 2000; Carrijo, 2013).
The symbiotic protist community of H. tenuis is incompletely described. Early morphological descriptions reported one species of Pseudotrichonympha, one species of Cononympha, and 1-5 species of Holomastigotoides (Mackinnon, 1926, 1927; de Mello, 1954). These three protist genera, all belonging to the phylum Parabasalia, are typical of Heterotermes and Coptotermes hosts and are restricted to the Rhinotermitidae (Yamin, 1979; Kitade, 2004; Jasso-Selles et al., 2017; Jasso-Selles et al., 2020). More recently, molecular investigations have documented two distinct phylotypes of Pseudotrichonympha (Saldarriaga et al., 2011), three distinct phylotypes of Holomastigotoides (Gile et al., 2018), and a phylotype related to Cthulhu macrofasciculumque from Prorhinotermes simplex (James et al., 2013). No molecular data from Cononympha from H. tenuis have been published to date.
In this study, we first revisited the hindgut protist community of H. tenuis in order to clarify its composition. Using a combination of light microscopy, 18S rRNA sequences from single isolated cells, and high-throughput amplicon sequencing of H. tenuis colonies collected across its range, we identified eight distinct species-level clades, belonging to four genera, which we refer to as phylotypes. We next examined the diversity and distribution of 18S rRNA amplicon sequence variants for each of the protist phylotypes in order to determine whether symbiotic protists might exhibit biogeographical patterns independent from those of their host.
Materials and Methods
Termite Collections and Barcoding
Live and RNA later-preserved termites were collected in Ecuador in 2016 under permit No 015 – 2016 – IC – FAU – FLO – DPAO - PNY, in French Guiana in 2019 under permit TREL1820249A/108, and in Panama in 2018 under permit SC/A-24-17. Ethanol-preserved specimens were provided by collections at the University of Florida and the Museu de Zoologia da USP (MZUSP), Brazil. Full accession and collection information for each specimen is provided in Supplementary Table 1.
Molecular barcodes for termite specimens were generated by amplifying and sequencing the mitochondrial large subunit 16S ribosomal RNA gene (mt16S), using the primers LRN 5′-CGC CTG TTT ATC AAA AAC AT-3′ and LRJ 5′-TTA CGC TGT TAT CCC TAA-3′ (Simon et al., 1994; Kambhampati and Smith, 1995). Template DNA was extracted from the excised hindgut of live workers or from the macerated abdomen of ethanol- or RNAlater-preserved workers using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. The PCR cycle included a 3 min denaturation at 95°C followed by 30 cycles of 95°C for 30 s, 46°C for 30 s, and 72°C for 60 s, and a final 10 min extension at 72°C. PCR products were purified using the GeneJET PCR Purification Kit (Thermo Fisher Scientific, Waltham, MA, United States) according to the manufacturer’s protocol and sequenced directly on both strands on an Applied Biosystems 3730 capillary sequencer.
Live Protist Isolation and Characterization
Live termite hindguts were removed and their contents suspended in Ringer’s solution (8.5 g NaCl, 0.2 g KCl, 0.2 g CaCl2, 0.1 g NaHCO3 per L, HiMedia Laboratories). Individual Pseudotrichonympha, Holomastigotoides, Cononympha, and Cthulhu cells were isolated by drawn-glass micropipette on an AxioVert inverted microscope and photographed with an Axiocam 105 color camera (Zeiss). Additional micrographs were taken on an AxioImager upright compound light microscope with an AxioCam 503 monochrome camera (Zeiss).
Each isolated cell was washed twice in fresh Ringer’s solution and transferred into a 0.5 ml tube for DNA extraction using the MasterPure DNA purification kit (Epicentre Biotechnologies) following the manufacturer’s protocol, except that purified DNA was resuspended in 5 μl of deionized, ELGA-purified water. The 18S rRNA gene was then amplified using a nested PCR approach. Primer pairs SpiroF1 5′-ATA CTT GGT CGA TCC TGC CAA GG-3′ and SpiroR1 5′-TGA TCC AAC GGC AGG TTC MCC TAC-3′ (Taerum et al., 2019) were used for the for the initial (outer) reaction and GGF 5′-CTT CGG TCA TAG ATT AAG CCA TGC-3′ and GGR 5′-CCT TGT TAC GAC TTC TCC TTC CTC-3′ (Gile et al., 2011) for the second (inner) reaction. PCR cycles for both primer pairs included a 3 min denaturation at 95°C, followed by 30 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 90 s, and a final 10 min extension at 72°C. Resulting PCR products were separated by gel electrophoresis and visualized with GelGreen stain (Biotium). Bands of the expected size (∼1500 base pairs) were excised from a 1% agarose gel, purified using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific), cloned using the TOPO TA cloning kit for sequencing (Invitrogen), and sequenced on both strands using the ABI 3730XL capillary sequencer.
Phylogenetic Analyses
We successfully amplified and sequenced the 18S rRNA gene from six individual Pseudotrichonympha cells, 10 Holomastigotoides cells, three Cononympha cells, and one pool of eight individually isolated Cthulhu cells. Between two and eight clones were sequenced from each cell, trimmed of vector, and assembled using Geneious R9 (Kearse et al., 2012). For preliminary analyses, all 116 new sequences were aligned with previously published Parabasalia 18S rRNA gene sequences using MAFFT v. 7.222 (Katoh and Standley, 2013). Ambiguously aligned sites were identified by eye and removed in AliView 1.17.1 (Larsson, 2014). A maximum likelihood (ML) phylogeny was computed using RAxML v. 8.0 (Stamatakis, 2014).
For the final parabasalian 18S rRNA phylogenetic analyses, we selected a subset of representative sequences from each protist phylotype in order to reduce redundancy. These sequences were submitted to GenBank under accessions MW353606-MW353628. New and previously published sequences from Honigbergiellida (Cthulhu and outgroup), Spirotrichonymphida (Holomastigotoides and Cononympha), and Trichonymphida (Pseudotrichonympha and outgroup) were aligned separately in order to maximize the number of alignable sites in each matrix. The final, manually trimmed, matrix sizes were: Honigbergiellida 18 taxa, 1389 sites; Spirotrichonymphea 31 taxa, 1471 sites; Trichonymphida (family Teranymphidae only) 31 taxa, 1404 sites. Bayesian and ML phylogenetic analyses were performed for each matrix using MrBayes v. 3.2.6 (Ronquist et al., 2012) and RAxML v. 8.0 (Stamatakis, 2014), respectively, under the GTR-Γ model, with support for each node assessed by 1,000 bootstrap replicates (ML) and posterior probability (Bayes). For the Bayesian analyses, two independent chains, sampled once every 100 generations, were run until they converged (the average standard deviation of partition frequency values between the chains dropped below 0.01). Convergence was reached after 20,000 generations for Honigbergiellida, 240,000 generations for Spirotrichonymphida, and 110,000 generations for Teranymphidae. The first 25% of trees were discarded as burn-in and majority rule consensus trees were computed from the remaining 300, 4600, and 1650 trees, respectively. We also computed ML trees from each of these data sets with all amplicon sequence variants (ASVs) from each taxon in order to visualize the phylogenetic diversity of the ASVs (see below for details).
For the host mt16S analyses, new and previously published sequences from Heterotermes species were aligned with MAFFT v. 7.222 (Katoh and Standley, 2013), which yielded a clean alignment of 67 taxa by 404 sites. This matrix was subjected to ML and Bayesian phylogenetic analyses under the GTR-Γ model of sequence evolution as above. New termite mt16S barcode sequences were submitted to GenBank under accession numbers MW345686-MW345724 (Supplementary Table 1).
Hindgut Community 18S rRNA Amplicon Sequencing
DNA was extracted from the whole gut contents of live termites or the macerated abdomens of termites preserved in ethanol or RNAlater (Thermo Fisher Scientific, Waltham, MA, United States) using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific, Waltham, MA, United States). Each DNA template was acquired from a single worker individual. Amplicon libraries were generated using a two-step PCR protocol with dual index sequencing (Kozich et al., 2013). The first PCR used gene-specific primers with Illumina adaptor sequences at their 5′ ends: nexF-ParaV45F 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3′ and nexF-ParaV45R 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G-3′ (Jasso-Selles et al., 2017). The second PCR attached indexing barcodes to both ends of each amplicon (Hamady et al., 2008). PCR programs included an initial 3 min denaturation at 95°C, then 35 cycles of 95°C for 30 s, 48°C (1st step) or 50°C (2nd step) for 30 s, and 72°C for 50 s, followed by a 10 min final extension at 72°C. All PCR reactions used EconoTaq 2x Master Mix (Lucigen) with 2.5 μl of 2 μM stock forward and reverse primers and 2.5 μl of undiluted template DNA in a 30 μl reaction. PCR products were purified using Ampure (Beckman Coulter) magnetic beads in combination with the Beckman Biomek NXp robot and quantified using Qubit broad range dsDNA fluorescent dye (Invitrogen) on a Biotek HT1 Plate Reader. After quantification, samples were pooled at approximately equivalent concentrations and sequenced on the Illumina MiSeq platform. This study includes samples run in 2017 using 2 × 300 paired-end chemistry and samples run in 2019 using 2 × 250 paired-end chemistry.
Amplicon sequence analyses were performed using QIIME 2 2020.2 (Bolyen et al., 2019). Casava 1.8 paired-end demultiplexed fastq files were imported via QIIME tools import. The demultiplexed reads were trimmed, merged, and denoised separately for each sequencing run using DADA2 (Callahan et al., 2016). The 2 × 250 samples did not require trimming. Both sequencing runs were merged into a single dataset after denoising. Samples consisting of fewer than 500 reads for the Parabasalia taxa of interest were discarded. Taxonomy was assigned using a naïve Bayes classifier implemented in QIIME 2 (Bokulich et al., 2018) trained on a manually curated Parabasalia 18S rRNA reference file of 1075 published and unpublished sequences, including all 116 18S rRNA clones from single cells sequenced in this study. Raw demultiplexed fastq files were submitted to NCBI SRA under bioproject PRJNA683896.
ASV Diversity and Distribution Analyses
Mean and maximum pairwise distances between amplicon sequence variants (ASVs) assigned to each protist phylotype and linear regressions of ASV diversity and uniqueness vs. protist prevalence were computed in R version 4.0.1 (R Core Team, 2020). Distributions of ASVs across sample locations were determined by sorting ASVs into individual datasets for each protist phylotype and for each geographical coordinate using QIIME 2 2020.2 (Bolyen et al., 2019). For Mantel tests, all genetic distance matrices (for protist 18S rRNA ASVs and host mt16S rRNA sequences) used uncorrected pairwise (“p”) distances with no model of evolution. In cases where multiple unique ASVs were present at the same coordinate, the ASV with the greatest read depth was selected to represent that coordinate in uncorrected pairwise distance matrices for each protist phylotype. Euclidean distance was used as the distance measure for the geographic distance matrices. All full and partial Mantel tests were conducted in R version 4.0.1, using packages vegan (Oksanen et al., 2019) and ape (Paradis and Schliep, 2019), with significance level determined from 999 permutations in each test.
