 Meiqing Yang1†
Meiqing Yang1† Guanglin He2,3,4†
Guanglin He2,3,4† Zheng Ren1
Zheng Ren1 Qiyan Wang1Yubo Liu1Hongling Zhang1Han Zhang1
Qiyan Wang1Yubo Liu1Hongling Zhang1Han Zhang1 Jing Chen1Jingyan Ji1
Jing Chen1Jingyan Ji1 Jing Zhao2,3
Jing Zhao2,3 Jianxin Guo2,3
Jianxin Guo2,3 Kongyang Zhu2,3
Kongyang Zhu2,3 Xiaomin Yang2,3
Xiaomin Yang2,3 Rui Wang2,3
Rui Wang2,3 Hao Ma2,3
Hao Ma2,3 Chuan-Chao Wang2,3,5,6*
Chuan-Chao Wang2,3,5,6* Jiang Huang1*
Jiang Huang1*- 1Department of Forensic Medicine, Guizhou Medical University, Guiyang, China
- 2Department of Anthropology and Ethnology, School of Sociology and Anthropology, Institute of Anthropology, Xiamen University, Xiamen, China
- 3State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, China
- 4Institute of Rare Diseases, West China Hospital of Sichuan University, Sichuan University, Chengdu, China
- 5State Key Laboratory of Marine Environmental Science, Xiamen University, Xiamen, China
- 6Institute of Artificial Intelligence, Xiamen University, Xiamen, China
Southern China was the original center of multiple ancestral populations related to modern Hmong-Mien, Tai-Kadai, Austroasiatic, and Austronesian people. More recent genetic surveys have focused on the fine-scale genetic structure and admixture history of southern Chinese populations, but the genetic formation and diversification of Hmong-Mien speakers are far from clear due to the sparse genetic sampling. Here, we reported nearly 700,000 single-nucleotide polymorphisms (SNPs) data from 130 Guizhou Miao and Yao individuals. We used principal component analysis, ADMIXTURE, f-statistics, qpAdm, phylogenetic tree, fineSTRUCTURE, and ALDER to explore the fine-scale population genetic structure and admixture pattern of Hmong-Mien people. The sharing allele patterns showed that our studied populations had a strong genetic affinity with ancient and modern groups from southern and southeastern East Asia. We identified one unique ancestry component maximized in Yao people, which widely existed in other Hmong-Mien-speaking populations in southern China and Southeast Asia and ancient samples of Guangxi. Guizhou Hmong-Mien speakers harbored the dominant proportions of ancestry related to southern indigenous East Asians and minor proportions of northern ancestry related to Yellow River farmers, suggesting the possibility of genetic admixture between Hmong-Mien people and recent southward Sino-Tibetan-related populations. Furthermore, we found a genetic substructure among geographically different Miao and Yao people in Leishan and Songtao. The Yao and Miao people in Leishan harbored more southern East Asian ancestry, but Miao in Songtao received more northern East Asian genetic influence. We observed high mtDNA but low Y-chromosome diversity in studied Hmong-Mien groups, supporting the role of sex-specific residence in influencing human genetic variation. Our data provide valuable clues for further exploring population dynamics in southern China.
Introduction
With the rapid development of molecular biology technologies and computational techniques, genetic history reconstruction on a fine scale of the modern and ancient populations in East Asia has been increasing. Previous studies have suggested that after the separation between northern and southern East Asians, southern East Asians were further divided into two genetic lineages geographically corresponding to the inland and coastal regions (Huang et al., 2020; Yang et al., 2020). Northern China is suggested to be the original center of modern Sino-Tibetan-speaking populations, while southern China was the homeland of multiple populations, including ancestral populations related to modern Hmong-Mien, Tai-Kadai, Austroasiatic, and Austronesian languages (Guo et al., 2019; Xu et al., 2019; Ning et al., 2020; Yang et al., 2020; Sun et al., 2021; Wang et al., 2021a; Chen et al., 2022; Liu et al., 2022). Archeological findings have demonstrated that Pre-Holocene hunter-gatherers near the middle and lower branches of the Yangtze River started domesticating rice more than 10,000 years ago, which contributed to the rise of southern Chinese civilizations. The ancient southern Chinese rice farmers finally have developed into several ancestral populations for different linguistic families in East and Southeast Asia, including Hmong-Mien, Austroasiatic, Tai-Kadai, and Austronesian (Zhao, 2010). Indeed, Recent genetic studies based on genome-wide data from modern (Huang et al., 2018; Ren et al., 2019; He et al., 2020, 2021a,2021b,2022a,2022b; Lu et al., 2020; Bin et al., 2021; Ma et al., 2021; Wang et al., 2022) and ancient populations (Yang et al., 2020; Wang et al., 2021c) have demonstrated complex genetic structure in southern China. Wang et al. (2022) have identified the genetic substructure between inland and coastal Tai-Kadai people. Other unique genetic ancestries were identified in geographically isolated island or boundary populations, such as Hainan Hlai, Fujian Tanka, and Yunnan Blang and Wa people (He et al., 2020, 2021c; Guo et al., 2021).
The Hmong-Mien-speaking populations are widely distributed in southern China and Southeast Asia. The Hmong-speaking and Mien-speaking groups are referred to as Miao and Yao, respectively (Schliesinger, 2000). There are multiple hypotheses about the origin and migration history of the proto-Hmong-Mien speakers. One hypothesis from the linguistic perspective stated that the Hmong-Mien-speaking groups had a common ancestor with Sino-Tibetan speakers at least 17,000–10,000 years ago. While archeologists and historians have suggested that the proto-Hmong-Mien populations might be related to the Neolithic cultures in the middle reach of the Yangtze River in southern China, including the Daxi Culture (6,500–5,300 BP) and the Qujialing Culture (5,000–4,600 BP) (Fei, 1999). In a sense, the study of ancient DNA from the Daxi Culture and Qujialing Culture would be of great importance to verify the possible affiliations of Hmong-Mien populations and these Neolithic cultures. Subsequently, some Proto-Hmong-Mien speakers expanded to the north and communicated with farming populations at the upper and middle of the Yellow River. Later, they migrated southward and became the ancestors of the present-day Miao. Some of these people migrated to the south and southeast and became the main source of the present-day Yao and She populations. After the Ming and Qing Dynasties (650–100 ybp), they gradually migrated southwest to Yunnan provinces and Southeast Asia, forming the present distribution pattern of Hmong-Mien speakers in southern China and Southeast Asia (Handel, 2012).
Previous genetic studies of Hmong-Mien speakers had focused on autosomal STR and InDel, paternal Y-chromosome, and maternal mitochondria DNA (mtDNA) lineage, which were limited in resolution to understand the fine genetic structure and admixture compared to the genome-wide data (Ren et al., 2018; Zhang et al., 2019; Tang et al., 2020). A previous study focused on mtDNA demonstrated that the maternal haplogroups of Hmong-Mien speakers were predominant in southern East Asia, including B, R9, N9a, and M7, suggesting their general southern China origin. Meantime, they indicated there was some extent of genetic differences between Miao and Yao populations (Wen et al., 2005; Lipson et al., 2018). In Southeast Asia, there were also Hmong-Mien-speaking people who migrated from southern China based on genetic evidence and cultural similarities (Liu et al., 2022). Thailand’s Hmong-Mien-speaking populations had high frequencies of northeast Asian paternal lineages but lower frequencies of Southeast Asian maternal lineages, which furtherly suggested that the ancestors of the Hmong-Mien groups had a genetic affinity with northeast Asians (Kutanan et al., 2020). Recent genetic studies also have revealed that Hmong and Mien had an additional genomic affinity to Tibeto-Burman and Tai-Kadai speakers (Huang et al., 2020). Simultaneously, the study also indicated a founding population called Pahng living in Hunan province from China substantially contributing to present-day Hmong-Mien speakers (Xia et al., 2019). Although more genetic surveys were conducted from South China, higher-density genome-wide SNP data from Hmong-Mien-speaking populations were still relatively limited (Luo et al., 2021; Liu et al., 2022; Tan et al., 2022). Several important questions of Hmong-Mien people remain to be solved in genetics: What’s the fine-scale genetic structure of Hmong-Mien people? Are there some genetic differences between geographically or ethnically different Hmong-Mien people (Miao and Yao)? What’s the genetic relationship between Hmong-Mien people and other geographically close modern and ancient Southern East Asians?
