 Gabriel C. Runte
Gabriel C. Runte Alex H. Smith
Alex H. Smith Holly V. Moeller
Holly V. Moeller Laura M. Bogar
Laura M. Bogar- Ecology, Evolution and Marine Biology, University of California, Santa Barbara, Santa Barbara, CA, United States
Host and symbiont diversity are inextricably linked across partnerships and ecosystems, with degree of partner reliance governing the strength of this correlation. In many forest soils, symbiotic ectomycorrhizal fungi coexist and compete with free-living saprotrophic fungi, with the outcomes of these interactions shaping resource availability and competitive outcomes for the trees aboveground. Traditional approaches to characterizing these communities rely on DNA sequencing of a ribosomal precursor RNA gene (the internal transcribed spacer region), but directly sequencing the precursor rRNA may provide a more functionally relevant perspective on the potentially active fungal communities. Here, we map ectomycorrhizal and saprotrophic soil fungal communities through a mixed hardwood-conifer forest to assess how above- and belowground diversity linkages compare across these differently adapted guilds. Using highly spatially resolved transects (sampled every 2 m) and well-mapped stands of varying host tree diversity, we sought to understand the relative influence of symbiosis versus environment in predicting fungal diversity measures. Canopy species in this forest included two oaks (Quercus agrifolia and Quercus douglasii) and one pine (Pinus sabiniana). At the scale of our study, spatial turnover in rRNA-based communities was much more predictable from measurable environmental attributes than DNA-based communities. And while turnover of ectomycorrhizal fungi and saprotrophs were predictable by the presence and abundance of different canopy species, they both responded strongly to soil nutrient characteristics, namely pH and nitrogen availability, highlighting the niche overlap of these coexisting guilds and the strong influence of aboveground plants on belowground fungal communities.
Introduction
Forest communities are shaped by symbiosis. Plant roots engage with staggeringly diverse communities of soil organisms: many of these interactions are symbiotic, and their effects on plant health and resource movement through soils can have profound impacts on the functioning of ecosystems (Baldrian, 2017; Crowther et al., 2019). One key symbiosis structuring many forests is the ectomycorrhizal association, an essential nutritional mutualism for roughly 60% of tree stems on the planet (Steidinger et al., 2019). The diversity of ectomycorrhizal fungal mutualists available to plant roots belowground has a powerful impact on the composition and structure of the plant community aboveground (Dickie et al., 2010; Gao et al., 2013; Koide et al., 2014). This partnership between plants and fungi also influences the structure and function of soil communities: Ectomycorrhizal fungi, with their reliable source of host carbon, can inhibit decomposition and mediate soil carbon storage by competing directly with saprotrophic fungi for soil resources (Gadgil and Gadgil, 1975; Averill and Hawkes, 2016), although this effect varies substantially based on the traits of the fungi and the environment in which they interact (Fernandez et al., 2020).
When sampled at a landscape scale, plant and fungal diversity are clearly linked. In forested systems, host specificity of the ectomycorrhizal community means that turnover in aboveground host tree community membership is mirrored belowground (Tedersoo et al., 2012; Moeller et al., 2015) and ectomycorrhizal fungal species richness is known to track the genus richness of available host plants (Gao et al., 2013). This allows for some predictive linkages between aboveground plant communities and belowground function: pine forests, for example, support a distinctive set of ectomycorrhizal fungi with conserved enzymatic capabilities (Talbot et al., 2013, 2014), and exhibit distinct soil chemistry and decomposition rates that are likely driven by these fungal associations. Yet aboveground-belowground linkages are complicated by environmental context and stochastic patterns of community assembly. Across the range of a single host tree species, the fungal community may turn over in response to climatological factors (Moeller et al., 2014; Bui et al., 2020) as well as dispersal limitation (Peay et al., 2010).
This context dependency becomes especially important when examining these above-belowground linkages at a fine spatial scale. Each tree influences and responds to a belowground rooting zone on the scale of meters, and is subject to habitat heterogeneity at that scale: it does not experience the habitat of an entire forest. The fungi, too, can extend from several centimeters to many meters belowground (Vincenot and Selosse, 2017), exposing them to substantial fine-scale variation in soil conditions. Competition among fungi for resource patches including decomposing organic matter (Fukami et al., 2010) and host root exudates (Smith et al., 2018) may also enhance community turnover at small scales (Pickles et al., 2012). Studies investigating the fine scale spatial turnover of fungal communities find that fungal community composition turns over on a scale ranging from centimeters to several meters (Lilleskov et al., 2004; Bahram et al., 2013), driven by factors such as host availability, host and fungal taxonomy, competition, and stochastic processes (Pickles et al., 2012; Bahram et al., 2013). Thus, communities of fungi and roots may be shaped strongly at the fine scale by factors that disappear when averaged across an entire landscape, obscuring the specific environmental conditions and species interactions that drive fungal diversity (Izzo et al., 2005).
Fine-scale coupling between belowground fungal communities and aboveground tree communities is shaped by how fungi interact with trees. Ectomycorrhizal fungi, reliant on host roots for carbon, might be expected to respond more strongly to host availability than saprotrophic fungi. Trees, however, have an enormous impact on the soil, in ways that go beyond carbon subsidies to their ectomycorrhizal partners. Litter inputs, for example, can vary both in quantity and in chemical quality across different host plants (Scott and Binkley, 1997). This variation could have profound effects on both ectomycorrhizal fungi and saprotrophic fungi, since both guilds must obtain nitrogen, phosphorus, and other resources from similar substrates; the outcomes of competition between these guilds may depend strongly on litter chemistry (Smith and Wan, 2019). Highlighting the powerful but divergent influences of tree identity on these guilds, Nguyen et al. (2016b) found that that host phylogenetic diversity can be an important influence on ectomycorrhizal fungal communities, while saprotrophs may be controlled more by chemical inputs such as plant leaf nitrogen and aboveground tree biomass. However, in other settings, ectomycorrhizal and saprotrophic fungi respond similarly to aboveground vegetation, revealing only subtle differentiation between the forces structuring these groups (Tedersoo et al., 2016; Hiiesalu et al., 2017). Both guilds of fungi should be influenced by aboveground plant communities, but in fundamentally different ways. It remains unclear when these distinct mechanisms should lead these guilds to respond similarly to aboveground plant communities, and when their responses should diverge on the basis of their different trophic strategies.
To understand how ectomycorrhizal and saprotrophic fungi respond to aboveground plant diversity, we must identify the fungi making up a given community. Fungal community characterization, and ectomycorrhizal characterization specifically, has traditionally relied on either sporocarp surveys or soil environmental DNA for the description of communities. As high-throughput sequencing technologies have become increasingly accessible, amplicon sequencing of the fungal internal transcribed spacer (ITS) region, a gene encoding a precursor component of mature ribosomal RNA, has become a standard for community characterization at the species level. Sequencing DNA, however, is an imperfect way to measure fungal communities: DNA can be preserved in soils for months to years (Nielsen et al., 2006), both as extracellular molecules sorbed to clays (Ogram et al., 1988; Morrissey et al., 2015), and within necromass and dormant propagules (Lennon and Jones, 2011; Carini et al., 2016). Fungal spores, too, can make it difficult to detect active community members using DNA, since fungi can disperse over great distances (Peay and Bruns, 2014; Horton, 2017), and their spores may persist in a dormant but intact state for many years (Bruns et al., 2009). To limit the influence of these forms of inconsistent data, researchers in the past have selected root tips from ectomycorrhizal plants and sequenced the individual fungi on each tip using Sanger sequencing (Moeller et al., 2014; Kumar and Satyanarayana, 2002; Janowski et al., 2019). This practice, though, is labor intensive and can be biased toward species with more conspicuous mycorrhizas. Additionally, saprotrophic fungi cannot be assessed in a parallel way because they cannot be isolated by visual identification of host association.
Ribosomal RNA-based community profiling can produce a more functionally relevant snapshot of microbial community composition (Dlott et al., 2015; Sorensen and Shade, 2020), allowing researchers to characterize which members of a community have the greatest protein synthesis potential – that is, the greatest number of ribosomes (Blazewicz et al., 2013). Thus, rRNA-based community profiling can reflect the potentially active, functional community at a site at the time of sampling, while DNA-based profiling of rRNA genes, like ITS, reveals all the fungi whose DNA is present at the site, whether or not they are active members of the community (Baldrian et al., 2012; Barnard et al., 2013; Liao et al., 2014; Žifčáková et al., 2016; Wutkowska et al., 2019). The ITS region is particularly well suited to capturing the truly active members of a fungal community, because it is a precursor rRNA molecule absent even from dormant spores (van der Linde and Haller, 2013). The ITS region itself is spliced out before a ribosome becomes active (Schoch et al., 2012), so any fungi with measurable ITS rRNA molecules are in the process of producing new ribosomes. These fungi are more likely to be active than fungi present only as DNA, since they are actively building the protein synthesis enzymes that drive their ecological functions.
Here, we use both precursor rRNA and DNA-based ITS communities to examine the connections between the spatial turnover in aboveground plant communities and the turnover in soil fungal communities, comparing ectomycorrhizal (symbiotic) and saprotrophic (free-living) fungi across four transects with varied aboveground plant community composition. Using high resolution sampling and mapping of above-ground canopy trees, we created a spatially explicit set of influences on each sequenced soil community. Each sample had a unique set of soil chemical characteristics and continuous ectomycorrhizal host availability data. We extracted and sequenced amplicons from both RNA and DNA in each soil sample in order to compare turnover at the meter scale within both potentially active (precursor rRNA-based) and more broadly inclusive DNA-based community profiles. We anticipated that host plant diversity would have a more pronounced effect on the potentially active, RNA-based community than the longer-term, DNA-based community, and that the RNA-based community composition would be more variable across our samples. We also hypothesized that ectomycorrhizal fungal communities would be much more responsive to the availability of host trees than saprotrophs, but that both guilds of fungi would respond to soil resource and chemical properties.