Results
Morphology of H. tenuis Hindgut Protist Symbionts
Using light microscopy, we observed the protists inhabiting the guts of live termites collected in Ecuador, Panama, and French Guiana. We identified four genera of protists, all belonging to the phylum Parabasalia: Pseudotrichonympha, Cthulhu, Cononympha, and Holomastigotoides (Figure 1). Each of these genera had been previously reported to inhabit the gut of H. tenuis, though Cononympha was previously known as Microspironympha or Spirotrichonympha (Mackinnon, 1926, 1927; de Mello, 1954; Saldarriaga et al., 2011; James et al., 2013). We did not observe any additional genera beyond those previously reported to occur in H. tenuis.
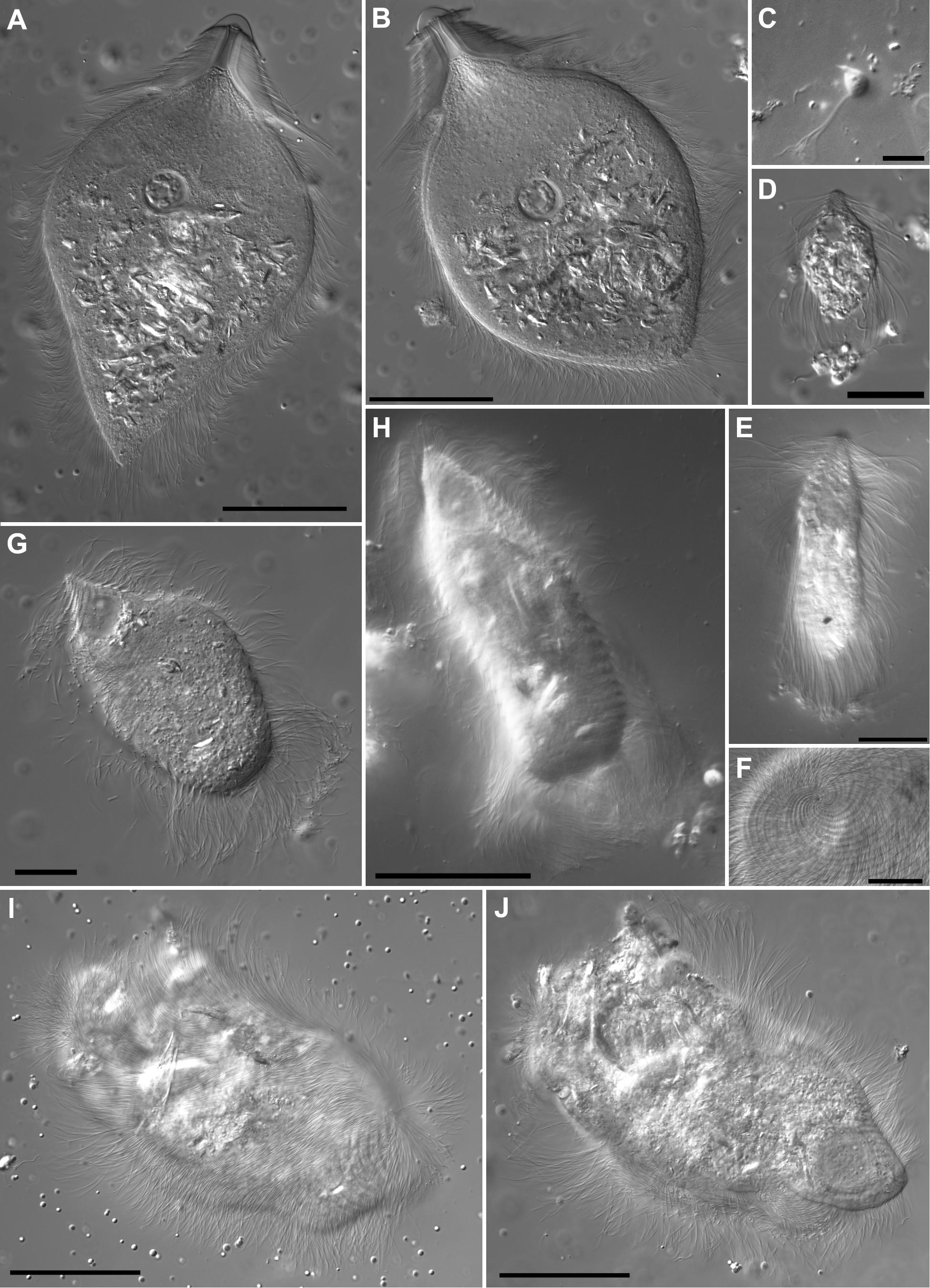
Figure 1. Differential interference contrast light micrographs of Heterotermes tenuis parabasalian hindgut symbionts. (A,B) Pseudotrichonympha cells (Trichonymphida) are characterized by a prominent rostral tube with apical cap or nose cone. Flagella of three distinct length classes cover the cell body, short flagella on the rostral area, long flagella form a fringe around the base of the rostral area, and medium length flagella cover the remainder of the cell body. Both cells have ingested many wood fragments (refractile material throughout cell body) and both are shown slightly flattened by the cover slip. (A) Smooth nose cone morphotype. (B) Morphotype with marginal ridge on nose cone. (C) Cthulhu cells (Honigbergiellida) are small and bear several apical flagella. (D,E) Cononympha cells (Spirotrichonymphida) with characteristic apical pseudorostrum in which flagellar bands form a spiral staircase-like structure. (D) Smaller morphotype with central to anterior nucleus location and detritus adhering to the posterior flagella. (E) Larger morphotype with more elongate shape and centrally located nucleus. (F–J) Holomastigotoides (Spirotrichonymphida). (F) Origin of spiraling flagellar bands at a cell apex. (G) Acuminate apex morphotype (type 3) with prominent anterior nucleus and posterior ingested wood particles. (H) Bottle-shape morphotype with anterior nucleus, ingested wood particles, and relatively long flagella. (I,J) Large amorphous morphotype cell in two optical sections. (I) Glancing surface section with spiral flagellar bands visible. (J). Deeper optical section with large anterior nucleus visible. Scale bars: (A,B,H,I,J) 50 μm; (D,E,F,G) 20 μm; (C) 10 μm.
Pseudotrichonympha (Trichonymphida) is a large, hypermastigote, wood-feeding protist. It has an anterior cell portion called a rostrum and a posterior portion called the cell body or post-rostral area. The rostrum has a smooth apical cap and an internal tube-like structure. Except for the apical cap, the entire cell is covered with flagella of three different lengths; short flagella on the rostrum, long flagella forming a fringe around the cell at the base of the rostrum, and medium-length flagella covering the rest of the cell (Grassi and Foà, 1911; Grassi, 1917; Saldarriaga et al., 2011). We observed two Pseudotrichonympha morphotypes in H. tenuis, one with a raised rim around the margin of its apical cap and one without (Figures 1A,B). Whether this characteristic represents a stable, heritable trait or a variable or artifactual one remains unknown. Previously published electron micrographs of Pseudotrichonympha from H. tenuis also showed a raised rim around the apical cap while light micrographs from the same study lacked this feature (Saldarriaga et al., 2011).
Cthulhu (Honigbergiellida) is a very small protist defined by its bundle of roughly 20 anterior flagella. This feature differentiates it from its relatives Cthylla and Hexamastix, which each have five anterior flagella (James et al., 2013). The number of flagella borne by the H. tenuis symbiont is difficult to capture in still micrographs of live specimens (Figure 1C), but video microscopy demonstrates that the number is more than 10 (Supplementary Video 1).
Cononympha (Spirotrichonymphida) is similar to Holomastigotoides in having spiral bands of flagella, but these come together at the cell apex to form a spiral staircase-like structure called a pseudorostrum with an internal axial tube-like structure called a columella (Koidzumi, 1921; Jasso-Selles et al., 2017). We observed two distinct Cononympha morphotypes inhabiting H. tenuis (Figures 1D,E). One type had rounded cells measuring less than 30 μm in length with a nucleus positioned near the anterior of the cell (Figure 1D), while the other type had larger, more elongated cells that measured 35–65 μm in length and a more centrally positioned nucleus (Figure 1E).
Holomastigotoides (Spirotrichonymphida) is also a wood-feeding, highly flagellated protist, but bears helical bands of flagella that originate at the cell apex and encircle the cell nearly to its posterior pole. Its nucleus is positioned anteriorly (Grassi and Foà, 1911; Grassi, 1917; Brugerolle and Lee, 2000). Within this morphological category, we observed a range of cell sizes and shapes, suggesting multiple Holomastigotoides species in H. tenuis (Figures 1F–J). One morphotype was characterized by very large cells, measuring 130–180 μm in length, with a flexible, irregular outline (Figures 1I,J). Other cells were smaller with an acutely pointed apex (Figure 1G) or an apex that tapered gradually to a point (Figure 1H) or a more rounded apex (not shown).
Molecular Phylogenetic Characterization of Individually Isolated Protist Cells
For molecular characterization of these symbionts, we isolated single protist cells and amplified, cloned, and sequenced their 18S rRNA genes. Cells were selected to represent the full range of morphological variability within each genus as much as possible. From 20 single cells, we successfully sequenced 116 clones, which clustered into eight groups of closely related sequences (species-level phylotypes) (Supplementary Table 2). We recovered two phylotypes from our isolated Pseudotrichonympha cells, one phylotype of Cthulhu, two phylotypes of Cononympha, and three phylotypes of Holomastigotoides, all of which branched with previously characterized congeners, consistent with our morphological identifications. Six of these phylotypes branched closely with previously published sequences from H. tenuis symbionts (Noda et al., 2007; Saldarriaga et al., 2011; James et al., 2013; Gile et al., 2018). We labeled our new sequences concordantly, so Pseudotrichonympha paulistana follows Saldarriaga et al., 2011, Pseudotrichonympha sp. follows Noda et al., 2007, and the three Holomastigotoides phylotypes follow previously published clones Ht1, Ht2, and Ht3 (Noda et al., 2007; Saldarriaga et al., 2011; Gile et al., 2018). The single Cthulhu phylotype did not require a distinguishing phylotype label. Cononympha phylotypes were numbered 1 and 2 arbitrarily but consistently.
In order to maximize the number of alignable sites, we carried out phylogenetic analyses separately for Pseudotrichonympha (Figure 2), Cthulhu (Figure 3), and the Spirotrichonymphida genera Holomastigotoides and Cononympha (Figure 4). The position of the two Pseudotrichonympha phylotypes was not resolved, though Pseudotrichonympha from Heterotermes hosts formed a supported clade (Figure 2). Our Cthulhu sequences were nearly identical to a previously published symbiont clone from H. tenuis collected in northern Colombia (James et al., 2013; Figure 3). This sequence (JX975351) was amplified from whole gut DNA, not an isolated cell, so it was not attributed to Cthulhu, though Cthulhu morphotypes were observed in H. tenuis in the same study (James et al., 2013). Our new sequence was amplified from a pool of eight Cthulhu cells isolated from H. tenuis collected in Ecuador (Figure 3). For Cononympha, the two phylotypes formed a clade with Cononympha aurea from Heterotermes aureus, the only Cononympha sequences so far available from any Heterotermes host (Jasso-Selles et al., 2017; Figure 4). Finally, two of the three Holomastigotoides phylotypes branched together with weak support while the third phylotype branched sister to the Holomastigotoides species from Heterotermes aureus. In contrast to Pseudotrichonympha, the Holomastigotoides species from Heterotermes hosts did not form a clade (Figure 4).