At present, Hmong-Mien speakers mainly dwell in Central-South and Southwest China, as well as sporadic distribution in mainland Southeast Asia after prehistoric and historic complex migration events. The heartland of Hmong-Mien-speaking populations was considered in the present-day Guizhou provinces in southwestern China. Guizhou province is an appropriate field to depict a fine-scale genetic landscape of Hmong-Mien speakers. However, no population genetic survey of Hmong-Mien speakers based on high-density SNPs was conducted systematically in the region. Thus, we conducted the population genetic survey based on the high-density genome-wide SNP data of the Guizhou Miao and Yao people to explore their fine-scale population structure and admixture history. We also combined our data with previously published modern and ancient data to investigate the special genetic substructure between Miao and Yao populations, as well as dissect the genetic relationships between Guizhou Miao and Yao with their neighboring populations. Furthermore, we used multiple models to explore the ancestral admixture history of the Hmong-Mien people.
Materials and Methods
Sample Collection, Genotyping, and Quality Control
We sampled the saliva of present-day Miao and Yao individuals from Guizhou Province in southwest China from three regions, namely, Libo, Leishan, and Songtao. The geographical coordinates of the sampling locations are shown in Supplementary Figure 1. We collected the samples from 58 Yao individuals from Libo (Yao_Libo), which was further divided into QingYao_Libo (n = 20), BaikuYao_Libo (n = 20), and ChangshanYao_Libo (n = 18) based on the difference of their cultures and dressing. We collected 77 Miao samples from Leishan (Miao_Leisan, n = 50) and Songtao (Miao_Songtao, n = 22). The study was reviewed and approved by the Medical Ethics Committee of Guizhou Medical University (approval number: 2019074) and Xiamen University (approval number: XDYX2019009). Besides, the protocol was following the recommendations provided by the revised Helsinki Declaration of 2000. The participants provided their written informed consent to participate in this study. These samples were collected randomly from unrelated participants whose parents and grandparents are indigenous people and have had a non-consanguineous marriage of the same ethnical group for at least three generations. Human genomic DNA was extracted using the PureLink Genomic DNA kit (Thermo Fisher Scientific) and genotyped by the Illumina WeGene arrays that contained about 692,462 SNPs. The quality control was conducted PLINK1.9 with the option “–missing 0.05 –HWE 0.0001” to calculate SNP calling rate. After SNP calling, SNPs that failed a missing threshold of 0.05 and an HWE test with an alpha level of 0.0001 were removed. The rates of sample missing data ranged from 0.001079 to 0.008514. We provided the sample missing data report in Supplementary Table 1A.
Relatedness Analysis and Data Merging
Before merging the data, we initially performed KING 2 (Manichaikul et al., 2010) to check family relationships by estimating kinship coefficients. We merged our 130 samples with previously published ancient and present-day populations from Human Origins Dataset (HO) and the 1240 K dataset from the Allen Ancient DNA Resource (AADR)1 by mergeit from EIGENSOFT (Patterson et al., 2006) with the strandcheck parameter setting Yes to remove the AT/CG SNPs. Human Origins Dataset (HO) and the 1240 K dataset both cover high-density SNPs, with 373,933 SNPs and 119,349 SNPs, respectively. And the HO dataset harbor more present-day populations than the 1240 K dataset, 1240 K dataset harbor more ancient populations than the HO dataset. We also added other recently published ancient East Asian populations into our analysis (Mccoll et al., 2018; Ning et al., 2020; Wang et al., 2021c). We finally generated two combined datasets used in subsequent analysis covering 72,532 (HO dataset) and 193,846 (1240 K dataset) SNPs. For principal component analysis (PCA) and Fst analysis, we used populations from the HO dataset. For ADMIXTURE, f-statistics, qpAdm, ALDER, and TREEMIX, we used populations from the 1240 K dataset and removed individuals within three levels of kinship among included studied individuals.
Principal Component Analysis
We carried out PCA using the smartpca program of the EIGENSOFT package (Patterson et al., 2006), projecting ancient samples onto axes generated by present-day samples. We used default parameters with numoutlieriter: 0 and lsqproject: YES for all present-day populations. The PCA focused on the Sino-Tibetan, Austronesian, Austroasiatic, Tai-Kadai, and Hmong-Mien speakers and ancients from East and Southeast Asia.
Admixture Analysis
To dissect the ancestry coefficients of the newly reported groups, we conducted ADMIXTURE (Alexander et al., 2009), a model-based maximum likelihood (ML) clustering algorithm approach, to estimate the ancestry of individuals and determine population structure through cross-validation. Given the strong linkage disequilibrium (LD) of SNPs, we used Plink (Chang et al., 2015) to prune them with the parameters “-indep-pairwise 200 25 0.4” before analyzing the admixture. We re-run ADMIXTURE 100 times with default 10-fold cross-validation (–CV = 10), varying the number of ancestral populations from K = 2 to K = 10 in bootstraps with different random seeds. Then, we chose the best run according to the highest log-likelihood. We used cross-validation to identify an “optimal” number of clusters that will be the lowest error values. Then, we implemented the AncestryPainter (Feng et al., 2018) to display population ancestry.
Neighbor-Joining Phylogenetic
We calculated the Fst indexes using the smartpca program of the EIGENSOFT package with default parameters (inbreed: YES; and fstonly: YES). Neighbor-Joining (N-J) phylogenetic relationship was constructed using MEGA4.0 (Tamura et al., 2007) based on the Fst value.
Admixture and Outgroup f3-Statistics
We employed the qp3pop program in ADMIXTOOLS to compute f3-statistics with default parameters (Patterson et al., 2012). We computed outgroup f3 -statistic values in the form of f3(X, Y; Mbuti), which quantifies shared genetic drift between populations X and Y since their separation from an outgroup Mbuti from Africa. Then, we used admixture-f3(X, Y; Targets) to evaluate the admixture signal in the newly studied Hmong-Mien speakers with different reference populations from East and Southeast Asia, where a significant negative-f3 value with a Z-score smaller than −3 denoted that the targeted population was an admixture between two parental populations of the first two positions (X and Y).
f4-Statistics
We employed the qpDstat program in ADMIXTOOLS to compute f4-statistics with default parameters (Patterson et al., 2012). The f4-statistics were calculated in the form of f4(Mbuti, Targets; X, Y) to test whether the newly targeted population shares excess gene flow with the X population compared with the Y population or shares more alleles with Y compared with X, where a significant negative Z-score value smaller than 3 denoted that the target populations shared more allele with X population comparing with Y populations. The f4-statistics with the form of f4(Mbuti, X; Target1, Target2) tested the genetic difference among the newly studied populations.
Maximum Likelihood Tree
To further explore the relationship between the newly studied group and modern southern Chinese and Southeast Asian populations, we constructed an ML tree via the TreeMix with the migration events from 1 to 10 to generate the admixture graph (Pickrell and Pritchard, 2012).
Admixture Date
We applied ALDER v.1.02 (Loh et al., 2013) to infer the possible date of admixture based on the exponential LD decay with considering a generation time of 29 years. The newly studied groups were included as the “admixpop” (admixed population) and the remaining present-day East and Southeast Asian populations dataset were used as the “refpops” (reference populations).
QpAdm Admixture Coefficient Estimation
We estimated the admixture proportion of the ancestral proportions of Hmong-Mien people using qpAdm implemented in ADMIXTOOLS (Patterson et al., 2012). We used one additional parameter of “allsnps: YES.” The right (outgroup) populations included Mbuti, Yana_UP, Ust_Ishim, Kolyma_M, Tianyuan, Bianbian, Mongolia_N_East, Bianbian, Liangdao, and Longlin.
FineSTRUCTURE Analysis Based on the Sharing Haplotypes
We phased 1,049 genomes from 47 populations using the SHAPEIT software with the default parameters (Browning et al., 2021) and calculated the sharing haplotypes using the ChromoPainterv2 software (Lawson et al., 2012). We explored the fine-scale populations based on the co-ancestry matrix using fineSTRUCTURE (version 4.0) (Lawson et al., 2012).
