 De-Long Guan
De-Long Guan Lu Zhao
Lu Zhao Yufei Li
Yufei Li Lian-Xi Xing
Lian-Xi Xing Huateng Huang
Huateng Huang Sheng-Quan Xu
Sheng-Quan Xu- 1College of Life Sciences, Shaanxi Normal University, Xi’an, China
- 2School of Public Health, Xi’an Jiaotong University, Xi’an, China
- 3College of Life Sciences, Northwest University, Xi’an, China
Conservation genomic resources over the past decade has drastically improved, since genomes can be used to predict diverse parameters vital to conservation management. Luehdorfia taibai is an endemic butterfly only found in restricted aeras in middle-west China and is critically endangered. It was classfied as a vunerlable (VN) species in the “China species red list.” Here we generated 34.38 Gb of raw DNA sequencing reads and obtained a high-qualified draft genome assembly of L. taibai. The final genome is ~683.3 Mb, with contig N50 size of 10.19 Mb. Further, 98.6% of single-copy orthologous genes have been recovered by BUSCO. An estimated 42.34% of the genome of L. taibai consists of repetitive elements. Combined with gene prediction and transcriptome sequencing, genome annotation produced 15,968 protein-coding genes. Additionally, a nearly 1:1 orthology ratio of syntenic blocks between L. taibai and its closest genome Luehdorfia chinensis suggested that the genome structures have not changed much after speciation. The genome of L. taibai have not undergone a whole genome duplication event. Population dynamics analyses indicates that L. taibai has an extremely low heterozygosity of 0.057%, and its population size has declined dramatically over the past 10 thousand years. Our study describes a draft genome assembly of the L. taibai, the first implication of this species. We consider the globally overexploited of the host plants is not the main reason to threaten L. taibai. The genome will provide advice for the conservation to the economically important Luehdorfia lineage and this specific species.
Introduction
Conservation genomics has rapidly developed in popularity over the past decade as genomic data has become increasingly valuable for answering conservation questions (Hohenlohe et al., 2021). By integrating high-quality reference genomes, scientists can gather detailed information about a species, such as its effective population size, genetic drift, and gene flow (Wright et al., 2020), providing essential benchmarks in assessing the protection status of species (Wu et al., 2020). However, the insects were less concerned than vertebrates, on which conservation management often focused (Podsiadlowski et al., 2021). With their high esthetical attractiveness, butterflies could serve as flagship species for conservation projects. Developing genome assemblies of endangered butterfly species could provide a more detailed understanding of their evolutionary history and contributes to their conservation.
The genus Luehdorfia, which belongs to the tribe Zerynthiini, is one of the rarest genera of butterflies in East Asia (Dong et al., 2016). The genus comprises only four species, two endemic to China (Luehdorfia chinensis and L. taibai); (Liu et al., 2013; Xing et al., 2014). L. taibai (NCBI txid: 367834) is a relatively recently established species [recognized in the 1994 (Chou, 1994)]. It has a restricted distribution range in the alpines of the Qinling Mountains in China (Chou, 1999; Guo et al., 2014). This species was categorized as vulnerable (VN) by the “China species red list.” A field survey in 2010 recorded less than 250 extant mature individuals on the south slopes of the Qinling Mountains (Xing et al., 2014; Dong et al., 2016). Further survey work in three consecutive summers from 2011 to 2013 recovered about 100∼200 larvas from six counties in Qinling Mountains per year and observed less than three mature individuals during the eclosion seasons per day (Guo, 2013). Such a small population size and low eclosion rate suggest this species faces a high threat of extinction and should be re-evaluated as “Endangered” (Dong et al., 2014; Guo et al., 2014; Fang et al., 2019). L. taibai is also classified as a species of “Beneficial or Have Important Economic and Scientific Research Value” by the National Forestry and Grassland Administration. In light of this threatened status, an effective conservation strategy is urgently needed (Dong et al., 2014; Guo et al., 2014). Here, we present a high-quality genome assembly of L. taibai, which sheds light on its demographic history and can serve as a critical resource for future population genomics research and conservation efforts.