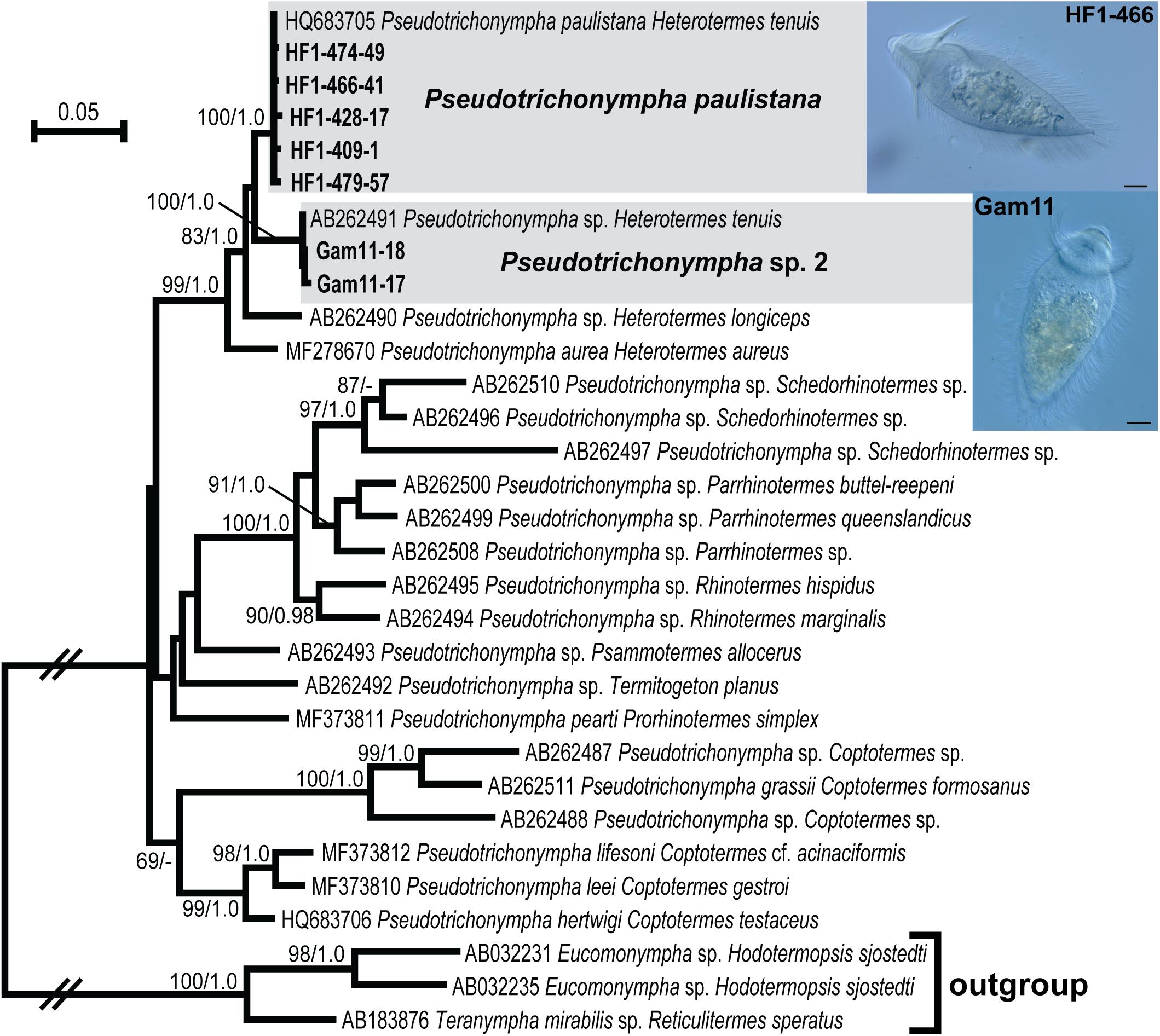
Figure 2. Maximum likelihood phylogeny of 18S ribosomal RNA gene sequences from Pseudotrichonympha with outgroup Eucomonympha and Teranympha (Teranymphidae: Trichonymphida). Protist species-level phylotypes from H. tenuis are indicated by gray boxes and new sequences obtained in this study are indicated by bold text. Support at nodes is given out of 100 bootstrap replicates computed by RAxML/Bayesian posterior probabilities if greater than 60/0.95. New sequences from H. tenuis symbionts are indicated by bold text. Select cells from which new sequences were derived are shown in micrographs inset at right; sequence names (tip labels) include the cell code. All scale bars: 20 μm.
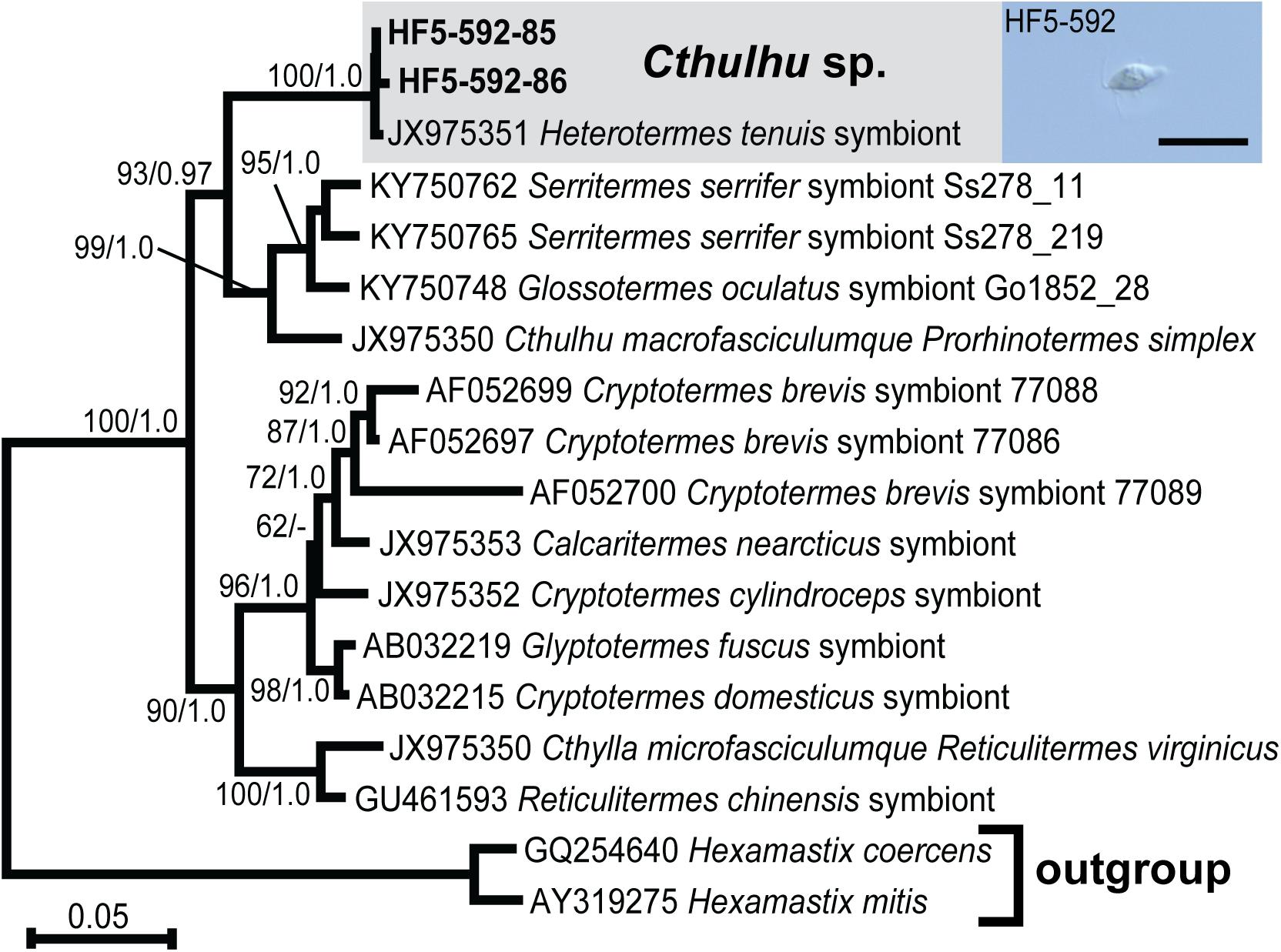
Figure 3. Maximum likelihood phylogeny of 18S ribosomal RNA gene sequences from Cthulhu species with Hexamastix sequences as outgroup. Protist species-level phylotypes from H. tenuis are indicated by gray boxes and new sequences obtained in this study are indicated by bold text. Support at nodes is given out of 100 bootstrap replicates computed by RAxML/Bayesian posterior probabilities if greater than 60/0.95. New sequences from H. tenuis symbionts are indicated by bold text. Inset DIC micrograph features one of the eight pooled Cthulhu cells that were combined to generate the template for these sequences. Scale bar: 20 μm.