Y-Chromosomal and mtDNA Haplogroup Assignment
We assigned the Y-chromosomal haplogroups following the International Society of Genetic Genealogy (ISOGG2) and mtDNA haplogroups with mtDNA tree Build 173 (Van Oven and Kayser, 2009).
Results
The Overall Pattern of Population Genetics Structure
We initially performed a PCA among 658 individuals, which included 102 Hmong-Mien speakers, 76 Tai-Kadai speakers, 51 Austronesian people, 50 Austroasiatic people, 164 Sinitic-Tibetan people, 81 Tungusic speakers, 42 ethnically unrecognized individuals, and 92 ancients from East and Southeast Asia (Supplementary Table 1B). Linguistically and geographically different people formed their unique genetic clusters (Figure 1A) and the clustering patterns correspond well with their current geographical locations and linguistics classification (Figure 1B) in the PC1-PC2 space. The newly studied Yao_Libo and Miao_Leishan formed the “Hmong-Mien Cline” along PC1, which has not been identified previously. Miao_Songtao was located at the intersection position between the genetic clines of Hmong-Mien and Sinitic groups, suggesting that Miao_Songtao people harbored more genetic influence from Han Chinese-related populations compared with Yao people. On the PC2 axis, Austronesian-, Austroasiatic-, Tai- Kadai-, and Sino-Tibetan-speaking groups could be distinguished and showed their specific cluster patterns. Interestingly, we identified a close genetic affinity between the ∼500-year-old GaoHuaHua sample from southern China and Yao people in the PCA, suggesting the long-term genetic stability in Yun-Gui Plateau and surrounding regions.

Figure 1. Genetic structure and population affinity of Hmong-Mien-speaking populations and their neighbors. (A) Principal component analysis among modern and ancient East Asians. Ancient individuals were projected onto modern backgrounds. (B) Model-based ADMIXTURE results among Chinese modern and ancient populations. (C) The geographical position of our studied populations and referenced populations. (D) Sharing genetic drift between Hmong-Mien people and other reference populations.
We further separated the genotype data into discrete ancestral components at an individual level using model-based clustering ADMIXTURE analysis. We run admixture from K = 2 to K = 10 and found the result at K = 5 with the lowest cross-validation error. At K = 5 (Figure 1C), ADMIXTURE recapitulated the previous observation of five ancestral components in East and Southeast populations. It is interesting that BaikuYao almost only harbored a pink ancestry component, which was also detected in Southeast Asian and East Asian populations. QingYao and ChangshanYao also possessed a significant proportion of pink ancestry components and a little purple ancestry component. The purple ancestry component was present in present-day Austronesian-, Austroasiatic-, and Tai-Kadai-speaking populations and ancient samples from southeastern and southern East Asia, as well as Southeast Asia. Leishan Miao could be approximately modeled as an admixture of a relatively high proportion of pink ancestry component and some purple, yellow, and green ancestry components, which is consistent with their intermediated position in the PCA. The green ancestry component was enriched in present-day population speaking Sino-Tibetan and ancient Neolithic people from the Yellow River Basin. The genetic profile of Songtao Miao was similar to Tai-Kadai-speaking groups, which were approximately modeled as an admixture of green, purple, pink, yellow, and litter blue components existing in the northern East Asians.
The Genetic Affinity Between the Studied Hmong-Mien Population and East or Southeast Asian Populations
We followingly calculated pairwise Fst between groups using smartpca and then reconstructed phylogenetic trees to explore the phylogenetic relationship between Hmong-Mien people and other Asians (Figure 2A). The N-J tree further showed that the genetic structure was consistent with the linguistic classification and geographical locations, as suggested by the PCA and ADMIXTURE clustering patterns. The newly studied Miao and Yao had a close relationship with the geographically adjacent Miao, as well as Vietnamese Hmong and PaThen who belong to the Hmong-Mien-language family. In addition, Miao_Songtao gathered together with the referenced Miao from southern China, but three Yao populations (BaikuYao_Libo, ChangshanYao_Libo, and QingYao_Libo) formed a tight cluster together showing a strong genetic affinity.
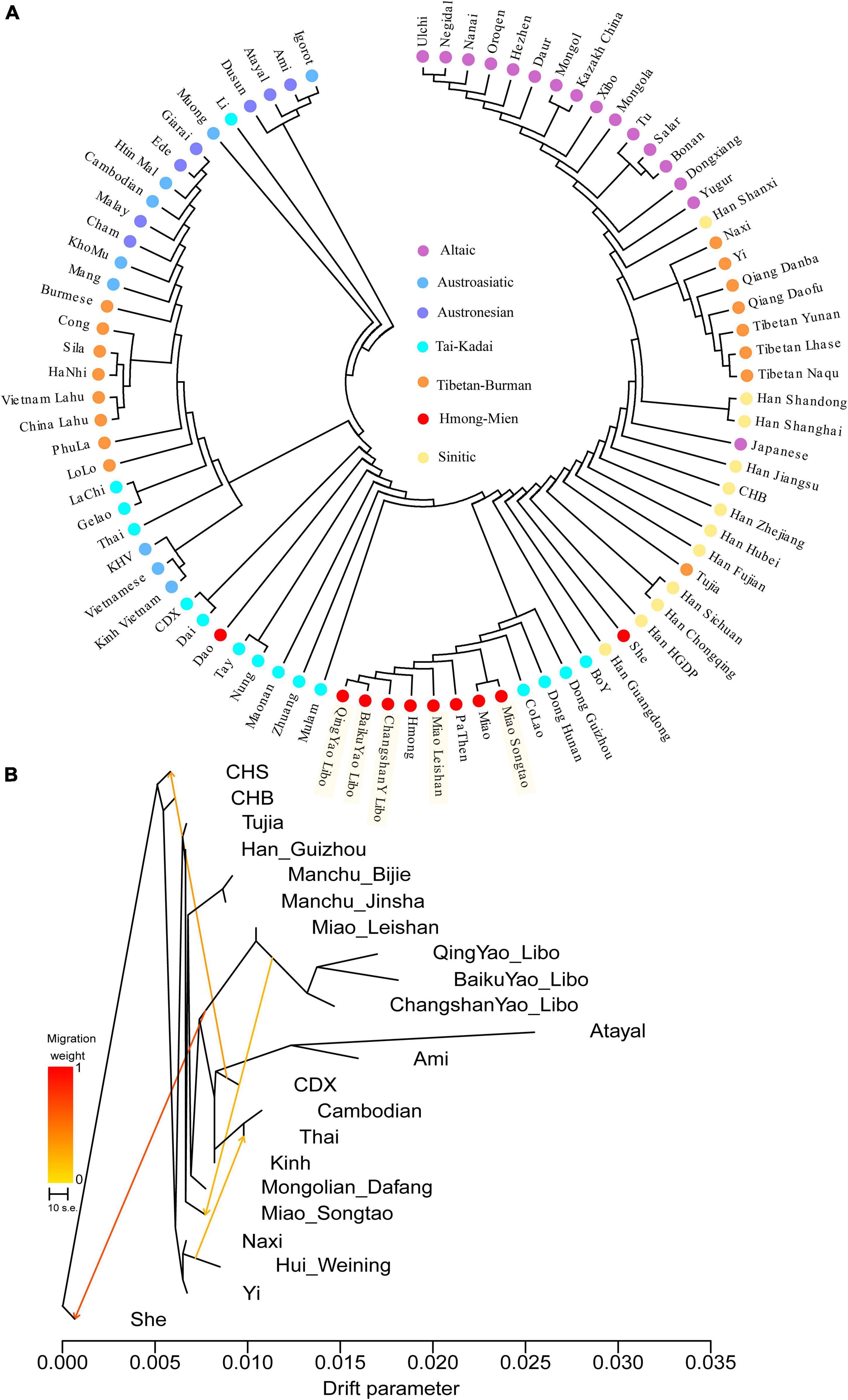
Figure 2. Phylogenetic relationships between our studied Hmong-Mien-speaking populations and other reference populations. Mega-based phylogenetic relationships without migration events (A) and TreeMix-based phylogenies with migration events (B).