Materials and methods
Sample collection and sequencing
Two L. taibai larvae and one adult individual were collected from Huxian County, Shaanxi province, China, in May 2019. All samples were immediately transferred into liquid nitrogen and stored for DNA/RNA extractions. We used one larva for DNA sequencing and reserved the other larva and the adult individual for transcriptome sequencing.
High-quality genomic DNA was extracted from the selected larva using the Qiagen DNAeasy Tissue kit. One library for nanopore sequencing was constructed with 50 μg DNA following the standard protocols. A total of ∼34.38 Gb of raw reads were produced, with a read N50 value of 33,504 bp. Another 5 μg DNA was used for short reads sequencing. One Illumina library was constructed according to the standard protocol and sequenced on the Illumina HiSeq X-ten platform (Nair et al., 2018), generating a total of 18.32 Gb raw data of 150 bp paired-end reads. Total RNA was isolated using the Qiagen RNeasy tissue kit for the other larva and the adult sample. After reverse transcription, two cDNA libraries were sequenced using the same Illumina platform. A total of 7.17 Gb of paired-end reads were generated. All sequencing was performed by Beijing Biomarker Biotechnology Co. Ltd (Beijing biomarker biotechnology co, LTD, Beijing, China).
Genome assembly and quality assessment
Long reads generated by nanopore sequencing were cleaned first. De novo genome assembly was carried out using the Nextdenovo v2.5.0 software with the read length cut-off and seed length cut-off value set to 12 and 20 Kb, respectively, Guiglielmoni et al. (2021). The raw assembly was polished using the Nexpolish v1.4.0 software (Hu et al., 2020) with Illumina short reads for three rounds. Then, the haplotigs were removed using PurgeHaplotigs (Roach et al., 2018) with default parameters. Finally, we assess the integrity of the genome assembly using the Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.2.3 (Simao et al., 2015) and Quast v5.2.0 (Gurevich et al., 2013) software.
Identification of repetitive elements, protein-coding gene prediction, and genome-guided transcriptome assembly
Repeatmasker v4.0.7 (Tarailo-Graovac and Chen, 2009) with the insect library of the Repbase and Repeatmodeler v1.0.8 (Flynn et al., 2020) were used to identify and mask repetitive elements on the genome assembly. Protein-coding genes were predicted with the masked genome assembly using a combination of ab initio and homology-based prediction methods. The transcriptomic data was initially used in PASA v2.5.1 (Haas et al., 2003) to obtain the top 500 gene models, which were then applied in Augustus v3.3.1 (Burge and Karlin, 1997) for ab initio prediction. For the homology-based prediction, annotated protein sequences of four closely related species (Papilio machaon, P. bianor, Kallima inachus, and Parnassius apollo) were downloaded from the Genbank and imported into GeneWise (Birney et al., 2004). Finally, we merged all predictions to produce a non-redundant raw gene set in Evidence Modeler (Haas et al., 2008). Functional annotation of the gene set was conducted by querying the protein sequences against the InterProscan database (Jones et al., 2014) with a customized searching script. The final gene set only retains the annotated genes. Based on this final gene set, genome-guided transcriptome assemblies were carried out using HiSat2 v2.1.3 (Kim et al., 2015) and Stringtie v1.3.5 (Pertea et al., 2015) with default parameters. Differentially expressed genes between two samples were identified using edgeR (R package; Robinson et al., 2010).