Materials and Methods
Sampling Site and Sample Collection
We conducted our study at the Sedgwick Reserve, part of the University of California Natural Reserve System, in Santa Barbara County, California in January, 2020. This southern California site experiences a Mediterranean climate of hot, dry summers and cool, wet winters (Davis et al., 2011), leading us to sample in the winter wet season when fungi could be expected to be most active. We selected four 30 m transects with varied aboveground plant community composition within 0.2 km of one another (Figure 1). The canopy species on our transects were gray pine (Pinus sabiniana, abbreviated PISA hereafter), blue oak (Quercus douglasii, abbreviated QUDO), and coast live oak (Quercus agrifolia, abbreviated QUAG). These two oak species are more phylogenetically distinct from the pine than one another, though blue oaks are deciduous and belong to the white oak group while coast live oaks are evergreen and belong to the red oak group (Plumb and Gomez, 1983). Previous studies have described distinct differences in the ectomycorrhizal (but not saprotrophic) communities associated with such phylogenetically diverged oaks (Morris et al., 2008), as well as between oaks and co-occurring pine species (Smith et al., 2009; Suz et al., 2017; Rasmussen et al., 2018).
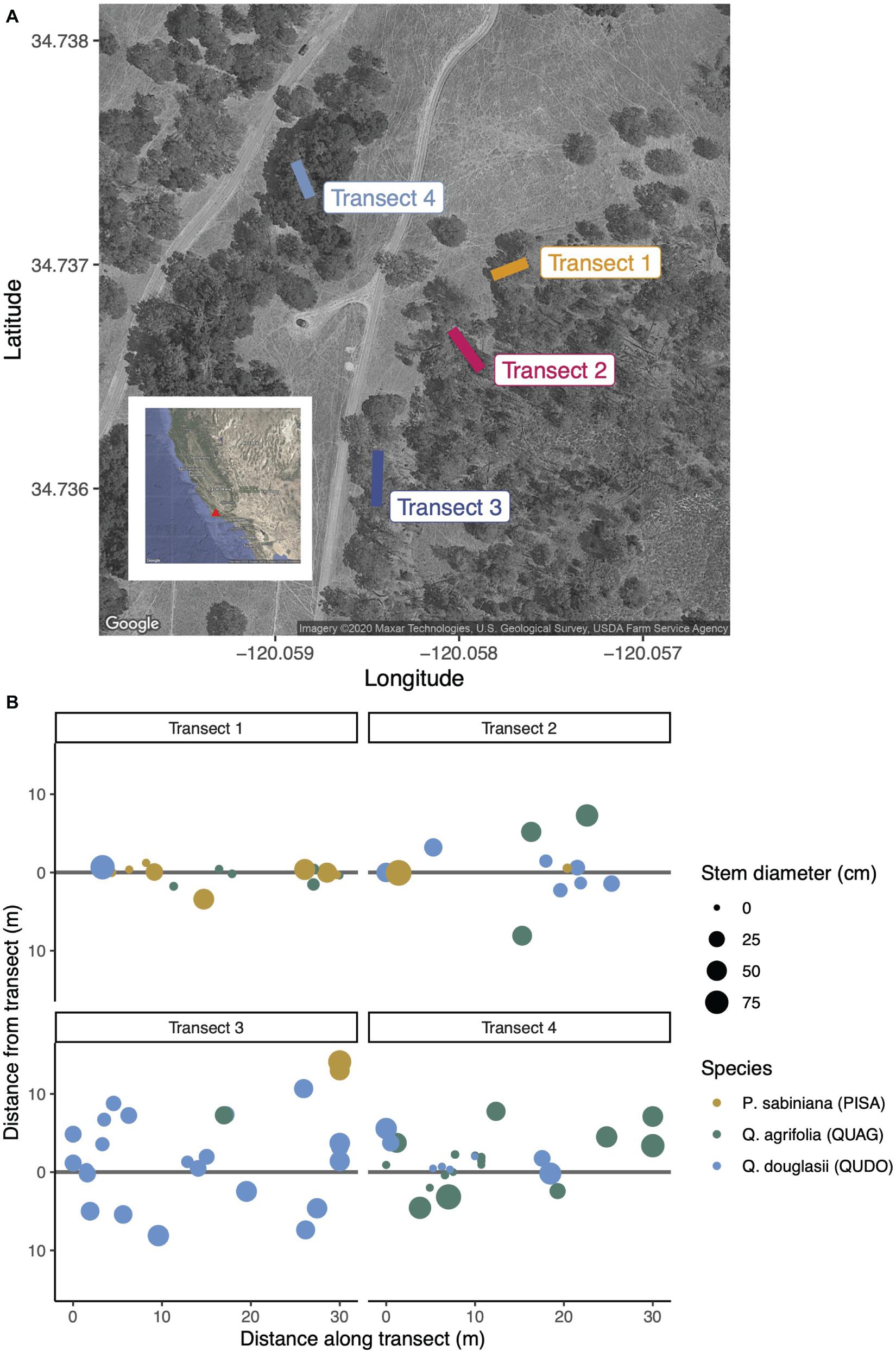
Figure 1. (A) Four 30 m transects were selected to capture different canopy communities across the mixed conifer-hardwood forest at Sedgwick Reserve in Santa Barbara County, CA, United States. The maximum distance between two transect endpoints (Transect 3 and Transect 4) is approximately 175 m. (B) Canopy communities were inventoried based on their minimum distance to the transect. The species surveyed were Pinus sabiniana (PISA, brown circles), Quercus agrifolia (QUAG, green circles), and Q. douglasii (QUDO, blue circles). Circle size is proportionate to stem diameter (DBH), and distance from the transect represents physical location of each tree.
Soils across the four transects derived from the same alluvium parent materials and were Chamise shaly loams (transects one, three, and half of two: Clayey-skeletal, mixed, active, thermic Ultic Palexerolls), Elder loams (transect four: Coarse-loamy, mixed, superactive, thermic Cumulic Haploxerolls), and Positas fine sandy loams (half of transect two: fine, smectitic, thermic Mollic Palexeralfs) (Soil Survey Staff, Natural Resources Conservation Service, and United States Department of Agriculture, 2020). We sampled each 30 m transect every 2 m (n = 16 soil cores per transect, including 0 and 30 m end points) by removing surface vegetation and using a hand auger to a depth of 25 cm. We emptied auger contents into an ethanol-sterilized bin and homogenized them by passing through a 4 mm soil sieve and mixing before collecting a sample for nucleic acid extraction. These samples were weighed into sterile 15 mL tubes (1–2 g soil per sample) and flash frozen on dry ice; samples were placed in a −80°C freezer within 24 h of collection. A separate 1-quart homogenized soil sample from the same core was placed in a clean plastic bag for chemical analysis and stored in a cooler with ice until placement in a 4°C refrigerator within 24 h.
We quantified aboveground plant community composition for each soil core by surveying host trees of all three species whose canopies extended to within 10 m of the transect (Day et al., 2010). For each individual tree, we measured diameter at breast height (DBH) and position relative to the transect (by measuring the distance of the tree to the nearest location on the transect tape to the nearest centimeter). This allowed us to create a high-resolution map of aboveground plant diversity. To compute the host influence on each soil core, we calculated the angular size of each tree relative to each sampling point (Hein et al., 2018), expressing the potential influence of each tree species as the percentage of each core’s 360° panoramic field of view that was occupied by that host species. If a host tree was too far from a given soil core to include that core in its rooting zone – that is, if the core was outside the tree’s sphere of influence – we excluded that tree from the calculation. We computed this distance as ½ of the tree’s maximum rooting diameter, calculated using a formula from a meta-analysis linking maximum rooting radius (r) to DBH (Day et al., 2010):
Soil Chemical Analysis
Within 1 week of sampling, we subsampled and weighed refrigerated soils, then dried them to constant mass in an oven at 45–60°C and reweighed them to determine percent moisture. The remainder of our soil analyses were performed at Brookside Laboratories Inc., on dried soil (OH, Untied States). Briefly, total carbon and total nitrogen were evaluated via dry combustion on an elementar vario EL cube and divided for site C:N ratios. Phosphorus was extracted via the Bray II method and other trace nutrients were extracted with Mehlich III extractions (data not shown). Soil pH was evaluated using a 1:1 soil to water slurry.
We compared soil properties between transects using analysis of variance (ANOVA) tests with a Tukey’s honest significant difference post-hoc test to assign significance groups. We used linear regression to evaluate correlation between tree-driven parameters and soil chemical properties, selecting our models based on lowest AIC score when grouping all transects’ data. From this point, we tested for Pearson’s correlation coefficient between selected parameters to choose which would be included in later analyses.
Nucleic Acid Extraction and Sequencing
We co-extracted RNA and DNA from 1 to 2 g of soil using the Qiagen RNeasy Powersoil Total RNA Kit and RNeasy Powersoil DNA Elution Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. We reverse transcribed cDNA from the RNA extracts using the Qiagen OneStep RT-PCR kit and used Promega GoTaq Green mastermix for the DNA extracts. We amplified all samples with the ITS1 primer pair ITS1F-KYO1 and ITS2-KYO1 (Toju et al., 2012) and the following thermal cycler steps: 95°C for 3 min, 35 rounds of 95°C for 30 s, 50°C for 30 s, 72°C for 30 s, then a final elongation step of 5 min at 72°C. We ligated Illumina indices with 3 min at 95°C, 35 cycles of 95°C for 30 s, 47°C for 30 s, 72°C for 30 s, then a final elongation step of 72°C for 5 min. Finally, we purified samples with AMPure XP beads and diluted them to 10 ng/uL before multiplexing all samples for sequencing in a single run. Amplicons were sequenced on the Illumina MiSeq platform with 250-bp paired-end reads at the California NanoSystems Institute (UC Santa Barbara).