Relatedness coefficient was applied to exclude the individuals within the three levels of kinship via KING (Supplementary Table 2), and we remained 103 individuals in the following formal f-statistics. We used the allele frequency-based method of the outgroup- f3(X, Studied; Mbuti) to measure the shared genetic drift. The results of outgroup-f3 (Figure 1D) showed that our targeted groups shared more genetic drift with the ∼500-year-old GaoHuaHua, followed by the Hmong-Mien populations Miao and She, Austronesian populations Ami and Atayal in southeastern East Asia, and the ancient people from Yellow River basin farmers (YR_LN/MN/BAIA).
We also performed f4(Mbuti, Studied; Ancients from Southern China, References) to test whether other populations shared any excess alleles with our newly studied populations when compared with the ancients from southern China (Supplementary Table 3). The ancients from southern China included BaBanQinCen and GaoHuaHua to represent the modern Tai-Kadai and Hmong-Mien groups, respectively. We found that our studied groups shared more alleles with ancient GaoHuaHua compared to the ancients or present-day groups in northern and southeast East Asia; shared more alleles with ancient BaBanQinCen compared to the ancients or present-day groups in northern China; shared more drift with present-day groups Ami compared with ancients or present-day groups in East and Southeast Asia; harbored more gene flow from ancient groups of Yellow River Basin compared with populations in northeastern China and Southeast Asia. In general, the results of PCA, ADMIXTURE, N-J phylogenetic tree, and f-statistic revealed that the newly studied Hmong-Mien populations had the closest relationship with the ancient GaoHuaHua and BaBanQinCen, and Austronesian-speaking Ami.
Genetic Substructure Among Guizhou Hmong-Mien-Speaking Populations
We additionally calculated f4-statistics in the form of f4(Mbuti, references; studied1, studied2) to explore whether there is a genetic substructure in studied Hmong-Mien-speaking populations (Supplementary Table 4). We found that Yao_Libo/Miao_Leishan shared more alleles with populations from southern China and Southeast Asia than Miao_Songtao with the Z-scores < –3 in the form f4(southern China/Southeast Asian, Mbuti; Yao/Miao_Leishan, Miao_Songtao). While Miao_Songtao harbored more Yellow River farmer-related ancestry than Yao/Miao_Leishan with statistically significant Z-scores (Z > 3) in the f4(Yellow River Basin farmers, Mbuti; Yao/Miao_Leishan, Miao_Songtao). These results suggested that there was no genetic substructure among three geographically different Yao populations and between Leishan Miao and Libo Yao. But there was a genetic difference between Songtao Miao and Leishan Miao.
We employed the TreeMix v1.13 software with migration edge weight to investigate the pattern of population splits and mixtures among selected East and Southeast Asian populations (Figure 2B). When we set the values of migration edge weight to four, the three Yao and Leishan Miao populations clustered together, and Songtao Miao clustered closely to Tai-Kadai and Austroasiatic speakers and populations from the same province. This fitted model highlighted the gene flow from Yao to Songtao Miao and She populations, suggesting that Yao and Leishan Miao could be used as the potential genetic source for the reconstruction of other Hmong-Mien-speaking groups.
Admixture Signatures and Admixture Dates
We performed admixture-f3 in the form (X, Y; Targeted) to detect admixture signals using all possible pairs of the source groups as populations X and Y (Supplementary Table 5). There were no reliable negative values of the Z-score (Z < –3) observed except for using Leishan Miao as the targeted population. Ancient GaoHuaHua from Guangxi combined with present-day Tai-Kadai (CDX)- and Sino-Tibetan (Han, Tujia, Naxi, Yi)-speaking populations, and Guizhou aborigines (Guizhou Han, Chuanqing, and Hui) living in southern and southeastern East Asia or ancient populations from Yellow River basin could generate the statistically negative f3 results for using Leishan Miao as the target. The positive result in f3 (X, Y; Miao_Songtao/Yao_Libo) suggested that Libo Yao and Songtao Miao might share a high degree of population-specific drift.
Estimating admixture time was important to understand the history of the admixed population. We investigated the approximate admixture date with pairwise modern populations from East and Southeast Asia based on the decay of admixture-induced linkage disequilibrium (Supplementary Table 6). We observed the estimated earliest admixture event that potentially gave rise to the newly studied Yao likely occurred 187 generations ago about 5,000 BP using Naxi and She as reference sources, which corresponds well to the time when the Qujialing culture was formed in the middle reaches of the Yangtze River. The earliest admixture of the Miao_Leishan occurred about 107 generations ago about 4,000 BP using southern Han and Chuanqing as reference sources. We note that the more accurate spatiotemporal landscape could be further validated directly via ancient DNA data in the future.
Two-Way Admixture Models With Multiple Ancestral Populations
We further applied qpAdm to estimate admixture proportions of the predefined ancestral populations. Considering both the south-north admixture signals observed in the newly studied groups, we used the Neolithic farmers from the Yellow River basin as the northern source and ancient populations from Fujian and Guangxi as the southeastern and southern sources, respectively, to estimate admixture proportions. In the two-way admixture models (Supplementary Table 7), we found that the Yao_Libo and Miao_Leishan possessed a higher proportion of ancestry related to Austronesian- (0.506–0.627) or proto-Tai-Kadai-speaking groups (0.697–0.830) from southern and southeastern China than the Neolithic farmers from the Yellow River basin (0.17–0.494), indicating that the Yao_Libo and Miao_Leishan populations were more affected by Tai_Kadai and Austronesian-speaking groups than Sino-Tibetan groups. Furthermore, we found Atayal from southeastern China-related ancestry existed in the Miao_Songtao group with a small percentage, while ancestry related to the later Neolithic farmers from the Yellow River reached a large percentage, suggesting that Miao_Songtao was less affected by the present-day groups from coastal southeastern East Asia. But we found a 1,300-year ancient LaCen sample from southern China contributed a higher percentage of ancestry to Miao_Songtao than the later Neolithic farmers from the Yellow River, suggesting that Miao_Songtao received more genetic influence from populations of inland southern East Asia.
Fine-Scale Population Structure Based on the Sharing Haplotype Confirmed the Unique Demographic History of Hmong-Mien People
To make a population comparison with modern and ancient East and Southeast Asian reference populations, we merged our newly generated data with the previously published population data genotyped via 1240-targeted sequencing or Human Origins array. There were 193,846 SNPs left in the 1240 K dataset and 72,532 SNPs left in the Human Origins dataset after the merging. To further validate the robustness and reliability of our identified specific genetic structure with a larger number of SNPs, we merged our data with our previously collected Chinese population data genotyped using the same chip (Wang et al., 2020, 2021b; Chen et al., 2021; He et al., 2021c,d; Liu et al., 2021; Yan et al., 2021; Yao et al., 2021). A total of 1,049 individuals from 47 populations were used in the PCA, ADMIXTURE, and fineSTRUCTURE analyses. Consistent with the results obtained based on the lower-density SNP data, we also observed one unique genetic cline of Hmong-Mien-speaking populations along PC2, which included our newly studied Miao and Yao and previously published Miao from Sichuan and Gejia and Dongjia people from Guizhou (Figure 3A). We also found significant genetic differences between the Guizhou Hmong-Mien people and Chongqing Miao. Model-based ADMIXTURE results with five redefined ancestral sources also identified one Hmong-Mien-specific ancestry component that widely existed in Yao, Miao, Gejia, Dongjia, and Xijia people (Figure 3B). We reconstructed the fine-scale population cluster patterns based on the pairwise coincidence matrix and found Hmong-Mien people from Guizhou formed one unique branch, consisting of Yao, Miao, Gejia, Dongjia, and Xijia people (Figure 4). In summary, our results based on the higher-density SNP dataset and sharing haplotypes further confirmed the specific genetic structure of Guizhou Hmong-Mien people.