Phylogenomic and comparative genomic analysis
Five closely related butterfly reference genomes—Luehdorfia chinensis, Papilio machaon, Papilio bianor, Parnassius apollo, and Parnassius orleans were downloaded from NCBI for identifying orthologous genes and gene families using Orthofinder v2.3.8 (Emms and Kelly, 2019). The first three species belong to the subfamily Parnassiidae, and the other two species belong to Papilioninae, another subfamily in Papilionidae (He et al., 2022). All single-copy orthologous genes shared across all six genomes were selected and aligned in MAFFT v7.4 (Katoh and Standley, 2013). With default settings, we used RaxML v8.2.12 (Stamatakis, 2014) to build a maximum-likelihood (ML) phylogeny with the concatenated sequences. Based on the ML tree topology, divergence times and nucleotide substitution rates were estimated using R8S v1.81 (Sanderson, 2003). The lowest chi-2 cross-validation score was used to select the best method in the calculation. From the website www.time-tree.org, we selected two calibration points: the divergence between Papilio machaon and P. bianor [20.3 Mya; (Condamine et al., 2012)] and that between Parnassius apollo and P. orleans [13.4 Mya; (Condamine et al., 2018)].
To examine patterns of genome evolution, we applied MCscanX (Wang et al., 2012) with default parameters to infer collinear syntenic blocks (defined as having at least five collinear genes, blast e-value set as 1e-10) within L. taibai and between L. taibai and its sister species, L. chinensis. The expansion and contraction of gene families were examined in CAFÉ v4.0 (Abramova et al., 2021) using the ultrametric tree derived in R8S.
Inference of demographic history
We inferred the L. taibai population size history using the Pairwise Sequentially Markovian Coalescent model [PSMC v0.6.5; Nadachowska-Brzyska et al. (2016)]. Illumina pair-end reads were mapped onto the genome assembly using bwa v0.7.17 mem (Li and Durbin, 2010). Genome consensus sequences based on the read alignments were generated with the mpileup utility in samtools v1.9 (Li et al., 2009) and the vcfutils.pl script from the psmc package. Then, we estimated the effective population size (Ne) using psmc with the “-p” option set to “28 × 2 + 3 + 5” as in a previous butterfly study (Yang et al., 2020). The result was scaled assuming a generation time of 1 year and a mutation rate of 3.59e-09 per site per generation—the rate estimated from our R8S analyses.
Results and discussions
De novo genome assembly
The genome of L. taibai was assembled into 232 contigs and had a size of 683.3 Mb. The de novo genome assembly is of high quality with N50 of 10.19 Mb, L50 of 24, and the most extended contig length of 26.79 Mb (Table 1). Over 99.9% of the nanopore reads can be mapped back to the assembly—most long reads were incorporated. Blasting (BLASTN) assembled contigs against the database of known sequencing adaptors did not find any potential matches. In addition, Quast analysis showed a high mapping rate to L. chinensis genomes (98.17% contigs can be aligned). BUSCO analysis revealed that 98.6% of the 5,286 expected Lepidoptera single-copy orthologous genes are complete on the assembled genome. Only 0.2% of duplicated BUSCOs indicate the absence of haplotigs. Mapping all Illumina short reads back to the genome shows one peak in the coverage depth distribution, confirming a neglectable proportion of haplotigs (Supplementary Figure 1). Furthermore, most of the short reads from transcriptome sequencing can be mapped to the genome assembly as well—the mapping rates of RNA-seq data of the adult and larval samples were 95.26 and 95.42%, respectively. Hence, our de novo genome assembly showed no obvious assembly error and is primarily complete regarding functional elements. Compared to the reported genome of L. chinensis (N50 of 2.39 Mb, and with 1,362 scaffolds), our genome assembly of L. taibai has higher connectivity and integrity. It could be a better reference for future studies on the genome evolution of the genus Luehdorfia.

Table 1. Summary statistics of the genome assembly of L. taibai.
Identification of repetitive elements and gene finding
In total, the repeat sequences comprised 42.34% (289.33 Mb) of the L. taibai genome. Interspersed Repeats occupied the most—282.34 Mb. Among them, the retroelements and long interspersed nuclear elements are the most abundant subtypes, accounting for 4.43% (30.29 Mb) and 3.80% (26.01 Mb) of the assembly. For non-coding RNAs, we identified 107 ribosomal RNA and 3918 transfer RNA sequences. A summary of the repeat annotations is provided in Supplementary Table 1.