Bioinformatics Pipeline
We processed amplicon sequences using the DADA2 pipeline (Callahan et al., 2016). To assess the broadest fungal community, we processed only forward sequences, as described in Pauvert et al. (2019). We removed adapter and primer sequences using the “cutadapt” package and removed chimeras before filtering (Martin, 2011). Our DADA2::filterandtrim settings removed any sequences with an “N” assignment and truncated reads at the first Q-score of 2 or lower, with a minimum read length of 50-bp. We chose not to rarefy our sequences due to the potential for false positives in differential abundance testing and our intention to use a consistent dataset through both richness and turnover (beta diversity) estimates (McMurdie and Holmes, 2014). We finally clustered our amplicon sequence variants (ASVs) at a 97-percent similarity threshold using the DECIPHER package before making taxon assignments on these operational taxonomic units (OTUs) with the UNITE database (Nilsson et al., 2019; Wright, 2020). We assigned fungal functional guilds using the FUNGuild database and retained OTUs called as either “probable” or “highly probable,” further filtering for any OTU with “Ectomycorrhizal” or “Saprotroph” in its guild assignment to be in that guild for our analyses (Nguyen et al., 2016a). Finally, we removed any samples with fewer than 500 reads after guild assignment and also removed any OTU with only a single read to maintain the power of later analyses to infer differences between samples.
Statistical Analyses
We compared DNA and RNA amplicon communities from the whole library and as subsets assigned (through FUNGuild) to ectomycorrhizal or saprotrophic fungal guilds. To understand patterns of diversity between transects and nucleic acid extracts, we assessed the whole library for species richness as the total number of OTUs and the Shannon’s diversity index using the vegan package in R. To quantify spatial community turnover (beta diversity), we also computed Bray–Curtis OTU community dissimilarities based on relative taxon abundance data with the distance function in the Phyloseq package (McMurdie and Holmes, 2013). We calculated differences across transects with linear regression and Tukey’s honest significant difference tests. Comparisons of DNA to RNA diversity metrics within transects were assessed with two-sample t-tests.
To visualize patterns of fungal community turnover as they related to tree identity, we created non-metric multidimensional scaling (NMDS) plots of Bray–Curtis dissimilarity using the metaMDS function in the vegan package, looking specifically at the ectomycorrhizal and saprotroph guilds across RNA- and DNA-extracted community profiles. We used permutational analysis of variance tests (PERMANOVA; adonis2 function in the vegan package) to quantify the influence of pine (PISA angular size) and oak (QUDO plus QUAG angular size) on community composition, grouping at the genus level to assess broader above-ground plant influence. In order to understand how much of the overall fungal community turnover could be attributed to forest canopy changes or soil core proximity, we employed Mantel tests on OTU and tree availability Bray–Curtis dissimilarity matrices using the mantel function in the vegan package.
To investigate how aboveground plant composition and soil chemistry interacted to drive fungal community turnover, we compared community dissimilarity matrices against environmental dissimilarity matrices including soil chemical data and canopy survey data, analyzing each of the same four groups as in the NMDS plots (ectomycorrhizal DNA, ectomycorrhizal RNA, saprotroph DNA, and saprotroph RNA). For these analyses, we used the bioenv function to select the subset of environmental factors that were most strongly correlated with community turnover. In total, the list of factors selected for one or more groups included PISA angular size, QUAG angular size, QUDO angular size, pH, nitrogen percentage, soil phosphorus concentration, litter depth, and C:N ratio. From this set of predictors, we removed correlated predictors (C:N ratio, correlated with PISA, and litter depth, correlated with pH) and used all remaining predictors in a PERMANOVA test of each group to assess relative predictive power between canopy and environmental factors. For these tests, we included transect as a random predictor.
Results
Site Properties and Aboveground Plant Diversity
Across our soil cores, we captured substantial diversity in tree species influence and soil chemistry (Figures 1B, 2). On average, cores in Transects 1 and 2 had the greatest influence by gray pine (PISA), with very limited to zero pine influence in Transects 3 and 4, respectively. Cores in Transects 1, 2, and 4 (but not 3) exhibited considerable influence of coast live oak (QUAG), and all transects had large amounts of blue oak (QUDO), though substantial within-transect variation existed. Soil chemistry varied subtly but significantly in our study. We found significantly lower phosphorus levels in Transects 1 and 3 than in Transects 2 and 4. Nitrogen was variable across transects (Figure 2E), with Transect 1 having the lowest percent nitrogen and Transect 3 having the highest. We found small but consistent transect-specific differences in pH, though generally all sites were near neutral (Figure 2F).
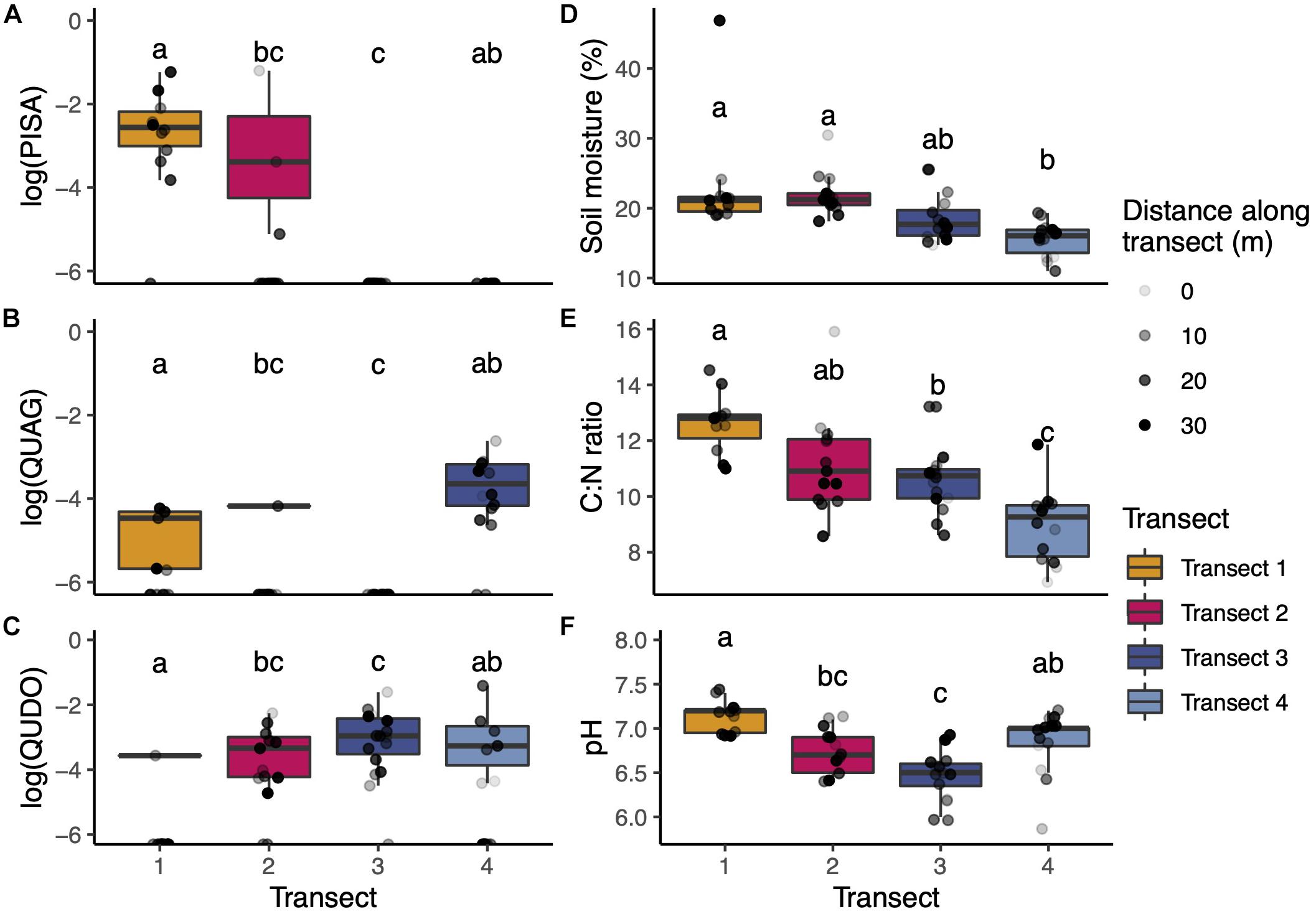
Figure 2. (A–C) Aboveground plant and soil properties of transects. Transects varied in the composition of their aboveground tree communities, measured in proportion of each soil core’s field-of-view represented by each host type (note log transformation of the y-axis). (D–F) Soil chemical properties were relatively consistent across transects with the greatest variation arising in C:N ratio (correlated with PISA). Boxplots span the 25th to 75th percentile with median values marked with the horizontal line. Whiskers depict at maximum ±1.5 times the IQR from the lower hinge. Data from individual soil cores are shown as points overlaid on each boxplot. Letters indicate statistically significant differences in transect means at the p < 0.05 level (ANOVA with Tukey’s HSD correction for multiple hypothesis tests).
Both pH and C:N ratio were strongly correlated with tree-driven factors. The influence of pine was correlated with C:N with a Pearson’s r-value of 0.491 and a p-value of p < 0.001. Similarly, deeper litter depth was correlated with lower pH with a Pearson’s r-value of 0.392 and a p-value of p < 0.01. To avoid including tightly correlated parameters in later analyses, we chose to drop C:N ratio and litter depth in favor of retaining PISA angular size and pH, respectively, in our PERMANOVA tests of community dissimilarity.