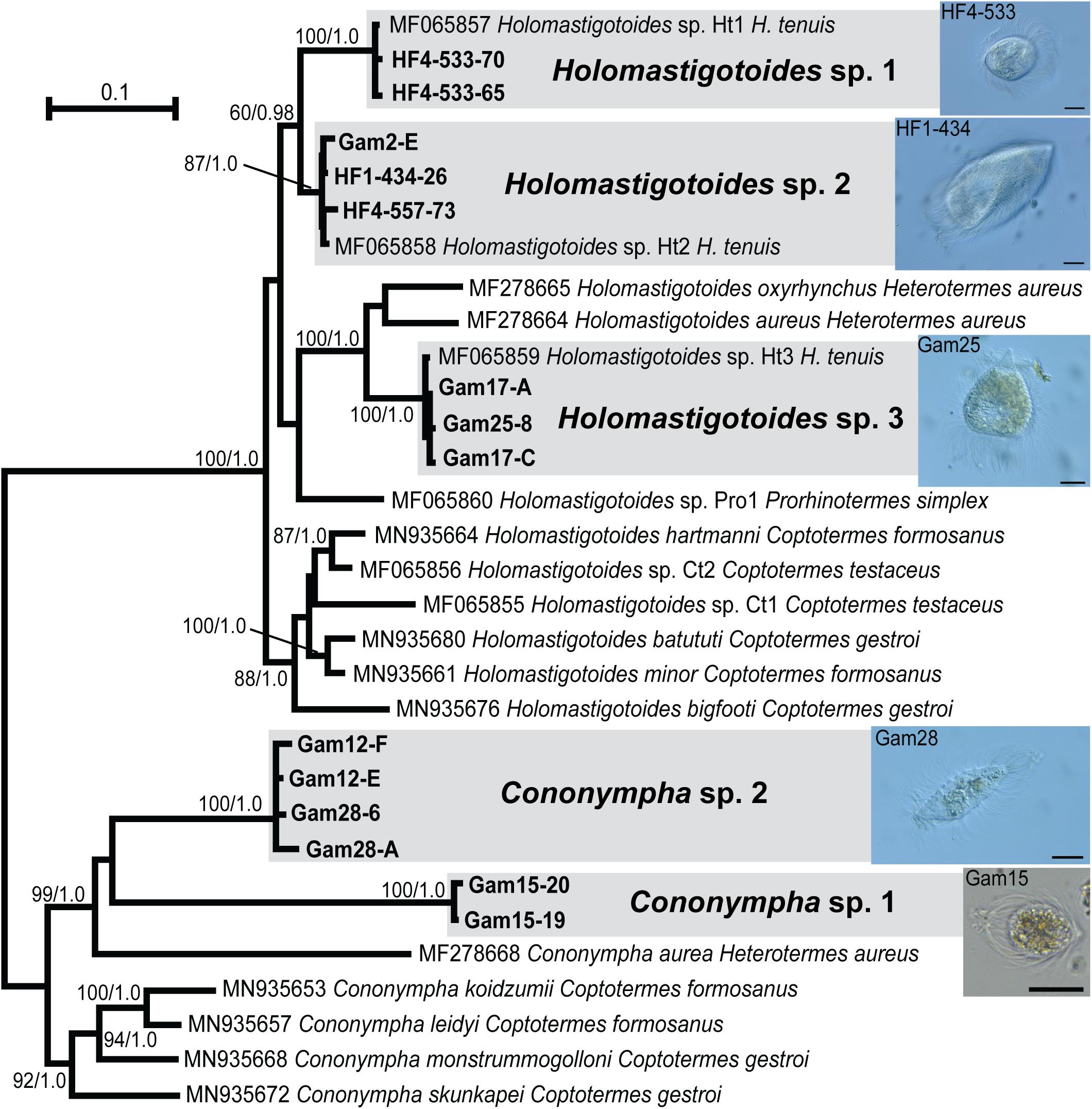
Figure 4. Unrooted maximum likelihood phylogeny of 18S ribosomal RNA gene sequences from Spirotrichonymphida genera Cononympha and Holo- mastigotoides, each serving as outgroup for the other. Protist species-level phylotypes from H. tenuis are indicated by gray boxes and new sequences obtained in this study are indicated by bold text. Support at nodes is given out of 100 bootstrap replicates computed by RAxML/Bayesian posterior probabilities if greater than 60/0.95. New sequences from H. tenuis symbionts are indicated by bold text. Select cells from which new sequences were derived are shown in micrographs inset at right; sequence names (tip labels) include the cell code. All scale bars: 20 μm.
Hindgut 18S rRNA Community Profiling
We also carried out 18S rRNA V4-V5 amplicon sequencing (metabarcoding) to characterize the hindgut parabasalian symbionts of H. tenuis sampled across much of its range. We included H. tenuis from four countries, French Guiana, Panama, Ecuador, and Brazil, with Brazilian samples coming from 10 states and both forest and savannah habitats (Figure 5). In total, we profiled the protist community of 95 individual worker termites from 39 distinct colonies, of which 16 were represented by a single individual (all ethanol-preserved museum specimens) and 21 had 2–4 individuals, while live collections from Ecuador and French Guiana had six and 20 individuals (biological replicates), respectively. In order to link each of these profiles to host genotype, we sequenced the mt16S of at least one individual from each colony. Our maximum likelihood phylogeny of these molecular barcodes recovered three distinct clades with moderate support (Figure 5), in agreement with a previous study (Carrijo et al., 2020).
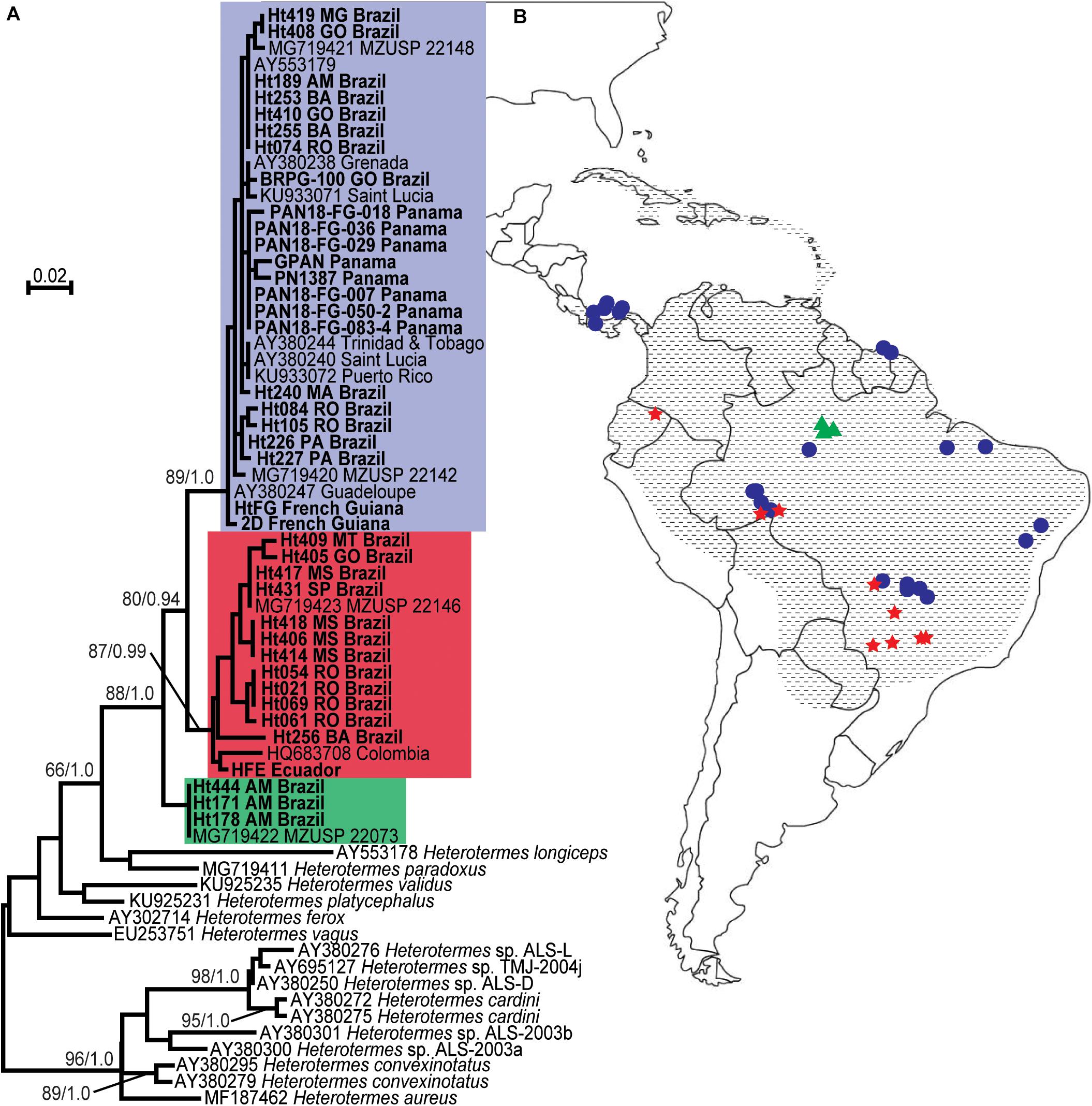
Figure 5. Collection locations and molecular identification of Heterotermes tenuis colonies. (A) Maximum likelihood phylogeny of Heterotermes partial mt16S gene sequences. Heterotermes tenuis sequences are indicated by colored boxes (top box is blue, middle is red, bottom is green); new sequences determined in this study are indicated by bold type. Support at nodes is given from 100 bootstrap replicates and Bayesian posterior probabilities where greater than 60/0.9. (B) Collection locations of H. tenuis colonies. Shapes indicate collection locations of samples included in this study, with color corresponding to the same haplotype group color scheme as in (A) (top blue haplotype = circles, middle red haplotype = stars, bottom green haplotype = triangles). Hatching indicates the geographical range of H. tenuis (Carrijo et al., 2020).
After trimming, merging, and denoising, we assigned taxonomy to our amplicon sequence variants (ASVs) using a custom reference database of full-length Parabasalia 18S rRNA sequences, including all 116 Sanger sequences generated in this study. All Parabasalia ASVs from the 95 samples were assignable to the eight phylotypes we characterized from single cells. Note that ASVs are equivalent to zero-radius OTUs; each has a unique sequence but no longer includes read abundance information. Together the ASVs represent the total sequence diversity present in the 18S rRNA amplicons. The number of ASVs per protist phylotype in our data set ranged from 17 (Cthulhu) to 136 (Holomastigotoides Ht2), for a total of 557 protist ASVs (Supplementary Table 3).
For a closer look at ASV phylogenetic diversity, we aligned all ASVs with the new and previously published 18S rRNA sequences used in the isolated cell analyses above (Figures 2–4) and computed phylogenetic trees (Supplementary Figures 1–4). All ASVs branched with the phylotypes to which they had been assigned, though they revealed considerably more sequence variability than the fewer single cell clones (Supplementary Figures 1–4). Particularly high levels of sequence divergence were exhibited by the two Cononympha phylotypes (Supplementary Figure 1, Supplementary Table 3), but because they lack clearly demarcated subclades, we maintain our interpretation of two phylotypes. In contrast, the ASVs clustering with Holomastigotoides Ht1 did form two supported subclades and therefore could alternatively be considered as two phylotypes, possibly representing two distinct species (Supplementary Figure 2). In keeping with this possibility, we noted that ASVs from these two subclades are nearly mutually exclusive in distribution, co-occurring in just one out of the nine colonies in which Ht1 was found, and suggesting they might belong to distinct cellular lineages. Because these subclades are very closely related and we did not isolate a representative cell from the additional subclade, we will consider Ht1 as one phylotype in this work. In sum, the ASV diversity across our data set supports eight protist species-level phylotypes as a reasonable accounting of the H. tenuis symbiont community. The 18S rRNA sequence variability within each of these protist phylotypes, including Ht1 (Supplementary Table 3), is consistent with that previously observed for named termite-associated protist species (Tai et al., 2014; Taerum et al., 2019, 2020; Jasso-Selles et al., 2020).
Distribution of Protist Phylotypes Across H. tenuis Colonies
Our sampled colonies contained different combinations of these eight phylotypes, with only two colonies harboring all eight phylotypes (Supplementary Table 1). None of the protist phylotypes was restricted to a single host haplotype group or a specific geographical region. In several colonies, we detected only two or three protist phylotypes. However, due to the destructive nature of our sampling, we were limited to just one termite individual from several of our museum collection colonies. This modest level of sampling increases the likelihood that protist species present in these samples’ colonies might not be detected by 18S rRNA profiling, whether due to biological variability among nestmates (e.g., in recently molted individuals) or technical limitations (e.g., biased sample degradation or allelic dropout during PCR). We therefore examined the prevalence of each protist phylotype across a reduced set of colonies for which at least two individual termites were sampled.