After masking repetitive sequences, we identified 15,968 protein-coding genes on the assembled genome. The mean lengths of the gene, coding DNA sequence (CDS), and intron were 12,582.95, 2,195.44, and 10,377.87 bp, respectively. The average number of CDS and exons per gene were 6.19 and 6.52, respectively. Gene functions are annotated based on protein domain conservation using Interproscan, which determines motifs and domains by querying protein sequences against 21 public databases, including the Pfam, PANTHER, Gene3D, and CDD. The InterProscan iprterm database annotated the most number of genes—83.69% (13,364) of the gene set, followed by PANTHER and Pfam, which were 78.85% (12,592) and 76.54% (12,222), respectively. In addition, 9,273 genes were annotated with gene ontology (GO) terms (see Supplementary Table 2 for a summary of the gene functional annotations).
The larval and adult samples expressed high proportions of genes—85.04% (13,580) and 83.52% (13,338) of the gene set, respectively. 12,600 genes were shared between samples, and 2,251 genes were differentially expressed (FDR adjusted P-value < 0.01). Enrichment analysis revealed several significantly enriched GO terms (see Supplementary Table 3 for a Table showing the results) among these differentially expressed genes. In particular, the most significantly enriched BP (Biological Pathway) terms include reproduction (GO:0000003), reproductive process (GO:0022414), and developmental process (GO:0032502), concordant with the different developmental stages of the two samples.
Phylogenetic analysis
Across the six butterfly genomes, OrthoFinder identified a total of 14,924 orthologous and/or paralogous groups of genes. Among them, 5,923 are single-copy orthologous genes shared by the six genomes. The concatenated alignment comprises 20,534,736 amino-acid sites. The derived ML tree is well-supported such that all bootstrapping values are 100% (Figure 1A). The time-calibrated tree shows that L. taibai and L. chinensis are sister species with a divergence time estimated at ∼1.76 Mya (Figure 1A). The clade of these two species clusters with the apollo butterflies (Genus Parnassius; divergence time around 29.49 Mya) and then with the swallowtail butterflies (Genus Papilio; divergence time around 46.60 Mya).