Belowground Fungal Diversity
We successfully extracted, amplified, and sequenced 61 of 64 DNA samples and 62 of 64 RNA samples. (The five unsequenced samples were randomly distributed among our study samples.) After filtering, our dataset contained just over 15.7 million reads, with an average of roughly 127,000 sequences per sample. We found 5,264 unique OTUs in 123 samples (see Supplementary Figure 1 for species accumulation curves). Of these OTUs, 3,192 were observed in the RNA samples, 4,153 were observed in the DNA samples, and 2,450 were observed in both. Subsetting our data to ectomycorrhizal and saprotroph OTUs reduced our OTU count to 430 and 396 OTUs and 1.85 million and 1.58 million reads, respectively (see Supplementary Figure 2 for a visualization of all fungi in dataset). RNA and DNA samples accounted for 46 and 54 percent of the total read counts, respectively. Species accumulation data suggest that read depth was closer to saturation in the RNA communities and that the subset of saprotrophic fungi were closer than either the ectomycorrhizal guild or the total community to saturating OTU richness (Supplementary Figure 1).
Operational taxonomic unit richness was stable across transects in the DNA communities, with no significant differences (p < 0.05), though Transect 3 had greater OTU abundance than the other transects in the RNA communities (p < 0.001, Figure 3A). DNA communities were generally more OTU-rich than their RNA counterparts, with significantly higher richness in Transects 1 and 4 (p < 0.05 and p < 0.001, respectively). Shannon diversity of DNA and RNA within transects was similar except for Transect 2 where the RNA community was significantly more diverse (p < 0.01) than in the DNA samples (Figure 3B). Richness and Shannon diversity trends were similar when rarefied data were analyzed with the most notable difference being a relative increase in richness in RNA samples versus DNA (Supplementary Figure 3). The most consistent difference in measured diversity was in fungal turnover (beta diversity). The DNA communities again were indistinguishable between transects but RNA varied widely (Figure 3C). Transect 1 had the highest spatial turnover (significantly higher than T2 and T3, p < 0.05 and p < 0.001, respectively) and Transect 3 the lowest (significantly lower than T1 and T4, p < 0.001 and p < 0.05, respectively). Across transects, each RNA community had significantly greater spatial turnover than the corresponding DNA community (p < 0.001 for all transects).
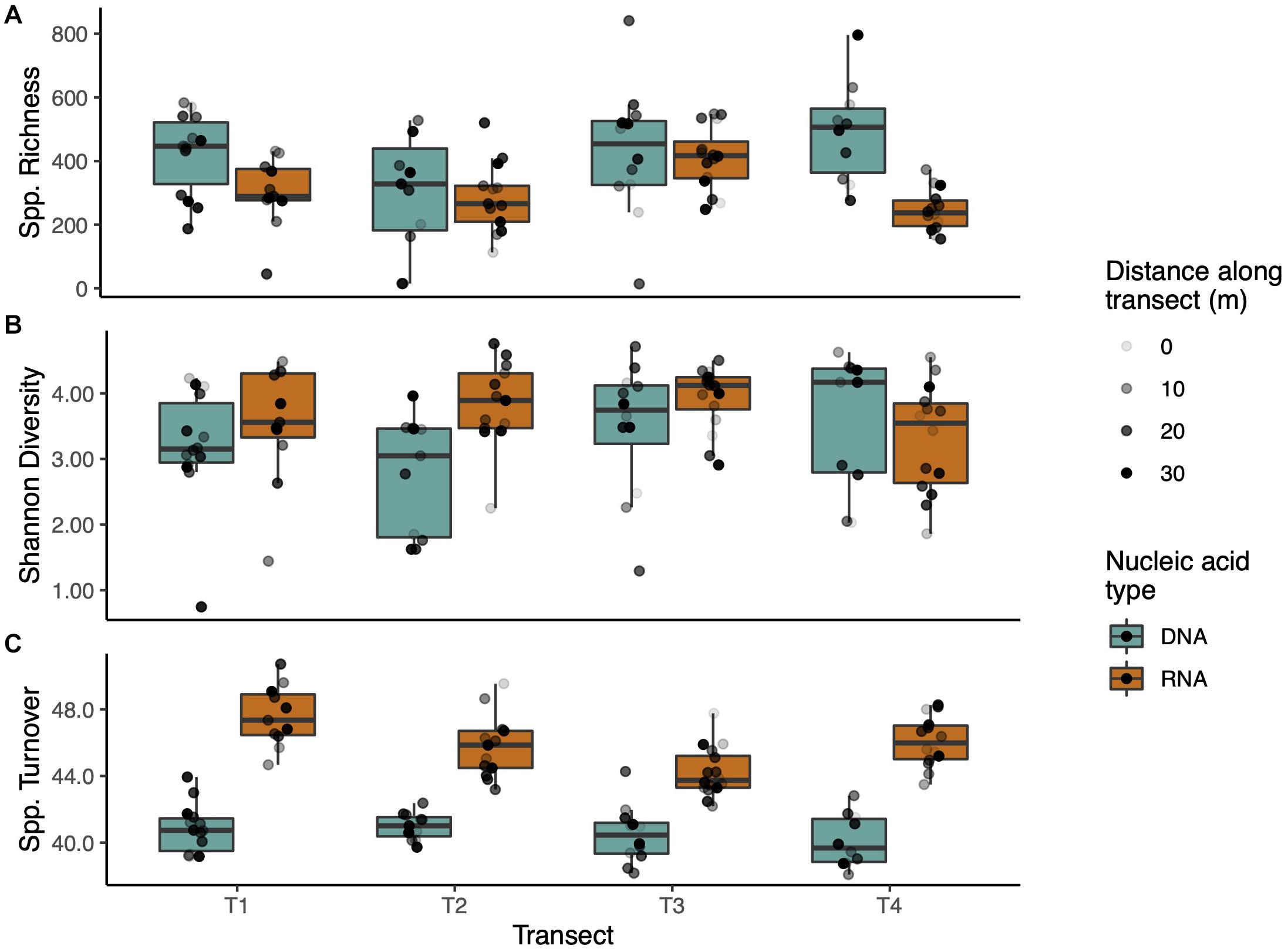
Figure 3. Alpha and beta diversity of transects. (A) Transects did not differ significantly in species richness, though DNA communities generally captured greater richness. (B) Shannon diversity was consistent among transects and nucleic acid types. (C) Species turnover (beta diversity) was much higher in RNA communities than DNA communities. Boxplots span the 25th to 75th percentile with median values marked with the horizontal line. Whiskers depict at maximum ±1.5 times the IQR from the lower hinge. Points are scattered individual data points shaded by distance along the transect.
Drivers of Spatial Turnover in Belowground Communities
In our NMDS plots, aboveground plant diversity drove turnover in belowground diversity in fungal RNA but not DNA communities (Figure 4 and Supplementary Figure 2). In the RNA samples, communities closer to a pine host separated from those closer to an oak host. Our host influence PERMANOVA revealed that, in the RNA communities, ectomycorrhizal fungal community turnover was significantly, but not strongly, predicted by both pine influence (pseudo-F = 2.794, R2 = 0.050, p < 0.001) and oak influence (pseudo-F = 1.658, R2 = 0.030, p < 0.05). The saprotrophic fungal community behaved similarly, although the influence of pine hosts was not statistically significant (pine: pseudo-F = 1.500, R2 = 0.027, p = 0.055; oak: pseudo-F = 2.201, R2 = 0.040, p < 0.01). Similar analysis of DNA-based communities did not reveal significant structuring by pine or oak host influence (Supplementary Table 1).
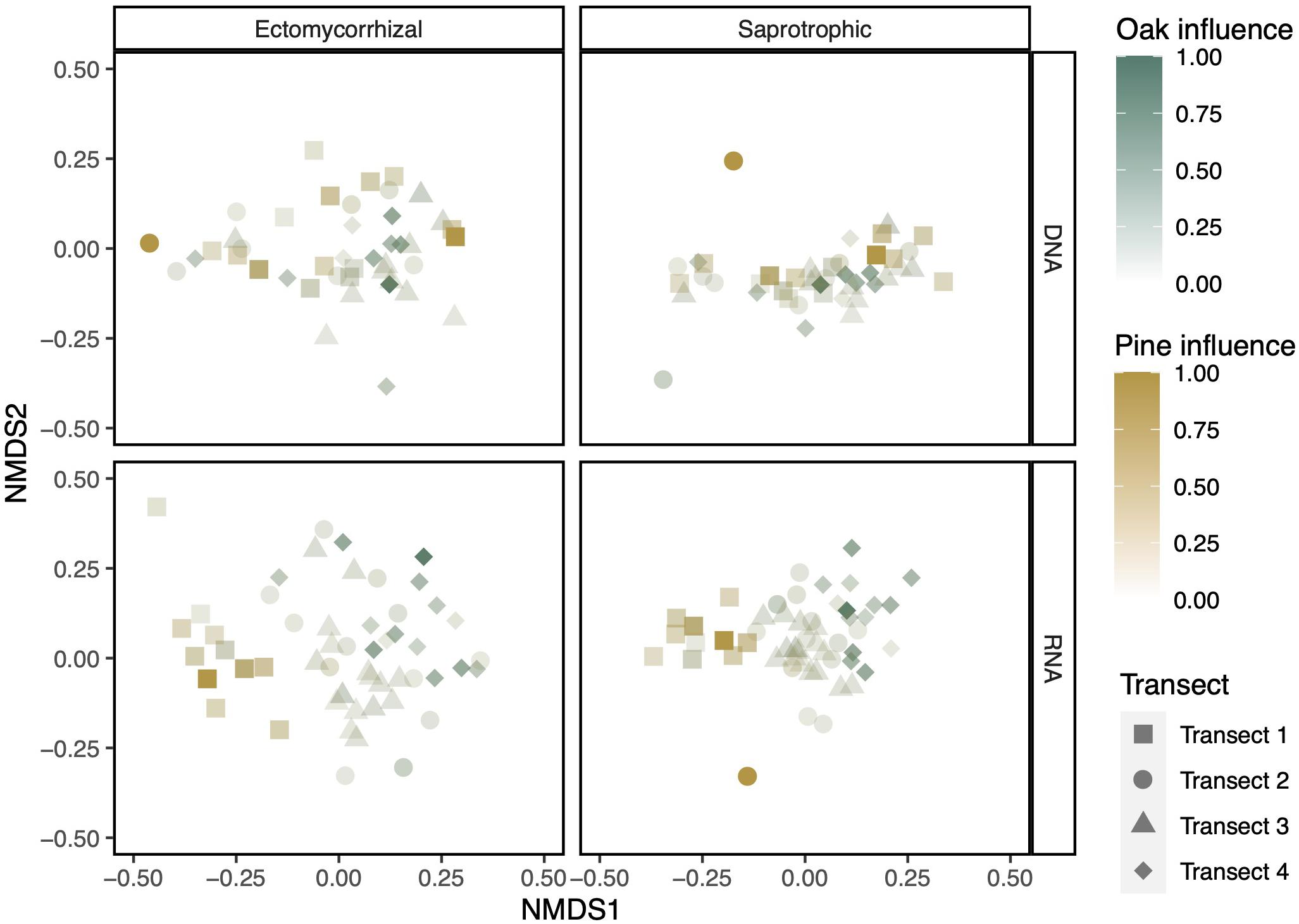
Figure 4. NMDS plots show a separation by aboveground influence in both ectomycorrhizal and saprotrophic communities when displaying RNA but not DNA community data. Colors are scaled to the maximum influence of each canopy genus in any soil core. NMDS plots were constructed using Bray–Curtis dissimilarity.