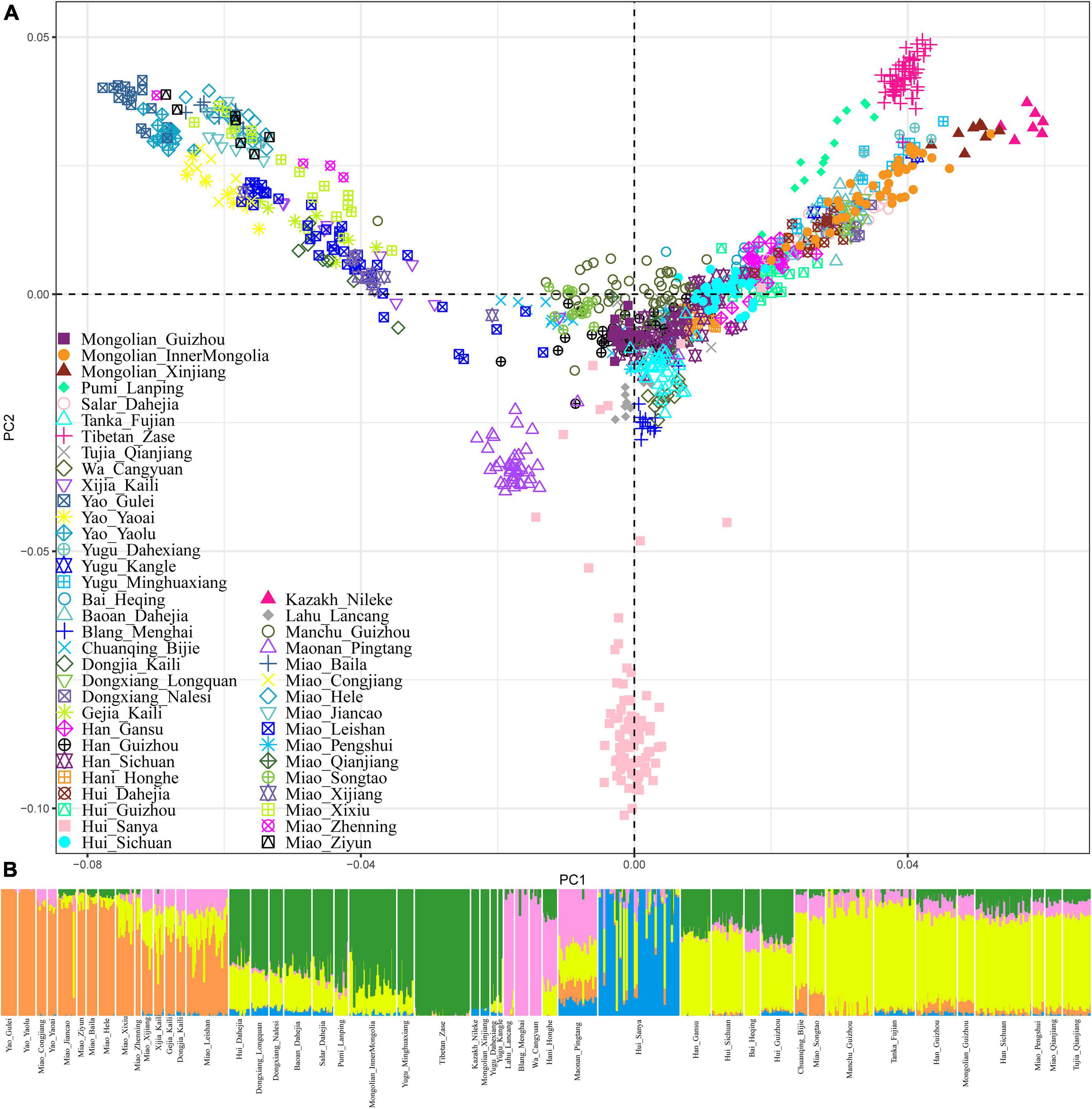
Figure 3. Genetic structure among 1,049 individuals from 47 ethnolinguistically different populations. (A) Principal component analysis results based on the high-density SNP data (533,938 SNPs). (B) Model-based ADMIXTURE results with five predefined ancestral sources.
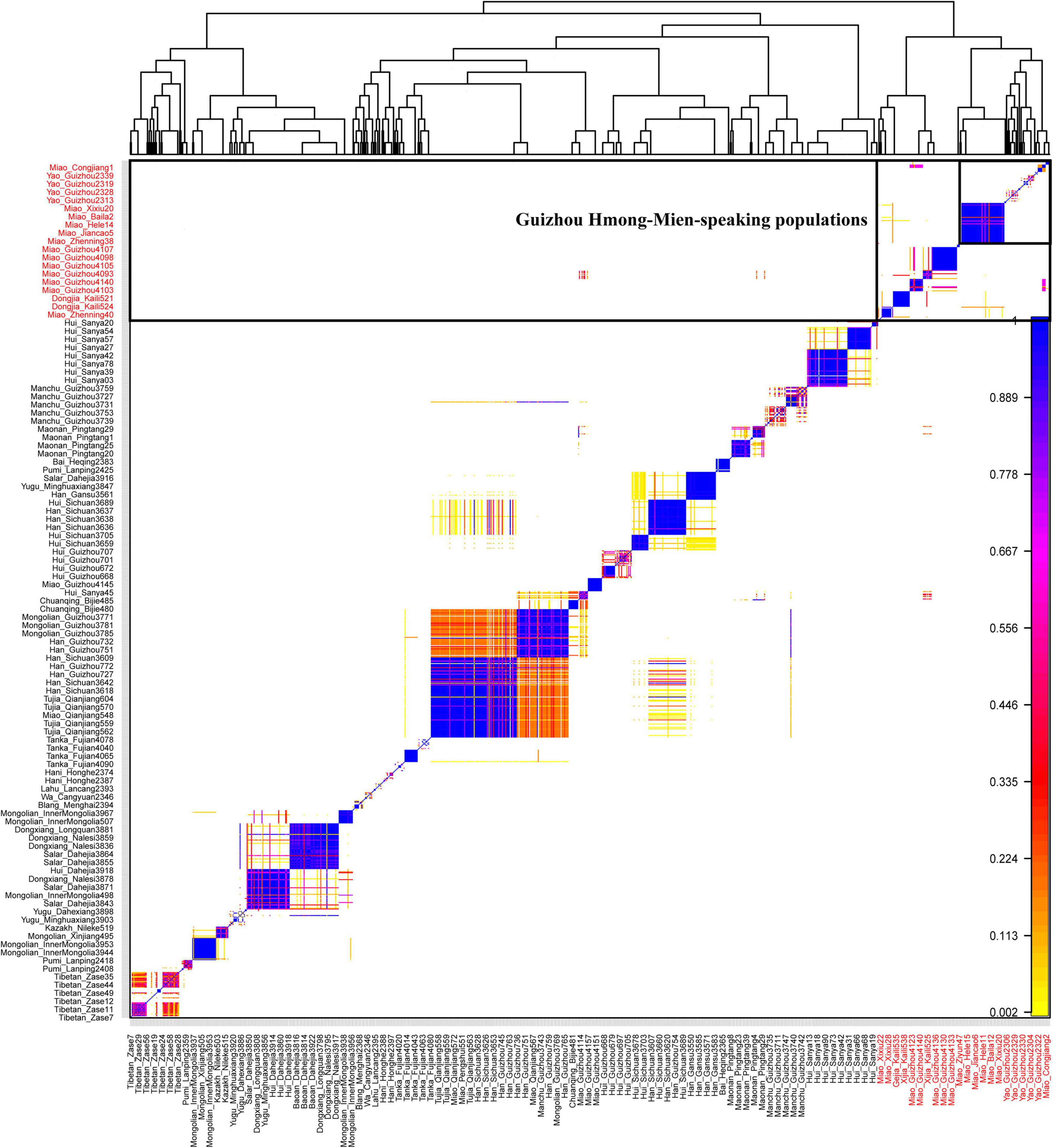
Figure 4. Fine-scale population structure based on the pairwise coincidence matrix among 1,049 Chinese individuals. Newly focused Hmong-Mien-speaking population is marked with red color.