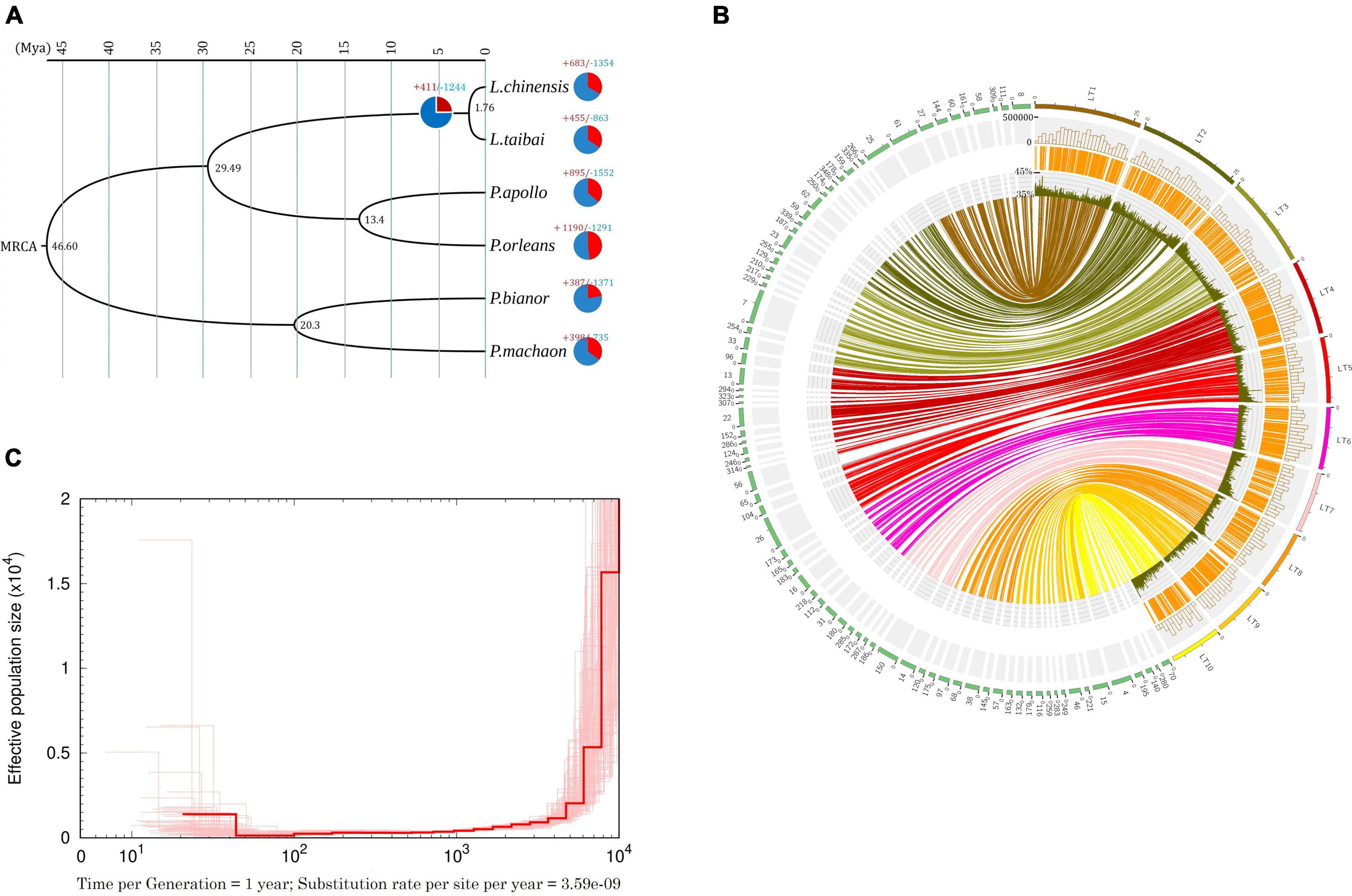
Figure 1. (A) Time-calibrated phylogeny of the six butterfly species based on single-copy orthologous genes. Numbers on the pie charts indicate the numbers of gene families that experienced expansion (red) or contractions (blue); the number beside each node denotes the estimated divergence time (million years ago). (B) Circos plot of syntenic blocks between L. taibai (longer colorful blocks on the right) and L. chinensis (shorter green blocks on the left) genome. Only the longest ten contigs in L. taibai are shown. The internal lines link the collinear gene pairs, and the outer circles (from inside outward) represent GC content (sliding window of 1 Mb), expressed gene location, and SNP density (sliding window of 1 Mb). The outermost numbers and strings represent the names of the contigs for L. taibai and the scaffold number for L. chinensis. (C) Estimated historical effective population size (Ne) of L. taibai with bootstrap results (thin lines).
Comparative genomics for L. taibai
Regarding gene families, L. taibai has 46 unique (254 genes), 455 expanded and 863 contracted gene families (Figure 1A). Functional enrichment analysis of the 211 significantly expanded (adjusted P-value < 0.01) gene families reveal several significantly enriched MF (Molecular Function) terms, including the heterocyclic compound binding (GO:1901363) and structural constituent of chromatin (GO:0030527). The most significantly enriched BP terms are the multi-organism process (GO:0051704) and metabolic process (GO:0008152; Supplementary Table 4). On the branch leading to L. taibai and L. chinensis, there are 411 predicted expanded and 1,244 contracted gene families. Several BP terms are enriched among the 63 significantly expanded gene families, including metabolic process (GO:0008152) and cellular process (GO:0009987).
Genome collinearity analyses inferred 10,260 collinear gene pairs from 369 syntenic blocks between L. taibai and L. chinensis. Each block’s average number of genes reached an astonishing value of 27.81. The overall genomic gene collinearity between L. taibai and L. chinensis revealed a nearly 1:1 orthology ratio, indicating similar genomic structures in these two species (Figure 1B).