In RNA communities, the spatial turnover in fungal community composition was significantly predicted by spatial turnover in aboveground plant community composition. This relationship between above- and below-ground diversity was strongest when analyzing all fungi in the data set (Mantel r = 0.25992, p < 0.01), although it was also significantly predictive for ectomycorrhizal fungi (r = 0.11021, p < 0.05) and showed a weaker trend for saprotrophic fungi (r = 0.1519, p = 0.056). The DNA fungal communities did not respond significantly to aboveground plant turnover (Supplementary Table 2). RNA communities from samples taken closer to one another were not significantly correlated in Mantel tests of any community subset (Supplementary Table 3). DNA-based fungal communities did not exhibit any significant spatial autocorrelation (Supplementary Table 3).
Examining spatial turnover with the PERMANOVA allowed us to identify significant drivers of community turnover in the RNA samples, but not in the DNA samples (Table 1). None of our predictors explained more than 5% of the variation in spatial turnover for RNA-based fungal communities, however, and the entire model only accounted for 20 and 25% of the variation in saprotrophic and ectomycorrhizal communities, respectively. RNA community turnover was significantly predicted in both saprotroph and ectomycorrhizal communities by pH and soil nitrogen levels (Table 1). Additionally, saprotroph RNA communities were significantly predicted by the influence of QUDO hosts, while ectomycorrhizal communities were correlated with PISA influence and soil phosphorus. A similar model with transect as a predictor (instead of a random effect) yielded similar results, although transect identity explained approximately 8% of the variation in all communities when included explicitly as a predictor, and effects of soil chemistry and aboveground plant community were less strong than in the main analysis (Supplementary Table 4).
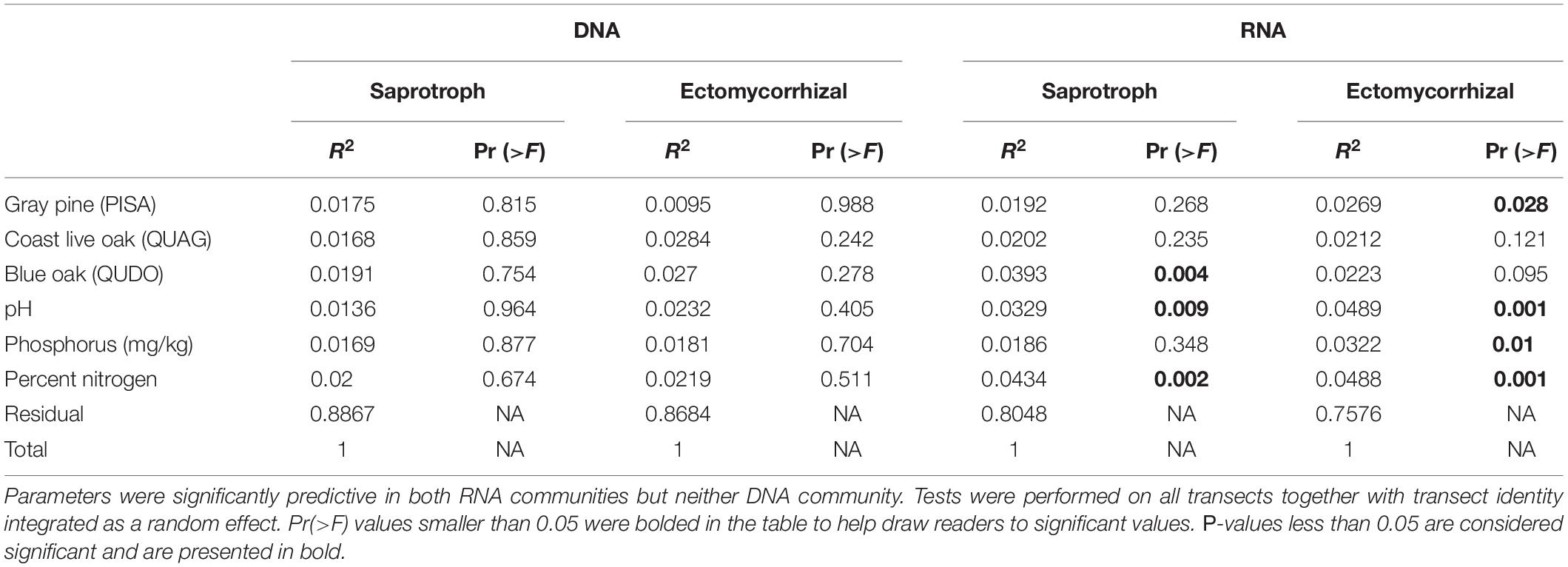
Table 1. PERMANOVA results show the overlap in predictive parameters between saprotrophic and ectomycorrhizal fungi.
Discussion
Understanding the ways in which aboveground diversity maps onto belowground patterns of spatial turnover in fungal communities is an important step toward predicting both the function and the vulnerabilities of forested ecosystems. We show here that both aboveground tree community composition and soil chemistry influence fungal communities regardless of fungal trophic mode, but that the chemical factors affecting symbiotic ectomycorrhizal fungi are not identical to those influencing free-living saprotrophs. These patterns were clearly discernible in the precursor rRNA communities, but were not significant for DNA communities.
Although both nucleic acid types allowed us to detect similar levels of fungal OTU richness, the precursor rRNA-based communities responded much more predictably to measured soil and plant factors than the DNA-based communities, which appeared to be mostly stochastic. This is likely due, at least in part, to the fact that precursor rRNA represents the metabolically active component of the fungal community (van der Linde and Haller, 2013; Wutkowska et al., 2019), and may therefore more closely mirror local environmental conditions. Supporting this possibility, other studies that have examined both nucleic acids in soils have also found that rRNA-based communities respond to a greater number of deterministic factors than DNA-based communities, or respond to those factors more strongly (Romanowicz et al., 2016; Lüneberg et al., 2018; Lasa et al., 2019; Meyer et al., 2019; Nawaz et al., 2019; Wutkowska et al., 2019). In some cases, like the present study, rRNA communities are the only ones in which deterministic effects on community structure are statistically detectable (Ragot et al., 2016; Gill et al., 2017). DNA- and precursor rRNA-based communities can exhibit more similar structures and environmental responses, but studies that find this tend to be conducted at a much larger spatial scale and to target substantially greater habitat diversity than ours did (Baldrian et al., 2012; Žifčáková et al., 2016). At the relatively fine spatial scale of our study, the potentially active fungal community (precursor rRNA-based) provided more useful ecological insight than the DNA-based community.
We found that soil pH, soil nutrients, and canopy species all had significant capacity to predict community turnover in the potentially active community for both saprotrophs and ectomycorrhizal fungi, but the variance explained by these factors was small (Table 1). This may be attributable to the fact that the levels of these factors did not, themselves, vary substantially at the relatively small spatial scale of this study. In this context, it is perhaps a testament to the importance of these soil factors that their influence was detectable even within this fairly limited range of values. Detecting these factors, which subtly but significantly influenced the structure of potentially active ectomycorrhizal and saprotrophic fungal communities, allowed us to disentangle how these distinct trophic guilds responded to their shared environment. We hypothesized that symbiotic ectomycorrhizal communities would track aboveground plant composition more closely than free-living saprotrophs, but this hypothesis was only partially supported by our precursor rRNA community data. In our study system, ectomycorrhizal community turnover was significantly predicted by pine influence, a result consistent with prior work on ectomycorrhizal root tips in mixed woodlands (Smith et al., 2009; Suz et al., 2017). The spatial turnover in saprotrophic fungi, by contrast, was significantly predicted by oaks (Table 1). Other belowground factors, such as nitrogen availability and pH, were significant predictors of both communities.
In part, effects on these two fungal guilds likely arise from the cascading effects of host trees on the soil community. While ectomycorrhizal host plants directly supply their belowground partners with carbon, they also indirectly influence the whole fungal community through root exudates and leaf litter (Aponte et al., 2010; Hugoni et al., 2018). For example, blue oak litter shapes the seasonality of nutrient deposition (Callaway and Nadkarni, 1991) and understory productivity (Callaway et al., 1991) in California woodlands. Indeed, in our study, we found that the pine availability correlated with soil C:N ratios, and the depth of surface litter correlated with soil pH. Soil pH is a dominant factor predicting soil microbial community structure (van der Linde et al., 2018), and soil C:N ratios indicate the relative cost of decomposition of soil organic matter and, thus, its bioavailability to free-living saprotrophs (Manzoni et al., 2010; Smith and Wan, 2019). The overlap in these resource-availability predictors between ectomycorrhizal fungi and saprotrophs underscores the niche similarity of these fungal guilds (Bödeker et al., 2016), despite their different trophic strategies. Our results emphasize the profound influence that aboveground tree communities can have on belowground fungal assemblages, both directly, through symbiotic interactions, and indirectly, by mediating soil chemistry with litter inputs and root exudates. Ectomycorrhizal and saprotrophic fungi share an environment and are affected by similar environmental factors, but these guilds perform fundamentally distinct ecosystem functions (Talbot et al., 2013; Averill et al., 2014; Lindahl and Tunlid, 2015; Bödeker et al., 2016). Understanding how these guilds will be differentially affected by ecological stressors will be important for predicting their contributions to ecosystem function moving forward.
The strength of host identity effects in our ecosystem is consistent with results from prior studies in California woodlands, particularly studies showing the effects of oak host identity using DNA-based sampling of ectomycorrhizal root tips (Morris et al., 2008; Smith et al., 2009). Further, ectomycorrhizal communities diverged more strongly between oak and pine hosts, than between oak species, consistent with the phylogenetic distances of the host trees (Tedersoo et al., 2013). Host effects may be particularly strong in these woodlands, where the water-limited environment creates a patchwork of trees and grass cover (Figures 1, 2; Borchert et al., 1991). This habitat mosaic results in greater spatial separation (and reduced rooting zone overlap) among host trees, potentially increasing our ability to detect differences in fungal community composition because of physical separation between hosts. In denser canopies, or more speciose systems, such signals might be more difficult to detect.
Despite the influences of these deterministic environmental factors on precursor rRNA-based communities, our analyses were unable to explain more than 25% of the variance in spatial turnover of potentially active fungal assemblages. The large amount of unaccounted-for variation likely stems from a combination of deterministic factors, such as unmeasured environmental variables and biotic interactions belowground, and the stochastic and partially stochastic processes well known to play important roles in community assembly, such as dispersal, ecological drift, and historical contingency (Peay and Bruns, 2014; Fukami, 2015; Vellend, 2016; Bogar and Peay, 2017). Although studies tend to agree that, for instance, pH and nitrogen are important in structuring fungal communities, it is not uncommon for much of the variance in fungal communities to remain unaccounted for even after modeling the influence of soil and plant traits, especially at relatively fine spatial scales (Romanowicz et al., 2016; Ren et al., 2018) or when focusing on functionally specific subsets of the total fungal community (Kivlin and Hawkes, 2016; Glassman et al., 2017; Daws et al., 2020). Because dispersal limitation likely played only a modest role in determining community membership at the fine spatial scale of our study (tens of meters) (Peay et al., 2012; Horton, 2017), it is especially striking that we found consistent, significant variation in the potentially active fungal communities predictable with soil and plant factors. Both tree identity and soil chemistry were significant factors structuring the active fungal communities in this study, even at a spatial scale where stochastic processes also likely exerted powerful influence.
Conclusion
In our study, active fungal communities, characterized with precursor rRNA (ITS), responded significantly to measured ecological factors, providing insight into the linkages between aboveground canopy composition and the community structure of symbiotic ectomycorrhizal and free-living saprotrophic fungi. DNA-based communities provided little ecological insight at this scale. The turnover of ectomycorrhizal and saprotrophic communities were differentially predicted by the availability of canopy species, though both were driven by similar soil properties. The overlap in predictive factors between the ectomycorrhizal and saprotrophic guilds reiterates their degree of niche overlap and the critical nature of symbiosis in structuring these communities. Our study suggests that, at relatively fine (meter) scales, precursor rRNA may allow researchers to identify ecological factors that affect fungal community function, while DNA would obscure these relationships. Future work should investigate whether precursor rRNA-based communities are, in fact, consistently more responsive to deterministic predictors than DNA-based communities, and at what spatial and temporal scales this is most true. In addition, manipulative experiments that isolate direct plant influences, such as symbiotic associations, from indirect plant effects on the soil environment, such as litter input, would provide valuable insight into the mechanisms driving differences between ectomycorrhizal and saprotrophic fungal community dynamics.
Data Availability Statement
All analyses were performed in R [version 3.6.3 (R Core Team, 2020)] using the R Studio interface [version 1.2.5001 (RStudio Team, 2020)]. Scripts and data are available on Github (https://github.com/gaberunte/spheres_of_influence). Data files not stored on Github (due to size constraints) are available upon request from GR. Sequences are available through GenBank with Bioproject accession numbers PRJNA684619 and SRR13253516–SRR13253632.
Author Contributions
LB, GR, and HM designed the study and led the sample collection effort. AS extracted all nucleic acids and prepared sequencing libraries with GR, and developed the host influence metric. LB and GR designed the analyses. GR conducted all data processing and statistical analyses. All authors contributed to manuscript preparation.
Funding
This work was supported by a UC Santa Barbara Faculty Research Grant (to HM) and an NSF Postdoctoral Research Fellowship in Biology (DBI-2011020 to LB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ean Eberhard, Sevan Esaian, Samantha Moore, and Oceana Tavasieff for their assistance with field sampling, and Natalie Chong, Mason Reynoso, and Brittney Straw for soil processing. Thanks to Glade Dlott for advice on study design, methods, and data interpretation. We are grateful to Oliver Chadwick, Kate McCurdy, and Frank Davis for their insights at Sedgwick Reserve. We acknowledge the use of the Biological Nanostructures Laboratory within the California NanoSystems Institute, supported by the University of California, Santa Barbara and the University of California, Office of the President. We also thank two reviewers and GC for feedback on earlier drafts of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.641732/full#supplementary-material
References
Aponte, C., García, L. V., Marañón, T., and Gardes, M. (2010). Indirect host effect on ectomycorrhizal fungi: leaf fall and litter quality explain changes in fungal communities on the roots of co-occurring mediterranean oaks. Soil Biol. Biochem. 42, 788–796. doi: 10.1016/j.soilbio.2010.01.014
Averill, C., and Hawkes, C. V. (2016). Ectomycorrhizal fungi slow soil carbon cycling. Ecol. Lett. 19, 937–947. doi: 10.1111/ele.12631
Averill, C., Turner, B. L., and Finzi, A. C. (2014). Mycorrhiza-mediated competition between plants and decomposers drives soil carbon storage. Nature 505, 534–545. doi: 10.1038/nature12901
Bahram, M., Kõljalg, U., Courty, P. E., Diédhiou, A. G., Kjøller, R., Põlme, S., et al. (2013). The distance decay of similarity in communities of ectomycorrhizal fungi in different ecosystems and scales. J. Ecol. 101, 1335–1344. doi: 10.1111/1365-2745.12120
Baldrian, P. (2017). Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol. Rev. 41, 109–130. doi: 10.1093/femsre/fuw040
Baldrian, P., Kolařík, M., Štursová, M., Kopecký, J., Valášková, V., Větrovský, T., et al. (2012). Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 6, 248–258. doi: 10.1038/ismej.2011.95
Barnard, R. L., Osborne, C. A., and Firestone, M. K. (2013). Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J. 7, 2229–2241. doi: 10.1038/ismej.2013.104
Blazewicz, S. J., Barnard, R. L., Daly, R. A., and Firestone, M. K. (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 7, 2061–2068. doi: 10.1038/ismej.2013.102
Bödeker, I. T. M., Lindahl, B. D., and Olson, Å, and Clemmensen, K. E. (2016). Mycorrhizal and saprotrophic fungal guilds compete for the same organic substrates but affect decomposition differently. Funct. Ecol. 30, 1967–1978. doi: 10.1111/1365-2435.12677
Bogar, L. M., and Peay, K. G. (2017). “Processes maintaining the coexistence of ectomycorrhizal fungi at a fine spatial scale,” in Biogeography of Mycorrhizal Symbiosis, ed. L. Tedersoo (Cham: Springer International Publishing), 79–105. doi: 10.1007/978-3-319-56363-3_4
Borchert, M., Davis, F. W., and Allen-Diaz, B. (1991). Environmental relationships of herbs in blue oak (Quercus douglasii) woodlands of central coastal California. Madroño 38, 249–266.
Bruns, T. D., Peay, K. G., Boynton, P. J., Grubisha, L. C., Hynson, N. A., Nguyen, N. H., et al. (2009). Inoculum potential of Rhizopogon spores increases with time over the first 4 yr of a 99-yr spore burial experiment. New Phytol. 181, 463–470. doi: 10.1111/j.1469-8137.2008.02652.x
Bui, A., Orr, D., Lepori-Bui, M., Konicek, K., Young, H. S., and Moeller, H. V. (2020). Soil fungal community composition and functional similarity shift across distinct climatic conditions. FEMS Microbiol. Ecol. 96:fiaa193. doi: 10.1093/femsec/fiaa193
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Callaway, R. M., and Nadkarni, N. M. (1991). Seasonal patterns of nutrient deposition in a Quercus douglasii woodland in central California. Plant Soil 137, 209–222. doi: 10.1007/bf00011199
Callaway, R. M., Nadkarni, N. M., and Mahall, B. E. (1991). Facilitation and interference of Quercus douglasii on understory productivity in central California. Ecology 72, 1484–1499. doi: 10.2307/1941122
Carini, P., Marsden, P. J., Leff, J. W., Morgan, E. E., Strickland, M. S., and Fierer, N. (2016). Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2:16242. doi: 10.1038/nmicrobiol.2016.242
Crowther, T. W., van den Hoogen, J., Wan, J., Mayes, M. A., Keiser, A. D., Mo, L., et al. (2019). The global soil community and its influence on biogeochemistry. Science 365:eaav0550. doi: 10.1126/science.aav0550
Davis, F. W., Tyler, C. M., and Mahall, B. E. (2011). Consumer control of oak demography in a mediterranean-climate savanna. Ecosphere 2:art108. doi: 10.1890/ES11-00187.1
Daws, S. C., Cline, L. A., Rotenberry, J., Sadowsky, M. J., Staley, C., Dalzell, B., et al. (2020). Do shared traits create the same fates? Examining the link between morphological type and the biogeography of fungal and bacterial communities. Fungal Ecol. 46:100948. doi: 10.1016/j.funeco.2020.100948
Day, S. D., Wiseman, P. E., Dickinson, S. B., and Harris, J. R. (2010). Contemporary concepts of root system architecture of urban trees. Arboric. Urban For. 36, 149–159.
Dickie, I. A., Bolstridge, N., Cooper, J. A., and Peltzer, D. A. (2010). Co-invasion by Pinus and its mycorrhizal fungi. New Phytol. 187, 475–484. doi: 10.1111/j.1469-8137.2010.03277.x
Dlott, G., Maul, J. E., Buyer, J., and Yarwood, S. (2015). Microbial rRNA: rDNA gene ratios may be unexpectedly low due to extracellular DNA preservation in soils. J. Microbiol. Methods 115, 112–120. doi: 10.1016/j.mimet.2015.05.027
Fernandez, C. W., See, C. R., and Kennedy, P. G. (2020). Decelerated carbon cycling by ectomycorrhizal fungi is controlled by substrate quality and community composition. New Phytol. 226, 569–582. doi: 10.1111/nph.16269
Fukami, T. (2015). Historical contingency in community assembly: integrating niches, species pools, and priority effects. Annu. Rev. Ecol. Evol. Syst. 46, 1–23. doi: 10.1146/annurev-ecolsys-110411-160340
Fukami, T., Dickie, I. A., Paula Wilkie, J., Paulus, B. C., Park, D., Roberts, A., et al. (2010). Assembly history dictates ecosystem functioning: evidence from wood decomposer communities. Ecol. Lett. 13, 675–684. doi: 10.1111/j.1461-0248.2010.01465.x
Gadgil, R. L., and Gadgil, P. D. (1975). Suppression of litter decomposition by mycorrhizal roots of Pinus radiata. N. Z. J. For. Sci. 5, 33–41.
Gao, C., Shi, N.-N., Liu, Y.-X., Peay, K. G., Zheng, Y., Ding, Q., et al. (2013). Host plant genus-level diversity is the best predictor of ectomycorrhizal fungal diversity in a Chinese subtropical forest. Mol. Ecol. 22, 3403–3414. doi: 10.1111/mec.12297
Gill, A. S., Lee, A., and McGuire, K. L. (2017). Phylogenetic and functional diversity of total (DNA) and expressed (RNA) bacterial communities in urban green infrastructure bioswale soils. Appl. Environ. Microbiol. 83:e00287-17. doi: 10.1128/AEM.00287-17
Glassman, S. I., Wang, I. J., and Bruns, T. D. (2017). Environmental filtering by pH and soil nutrients drives community assembly in fungi at fine spatial scales. Mol. Ecol. 26, 6960–6973. doi: 10.1111/mec.14414
Hein, A. M., Gil, M. A., Twomey, C. R., Couzin, I. D., and Levin, S. A. (2018). Conserved behavioral circuits govern high-speed decision-making in wild fish shoals. Proc. Natl. Acad. Sci. U.S.A. 115, 12224–12228. doi: 10.1073/pnas.1809140115
Hiiesalu, I., Bahram, M., and Tedersoo, L. (2017). Plant species richness and productivity determine the diversity of soil fungal guilds in temperate coniferous forest and bog habitats. Mol. Ecol. 26, 4846–4858. doi: 10.1111/mec.14246
Horton, T. R. (2017). “Spore dispersal in ectomycorrhizal fungi at fine and regional Scales,” in Biogeography of Mycorrhizal Symbiosis Ecological Studies, ed. L. Tedersoo (Cham: Springer International Publishing), 61–78. doi: 10.1007/978-3-319-56363-3_3
Hugoni, M., Luis, P., Guyonnet, J., and Haichar, F. el Z. (2018). Plant host habitat and root exudates shape fungal diversity. Mycorrhiza 28, 451–463. doi: 10.1007/s00572-018-0857-5
Izzo, A., Agbowo, J., and Bruns, T. D. (2005). Detection of plot-level changes in ectomycorrhizal communities across years in an old-growth mixed-conifer forest. New Phytol. 166, 619–630. doi: 10.1111/j.1469-8137.2005.01354.x
Janowski, D., Wilgan, R., Leski, T., Karliński, L., and Rudawska, M. (2019). Effective molecular identification of ectomycorrhizal fungi: revisiting DNA isolation methods. Forests 10:218. doi: 10.3390/f10030218
Kivlin, S. N., and Hawkes, C. V. (2016). Tree species, spatial heterogeneity, and seasonality drive soil fungal abundance, richness, and composition in Neotropical rainforests. Environ. Microbiol. 18, 4662–4673. doi: 10.1111/1462-2920.13342
Koide, R. T., Fernandez, C., and Malcolm, G. (2014). Determining place and process: functional traits of ectomycorrhizal fungi that affect both community structure and ecosystem function. New Phytol. 201, 433–439. doi: 10.1111/nph.12538
Kumar, S., and Satyanarayana, T. (2002). “Isolation of ectomycorrhizal fungi: methods and techniques,” in Techniques in Mycorrhizal Studies, eds K. G. Mukerji, C. Manoharachary, and B. P. Chamola (Dordrecht: Springer Netherlands), 133–142. doi: 10.1007/978-94-017-3209-3_7
Lasa, A. V., Fernández-González, A. J., Villadas, P. J., Toro, N., and Fernández-López, M. (2019). Metabarcoding reveals that rhizospheric microbiota of Quercus pyrenaica is composed by a relatively small number of bacterial taxa highly abundant. Sci. Rep. 9:1695. doi: 10.1038/s41598-018-38123-z
Lennon, J. T., and Jones, S. E. (2011). Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 9, 119–130. doi: 10.1038/nrmicro2504
Liao, H.-L., Chen, Y., Bruns, T. D., Peay, K. G., Taylor, J. W., Branco, S., et al. (2014). Metatranscriptomic analysis of ectomycorrhizal roots reveals genes associated with Piloderma-Pinus symbiosis: improved methodologies for assessing gene expression in situ. Environ. Microbiol. 16, 3730–3742. doi: 10.1111/1462-2920.12619
Lilleskov, E. A., Bruns, T. D., Horton, T. R., Taylor, D., and Grogan, P. (2004). Detection of forest stand-level spatial structure in ectomycorrhizal fungal communities. FEMS Microbiol. Ecol. 49, 319–332. doi: 10.1016/j.femsec.2004.04.004
Lindahl, B. D., and Tunlid, A. (2015). Ectomycorrhizal fungi–potential organic matter decomposers, yet not saprotrophs. New Phytol. 206, 1443–1447. doi: 10.1111/nph.13201
Lüneberg, K., Schneider, D., Siebe, C., and Daniel, R. (2018). Drylands soil bacterial community is affected by land use change and different irrigation practices in the Mezquital Valley, Mexico. Sci. Rep. 8, 1413. doi: 10.1038/s41598-018-19743-x
Manzoni, S., Trofymow, J. A., Jackson, R. B., and Porporato, A. (2010). Stoichiometric controls on carbon, nitrogen, and phosphorus dynamics in decomposing litter. Ecol. Monogr. 80, 89–106. doi: 10.1890/09-0179.1
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17, 10–12. doi: 10.14806/ej.17.1.200
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
McMurdie, P. J., and Holmes, S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10:e1003531. doi: 10.1371/journal.pcbi.1003531
Meyer, K. M., Petersen, I. A. B., Tobi, E., Korte, L., and Bohannan, B. J. M. (2019). Use of RNA and DNA to identify mechanisms of bacterial community homogenization. Front. Microbiol. 10:2066. doi: 10.3389/fmicb.2019.02066
Moeller, H. V., Dickie, I. A., Peltzer, D. A., and Fukami, T. (2015). Mycorrhizal co-invasion and novel interactions depend on neighborhood context. Ecology 96, 2336–2347. doi: 10.1890/14-2361.1
Moeller, H. V., Peay, K. G., and Fukami, T. (2014). Ectomycorrhizal fungal traits reflect environmental conditions along a coastal California edaphic gradient. FEMS Microbiol. Ecol. 87, 797–806. doi: 10.1111/1574-6941.12265
Morris, M. H., Smith, M. E., Rizzo, D. M., Rejmánek, M., and Bledsoe, C. S. (2008). Contrasting ectomycorrhizal fungal communities on the roots of co-occurring oaks (Quercus Spp.) in a California Woodland. New Phytol. 178, 167–176. doi: 10.1111/j.1469-8137.2007.02348.x
Morrissey, E. M., McHugh, T. A., Preteska, L., Hayer, M., Dijkstra, P., Hungate, B. A., et al. (2015). Dynamics of extracellular DNA decomposition and bacterial community composition in soil. Soil Biol. Biochem. 86, 42–49. doi: 10.1016/j.soilbio.2015.03.020
Nawaz, A., Purahong, W., Herrmann, M., Küsel, K., Buscot, F., and Wubet, T. (2019). DNA- and RNA- derived fungal communities in subsurface aquifers only partly overlap but react similarly to environmental factors. Microorganisms 7:341. doi: 10.3390/microorganisms7090341
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016a). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Nguyen, N. H., Williams, L. J., Vincent, J. B., Stefanski, A., Cavender-Bares, J., Messier, C., et al. (2016b). Ectomycorrhizal fungal diversity and saprotrophic fungal diversity are linked to different tree community attributes in a field-based tree experiment. Mol. Ecol. 25, 4032–4046. doi: 10.1111/mec.13719
Nielsen, K. M., Calamai, L., and Pietramellara, G. (2006). “Stabilization of extracellular DNA and proteins by transient binding to various soil components,” in Nucleic Acids and Proteins in Soil Soil Biology, eds P. Nannipieri and K. Smalla (Berlin: Springer), 141–157. doi: 10.1007/3-540-29449-X_7
Nilsson, R. H., Larsson, K.-H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Ogram, A., Sayler, G. S., Gustin, D., and Lewis, R. J. (1988). DNA adsorption to soils and sediments. Environ. Sci. Technol. 22, 982–984. doi: 10.1021/es00173a020
Pauvert, C., Buée, M., Laval, V., Edel-Hermann, V., Fauchery, L., Gautier, A., et al. (2019). Bioinformatics matters: the accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecol. 41, 23–33. doi: 10.1016/j.funeco.2019.03.005
Peay, K. G., and Bruns, T. D. (2014). Spore dispersal of basidiomycete fungi at the landscape scale is driven by stochastic and deterministic processes and generates variability in plant-fungal interactions. New Phytol. 204, 180–191. doi: 10.1111/nph.12906
Peay, K. G., Garbelotto, M., and Bruns, T. D. (2010). Evidence of dispersal limitation in soil microorganisms: isolation reduces species richness on mycorrhizal tree islands. Ecology 91, 3631–3640. doi: 10.1890/09-2237.1
Peay, K. G., Schubert, M. G., Nguyen, N. H., and Bruns, T. D. (2012). Measuring ectomycorrhizal fungal dispersal: macroecological patterns driven by microscopic propagules. Mol. Ecol. 21, 4122–4136. doi: 10.1111/j.1365-294X.2012.05666.x
Pickles, B. J., Genney, D. R., Anderson, I. C., and Alexander, I. J. (2012). Spatial analysis of ectomycorrhizal fungi reveals that root tip communities are structured by competitive interactions. Mol. Ecol. 21, 5110–5123. doi: 10.1111/j.1365-294X.2012.05739.x
Plumb, T. R., and Gomez, A. P. (1983). Five Southern California Oaks: Identification and Postfire Management. PSW-GTR-71. Berkeley, CA: U.S. Department of Agriculture, Forest Service, Pacific Southwest Forest and Range Experiment Station, doi: 10.2737/PSW-GTR-71
R Core Team (2020). R: a Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ragot, S. A., Huguenin-Elie, O., Kertesz, M. A., Frossard, E., and Bünemann, E. K. (2016). Total and active microbial communities and phoD as affected by phosphate depletion and pH in soil. Plant Soil 408, 15–30. doi: 10.1007/s11104-016-2902-5
Rasmussen, A. L., Busby, R. R., and Hoeksema, J. D. (2018). Host preference of ectomycorrhizal fungi in mixed pine–oak woodlands. Can. J. For. Res. 48, 153–159. doi: 10.1139/cjfr-2017-0227
Ren, B., Hu, Y., Chen, B., Zhang, Y., Thiele, J., Shi, R., et al. (2018). Soil pH and plant diversity shape soil bacterial community structure in the active layer across the latitudinal gradients in continuous permafrost region of Northeastern China. Sci. Rep. 8:5619. doi: 10.1038/s41598-018-24040-8
Romanowicz, K. J., Freedman, Z. B., Upchurch, R. A., Argiroff, W. A., and Zak, D. R. (2016). Active microorganisms in forest soils differ from the total community yet are shaped by the same environmental factors: the influence of pH and soil moisture. FEMS Microbiol. Ecol. 92:fiw149. doi: 10.1093/femsec/fiw149
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Scott, N. A., and Binkley, D. (1997). Foliage litter quality and annual net N mineralization: comparison across North American forest sites. Oecologia 111, 151–159. doi: 10.1007/s004420050219
Smith, G. R., Steidinger, B. S., Bruns, T. D., and Peay, K. G. (2018). Competition-colonization tradeoffs structure fungal diversity. ISME J. 12, 1758–1767. doi: 10.1038/s41396-018-0086-0
Smith, G. R., and Wan, J. (2019). Resource−ratio theory predicts mycorrhizal control of litter decomposition. New Phytol. 223, 1595–1606. doi: 10.1111/nph.15884
Smith, M. E., Douhan, G. W., Fremier, A. K., and Rizzo, D. M. (2009). Are true multihost fungi the exception or the rule? Dominant ectomycorrhizal fungi on Pinus sabiniana differ from those on co-occurring Quercus species. New Phytol. 182, 295–299. doi: 10.1111/j.1469-8137.2009.02801.x
Soil Survey Staff, Natural Resources Conservation Service, and United States Department of Agriculture (2020). Web Soil Survey. Available online at: http://websoilsurvey.sc.egov.usda.gov/ (Accessed December 3, 2020)
Sorensen, J. W., and Shade, A. (2020). Dormancy dynamics and dispersal contribute to soil microbiome resilience. Philos. Trans. R. Soc. B Biol. Sci. 375:20190255. doi: 10.1098/rstb.2019.0255
Steidinger, B. S., Crowther, T. W., Liang, J., Van Nuland, M. E., Werner, G. D. A., Reich, P. B., et al. (2019). Climatic controls of decomposition drive the global biogeography of forest-tree symbioses. Nature 569, 404–408. doi: 10.1038/s41586-019-1128-0
Suz, L. M., Kallow, S., Reed, K., Bidartondo, M. I., and Barsoum, N. (2017). Pine mycorrhizal communities in pure and mixed pine-oak forests: abiotic environment trumps neighboring oak host effects. For. Ecol. Manag. 406, 370–380. doi: 10.1016/j.foreco.2017.09.030
Talbot, J. M., Bruns, T. D., Smith, D. P., Branco, S., Glassman, S. I., Erlandson, S., et al. (2013). Independent roles of ectomycorrhizal and saprotrophic communities in soil organic matter decomposition. Soil Biol. Biochem. 57, 282–291. doi: 10.1016/j.soilbio.2012.10.004
Talbot, J. M., Bruns, T. D., Taylor, J. W., Smith, D. P., Branco, S., Glassman, S. I., et al. (2014). Endemism and functional convergence across the North American soil mycobiome. Proc. Natl. Acad. Sci. U.S.A. 111, 6341–6346. doi: 10.1073/pnas.1402584111
Tedersoo, L., Mett, M., Ishida, T. A., and Bahram, M. (2013). Phylogenetic relationships among host plants explain differences in fungal species richness and community composition in ectomycorrhizal symbiosis. New Phytol. 199, 822–831. doi: 10.1111/nph.12328
Tedersoo, L., Bahram, M., Cajthaml, T., Põlme, S., Hiiesalu, I., Anslan, S., et al. (2016). Tree diversity and species identity effects on soil fungi, protists and animals are context dependent. ISME J. 10, 346–362. doi: 10.1038/ismej.2015.116
Tedersoo, L., Bahram, M., Toots, M., Diédhiou, A. G., Henkel, T. W., Kjøller, R., et al. (2012). Towards global patterns in the diversity and community structure of ectomycorrhizal fungi. Mol. Ecol. 21, 4160–4170. doi: 10.1111/j.1365-294X.2012.05602.x
Toju, H., Tanabe, A. S., Yamamoto, S., and Sato, H. (2012). High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS One 7:e40863. doi: 10.1371/journal.pone.0040863
van der Linde, S., and Haller, S. (2013). Obtaining a spore free fungal community composition. Fungal Ecol. 6, 522–526. doi: 10.1016/j.funeco.2013.10.001
van der Linde, S., Suz, L. M., Orme, C. D. L., Cox, F., Andreae, H., Asi, E., et al. (2018). Environment and host as large-scale controls of ectomycorrhizal fungi. Nature 558, 243–248. doi: 10.1038/s41586-018-0189-9
Vellend, M. (2016). The Theory of Ecological Communities. Princeton, NJ: Princeton University Press.
Vincenot, L., and Selosse, M.-A. (2017). “Population biology and ecology of ectomycorrhizal fungi,” in Biogeography of Mycorrhizal Symbiosis Ecological Studies, ed. L. Tedersoo (Cham: Springer International Publishing), 39–59. doi: 10.1007/978-3-319-56363-3_2
Wright, E. (2020). DECIPHER: Tools for Curating, Analyzing, and Manipulating Biological Sequences. Bioconductor Version: Release (3.12). doi: 10.18129/B9.bioc.DECIPHER
Wutkowska, M., Vader, A., Mundra, S., Cooper, E. J., and Eidesen, P. B. (2019). Dead or alive; or does it really matter? Level of congruency between trophic modes in total and active fungal communities in high arctic soil. Front. Microbiol. 9:3243. doi: 10.3389/fmicb.2018.03243
Keywords: aboveground-belowground linkages, ectomycorrhizal fungi, fungal trophic mode, neighbor effects, rDNA, saprotrophic fungi, spatial community turnover
Citation: Runte GC, Smith AH, Moeller HV and Bogar LM (2021) Spheres of Influence: Host Tree Proximity and Soil Chemistry Shape rRNA, but Not DNA, Communities of Symbiotic and Free-Living Soil Fungi in a Mixed Hardwood-Conifer Forest. Front. Ecol. Evol. 9:641732. doi: 10.3389/fevo.2021.641732
Received: 14 December 2020; Accepted: 28 May 2021;
Published: 29 June 2021.
Edited by:
Guillaume Chomicki, The University of Sheffield, United KingdomReviewed by:
Roland Mrmeisse, UMR 7205 Institut de Systématique, Evolution, Biodiversité (ISYEB), FranceMatthew Edward Smith, University of Florida, United States
Copyright © 2021 Runte, Smith, Moeller and Bogar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel C. Runte, Z2FiZS5ydW50ZUBsaWZlc2NpLnVjc2IuZWR1