Paternal and Maternal Genetic Profile
Y-chromosome and mtDNA are powerful tools in reconstructing the evolutionary history of human populations (Wallace et al., 1999; Cavalli-Sforza and Feldman, 2003). We successfully classified uniparental Y-chromosome and mtDNA haplogroups for 62 males and 68 females (Supplementary Table 8). The maternal lineages of Yao_Libo, Miao_Leishan, and Miao_Songtao were diverse with the major lineages belonging to the haplogroups of A, B5, C4, D4, D5, D6, F1, F3, M7, M8, N, R9, and R1. In the Yao_Libo population, matrilineal subhaplogroup D4 was the most dominant maternal haplogroup with a frequency of 36.21%, followed by B5 (27.59%). Haplogroup B4 was commonly found in the Miao_Leishan population with a frequency of 20%, followed by the D4 and F1 (14%). In the Miao_Songtao population, A, D4, and F1 were detected at frequencies of 22.73%, 18.18%, and 18.18%, respectively. In total, the haplogroup D4 appeared in the studied Hmong-Mien-speaking populations with the highest frequency.
From the Y-chromosome perspective, the Yao_Libo population has one dominant haplogroup O2a2a1a2a1a2 with a frequency of 90%. The haplogroup O2a2a1a2a1a2 was also the most prevailing lineage in Miao_Leishan (48.28%), and O1a1a was the most dominant lineage in Miao_Songtao (66.67%). The newly studied Hmong-Mien populations harbored O1a and O2a, which were special haplogroups in Austronesian and Tai-Kadai groups. Previous studies revealed that O2a2a1a2a1a2 (O-N5) was most prevalent in Hmong-Mien populations, while the coalescent ages were ∼2.45 kya (Kutanan et al., 2020). The newly studied populations Yao_Libo and Miao_Leishan possessed high frequencies of haplogroup O2a2a1a2a1a2, suggesting that they were typical Hmong-Mien groups, while Songtao Miao showed an affinity with Tai-Kadai populations with high frequencies of haplogroup O1a1a (Deng et al., 2004).
We observed high mtDNA but low Y-chromosome diversity in studied Hmong-Mien groups. One possible explanation is the higher rate of female vs. male migration due to the widespread phenomenon of patrilocality (Burton et al., 1996; Seielstad et al., 1998), in which males stay in their birthplace and females move to their mate’s residence after marriage. The sex-specific migration has shaped the genetic profiles of mtDNA and Y-chromosome in Hmong-Mien populations.
Discussion
Hmong-Mien is a language family spoken mainly in southern China and Southeast Asia. The origin and dispersal of Hmong-Mien languages and populations are far from clear due to sparse genetic sampling. We here genotyped and analyzed genome-wide SNP data of 130 individuals from Hmong-Mien-speaking populations collected from Guizhou province in southwest China to shed more light on the genetic formation and diversification of Hmong-Mien people.
The Unique Genetic Profile of Hmong-Mien Speakers
We observed a unique genetic profile of the newly studied Hmong-Mien speakers compared with other East and Southeast Asians. The Hmong-Mien-speaking populations in Guizhou did not overlap with other populations but formed a specific genetic cline with Hmong people from Southeast Asia in the PCA plots. We also observed a unique ancestry component in Hmong-Mien-related ancient samples in the ADMIXTURE analysis. The sharing haplotype also detected the Hmong-Mien people in Guizhou had a special genetic structure similar to the Hmong-Mien population in Chongqing, Guangxi, and Hunan in previous studies from southern China.
The Genetic Affinity With Southern East Asians
There are two main hypotheses about the origin and migration history of Hmong-Mien speakers. One hypothesis from the linguistic perspective stated that the Hmong-Mien-speaking groups had a common ancestor with Sino-Tibetan speakers. While the other hypothesis from archeology and history suggested that the proto-Hmong-Mien populations might be related to the Neolithic cultures in the middle reach of the Yangtze River in southern China, including the Daxi Culture (6,500–5,300 BP) and the Qujialing Culture (5,000–4,600 BP) (Fei, 1999). Although we do not have ancient DNA from the Daxi Culture and Qujialing Culture, our results supported that the newly studied Hmong-Mien groups were genetically closely related to populations from southern China. For example, the quantitative analyses via f-statistics and qpAdm further confirmed that the newly studied Yao and Leishan Miao have a strong affinity with Guizhou aborigines, Hmong-Mien, and Ami-related populations. Songtao Miao has a strong affinity with Guizhou aborigines, Tai-Kadai, and Austronesian-related peoples. We also detected a minor genetic influence from the Neolithic Yellow River farmers in Hmong-Mien speakers, which we suspected was most likely introduced during the southward migration of Sino-Tibetan populations.
The Genetic Substructure of Hmong-Mien Speakers
We found that Libo Yao groups (ChangshanYao, QingYao, and BaikuYao) in Guizhou were genetically homogenous, which is consistent with their cultural similarity. While the Miao_Leishan and Yao_Libo also were genetically homogenous, there was a genetic substructure within the Miao groups. The f-statistics showed that the different levels of the northern East Asian-related gene flow might lead to the heterogeneity between Miao_Songtao and Miao_Leishan. Furthermore, we confirmed that the newly studied Yao_Libo groups were more suitable to be the representative Hmong-Mien ancestry than the ∼500-year-old GaoHuaHua.
Conclusion
We presented the first batch of genome-wide SNP data of Hmong-Mien-speaking Miao and Yao from Guizhou province, southwestern China. We identified one unique genetic ancestry maximized in our studied Hmong-Mien speakers, which was different from the previously genetically documented “Sino-Tibetan” cline in northern China and “southern China and Southeast Asian” cluster in southern China and Southeast Asia. Guizhou Yao and Miao people not only harbored genetic influence from ancient southern East Asians but also absorbed additional gene fluxes from Sino-Tibetan-related ancestry. Our results suggested that there was a genetic substructure between Miao_Leishan and Miao_Songtao people, in which Han-related ancestry has greatly influenced the gene pool of the Songtao Miao but the Austronesian and Tai-Kadai-related ancestry contributed more to Yao and Leishan Miao. Importantly, we identified the stronger genetic affinity between Hmong-Mien-speaking Yao and ∼500-year-old GaoHuaHua sample, suggesting a long-term genetic stability in the Yun-Gui Plateau and surrounding regions. We observed high mtDNA but low Y-chromosome diversity in studied Hmong-Mien groups, which was likely caused by the higher migration rate of females in practicing patrilocal residence.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://doi.org/10.5281/zenodo.5821037, doi: 10.5281/zenodo.5821037.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Guizhou Medical University (approval number: 2019074) and Xiamen University (Approval Number: XDYX2019009). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
C-CW and JH designed this study. MY and C-CW wrote the manuscript. ZR, QW, YL, HoZ, HaZ, JC, JJ, and JH collected the samples and conducted the experiment. MY, JZ, GH, JG, KZ, XY, RW, HM, and C-CW analyzed the data. All authors reviewed the manuscript.
Funding
This study was funded by the Guizhou Province Education Department, Characteristic Region Project, Qian Education KY No. (2021)065, the Guizhou Province Engineering Technology Research Center Project [Qian High-Tech of Development and Reform Commission No. (2016)1345], the Guizhou Scientific Support Project [Qian Science Support (2019) 2825], the Guizhou “Hundred” innovative talents project [Qian Science Talent Platform (2020) 6012], the Guizhou Scientific Support Project [Qian Science Support (2020) 4Y057], the Guizhou Science Project [Qian Science Foundation (2020) 1Y353], the Guizhou Medical University Academic Sprout Cultivation Project [Qian Science Platform Talent (2018)5779-X], and the Opening Foundation of Shanghai Key Lab of Forensic Medicine, Key Lab of Forensic Science, Ministry of Justice, China (Academy of Forensic Science) (KF202009), the Major Project of National Social Science Foundation of China granted to C-CW (21&ZD285), the “Double First-Class University Plan” key construction project of Xiamen University (the origin and evolution of East Asian populations and the spread of Chinese civilization, 0310/X2106027), the Nanqiang Outstanding Young Talents Program of Xiamen University (X2123302), the Major Project of National Social Science Foundation of China (2021MZD014), and the European Research Council (ERC) grant (ERC-2019-ADG-883700-TRAM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
S. Fang and Z. Xu from Information and Network Center of Xiamen University are acknowledged for their help in the high-performance computing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.849195/full#supplementary-material
Supplementary Figure 1 | Geographical distribution of Guizhou Miao and Yao in this study.