Genetic diversity and demographic history of L. taibai
The assembled L. taibai’s genome has 395,579 heterozygous sites, corresponding to a heterozygosity rate of 0.057%. This heterozygosity is extremely low compared to other species in the Papiliondae family. For example, Papilio bianor, a common species with effective population size (Ne) size over 10 million, has a heterozygosity of 1.81%. This low heterozygosity rate is comparable to the Gaint Panda (0.049%), a famous animal with worldwide conservation interest (Westbury et al., 2018).
The PSMC analysis indicates that around 10 thousand years ago, the effective population size (Ne) of L. taibai experienced a rapid decline and then stayed at a deficient level ever since (Figure 1C). This pattern is contrary to common ideas about why L. taibai became an endangered species. This species currently only oviposits on Saruma henryi Oliv., a species used in traditional Chinese herbal medicine. Over the past decades, this host species has undergone excessive exploration, which is considered the leading cause of the population decline in L. taibai (Zhou et al., 2010). If the over-exploitation of host plants is the primary reason, we would only observe a sharp population size decline in recent history, just as in the walrus (Shafer et al., 2015), and whales (Morin et al., 2021). The rapid decline of L. taibai occurred about 7000∼10,000 years ago long predated any anthropogenic activities on their host plants. Also, a diet experiment on L. taibai showed that under starvation, these butterflies will alternate and expand host plants (Guo, 2013). That is, a shortage of S. henryi might not necessarily cause the butterfly population to collapse.
Nevertheless, geological analyses of the local climate history of the Qinling Mountains showed that the local temperature significantly raised in the early Holocene (Fang and Hou, 2011; Li et al., 2015). The time coincided with the population decline we observed in L. taibai (Figure 1C). L. taibai lives at mid-high altitudes (above 1,500 m ASL). It is likely that this butterfly has adapted to a cold environment for most of their life cycle. The rising temperature could severely impede the growth and productivity of the butterfly, leading to population decline. If the main reason for L. taibai’s low effective population size was climate change, the effect of it should be the focus of future conservation and population management.
Conclusion
Here, we have assembled and annotated the genome of Luehdorfia taibai using a combination of Nanopore long-read and Illumina short-read sequencing. This is the first such effort for this species and the genus Luehdorfia. The extremely low heterozygosity of L. taibai and its demographic history suggest that this species should be a priority for conservation management, and conservation efforts should focus on the impact of climate change.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank under BioProject accession numbers: PRJNA615396 and PRJNA615348.
Author contributions
D-LG and LZ performed the bioinformatics analyses and wrote the manuscript. YL and L-XX collected and identified L. taibai samples for this research. HH and S-QX conceived the study. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Fundamental Research Funds for the Central Universities (GK201903063, GK202105003, and TD2020041Y). This work was partly supported by the National Natural Science Foundation of China (No. 31872273).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.955246/full#supplementary-material
Supplementary Figure 1 | Depth distribution of Illumina short reads mapped to the genome assembly of L. taibai.
Supplementary Table 1 | A summary of the repeat annotations in L. taibai.
Supplementary Table 2 | Functional annotation results from the Interproscan databases.
Supplementary Table 3 | GO enrichment for differently expressed genes in larval and adult samples of L. taibai.
Supplementary Table 4 | GO enrichment for gene families significantly expanded in L. taibai.
Abbreviations
BUSCO, Benchmarking Universal Single-Copy Orthologs; GO, gene ontology; LINE, long interspersed nuclear elements; WGD, whole genome duplication; Ka, non-synonymous substitution rate; Ks, synonymous substitution rate; PSMC, pairwise sequentially Markovian coalescent.
References
Abramova, A., Osińska, A., Kunche, H., Burman, E., and Bengtsson-Palme, J. (2021). CAFE: A software suite for analysis of paired-sample transposon insertion sequencing data. Bioinformatics 37, 121–122. doi: 10.1093/bioinformatics/btaa1086
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and Genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Chou, I. (1994). Monographia rhopalocerorum sinensium (Monograph of Chinese butterflies) Henan, Vol. 1. Setúbal: Scientific and Technological Publishing House, 408.