Holomastigotoides Ht2 was the most prevalent phylotype, present in 22 out of 23 colonies (96%). Next most common were Pseudotrichonympha paulistana and Pseudotrichonympha sp., each present in 21 of the 31 colonies (91%). Only one colony harbored neither Pseudotrichonympha species: our live colony from Panama that was in visible decline in the lab. While we did isolate a single Pseudotrichonympha cell (Gam11) from this colony shortly after collection, by the time we collected gut samples for 18S rRNA profiling no Pseudotrichonympha species were apparent by light microscopy. We have also observed Pseudotrichonympha aurea disappear from declining lab colonies of Heterotermes aureus (Jasso-Selles et al., 2017). The next most prevalent phylotypes were Cononympha sp. 1 and Cononympha sp. 2, occurring in 17 and 19 out of 23 colonies, respectively (74% and 83%). The phylotypes with the patchiest distributions were Cthulhu and Holomastigotoides Ht1, present in six and four colonies, respectively (26% and 17%, Supplementary Table 4).
Because differences in preservation mode among samples (i.e., ethanol, RNAlater, live) might lead to biased detection of protist phylotypes (Hammer et al., 2015), we also examined protist phylotype prevalence across each sample preservation mode separately (Supplementary Table 4). The ethanol-only set showed very similar values to those observed in the 2 + termite subset, with the same ranked order of protist phylotypes by proportion of colonies in which they were detected (i.e., Ht2 > Pseudotrichonympha spp. > Cononympha sp. 2 > Cononympha sp. 1 > Ht3 > Cthulhu > Ht1). However, the RNAlater (n = 7) and live (n = 3) subsets deviated from this trend in some respects. Most notably, both Cononympha species were detected in all of the non-ethanol-preserved samples, suggesting that ethanol samples might be biased toward faster degradation of Cononympha sequences. Holomastigotoides Ht3 was also more prevalent in these sample types than in ethanol, present in six out of seven RNAlater colonies and all three live colonies, while Cthulhu was completely absent from RNAlater colonies and present in one out of three live colonies. Without further sampling it is impossible to determine whether these differences reflect technical bias or true biological differences in the sampled colonies. Note that six out of seven RNAlater colonies and one out of three live colonies come from Panama, as opposed to just one out of 29 ethanol-preserved colonies.
The most common number of protist phylotypes recovered across our 2 + termite colonies is six (seen in 9 out of 23 colonies), and these communities generally consist of both Pseudotrichonympha species, Holomastigotoides Ht2 and Ht3, and both Cononympha species. In other words, they most commonly lack Cthulhu and Holomastigotoides Ht1 (Supplementary Table 1). The next most common number of protist phylotypes is five (5 out of 23), and these colonies are most often additionally missing one of the Cononympha species or Holomastigotoides Ht3. The seven- and eight-protist phylotype colonies (1 and 2 out of 23) add one or both of the rarer Cthulhu and Holomastigotoides Ht1. From these observations, we qualitatively conclude that the “typical” hindgut protist community of H. tenuis consists of Pseudotrichonympha paulistana and/or Pseudotrichonympha sp., Holomastigotoides Ht2, another of the H. tenuis-associated Holomastigotoides species, and at least one of the H. tenuis-associated Cononympha species.
Diversity and Phylogeography of Protist 18S rRNA Sequence Variants
There is considerable diversity among 18S rRNA amplicon sequence variants (ASVs) within each protist phylotype, and the level of diversity itself varies among phylotypes (Supplementary Figures 1–4, Supplementary Table 3). This is consistent with the intragenomic sequence variability of 18S rRNA genes observed in single cell PCR (e.g., Saldarriaga et al., 2011; Taerum et al., 2018). We recovered between 17 (Cthulhu) and 136 (Holomastigotoides Ht2) unique ASVs for each protist phylotype across our data set. However, the number of unique ASVs per phylotype correlated strongly with the prevalence of each phylotype across our data set (R2 = 0.84), suggesting that the observed among-phylotype variability in number of unique ASVs is driven by sampling depth rather than interspecific biological differences (Supplementary Table 3). Similarly, the proportion of ASVs that were unique to a single location varied across phylotypes, and these values were also correlated with phylotype prevalence across samples, though weakly (R2 = 0.49, Supplementary Table 3).
Looking instead at the sequence diversity among ASVs, the Cononympha phylotypes stand out as the most diverse, with mean/maximum uncorrected pairwise distance values of 0.02/0.12 for Cononympha sp. 1 and 0.05/0.13 for Cononympha sp. 2 (Supplementary Table 3). Even Holomastigotoides Ht1, which we suspect might reasonably represent two species, has lower ASV diversity, at 0.02/0.04, though still roughly twice as high as Holomastigotoides Ht3 (0.01/0.02) and Holomastigotoides Ht2 (0.00/0.02) (Supplementary Table 3).
We next asked whether the genetic distances among protist ASVs correlate with geographical distances using Mantel tests (Table 1). Mantel tests compare two distance matrices (i.e., uncorrected pairwise distances for all ASVs and geographical distances between sample locations) and compute a test statistic from cross products of the two matrices. Significance can be assessed by comparison to a distribution of test statistics from permutated data (Diniz-Filho et al., 2013; Buttigieg and Ramette, 2014). Note that while biased sample degradation would be expected to impact the inferred presence or absence of a phylotype, it would not be expected to impact the sequence of nucleotides in an amplified fragment. Our Mantel tests therefore include ASVs from all 39 colonies sampled in this study.
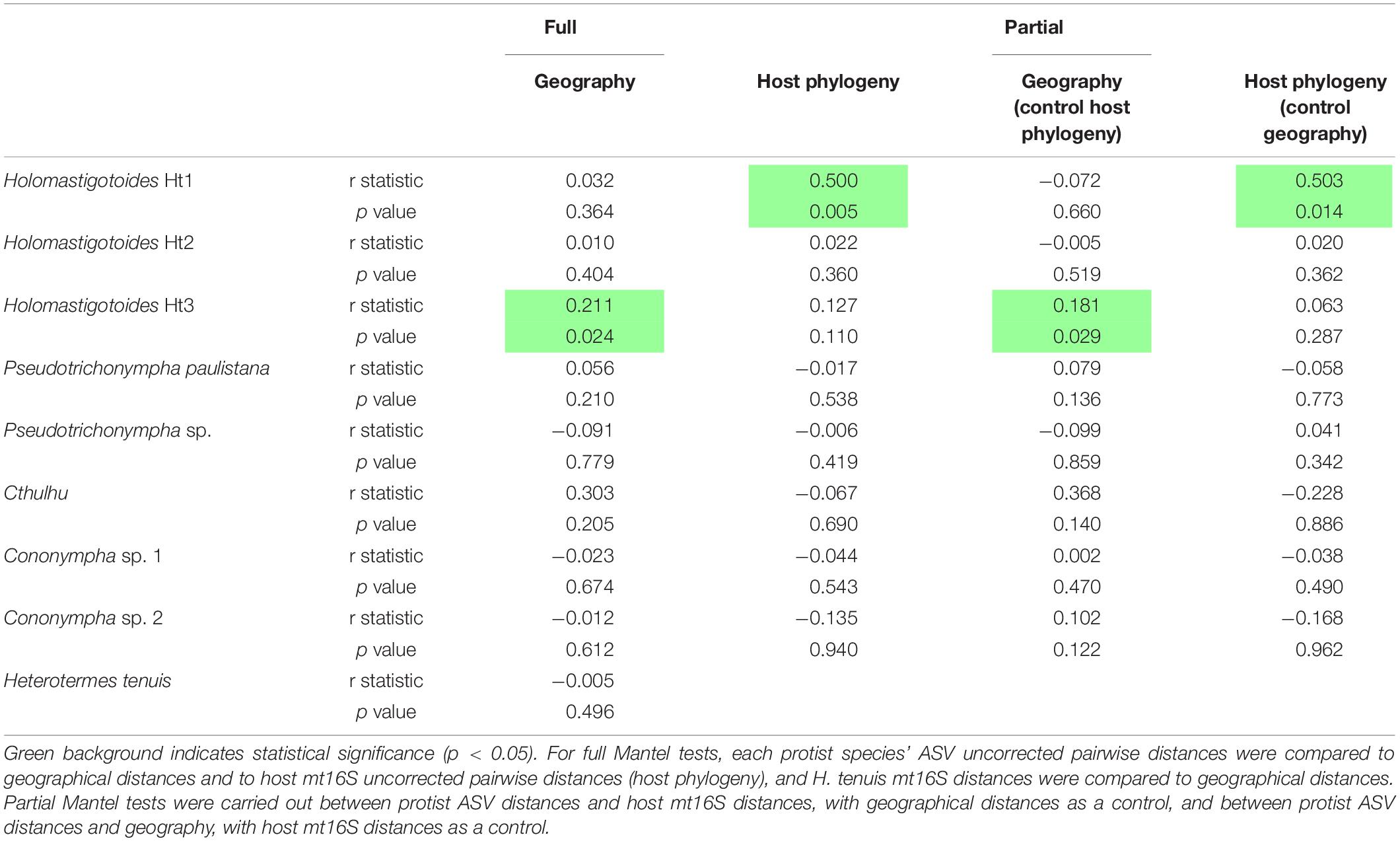
Table 1. Mantel test results (coefficient of correlation, R, and p-values).
For nearly all protist species, there was no correlation between geographical distance and genetic distance of ASVs. Only one species, Holomastigotoides Ht3, showed a weakly significant correlation between the genetic distances among its ASVs and the geographical distances between their sample locations (r = 0.211, p = 0.024; Table 1). These results indicate an overall lack of geographical pattern in the protist ASV sequence diversity in our data set.
An important caveat to this test is that protist biogeography depends on host biogeography, and host biogeography might be correlated with host phylogeny. We therefore carried out a series of Mantel tests to examine this relationship further. First, we compared host mt16S distances to host geographical distances and found no significant correlation, suggesting that host phylogeny should not be a confounding factor in this case. Next, we computed partial Mantel tests between protist ASV genetic and geographical distances while controlling for host phylogeny, and again found only a weakly significant correlation between Holomastigotoides Ht3 genetic and geographical distances (r = 0.181, p = 0.029; Table 1).
We also checked for correlation between protist ASV distances and host mt16S distances. Although host phylogeny is known to impact protist phylogeny and community composition in termites (Tai et al., 2015), it is unknown whether this influence manifests or is detectable at the host population level. In both full and partial Mantel tests, there was no detectable correlation between host and protist genetic distances, except for one species: Holomastigotoides Ht1, whose genetic distance showed a significant correlation with that of the host genetic distance (full test r = 0.500, p = 0.005; partial test controlling for geography r = 0.503, p = 0.014; Table 1).
Discussion
Drivers of Protist Diversification
In this study, we asked whether the considerable intraspecific variation in protist ASVs exhibits any patterns with respect to geography. Because protist communities are reported to be quite consistent across their termite host species, it is perhaps unsurprising that there was no significant correlation between most of the protists’ 18S rRNA ASV sequence diversification and their geographical or host phylogenetic distances. This suggests that there is as much ASV diversity within samples and/or between close (geographically or host phylogenetically) samples as there is across more distant samples. However, we did detect a weakly significant correlation between geographical and ASV pairwise distances of Holomastigotoides Ht3 (Table 1). This phylotype exhibits considerable shared sequence divergence (i.e., long branches) among subsets of ASVs, though their phylogenetic relationships are not resolved (Supplementary Figure 2). The most divergent Holomastigotoides Ht3 ASVs are closely related to our single-cell 18S rRNA sequences from Panama while the rest of the ASVs mainly come from Brazilian samples. This appears to be driving the correlation between ASV and geographical distances, which are greatest between Panama and southern and eastern Brazil.