Footnotes
References
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Bin, X., Wang, R., Huang, Y., Wei, R., Zhu, K., Yang, X., et al. (2021). genomic insight into the population structure and admixture history of tai-kadai-speaking sui people in Southwest China. Front. Genet. 12:735084. doi: 10.3389/fgene.2021.735084
Browning, B. L., Tian, X., Zhou, Y., and Browning, S. R. (2021). Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 108, 1880–1890. doi: 10.1016/j.ajhg.2021.08.005
Burton, M. L., Moore, C. C., Whiting, J. W. M., and Romney, A. K. (1996). Regions based on social structure. Curr. Anthropol. 37, 87–123.
Cavalli-Sforza, L. L., and Feldman, M. W. (2003). The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 33, 266–275. doi: 10.1038/ng1113
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi: 10.1186/s13742-015-0047-8
Chen, J., He, G., Ren, Z., Wang, Q., Liu, Y., Zhang, H., et al. (2021). Genomic insights into the admixture history of mongolic- and tungusic-speaking populations From Southwestern East Asia. Front. Genet. 12:685285. doi: 10.3389/fgene.2021.685285
Chen, J., He, G., Ren, Z., Wang, Q., Liu, Y., Zhang, H., et al. (2022). Fine-scale population admixture landscape of tai-kadai-speaking maonan in Southwest China inferred From genome-wide SNP data. Front. Genet. 13:815285. doi: 10.3389/fgene.2022.815285
Deng, W., Shi, B., He, X., Zhang, Z., Xu, J., Li, B., et al. (2004). Evolution and migration history of the Chinese population inferred from Chinese Y-chromosome evidence. J. Hum. Genet. 49, 339–348. doi: 10.1007/s10038-004-0154-3
Fei, X. T. (1999). The Pattern of Diversity in Unity of the Chinese Nation. Beijing: Central University for Nationalities Press.
Feng, Q., Lu, D., and Xu, S. (2018). AncestryPainter: a graphic program for displaying ancestry composition of populations and individuals. Genomics Proteomics Bioinformatics 16, 382–385. doi: 10.1016/j.gpb.2018.05.002
Guo, J., Wang, W., Zhao, K., Li, G., He, G., Zhao, J., et al. (2021). Genomic insights into Neolithic farming-related migrations in the junction of east and southeast Asia. Am. J. Biol. Anthropol. 177, 328–342.
Guo, J., Xu, B., Li, L., He, G., Zhang, H., Cheng, H. Z., et al. (2019). Paternal Y chromosomal genotyping reveals multiple large-scale admixtures in the formation of Lolo-Burmese-speaking populations in southwest China. Ann. Hum. Biol. 46, 581–588. doi: 10.1080/03014460.2019.1698655
He, G., Li, Y., Zou, X., Yeh, H. Y., Tang, R., Wang, P., et al. (2022a). The northern gene flow into southeastern East Asians inferred from genome-wide array genotyping. J. Syst. Evol. [Preprint]. doi: 10.1111/jse.12826
He, G. L., Wang, M. G., Zou, X., Yeh, H. Y., Liu, C. H., Liu, C., et al. (2022b). Extensive ethnolinguistic diversity at the crossroads of North China and South Siberia reflects multiple sources of genetic diversity. J. Syst. Evol. [Preprint]. doi: 10.1111/jse.12827
He, G., Li, Y. X., Wang, M. G., Zou, X., Yeh, H. Y., Yang, X. M., et al. (2021a). Fine-scale genetic structure of Tujia and central Han Chinese revealing massive genetic admixture under language borrowing. J. Syst. Evol. 59, 1–20.
He, G., Liu, J., Wang, M., Zou, X., Ming, T., Zhu, S., et al. (2021b). Massively parallel sequencing of 165 ancestry-informative SNPs and forensic biogeographical ancestry inference in three southern Chinese Sinitic/Tai-Kadai populations. Forensic Sci. Int. Genet. 52:102475. doi: 10.1016/j.fsigen.2021.102475
He, G., Zhang, Y., Wei, L.-H., Wang, M., Yang, X., Guo, J., et al. (2021c). The genomic formation of Tanka people, an isolated “Gypsies in water” in the coastal region of Southeast China. Am. J. Biol. Anthropol. [Preprint]. doi: 10.1002/ajpa.24495
He, G. L., Wang, M. G., Li, Y. X., Zou, X., Yeh, H. Y., Tang, R. K., et al. (2021d). Fine-scale north-to-south genetic admixture profile in Shaanxi Han Chinese revealed by genome-wide demographic history reconstruction. J. Syst. Evol. [Preprint]. doi: 10.1111/jse.12715
He, G., Wang, Z., Guo, J., Wang, M., Zou, X., Tang, R., et al. (2020). Inferring the population history of Tai-Kadai-speaking people and southernmost Han Chinese on Hainan Island by genome-wide array genotyping. Eur. J. Hum. Genet. 28, 1111–1123. doi: 10.1038/s41431-020-0599-7
Huang, X., Xia, Z.-Y., Bin, X., He, G., Guo, J., Lin, C., et al. (2020). Genomic insights into the demographic history of Southern Chinese. bioRxiv [Preprint]. doi: 10.1101/2020.11.08.373225
Huang, X., Zhou, Q., Bin, X., Lai, S., Lin, C., Hu, R., et al. (2018). The genetic assimilation in language borrowing inferred from Jing People. Am. J. Phys. Anthropol. 166, 638–648. doi: 10.1002/ajpa.23449
Kutanan, W., Shoocongdej, R., Srikummool, M., Hubner, A., Suttipai, T., Srithawong, S., et al. (2020). Cultural variation impacts paternal and maternal genetic lineages of the Hmong-Mien and Sino-Tibetan groups from Thailand. Eur. J. Hum. Genet. 28, 1563–1579. doi: 10.1038/s41431-020-0693-x
Lawson, D. J., Hellenthal, G., Myers, S., and Falush, D. (2012). Inference of population structure using dense haplotype data. PLoS Genet. 8:e1002453. doi: 10.1371/journal.pgen.1002453
Lipson, M., Cheronet, O., Mallick, S., Rohland, N., Oxenham, M., Pietrusewsky, M., et al. (2018). Ancient genomes document multiple waves of migration in Southeast Asian prehistory. Science 361, 92–95. doi: 10.1126/science.aat3188
Liu, Y., Xie, J., Wang, M., Liu, C., Zhu, J., Zou, X., et al. (2022). Genomic insights into the population history and biological adaptation of Southwestern Chinese Hmong–Mien People. Front. Genet. 12:815160.
Liu, Y., Yang, J., Li, Y., Tang, R., Yuan, D., Wang, Y., et al. (2021). Significant East Asian affinity of the sichuan hui genomic structure suggests the predominance of the cultural diffusion model in the genetic Formation Process. Front. Genet. 12:626710. doi: 10.3389/fgene.2021.626710
Loh, P. R., Lipson, M., Patterson, N., Moorjani, P., Pickrell, J. K., Reich, D., et al. (2013). Inferring admixture histories of human populations using linkage disequilibrium. Genetics 193, 1233–1254. doi: 10.1534/genetics.112.147330
Lu, J., Zhang, H., Ren, Z., Wang, Q., Liu, Y., Li, Y., et al. (2020). Genome-wide analysis of unrecognised ethnic group Chuanqing people revealing a close affinity with Southern Han Chinese. Ann. Hum. Biol. 47, 465–471. doi: 10.1080/03014460.2020.1782470
Luo, T., Wang, R., and Wang, C. C. (2021). Inferring the population structure and admixture history of three Hmong-Mien-speaking Miao tribes from southwest China based on genome-wide SNP genotyping. Ann. Hum. Biol. 48, 418–429. doi: 10.1080/03014460.2021.2005825
Ma, B., Chen, J., Yang, X., Bai, J., Ouyang, S., Mo, X., et al. (2021). The genetic structure and East-West population admixture in Northwest China inferred from genome-wide array genotyping. Front. Genet. 12:795570. doi: 10.3389/fgene.2021.795570
Manichaikul, A., Mychaleckyj, J. C., Rich, S. S., Daly, K., Sale, M., and Chen, W. M. (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. doi: 10.1093/bioinformatics/btq559
Mccoll, H., Racimo, F., Vinner, L., Demeter, F., Gakuhari, T., Moreno-Mayar, J. V., et al. (2018). The prehistoric peopling of Southeast Asia. Science 361, 88–92. doi: 10.1126/science.aat3628
Ning, C., Li, T. J., Wang, K., Zhang, F., Li, T., Wu, X. Y., et al. (2020). Ancient genomes from northern China suggest links between subsistence changes and human migration. Nat. Commun. 11:2700. doi: 10.1038/s41467-020-16557-2
Patterson, N., Moorjani, P., Luo, Y. T., Mallick, S., Rohland, N., Zhan, Y. P., et al. (2012). Ancient admixture in human history. Genetics 192:1065.