Chou, I. (1999). Monographia rhopalocerorum sinensium, Revised Edn. Zhengzhou: Henan Scientific and Technological Publishing House.
Condamine, F. L., Rolland, J., Höhna, S., Sperling, F. A. H., and Sanmartín, I. (2018). Testing the role of the red queen and court jester as drivers of the macroevolution of apollo butterflies. Syst. Biol. 67, 940–964. doi: 10.1093/sysbio/syy009
Condamine, F. L., Sperling, F. A., Wahlberg, N., Rasplus, J. Y., and Kergoat, G. J. (2012). What causes latitudinal gradients in species diversity? Evolutionary processes and ecological constraints on swallowtail biodiversity. Ecol. Lett. 15, 267–277. doi: 10.1111/j.1461-0248.2011.01737.x
Dong, S., Jiang, G., and Hong, F. (2014). Advances in conservation biology of the rare and threatened butterfly genus Luehdorfia(Lepidoptera: Papilionidae). Chin. J. Appl. Environ. Biol. 20, 1139–1144.
Dong, Y., Zhu, L. X., Wang, C. B., Zhang, M., and Ding, P. P. (2016). The complete mitochondrial genome of Luehdorfia chinensis Leech (Lepidoptera: Papilionidae) from China. Mitochondrial DNA B Resour. 1, 198–199. doi: 10.1080/23802359.2016.1155084
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Fang, L. J., Zhang, Y. L., Gao, K., Ding, C. P., and Zhang, Y. J. (2019). Butterfly communities along the Heihe River Basin in Shaanxi Province, a biodiversity conservation priority area in China. J. Insect Conserv. 23, 873–883. doi: 10.1007/s10841-019-00184-4
Fang, X., and Hou, G. (2011). Synthetically reconstructed holocene temperature change in China. Sci. Geogr. Sin. 31, 385–393.
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Guiglielmoni, N., Houtain, A., Derzelle, A., Van Doninck, K., and Flot, J. F. (2021). Overcoming uncollapsed haplotypes in long-read assemblies of non-model organisms. BMC Bioinformatics 22:303. doi: 10.1186/s12859-021-04118-3
Guo, Z., Gao, K., Li, X., and Zhang, Y. (2014). Study on the bionomics and habitat of Luehdorfia taibai (Lepidoptera : Papilionidae). Acta Ecol. Sin. 34, 6943–6953.
Guo, Z.-Y. (2013). The conservation biology of the endangered butterfly Luehdorfia taibai. Xianyang: Northwest A&F University.
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K. Jr., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
He, J. W., Zhang, R., Yang, J., Chang, Z., Zhu, L. X., Lu, S. H., et al. (2022). High-quality reference genomes of swallowtail butterflies provide insights into their coloration evolution. Zool. Res. 43, 367–379. doi: 10.24272/j.issn.2095-8137.2021.303
Hohenlohe, P. A., Funk, W. C., and Rajora, O. P. (2021). Population genomics for wildlife conservation and management. Mol. Ecol. 30, 62–82. doi: 10.1111/mec.15720
Hu, J., Fan, J., Sun, Z., and Liu, S. (2020). NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255. doi: 10.1093/bioinformatics/btz891
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Li, F., Hou, G., Chongyi, E., and Jiang, Y. (2015). Integrated reconstruction of the holocene temperature series of Qinghai-Tibet plateau. Arid Zone Res. 32, 716–725.
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, G., Jiang, G. F., Pang, H. C., and Hong, F. (2013). The mitochondrial genome of the Chinese special butterfly Luehdorfia chinensis Leech (Lepidoptera: Papilionidae). Mitochondrial DNA 24, 211–213. doi: 10.3109/19401736.2012.748043
Morin, P. A., Archer, F. I., Avila, C. D., Balacco, J. R., Bukhman, Y. V., Chow, W., et al. (2021). Reference genome and demographic history of the most endangered marine mammal, the vaquita. Mol. Ecol. Resour. 21, 1008–1020. doi: 10.1111/1755-0998.13284
Nadachowska-Brzyska, K., Burri, R., Smeds, L., and Ellegren, H. J. M. E. (2016). PSMC-analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol. Ecol. 25, 1058–1072. doi: 10.1111/mec.13540
Nair, S. S., Luu, P. L., Qu, W., Maddugoda, M., Huschtscha, L., Reddel, R., et al. (2018). Guidelines for whole genome bisulphite sequencing of intact and FFPET DNA on the Illumina HiSeq X Ten. Epigenetics Chromatin 11:24. doi: 10.1186/s13072-018-0194-0
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Podsiadlowski, L., Tunström, K., Espeland, M., and Wheat, C. W. (2021). The genome assembly and annotation of the Apollo butterfly Parnassius apollo, a flagship species for conservation biology. Genome Biol. Evol. 13:evab122. doi: 10.1093/gbe/evab122
Roach, M. J., Schmidt, S. A., and Borneman, A. R. (2018). Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19:460. doi: 10.1186/s12859-018-2485-7
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Sanderson, M. J. (2003). r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
Shafer, A. B., Gattepaille, L. M., Stewart, R. E., and Wolf, J. B. (2015). Demographic inferences using short-read genomic data in an approximate Bayesian computation framework: In silico evaluation of power, biases and proof of concept in Atlantic walrus. Mol. Ecol. 24, 328–345. doi: 10.1111/mec.13034
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4:Unit 4.10. doi: 10.1002/0471250953.bi0410s25
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Westbury, M. V., Hartmann, S., Barlow, A., Wiesel, I., Leo, V., Welch, R., et al. (2018). Extended and continuous decline in effective population size results in low genomic diversity in the world’s rarest hyena species, the brown hyena. Mol. Biol. Evol. 35, 1225–1237. doi: 10.1093/molbev/msy037
Wright, B. R., Farquharson, K. A., McLennan, E. A., Belov, K., Hogg, C. J., and Grueber, C. E. (2020). A demonstration of conservation genomics for threatened species management. Mol. Ecol. Resour. 20, 1526–1541. doi: 10.1111/1755-0998.13211
Wu, M. Y., Low, G. W., Forcina, G., van Grouw, H., Lee, B. P. Y., Oh, R. R. Y., et al. (2020). Historic and modern genomes unveil a domestic introgression gradient in a wild red junglefowl population. Evol. Appl. 13, 2300–2315. doi: 10.1111/eva.13023
Xing, L. X., Li, P. F., Wu, J., Wang, K., and You, P. (2014). The complete mitochondrial genome of the endangered butterfly Luehdorfia taibai Chou (Lepidoptera: Papilionidae). Mitochondrial DNA 25, 122–123. doi: 10.3109/19401736.2013.800506
Yang, J., Wan, W., Xie, M., Mao, J., Dong, Z., Lu, S., et al. (2020). Chromosome-level reference genome assembly and gene editing of the dead-leaf butterfly Kallima inachus. Mol. Ecol. Resour. 20, 1080–1092. doi: 10.1111/1755-0998.13185
Keywords: China species red list, genome assembly, genome annotation, conservation management, Luehdorfia taibai
Citation: Guan D-L, Zhao L, Li Y, Xing L-X, Huang H and Xu S-Q (2022) Genome assembly of Luehdorfia taibai, an endangered butterfly endemic to Qinling Moutains in China with extremely small populations. Front. Ecol. Evol. 10:955246. doi: 10.3389/fevo.2022.955246
Received: 28 May 2022; Accepted: 19 July 2022;
Published: 04 August 2022.
Edited by:
Yongjie Wu, Sichuan University, ChinaReviewed by:
Arong Luo, Institute of Zoology (CAS), ChinaXiangqun Yuan, Northwest A&F University, China
Copyright © 2022 Guan, Zhao, Li, Xing, Huang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huateng Huang, aHVhbmdodWF0ZW5nQHNubnUuZWR1LmNu; Sheng-Quan Xu, eHVzaGVuZ3F1YW5Ac25udS5lZHUuY24=