The other significant correlation in our data set was between host mt16S pairwise distances and the pairwise distances of ASVs from Holomastigotoides Ht1. This is the protist phylotype for which the ASVs formed two supported, sister subclades in phylogenetic analyses (Supplementary Figure 2). As mentioned previously, these ASV subclades had a nearly mutually exclusive distribution across host colonies, making it unlikely that they co-exist in a single genome. While both subclades were found in samples from both the red and blue host haplotype groups (Figure 5, Supplementary Table 1), they were differentially distributed among the haplotype subclades. In particular, host colonies HFE, Ht061, and Ht069, which branch relatively deeply in the red haplotype group, harbor one of the Holomastigotoides Ht1 subclades, while host colonies Ht409, Ht431, and Ht406 in the crown clade of the red haplotype group harbor the other subclade of Holomastigotoides Ht1. This differential distribution of ASV subclades within the red host haplotype group appears to be driving the correlation between ASV and host mt16S distances.
Whether or not these correlations reveal incipient speciation in Holomastigotoides Ht1 or Ht3, they certainly highlight plausible scenarios by which termite-associated protists might speciate independently of their hosts or of each other (Figure 6). Such examples are welcome because the coevolutionary history of termites and their protists is too complex to be explained by co-speciation alone. In the case of Holomastigotoides Ht3, whose ASV genetic distances correlate with geographical distances, a divergent subset of ASVs is found almost exclusively in samples from Panama, on the margin of its mainland range. This pattern is reminiscent of peripatric speciation, in which a small marginal population becomes isolated from the parent population and diverges by genetic drift (Mayr, 1954). Although the host genetic distances do not show this pattern, Holomastigotoides Ht3 in Panama would be isolated from its conspecifics if they were missing from contiguous host populations toward the mainland. As our data suggest, the lack of Holomastigotoides Ht3 in certain host colonies is not only plausible but common.
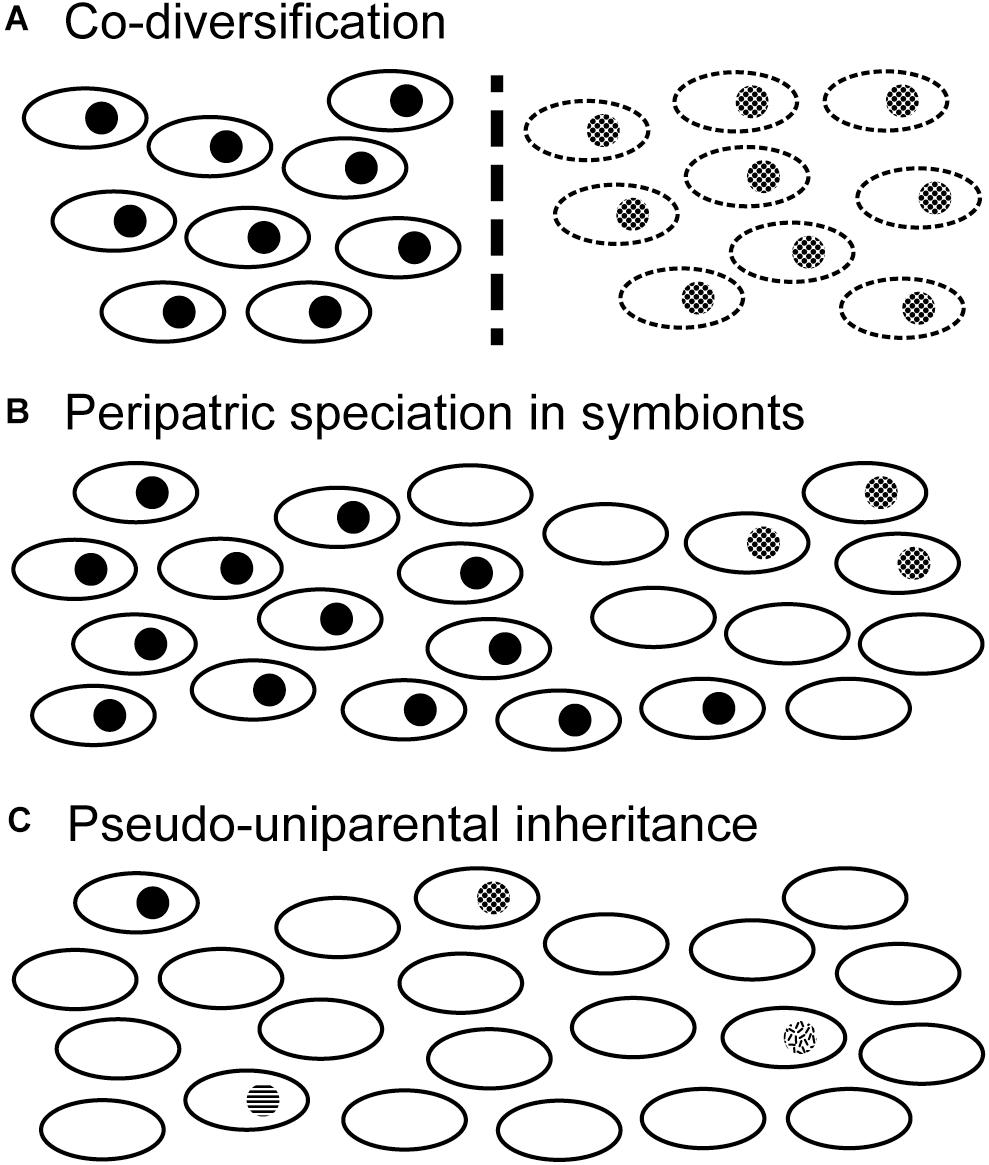
Figure 6. Scenarios of evolutionary diversification in termite-specific protists. Open black ovals represent termite colonies while filled circles indicate one of their symbiotic protist species. Note that termite-specific protists are biparentally inherited and do not persist in the environment outside their hosts. (A) Co-diversification of host and symbiont species. If host populations become separated by some barrier to gene flow (dashed vertical line), symbiont populations will also be separated. Over time protists and hosts can co-speciate. (B) If certain host colonies lose the symbiont species (empty ovals), they can form a barrier isolating two populations of symbionts. Symbiont populations can then diverge from one another in the absence of host diversification. This might occur more readily on the margins of a host species’ range, in a process reminiscent of peripatric speciation. Note that other symbiont species occupying the same host species could remain unaffected. (C) If a symbiont species becomes rare due to loss from many host colonies, its inheritance becomes nearly uniparental: any host colony harboring that symbiont species would have inherited it from just one parent (i.e., king or queen), unless both parents came from the same colony. In this pseudo-uniparental inheritance scenario, symbiont genetic diversification is more likely to be congruent with that of the uniparentally inherited mitochondrial genome.
Holomastigotoides Ht1 is the only H. tenuis symbiont whose ASV genetic distances are significantly correlated with the mt16S genetic distances of its host. While co-phylogeny at the genus level has been demonstrated between Pseudotrichonympha and its rhinotermitid hosts (Noda et al., 2007), incomplete protist 18S rRNA lineage sorting would be expected to obscure co-diversification in recently diverged host lineages (Maddison, 1997). Indeed, there is no detectable protist diversification in symbionts of recently diverged Zootermopsis species (Taerum et al., 2018), and evidence of incomplete lineage sorting has been reported from Teranympha symbionts of Reticulitermes (Noda et al., 2018). So why is host/symbiont co-diversification detectable in Holomastigotoides Ht1? We suspect that the cause is the patchy distribution of this protist phylotype. Because Holomastigotoides Ht1 is more commonly absent than present among our sampled H. tenuis colonies, a host lineage lacking Holomastigotoides Ht1 is unlikely to gain it by mating with a different host lineage, and daughter colonies are unlikely to inherit Holomastigotoides Ht1 from both parents, unless both parents belong to the same host lineage. In this way, protist populations could become isolated even while their hosts maintain gene flow. Protist rarity would therefore strengthen the congruence between maternally inherited mt16S distances and biparentally inherited protist ASVs. Another consequence of protist rarity could be the pruning of ASV diversity whenever a protist species fails to be transmitted to a daughter colony. This would further accelerate the ASV lineage sorting process.
Variability of Protist Community Composition
From early morphology-based observations it became a paradigm that symbiont communities are highly consistent across populations of a termite species with only occasional exceptions where a colony might be lacking one of the expected protist species (Kirby, 1937, 1949). This assumption was first directly tested in Japanese Reticulitermes species, in which colonies missing protist species were found to be common (Kitade and Matsumoto, 1993). For example, 10 out of 41 Reticulitermes speratus nests were found to be missing one or two of the 15 identified protist morphospecies (Kitade and Matsumoto, 1993). A similar prevalence of missing protists was seen in Hodotermopsis sjostedti, in which 13 out of 34 nests were missing one or two protist morphospecies (Kitade et al., 2012). These relatively high-variability termite species stand in contrast with Coptotermes formosanus, in which all 11 nests investigated contained all three of the recognized morphospecies, though an unidentified small trichomonad flagellate was also present in three of the colonies examined (Kitade et al., 2013). In our study, 21 out of 23 H. tenuis colonies were missing at least one of the eight protist species, 20 were missing two or more, and 11 were missing at least three species. By these metrics, the H. tenuis protist community has far higher inter-colony variability than any other reported to date. In H. tenuis, colonies missing protist species are the norm rather than the exception.
An important caveat to this comparison is the difference in methods. First, our combined morphological and molecular approach likely detects more species than a purely morphological approach, providing more opportunity to detect missing species. For example, the protist community of C. formosanus was long considered to consist of three species, Pseudotrichonympha grassii, Holomastigotoides hartmanni, and Cononympha (Spirotrichonympha) leidyi (Koidzumi, 1921), but molecular studies have detected an additional Holomastigotoides and an additional Cononympha species (Jasso-Selles et al., 2020; Nishimura et al., 2020). Thus, although the morphology-based study of C. formosanus found all three of the traditional morphospecies in all 11 colonies examined, the absence of a Holomastigotoides and/or Cononympha species could have gone unnoticed (Kitade et al., 2013). Next, molecular approaches have different biases. Amplicon sequencing may fail to detect some protist species, particularly those present in low abundance (Michaud et al., 2020). On the other hand, index hopping may result in protist species being erroneously detected in samples where they are absent (van der Valk et al., 2020). By considering only the colonies for which we sampled at least two termite individuals, and by employing dual indexing in our experimental design, we aimed to mitigate both of these potential biases, but we still likely detected more inter-colony variability than a morphological study alone would have.
Conclusion
Here, we have characterized the H. tenuis symbiont community as consisting of eight protist phylotypes. By combining 18S rRNA data from single isolated cells with high-throughput amplicon sequencing, we were able to confidently delimit these phylotypes and investigate their distribution across much of the geographical range of H. tenuis. None of the protist phylotypes was restricted to a single host haplotype group or a specific geographical region. Instead, we found that the protist symbiont community of H. tenuis is highly variable, with only two colonies harboring all eight phylotypes, and the vast majority of colonies missing at least two phylotypes. This result should be interpreted with caution due to limitations in our level of sampling, but it is so extreme that even a heavy bias, once corrected, would leave H. tenuis with the title of most variable protist community. We also investigated each phylotype’s genetic diversity across host colonies and found an overall lack of correlation with geographical or host phylogenetic distances, unsurprisingly for samples from a single host species. However, the two exceptions highlighted theoretical mechanisms of protist speciation that could work within the context of a diverse and widespread host species such as H. tenuis. These mechanisms bring a new perspective to the complex patterns of coevolution between termites and their protist symbionts.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the manuscript/Supplementary Material.
Author Contributions
DJ-S and GG performed microscopy. DJ-S performed single cell isolation. FM, NC, DJ-S, JS, and AR performed molecular work. FM, NC, and GG performed data analyses. PS, JŠ, DS-D, RS, and TC performed termite collection, maintenance, and preservation. GG performed study conception and manuscript writing. FM, NC, DJ-S, JS, AR, PS, JŠ, DS-D, RS, TC, and GG performed manuscript editing and improvement. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the School of Life Sciences and College of Liberal Arts and Sciences at Arizona State University (startup grant to GG). TC received financial support from the São Paulo Research Foundation, Brazil (FAPESP), through grants 13/03767-0 and 13/20247-0 to TC. Samples from Brazil were collected under the SISBIO license no. 40673-9 (Brazilian Environmental Ministry – MMA). JS was supported by the Internal Grant Agency of Faculty of Tropical AgriSciences, CZU (project number 20205014).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Mikaela Garcia, Xyonane Segovia, and Shaurya Aggarwal for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.640625/full#supplementary-material
Supplementary Figure 1 | Unrooted maximum likelihood phylogenetic tree of Cononympha 18S rRNA gene sequences from isolated cells and from amplicon profiling of termite gut contents. Nodes are intentionally compressed to show backbone structure. Isolated cell sequences determined in this study are nearly full length (∼1500 bp) and are indicated by red font. ASV sequences from amplicon profiling comprise only the V4–V5 regions of the 18S rRNA gene (∼450 bp) and are indicated by qiime2-generated sequence IDs in black text.
Supplementary Figure 2 | Unrooted maximum likelihood phylogenetic tree of Holomastigotoides 18S rRNA gene sequences from isolated cells and from amplicon profiling of termite gut contents. Nodes are intentionally compressed to show backbone structure. Isolated cell sequences determined in this study are nearly full length (∼1500 bp) and are indicated by red font. ASV sequences from amplicon profiling comprise only the V4–V5 regions of the 18S rRNA gene (∼450 bp) and are indicated by qiime2-generated sequence IDs in black text.
Supplementary Figure 3 | Unrooted maximum likelihood phylogenetic tree of Cthulhu 18S rRNA gene sequences from isolated cells and from amplicon profiling of termite gut contents. Isolated cell sequences determined in this study are nearly full length (∼1500 bp) and are indicated by red font. ASV sequences from amplicon profiling comprise only the V4–V5 regions of the 18S rRNA gene (∼450 bp) and are indicated by qiime2-generated sequence IDs in black text.
Supplementary Figure 4 | Unrooted maximum likelihood phylogenetic tree of Pseudotrichonympha 18S rRNA gene sequences from isolated cells and from amplicon profiling of termite gut contents. Nodes are intentionally compressed to show backbone structure. Isolated cell sequences determined in this study are nearly full length (∼1500 bp) and are indicated by red font. ASV sequences from amplicon profiling comprise only the V4–V5 regions of the 18S rRNA gene (∼450 bp) and are indicated by qiime2-generated sequence IDs in black text.
Supplementary Table 1 | Termite collection data, number of individuals assayed per collection, and protist taxonomy assignment of 18S rRNA ASV reads.
Supplementary Table 2 | Isolated cell codes, localities, length measurements, and number of clones sequenced.
Supplementary Table 3 | Diversity and distribution metrics of ASVs for each protist phylotype.
Supplementary Table 4 | Protist phylotype prevalence across samples.
Supplementary Video 1 | Live Cthulhu cell in motion displaying > 10 flagella in an anterior bundle. Scale bar 10 μm.
References
Ackerman, I. L., Constantino, R., Gauch, H. G. Jr., Lehmann, J., Riha, S. J., and Fernandes, E. C. M. (2009). Termite (Insecta: Isoptera) species composition in a primary rain forest and agroforests in central Amazonia. Biotropica 41, 226–233. doi: 10.1111/j.1744-7429.2008.00479.x
Batista-Pereira, L. G., Santos, M. G., Corrêa, A. G., Fernandes, J. B., Dietrich, C. R. R., Pereira, D. A., et al. (2004). Electroantennographic responses of Heterotermes tenuis (Isoptera: Rhinotermitidae) to synthetic (3Z,6Z,8E)-dodecatrien-1-ol. J. Braz. Chem. Soc. 15, 372–377.
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bourguignon, T., Lo, N., Dietrich, C., Šobotník, J., Sidek, S., Roisin, Y., et al. (2018). Rampant host switching shaped the termite gut microbiome. Curr. Biol. 28, 649–654.e2. doi: 10.1016/j.cub.2018.01.035
Brossette, L., Meunier, J., Dupont, S., Bagnères, A. G., and Lucas, C. (2019). Unbalanced biparental care during colony foundation in two subterranean termites. Ecol. Evol. 9, 192–200. doi: 10.1002/ece3.4710
Brugerolle, G., and Lee, J. J. (2000). “Phylum Parabasalia,” in An Illustrated Guide to the Protozoa, eds J. J. Lee, G. F. Leedale, and P. Bradbury (Lawrence, KS: Allen Press), 1196–1250.
Brune, A., and Dietrich, C. (2015). The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annu. Rev. Microbiol. 69, 145–166. doi: 10.1146/annurev-micro-092412-155715
Buttigieg, P. L., and Ramette, A. (2014). A guide to statistical analysis in microbial ecology: a community-focused, living review of multivariate data analyses. FEMS Microbiol. Ecol. 90, 543–550. doi: 10.1111/1574-6941.12437
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Carrijo, T. F. (2013). Estudo da Termitofauna (Insecta, Isoptera) da Região do Alto Rio Madeira, Rondônia. Ph.D thesis, Universidade de São Paulo, Ribeirão Preto.
Carrijo, T. F., Pontes-Nogueira, M., Santos, R. G., Morales, A. C., Cancello, E. M., and Scheffrahn, R. H. (2020). New World Heterotermes (Isoptera, Rhinotermitidae): molecular phylogeny, biogeography and description of a new species. Syst. Entomol. 45, 527–539. doi: 10.1111/syen.12412
Constantino, R. (1998). Catalog of the living termites of the New World (Insecta: Isoptera). Arq. Zool. 35, 135–231.
Constantino, R. (2000). Key to the soldiers of South American Heterotermes with a new species from Brazil (Isoptera: Rhinothermitidae). Insect Syst. Evol. 31, 463–472.
Constantino, R. (2002). The pest termites of South America: taxonomy, distribution and status. J. Appl. Entomol. 126, 355–365. doi: 10.1046/j.1439-0418.2002.00670.x
Davies, R. G., Hernández, L. M., Eggleton, P., Didham, R. K., Fagan, L. L., and Winchester, N. N. (2003). Environmental and spatial influences upon species composition of a termite assemblage across Neotropical forest islands. J. Trop. Ecol. 19, 509–524. doi: 10.1017/S0266467403003560
de Mello, I. F. (1954). Contribution à l’étude des microparasites des termites brésiliens. Flagellés du contenu intestinal d’Heterotermes tenuis. Mem. Inst. Oswaldo Cruz 52, 17–51.
Diniz-Filho, J. A. F., Soares, T. N., Lima, J. S., Dobrovolski, R., Landeiro, V. L., Telles, M. P., et al. (2013). Mantel test in population genetics. Genet. Mol. Biol. 36, 475–485. doi: 10.1590/S1415-47572013000400002
Dropkin, V. H. (1946). The use of mixed colonies of termites in the study of host-symbiont relations. J. Parasitol. 32, 247–251.
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects – diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Gile, G. H., James, E. R., Scheffrahn, R. H., Carpenter, K. J., Harper, J. T., and Keeling, P. J. (2011). Molecular and morphological analysis of the family Calonymphidae with a description of Calonympha chia sp. nov., Snyderella kirbyi sp. nov., Snyderella swezyae sp. nov. and Snyderella yamini sp. nov. Int. J. Syst. Evol. Microbiol. 61, 2547–2558. doi: 10.1099/ijs.0.028480-0
Gile, G. H., James, E. R., Tai, V., Harper, J. T., Merrell, T. L., Boscaro, V., et al. (2018). New species of Spirotrichonympha from Reticulitermes and the relationships among genera in Spirotrichonymphea (Parabasalia). J. Eukaryot. Microbiol. 65, 159–169. doi: 10.1111/jeu.12447
Grassi, B., and Foà, A. (1911). Intorno ai protozoi dei termitidi. Rend. R. Accad. Lincei Cl. Sci. Fis. Mat. E Nat. 5, 725–741.
Haifig, I., Costa-Leonardo, A. M., and Marchetti, F. F. (2008). Effects of nutrients on feeding activities of the pest termite Heterotermes tenuis (Isoptera: Rhinotermitidae). J. Appl. Entomol. 132, 497–501. doi: 10.1111/j.1439-0418.2008.01288.x
Hamady, M., Walker, J. J., Harris, J. K., Gold, N. J., and Knight, R. (2008). Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5, 235–237. doi: 10.1038/nmeth.1184
Hammer, T. J., Dickerson, J. C., and Fierer, N. (2015). Evidence-based recommendations on storing and handling specimens for analyses of insect microbiota. PeerJ 3:e1190. doi: 10.7717/peerj.1190
Hammer, T. J., Sanders, J. G., and Fierer, N. (2019). Not all animals need a microbiome. FEMS Microbiol. Lett. 366:fnz117. doi: 10.1093/femsle/fnz117
James, E. R., Okamoto, N., Burki, F., Scheffrahn, R. H., and Keeling, P. J. (2013). Cthulhu macrofasciculumque n. g., n. sp. and Cthylla microfasciculumque n. g., n. sp., a newly identified lineage of parabasalian termite symbionts. PLoS One 8:e58509. doi: 10.1371/journal.pone.0058509
Jasso-Selles, D. E., De Martini, F., Freeman, K. D., Garcia, M. D., Merrell, T. L., Scheffrahn, R. H., et al. (2017). The parabasalid symbiont community of Heterotermes aureus: molecular and morphological characterization of four new species and reestablishment of the genus Cononympha. Eur. J. Protistol. 61, 48–63. doi: 10.1016/j.ejop.2017.09.001
Jasso-Selles, D. E., de Martini, F., Velenovsky, J. F., Mee, E. D., Montoya, S. J., Hileman, J. T., et al. (2020). The complete protist symbiont communities of Coptotermes formosanus and Coptotermes gestroi: morphological and molecular characterization of five new species. J. Eukaryot. Microbiol. 67, 626–641. doi: 10.1111/jeu.12815
Kambhampati, S., and Smith, P. T. (1995). PCR primers for the amplification of four insect mitochondrial gene fragments. Insect Mol. Biol. 4, 233–236. doi: 10.1111/j.1365-2583.1995.tb00028.x
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kirby, H. (1937). Host-parasite relations in the distribution of protozoa in termites. Univ. Calif. Publ. Zool. 41, 189–211.
Kirby, H. (1949). Systematic differentiation and evolution of flagellates in termites. Rev. Soc. Mex. Hist. Nat. 10, 57–79.
Kitade, O. (2004). Comparison of symbiotic flagellate faunae between termites and a wood-feeding cockroach of the genus Cryptocercus. Microb. Environ. 19, 215–220. doi: 10.1264/jsme2.19.215
Kitade, O., Hayashi, Y., and Noda, S. (2013). Symbiotic protist communities in the termite Coptotermes formosanus in Japan and a comparison of community structures between workers and soldiers. Jpn. J. Protozool. 46, 21–29. doi: 10.18980/JJPROTOZOOL.46.1-2_21
Kitade, O., Hayashi, Y., Takatsuto, K., and Matsumoto, T. (2012). Variation and diversity of symbiotic protist composition in the damp-wood termite Hodotermopsis sjoestedti. Jpn. J. Protozool. 45, 29–36. doi: 10.18980/JJPROTOZOOL.45.1-2_29
Kitade, O., and Matsumoto, T. (1993). Symbiotic protistan faunae of Reticulitermes (Isoptera: Rhinotermitidae) in the Japan archipelago. Sociobiology 23, 135–153.
Koidzumi, M. (1921). Studies on the intestinal protozoa found in the termites of Japan. Parasitology 13, 235–309.
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Krishna, K., Grimaldi, D. A., Krishna, V., and Engel, M. S. (2013). Treatise on the Isoptera of the world. Bull. Am. Mus. Nat. Hist. 377, 623–973. doi: 10.1206/377.3
Kwong, W. K., Medina, L. A., Koch, H., Sing, K. W., Soh, E. J. Y., Ascher, J. S., et al. (2017). Dynamic microbiome evolution in social bees. Sci. Adv. 3:e1600513. doi: 10.1126/sciadv.1600513
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278. doi: 10.1093/bioinformatics/btu531
Light, S., and Sanford, M. (1928). Experimental transfaunation of termites. Univ. Calif. Publ. Zool. 31, 269–274.
Mackinnon, D. (1926). Observations on Trichonymphids. I. The nucleus and axostyle of Holomastigotoides hemigymnum Grassi (?). Q. J. Micr. Sci. 70, 173–191.
Mackinnon, D. L. (1927). Observations on Trichonymphids. II. The structure of Microspironympha elegans, n. sp. Q. J. Microsc. Sci. 71, 47–56.
Maddison, W. P. (1997). Gene trees in species trees. Syst. Biol. 46, 523–536. doi: 10.1093/sysbio/46.3.523
Mathews, A. G. A. (1977). Studies on Termites From the Mato Grosso State, Brazil. Rio de Janeiro: Academia Brasileira De Ciências.
Mayr, E. (1954). “Change of genetic environment and evolution,” in Evolution as a Process, eds J. Huxley, A. C. Hardy, and E. B. Ford (London: Unwin Brothers), 157–180.
Michaud, C., Hervé, V., Dupont, S., Dubreuil, G., Bézier, A. M., Meunier, J., et al. (2020). Efficient but occasionally imperfect vertical transmission of gut mutualistic protists in a wood-feeding termite. Mol. Ecol. 29, 308–324. doi: 10.1111/mec.15322
Nalepa, C. A. (2015). Origin of termite eusociality: trophallaxis integrates the social, nutritional, and microbial environments. Ecol. Entomol. 40, 323–335. doi: 10.1111/een.12197
Nishimura, Y., Otagiri, M., Yuki, M., Shimizu, M., Inoue, J., Moriya, S., et al. (2020). Division of functional roles for termite gut protists revealed by single-cell transcriptomes. ISME J. 14, 2449–2460. doi: 10.1038/s41396-020-0698-z
Noda, S., Kitade, O., Inoue, T., Kawai, M., Kanuka, M., Hiroshima, K., et al. (2007). Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mol. Ecol. 16, 1257–1266. doi: 10.1111/j.1365-294X.2006.03219.x
Noda, S., Shimizu, D., Yuki, M., Kitade, O., and Ohkuma, M. (2018). Host-symbiont cospeciation of termite-gut cellulolytic protists of the genera Teranympha and Eucomonympha and their Treponema endosymbionts. Microb. Environ. 33, 26–33. doi: 10.1264/jsme2.me17096
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). vegan: Community Ecology Package. Available online at: https://cran.r-project.org/package=vegan (accessed May 2020).
Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528.
R Core Team (2020). R: A Language and Environment for Statistical Computing. Available online at: https://www.r-project.org/ (accessed May 2020).
Radek, R., Meuser, K., Strassert, J. F. H., Arslan, O., Teßmer, A., Šobotník, J., et al. (2018). Exclusive gut flagellates of Serritermitidae suggest a major transfaunation event in lower termites: description of Heliconympha glossotermitis gen. nov. spec. nov. J. Eukaryot. Microbiol. 65, 77–92. doi: 10.1111/jeu.12441
Ravenscraft, A., Berry, M., Hammer, T., Peay, K., and Boggs, C. (2019). Structure and function of the bacterial and fungal gut microbiota of Neotropical butterflies. Ecol. Monogr. 89:e01346. doi: 10.1002/ecm.1346
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Saldarriaga, J. F., Gile, G. H., James, E. R., Horák, A. A. A., Scheffrahn, R. H., and Keeling, P. J. (2011). Morphology and molecular phylogeny of Pseudotrichonympha hertwigi and Pseudotrichonympha paulistana (Trichonymphea, Parabasalia) from Neotropical rhinotermitids. J. Eukaryot. Microbiol. 58, 487–496. doi: 10.1111/j.1550-7408.00575.x
Sanders, J. G., Powell, S., Kronauer, D. J. C., Vasconcelos, H. L., Frederickson, M. E., and Pierce, N. E. (2014). Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol. Ecol. 23, 1268–1283. doi: 10.1111/mec.12611
Shelomi, M., Lo, W. S., Kimsey, L. S., and Kuo, C. H. (2013). Analysis of the gut microbiota of walking sticks (Phasmatodea). BMC Res. Notes 6:368. doi: 10.1186/1756-0500-6-368
Shimada, K., Lo, N., Kitade, O., Wakui, A., and Maekawa, K. (2013). Cellulolytic protist numbers rise and fall dramatically in termite queens and kings during colony foundation. Eukaryot. Cell 12, 545–550. doi: 10.1128/EC.00286-12
Simon, C., Frati, F. F. F., Beckenbach, A., Crespi, B., Liu, H., Flook, P., et al. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701. doi: 10.1080/17470210902990829
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Taerum, S. J., De Martini, F., Liebig, J., and Gile, G. H. (2018). Incomplete co-cladogenesis between Zootermopsis termites and their associated protists. Environ. Entomol. 47, 184–195. doi: 10.1093/ee/nvx193
Taerum, S. J., Jasso-Selles, D. E., Hileman, J. T., de Martini, F., Mizumoto, N., and Gile, G. H. (2020). Spirotrichonymphea (Parabasalia) symbionts of the termite Paraneotermes simplicicornis. Eur. J. Protistol. 76:125742. doi: 10.1016/j.ejop.2020.125742
Taerum, S. J., Jasso-Selles, D. E., Wilson, M., Ware, J. L., Sillam−Dussès, D., Šobotník, J., et al. (2019). Molecular identity of Holomastigotes (Spirotrichonymphea, Parabasalia) with descriptions of Holomastigotes flavipes n. sp. and Holomastigotes tibialis n. sp. J. Eukaryot. Microbiol. 66, 882–891. doi: 10.1111/jeu.12739
Tai, V., Gile, G. H., Pan, J., James, E. R., Carpenter, K. J., Scheffrahn, R. H., et al. (2014). The phylogenetic position of Kofoidia loriculata (Parabasalia) and its implications for the evolution of the Cristamonadea. J. Eukaryot. Microbiol. 62, 255–259. doi: 10.1111/jeu.12163
Tai, V., James, E. R., Nalepa, C. A., Scheffrahn, R. H., Perlman, S. J., and Keeling, P. J. (2015). The role of host phylogeny varies in shaping microbial diversity in the hindguts of lower termites. Appl. Environ. Microbiol. 81, 1059–1070. doi: 10.1128/AEM.02945-14
van der Valk, T., Vezzi, F., Ormestad, M., Dalén, L., and Guschanski, K. (2020). Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies. Mol. Ecol. Resour. 20, 1171–1181. doi: 10.1111/1755-0998.13009
Yamin, M. (1979). Flagellates of the orders Trichomonadida Kirby, Oxymonadida Grassé, and Hypermastigida Grassi and Foà reported from lower termites (Isoptera families Mastotermitidae, Kalotermitidae, Hodotermitidae, Termopsidae, Rhinotermitidae, and Serritermitidae) and from the wood-feeding roach Cryptocercus (Dictyoptera: Cryptocercidae). Sociobiology 4, 1–120.
Keywords: coevolution, microbiome, termite, Cononympha, Pseudotrichonympha, Cthulhu, Rhinotermitidae
Citation: De Martini F, Coots NL, Jasso-Selles DE, Shevat J, Ravenscraft A, Stiblík P, Šobotník J, Sillam-Dussès D, Scheffrahn RH, Carrijo TF and Gile GH (2021) Biogeography and Independent Diversification in the Protist Symbiont Community of Heterotermes tenuis. Front. Ecol. Evol. 9:640625. doi: 10.3389/fevo.2021.640625
Received: 11 December 2020; Accepted: 01 March 2021;
Published: 19 March 2021.
Edited by:
Daniel Aguilera-Olivares, University of Concepción, ChileReviewed by:
Brittany Faye Peterson, Southern Illinois University Edwardsville, United StatesFranck Dedeine, Université de Tours, France
Copyright © 2021 De Martini, Coots, Jasso-Selles, Shevat, Ravenscraft, Stiblík, Šobotník, Sillam-Dussès, Scheffrahn, Carrijo and Gile. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gillian H. Gile, Z2dpbGVAYXN1LmVkdQ==
†These authors have contributed equally to this work and share first authorship
‡ORCIDs: Alison Ravenscraft, orcid.org/0000-0003-4974-4804; Petr Stiblík, orcid.org/0000-0001-6141-5603; Jan Šobotník, orcid.org/0000-0002-8581-637X; David Sillam-Dussès, orcid.org/0000-0001-5027-8703; Tiago F. Carrijo, orcid.org/0000-0001-6308-7252; Gillian H. Gile, orcid.org/0000-0003-3955-0942
§Present address: Francesca De Martini, Life Science Department, Mesa Community College, Mesa, AZ, United States