Patterson, N., Price, A. L., and Reich, D. (2006). Population structure and eigenanalysis. PLoS Genet. 2:e190. doi: 10.1371/journal.pgen.0020190
Pickrell, J. K., and Pritchard, J. K. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8:e1002967. doi: 10.1371/journal.pgen.1002967
Ren, Z., Guo, J., He, G., Zhang, H., Zou, X., Zhang, H., et al. (2019). Forensic genetic polymorphisms and population structure of the Guizhou Bouyei people based on 19 X-STR loci. Ann. Hum. Biol. 46, 574–580. doi: 10.1080/03014460.2019.1697362
Ren, Z., Yang, M. Q., Zhang, H. L., Dai, J. L., Liu, Y. B., Wang, Q. Y., et al. (2018). Population genetic data of 22 autosomal STRs in the Guizhou Miao population, southwestern China. Forensic Sci. Int. Genet. 32, E7–E8. doi: 10.1016/j.fsigen.2017.10.007
Schliesinger, J. (2000). Ethnic Groups of Thailand: Non-Tai-Speaking Peoples. Bangkok: White Lotus Press.
Seielstad, M., Minch, E., and Cavalli-Sforza, L. (1998). Genetic evidence for a higher female migration rate in humans. Nat. Genet. 20, 278–280. doi: 10.1038/3088
Sun, J., Li, Y. X., Ma, P. C., Yan, S., Cheng, H. Z., Fan, Z. Q., et al. (2021). Shared paternal ancestry of Han, Tai-Kadai-speaking, and Austronesian-speaking populations as revealed by the high resolution phylogeny of O1a-M119 and distribution of its sub-lineages within China. Am. J. Phys. Anthropol. 174, 686–700. doi: 10.1002/ajpa.24240
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Tan, H., Wang, R., and Wang, C. C. (2022). Fine-scale genetic profile and admixture history of two Hmong-Mien–speaking Miao tribes from Southwest China inferred from genome-wide data. Hum. Biol. Open Access Pre Prints 196. Available online at: https://digitalcommons.wayne.edu/humbiol_preprints/196
Tang, J., Yang, M. Q., Wang, X. J., Wang, Q., Wang, Q. Y., Zhang, H. L., et al. (2020). Genetic structure and forensic characterisation of 36 Y-chromosomal STR loci in Hmong-Mien-speaking Miao population. Ann. Hum. Biol. 47, 541–548. doi: 10.1080/03014460.2020.1788159
Van Oven, M., and Kayser, M. (2009). Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 30, E386–E394. doi: 10.1002/humu.20921
Wallace, D. C., Brown, M. D., and Lott, M. T. (1999). Mitochondrial DNA variation in human evolution and disease. Gene 238, 211–230.
Wang, C. C., Yeh, H. Y., Popov, A. N., Zhang, H. Q., Matsumura, H., Sirak, K., et al. (2021a). Genomic insights into the formation of human populations in East Asia. Nature 591, 413–419. doi: 10.1038/s41586-021-03336-2
Wang, T., Wang, W., Xie, G., Li, Z., Fan, X., Yang, Q., et al. (2021c). Human population history at the crossroads of East and Southeast Asia since 11,000 years ago. Cell 184:e3821. doi: 10.1016/j.cell.2021.05.018
Wang, M., Yuan, D., Zou, X., Wang, Z., Yeh, H. Y., Liu, J., et al. (2021b). Fine-scale genetic structure and natural selection signatures of Southwestern Hans inferred from patterns of genome-wide allele, haplotype, and haplogroup lineages. Front. Genet. 12:727821. doi: 10.3389/fgene.2021.727821
Wang, M., He, G., Zou, X., Chen, P., Wang, Z., Tang, R., et al. (2022). Reconstructing the genetic admixture history of Tai-Kadai and Sinitic people: insights from genome-wide SNP data from South China. J. Syst. Evol. [Preprint]. doi: 10.1111/jse.12825
Wang, Q., Zhao, J., Ren, Z., Sun, J., He, G., Guo, J., et al. (2020). Male-dominated migration and massive assimilation of indigenous East Asians in the formation of muslim hui people in Southwest China. Front. Genet. 11:618614. doi: 10.3389/fgene.2020.618614
Wen, B., Li, H., Gao, S., Mao, X., Gao, Y., Li, F., et al. (2005). Genetic structure of Hmong-Mien speaking populations in East Asia as revealed by mtDNA lineages. Mol. Biol. Evol. 22, 725–734. doi: 10.1093/molbev/msi055
Xia, Z.-Y., Yan, S., Wang, C.-C., Zheng, H.-X., Zhang, F., Liu, Y.-C., et al. (2019). Inland-coastal bifurcation of southern East Asians revealed by Hmong-Mien genomic history. bioRxiv [Preprint]. doi: 10.1101/730903
Xu, B., Guo, J., Huang, Y., Chen, X., Deng, X., and Wang, C. C. (2019). The paternal genetic structure of Jingpo and Dai in southwest China. Ann. Hum. Biol. 46, 279–283. doi: 10.1080/03014460.2019.1624821
Yan, L., Jie, X., Mengge, W., Changhui, L., Jingrong, Z., Xing, Z., et al. (2021). Genomic insights into the population history and biological adaptation of Southwestern Chinese Hmong-Mien people. Front. Genet. [Preprint]. doi: 10.3389/fgene.2021.815160
Yang, M. A., Fan, X., Sun, B., Chen, C., Lang, J., Ko, Y. C., et al. (2020). Ancient DNA indicates human population shifts and admixture in northern and southern China. Science 369, 282–288. doi: 10.1126/science.aba0909
Yao, H., Wang, M., Zou, X., Li, Y., Yang, X., Li, A., et al. (2021). New insights into the fine-scale history of western-eastern admixture of the northwestern Chinese population in the Hexi Corridor via genome-wide genetic legacy. Mol. Genet. Genomics 296, 631–651. doi: 10.1007/s00438-021-01767-0
Zhang, H., He, G. L., Guo, J. X., Ren, Z., Zhang, H. L., Wang, Q. Y., et al. (2019). Genetic diversity, structure and forensic characteristics of Hmong-Mien-speaking Miao revealed by autosomal insertion/deletion markers. Mol. Genet. Genomics 294, 1487–1498. doi: 10.1007/s00438-019-01591-7
Keywords: Hmong-Mien people, Miao and Yao, genetic structure, admixture, genome-wide SNP data
Citation: Yang M, He G, Ren Z, Wang Q, Liu Y, Zhang H, Zhang H, Chen J, Ji J, Zhao J, Guo J, Zhu K, Yang X, Wang R, Ma H, Wang C-C and Huang J (2022) Genomic Insights Into the Unique Demographic History and Genetic Structure of Five Hmong-Mien-Speaking Miao and Yao Populations in Southwest China. Front. Ecol. Evol. 10:849195. doi: 10.3389/fevo.2022.849195
Received: 05 January 2022; Accepted: 09 May 2022;
Published: 09 June 2022.
Edited by:
Lian Deng, Fudan University, ChinaReviewed by:
Min-Sheng Peng, Kunming Institute of Zoology (CAS), ChinaMagdalena Gayà-Vidal, Research Center in Biodiversity and Genetic Resources (CIBIO-InBIO), Portugal
Copyright © 2022 Yang, He, Ren, Wang, Liu, Zhang, Zhang, Chen, Ji, Zhao, Guo, Zhu, Yang, Wang, Ma, Wang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuan-Chao Wang, d2FuZ0B4bXUuZWR1LmNu; Jiang Huang, bW1tX2hqQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship