 Florian Wimmers
Florian Wimmers Gerty Schreibelt
Gerty Schreibelt Annette E. Sköld
Annette E. Sköld Carl G. Figdor
Carl G. Figdor I. Jolanda M. De Vries1,2*
I. Jolanda M. De Vries1,2*- 1Department of Tumor Immunology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, Netherlands
- 2Department of Medical Oncology, Radboud University Medical Center, Nijmegen, Netherlands
Dendritic cell (DC)-based immunotherapy employs the patients’ immune system to fight neoplastic lesions spread over the entire body. This makes it an important therapy option for patients suffering from metastatic melanoma, which is often resistant to chemotherapy. However, conventional cellular vaccination approaches, based on monocyte-derived DCs (moDCs), only achieved modest response rates despite continued optimization of various vaccination parameters. In addition, the generation of moDCs requires extensive ex vivo culturing conceivably hampering the immunogenicity of the vaccine. Recent studies, thus, focused on vaccines that make use of primary DCs. Though rare in the blood, these naturally circulating DCs can be readily isolated and activated thereby circumventing lengthy ex vivo culture periods. The first clinical trials not only showed increased survival rates but also the induction of diversified anti-cancer immune responses. Upcoming treatment paradigms aim to include several primary DC subsets in a single vaccine as pre-clinical studies identified synergistic effects between various antigen-presenting cells.
Introduction
Melanoma is a malignant transformation of melanocytes – the pigment producing cells of the epidermis – and the most aggressive cancer of the skin (1). Over the past years, the number of melanoma incidences rose worldwide and reached 232,130 diagnosed cases in 2012 (2–4). Once melanoma patients develop metastatic disease, life expectancy drops and survival rates are low (1, 5, 6). Traditional treatment methods focus on chemotherapy and radiation therapy, which are highly invasive and often fail to induce objective clinical response (6).
Novel treatment strategies focus on melanoma patients that carry an activating mutation in protein kinases involved in MAPK or AKT signaling (7). Recently approved small molecule inhibitors, such as vemurafenib, allow specific targeting of these mutated kinases and lead to rapid tumor regression and prolonged survival in treated patients (7–9). However, due to the prompt development of resistance in many cases, and major cutaneous side effects, including the induction of neoplastic lesions, small molecule inhibitors are so far of limited clinical use (6, 8).
As pharmacological treatment paradigms fail to induce lasting responses, researchers, clinicians, and patients turn to immunotherapy, which – due to major advances – was recently declared as breakthrough of the year 2013 by scientific journal Science (10).
The ability of the immune system to fight tumors was first described by William B. Coley, who in the nineteenth century observed cancer regression in patients suffering from inoperable sarcoma after injecting bacterial toxins into neoplastic lesions (11). Today, cytotoxic CD8+ T lymphocytes (CTLs) are considered to be the fundamental mediators of anti-cancer immunity (12–16). In vitro experiments and studies in mice showed that CTLs are able to specifically target cancerous cells and destroy them by inducing apoptosis (12, 13, 17). Clinical evidence confirmed the importance of CTLs in patients suffering from melanoma and other cancers, as infiltrating CD8+ T cells found in tumor biopsies were strongly associated with improved life expectancy (18–20). Furthermore, melanoma patients with tumor-specific T cells in peripheral blood displayed increased clinical response rates (21). Immunotherapy hence aims to induce a potent and lasting T cell response against malignant cells.
One approach to potentiate the patient’s own immune response is to prolong the activity phase of the T cell response. Immunomodulatory drugs, such as the CTLA-4-blocking antibody ipilimumab or the PD-1-blocking antibody nivolumab, aim to unleash the patients’ natural anti-cancer T cell responses by interfering with inhibitory pathways (22–27). Neoplastic cells frequently exploit, e.g., the PD-1 pathways to suppress the immune system leading to immune escape and disease progression (28, 29). Notably, ipilimumab was the first treatment agent to provide survival benefit for patients suffering from melanoma and is now standard treatment for this type of cancer (10, 26, 28). Although only effective in a minority of patients, ipilimumab frequently induces objective responses that are remarkably long lasting (26, 30). Due to their broad mechanism of action, immunomodulatory antibodies can, however, cause severe and potentially fatal side effects by activating autoreactive T cells. Patients with, e.g., skin rash, colitis, hypophysitis, or high-grade hepatic adverse events were reported (6, 30). To overcome these side effects, targeted therapies that only activate cancer-specific T cells are desired.
Specific T cell responses are naturally induced by dendritic cells (DCs) (31, 32). DCs are professional antigen-presenting cells (APCs) that sample the body for antigens and danger signals derived from pathogens or tumors (33). After encountering such signals, DCs become activated and migrate to the lymph node, where they activate naïve T cells to become CTLs or helper T cells (32, 33). Due to their great regulatory capacities and outstanding ability to activate antigen-specific T cells, DCs have become an attractive target in several immunotherapeutic approaches in cancer.
Cellular vaccination therapies were developed in the mid 1990s, when new laboratory techniques allowed the enrichment of DCs from peripheral blood (34–37). Murine DCs were isolated from peripheral blood by density gradients, loaded ex vivo with tumor antigens, and injected back into the blood (17, 38). This technique was rapidly transferred to the clinical setting when in 1996 pioneer Frank Hsu treated patients suffering from B-cell lymphoma with autologous, antigen-loaded DCs (39). Strikingly, clinical response could be detected in a majority of patients, kickstarting the field of therapeutic DC vaccination (Figure 1).
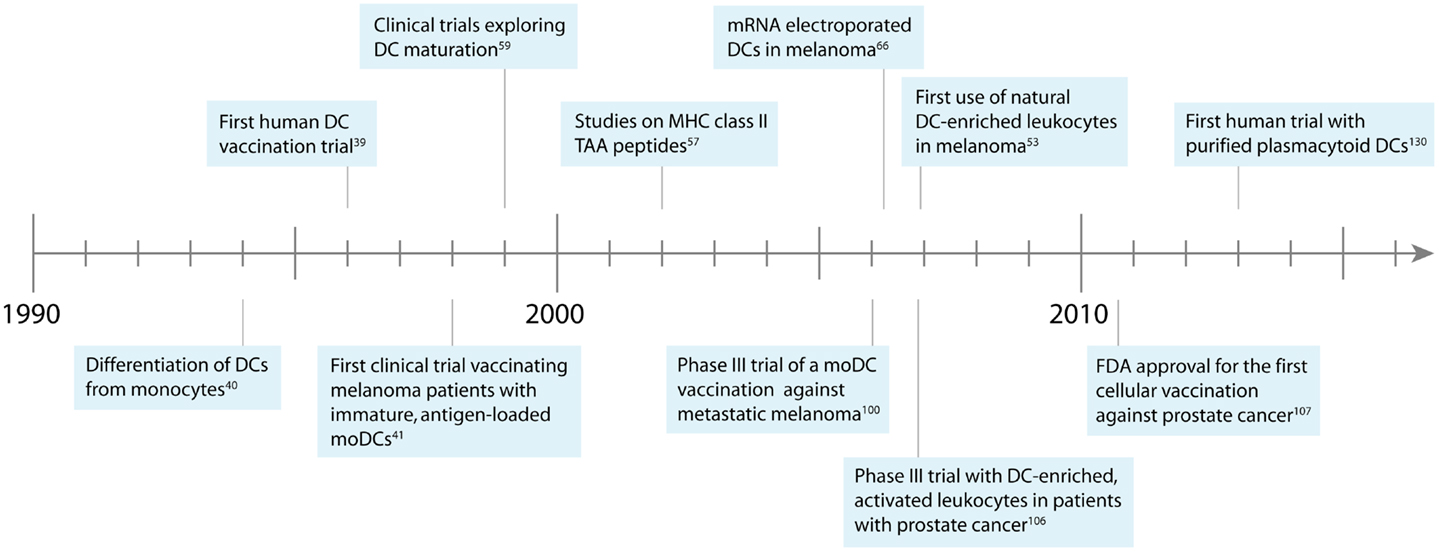
Figure 1. Development of DC-based immunotherapy against melanoma.
However, only after Sallusto and colleagues discovered a method to differentiate DCs from monocytes in vitro, sufficient cellular material was available to start clinical trials that went further than pure proof of principal (40). Following this development, Nestle and colleagues conducted the first DC vaccination trail in melanoma patients in 1998 (41). In this study, the group isolated autologous monocytes from peripheral blood of the patients and generated DCs ex vivo. Monocyte-derived DCs (moDCs) were subsequently pulsed with tumor-associated antigen (TAA) peptides or tumor lysate, and injected into the lymph nodes of the patients to activate the immune system. The results of this study were promising, as complete and partial responses could be observed in a number of patients. Furthermore, tumor-specific T cells were found in vaccinated patients, indicating the induction of a melanoma-specific immune response.
In the following years, a considerable number of phase I/II clinical trials explored the impact of various vaccination parameters on the treatment outcome. In this review, we will give an overview of the major advances in the field of therapeutic DC vaccination against melanoma since the initial study by Nestle. Further, we will highlight current developments focusing on natural DC subsets and their impact on immunotherapy, and we will conclude with an outlook on future vaccination strategies including the synergistic effects of DC subsets.
Maturation of DCs
A major disadvantage of the DC vaccination protocol employed by Nestle et al. was the lack of activation signals. After differentiation, most moDCs possess an immature phenotype, which is dominated by high antigen uptake capabilities and poor T cell stimulatory abilities (42–45). Activation of DCs leads to the development of a mature phenotype characterized by upregulation of co-stimulatory molecules, major histocompatibility complex (MHC) molecules, and certain chemokine receptors (33, 46, 47). Especially the latter is of great importance for vaccination efficacy, as expression of the chemokine receptor CCR7 promotes the migration of injected DCs to the lymph nodes where the activation of T- and B-cells occurs (42, 47, 48). In addition to their inferior stimulatory capabilities, immature DCs were shown to induce antigen-specific tolerance, proposing that injection without activation signals is not only ineffective but also potentially detrimental (49).
While in vivo maturation signals primarily come from contact with pathogens or tissue injury, immature DCs can be matured by incubation with pathogen recognition receptor (PRR) agonists or cytokines such as TNF-α, and prostaglandin E2 (50, 51). In a clinical setting, CD40 ligation has also been used for DC activation (52, 53).
In 2003, a phase I/II clinical trial treating stage IV metastatic melanoma patients with autologous, antigen-loaded moDCs confirmed the superiority of mature DCs to induce strong immunity, as the immunological response against both included TAAs and the control antigen keyhole limpet hemocyanin (KLH) was improved in the majority of patients treated with mature DCs, as opposed to immature DCs (54). Strikingly, tumor regression could only be observed in patients of the mature DC arm, indicating that activating DCs prior injection improves clinical response as well. Other groups that employed modified maturation cocktails made the similar observations that DC maturation is necessary for the induction of a superior immune response (55–59). These results confirmed in a clinical setting what was already known for in vitro models: infused DCs need to express potent stimulatory molecules to generate a strong T cell response, especially when presenting cancer antigens with low immunogenicity. Nevertheless, as proper homing to the lymph nodes is a prerequisite for DC-mediated T cell activation, upregulation of CCR7 may also partly explain the observed differences (42).
Route of Administration
In addition to maturation-induced upregulation of CCR7, the route of administration has a major impact on the migration of DCs to the T cell rich zones in the lymph nodes (42). Since intravenously (i.v.) injected, ex vivo generated DCs fail to induce potent skin-homing T cells in mice and appeared to be less efficient in inducing TH1 responses in humans, previous clinical trials focused on subcutaneous or intradermal (i.d.) administration of the vaccines (60–62). However, using 111In-labeling and scintigraphy, we could show that most of the injected DCs remain at the injection site, where they rapidly die to be phagocytosed by macrophages (42, 63, 64). Pretreatment of the skin with cytokines, toll-like receptor (TLR) ligands, or activated DCs did not lead to increased migration (64). Interestingly, Aarntzen et al. identified the number of injected DCs as an important factor for migration as a low cell density at the injection site correlated with high migration efficiency (64).
To further improve migration of DCs to lymph nodes and enhance the induced immune responses, different routes of administration have been explored in various studies (65, 66). Direct injection of DCs into the lymphatic system of the skin appeared to be a promising approach, as it ensures that most of the DCs reach the T- and B-cell rich zones of the lymph nodes. To test this hypothesis, our group conducted a phase I/II clinical trial and vaccinated melanoma patients with ex vivo generated, antigen-loaded, mature moDCs that were injected either intranodally or intradermally (65). Although intranodal vaccination led to increased DC migration to efferent lymph nodes, no difference in the frequency of tetramer-specific T cells could be detected. Furthermore, melanoma-specific T cells induced by i.d. vaccination turned out to be more functional, which might be caused by bystander activation of APCs at the injection site. Similar results have been found by Kyte et al. using mRNA transfected moDCs (66). Taking the complicated procedure of intranodal vaccination into account, intradermal injection of DCs appears to be the optimal route of administration in case of sufficient cellular material.
T Cell Help
In the late 90s several groups independently discovered that, in absence of a strong inflammatory stimulus, DCs need to interact with CD4+ T cells to induce potent cytotoxic CD8+ T cells – a process called DC licensing (67–70). These findings, together with other important discoveries in the early 2000s, shifted the focus of therapeutic anti-cancer vaccination toward the CD4+ T cells and the impact of helper responses (71–73). Besides licensing DCs, T cell help plays a crucial role in memory generation and maintenance as well as affinity maturation of tumor-specific antibodies (72, 74, 75). Additionally, CD4+ T cells were shown to activate the innate immune system, to enhance the cytolytic function of macrophages, to induce senescence in malignant cells, and to destroy neoplastic cells directly (76, 77). The latter is of particular importance in the melanoma setting, where transformed melanocytes tend to constitutively express MHC class II molecules (78, 79). In particular, TH1 cells appear to be associated with favorable clinical outcome and overall survival (80). Despite this knowledge, integration of CD4+ T cell help in clinical trials was hampered due to the lack of defined TAA peptides binding to MHC class II molecules. To partly overcome this limitation, DCs were pulsed with unrelated antigens such as KLH or tetanus toxoid. The CD4+ T cells generated against these antigens were supposed to secrete interleukin (IL)-2 and pro-inflammatory cytokines, and to further activate the injected DCs, leading to an improved priming of cancer-specific CTLs (81). Whether or not the antigen-independent CD4+ T cell help had a strong effect on T cell priming could however not been definitely proven.
This changed when several groups characterized immunogenic melanoma-associated MHC class II epitopes of the tumor antigens gp100 and tyrosinase leading to a comparative study of melanoma patients treated with moDCs pulsed with both MHC class I and class II epitopes or MHC class I epitopes alone (79, 82–84). Analysis of patient samples showed that the simultaneous administration of TAAs restricted to both MHC classes lead to a broader anti-cancer T cell response with higher functionality compared to patients who received DCs loaded with epitopes for MHC class I only (79). Importantly, the tumor-specific CD4+ T cells were Foxp3 negative and displayed a TH1 phenotype, indicating that the vaccination did not induce regulatory T cells. This trend was reflected in the clinical response, as patients of the MHC class I/II arm showed increased progression free and overall survival, whereas no clinical benefit could be detected in patients of the MHC class I arm. The results thus indicate that antigen-specific CD4+ T cell help is indeed beneficial for the induction of a strong cancer-specific immune response, which is in line with a number of other studies (57, 85).
Antigen Loading and Heteroclitic Peptides
Antigen loading was revolutionized when clinical grade mRNA electroporated moDCs became available. MRNAs coding for full-length TAA proteins containing multiple immunogenic epitopes were synthesized and used to transfect DCs (86, 87). In this approach, the transfected DCs translate the injected mRNA into full-length proteins, which are subsequently degraded by the proteasome and presented on MHC class I molecules (86). Adding an MHC class II targeting tag to the mRNA leads to the transport of translated proteins to exosomes and presentation on MHC class II molecules, necessary for priming CD4+ T cells (88, 89). Using electroporated DCs, several problems were solved: due to the presence of multiple immunogenic epitopes within the same protein, CD8+ and CD4+ T cells could be stimulated at the same time, and the induced immune responses became broader. The same effect rendered human leukocyte antigen (HLA)-restriction obsolete, as the various epitopes contained in each protein are able to bind to different HLA molecules. This made the enrollment of a much larger number of melanoma patients possible and increased the number of individuals potentially benefiting from this treatment (90, 91). These improvements however come with the price of reduced viability, which can turn into a serious problem when cellular material is scarce (92).
Studies using electroporated moDCs conducted by our group and others indeed showed the induction of specific CD4+ and CD8+ T cells in patients suffering from metastatic melanoma (63, 90, 91, 93). Interestingly, T cells specific for epitopes different from the TAA peptides employed in previous vaccines were readily detected in a number of patients, thus indicating an increased breadth of the immune response (93).
Soon after the first studies with electroporated moDCs were published, Bonehill et al. simplified the loading and activation process for moDCs distinctly. In their approach, they transfected DCs with mRNA, not only coding for TAA proteins, but also for the maturation-inducing molecules, CD40L and caTLR4 (constitutively active form of TLR4), as well as the T cell co-stimulatory molecule, CD70. This led to prolonged and enhanced maturation of DCs (90, 94, 95).
In parallel to the development of mRNA-based DC vaccines, various groups tried to improve the immunogenicity of the traditional peptide-pulsing approach to load DCs. Using rational design, researchers modified known TAA peptides by replacing single amino acids to improve binding to the MHC groove creating so called heteroclitic peptides (96–98). Due to tighter binding, heteroclitic peptides are presented for an extended time period, supposedly leading to stronger T cell activation. However, whereas many pre-clinical studies showed increased immunogenicity in vitro, clinical trials directly comparing modified and wild type peptides failed to measure any positive effect of heteroclitic peptides and even showed decreased frequencies of TAA-specific T cells in some patients (98). It appeared that the modified epitopes differed too much from the wild type peptide leading to the induction of T cells that were unable to detect endogenously presented antigens (99).
In summary, the development of mRNA electroporated moDCs simplified anti-cancer immunotherapy significantly as transfection of DCs not only induces a broad, HLA-independent CD4+ and CD8+ immune response but also reduces the time and costs for vaccine preparation. In contrast, heteroclitic peptides failed to prove superior immunogenicity in immunotherapy against melanoma.
Efficacy of DC Immunotherapy
Although various vaccination parameters could be optimized and lasting responses were observed in selected patients, so far none of the conducted clinical trials using moDCs could demonstrate statistically supportable evidence for survival benefits in vaccinated patients. This became especially evident when in 2006 Schadendorf et al. published the first and so far only randomized phase III trial designed to demonstrate the clinical efficacy of moDC therapy in melanoma patients (100). The study was aborted early, as the Data Safety and Monitoring Board did not expect the group to reach the study goal. Analysis of the preliminary data could demonstrate the induction of an anti-cancer immune response in various patients but failed to show improved overall survival. Further, objective response was lower in the group of patients treated with DC vaccination as opposed to chemotherapy with dacarbazine (DTIC); thus no clinical benefit of moDC therapy could be detected.
One explanation for the observed lack of clinical response could be the inferior capacity of moDCs to induce effective anti-cancer immunity. However, as the study was already initiated in 1999 – thus only 1 year after the publication of the first phase I trial on moDC-based vaccines in melanoma by Nestle et al. – many of the aforementioned developments, including proper maturation of DCs, were not yet translated to the clinics (54, 100–103). Furthermore, several studies suggest that the employed maturation cocktail based on pro-inflammatory cytokines might not have been optimal for the induction of a strong anti-cancer immune response (51). DCs solely activated by these cytokines show only limited capabilities to produce polarizing cytokines that further decrease soon after activation – a phenomenon called exhaustion (51, 104, 105). At the time of injection, DCs thus might have possessed only limited capabilities to induce TH1 cells and CTLs. Additionally, the employed clinical protocols were not suited for multicenter trials leading to highly variable maturation levels and low numbers of generated DCs (100).
Interestingly, in the same year as Schadendorf et al. published their moDC study, Small et al. presented the results of a placebo-controlled phase III trial on DC-based immunotherapy in patients with metastatic asymptomatic hormone refractory prostate cancer (106). In contrast to Schadendorf et al., the authors employed a heterogeneous mixture of readily isolated leukocytes enriched for naturally circulating DCs by gradient centrifugation, thus avoiding long term in vitro culture. The leukocytes were activated and antigen-loaded using a recombinant fusion protein consisting of granulocyte-macrophage colony-stimulating factor and the TAA protein prostatic acid phosphatase. The prepared leukocytes were subsequently injected i.v. – <48 h after isolation. Strikingly, significantly increased overall survival and prolonged time to disease progression could be observed among patients of the treatment arm, thereby proving clinical efficacy of DC-based immunotherapy. Together with supporting studies, these results finally led to the first FDA approval for a cell-based therapy, Provenge®, in 2010 (107).
Naturally Circulating DCs
Inspired by the promising results of the Provenge® trial, we postulated that purified naturally circulating DCs would be superior in anti-cancer immunotherapy against melanoma (51). Not only are these DCs efficient in generating CTLs, they can also be readily isolated from the blood (108, 109). This allows immediate activation and antigen loading, thus avoiding long incubation periods and enabling robust standardization for use in multicenter trials. Therefore, natural DCs, despite their rare occurrence in peripheral blood, display various advantages over moDCs that are making them an attractive target for anti-cancer therapy.
Human naturally circulating DCs can be divided into two main subsets: plasmacytoid DCs (pDCs) and myeloid DCs (mDCs), each with distinct phenotype and function during the immune response (Figure 2) (110). MDCs can be further subdivided in CD1c+ (BDCA1) DCs, CD141+ (BDCA3) DCs, and CD16+ cells, where the latter are considered to be more monocyte-like (111–115). MDCs are specialized in immunity against fungi and bacteria and have an enhanced ability to sense tissue injuries (110, 111). They are able to capture environmental- and cell-associated antigens and show high phagocytic activity (116).
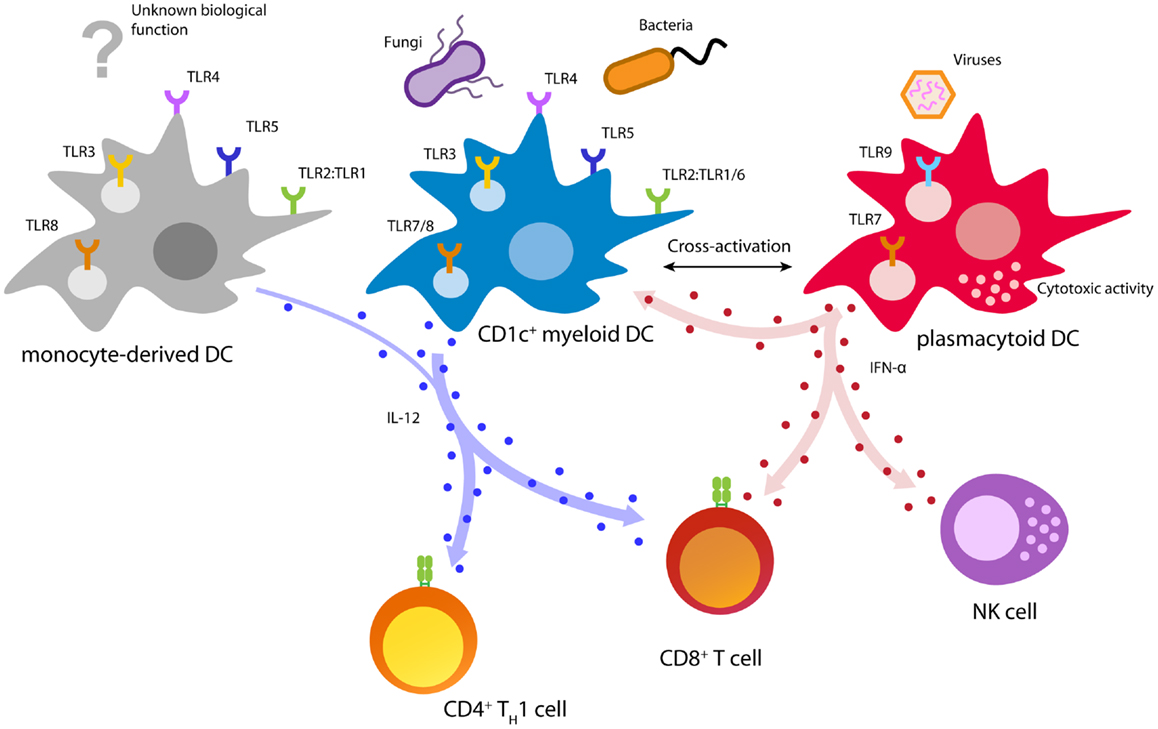
Figure 2. Biology of immunotherapy-relevant human DC subsets. Depicted are major DC functions relevant for pathogen recognition and DC activation, T cell priming, and anti-cancer immunity.
CD141+ DCs are specialized in the detection and uptake of necrotic cells and excel in cross-presenting these antigens to T cells (117–120). Remarkably, CD141+ DCs uniquely express the C-type lectin CLEC9A (DNGR-1), which allows sensing of damaged cells by binding to exposed actin filaments (121, 122). In addition, CD141+ DCs can be activated using a distinct set of TLRs including TLR1, 2, 3, 6, and 8 (117, 123). Especially, TLR3 is strongly expressed and leads to upregulation of co-stimulatory molecules, as well as the secretion of pro-inflammatory cytokines and chemokines (117, 123). Upon activation, CD141+ DCs are able to secrete IFN-γ and IL-12, which allows the effective induction of TH1 and CTL responses (117, 119). However, due to the limited availability in blood and lack of GMP-grade isolation reagents, CD141+ DCs are currently not feasible for cellular immunotherapy. Several developments focusing on improved isolation and culturing, nevertheless, might allow their employment in future DC vaccination.
CD1c+ DCs are responsive to a great variety of microbial and fungal stimuli (124). Triggering of TLRs 1/2/6 by bacterial ligands leads to the activation of CD1c+ DCs and secretion of large amounts of the TH1-skewing cytokine IL-12 (123, 125, 126). Due to their potent antigen processing and presentation machinery, activated CD1c+ DCs are able to induce TH1 cells and cytotoxic T cells leading to a potent cellular immune response (108, 112, 117, 123, 126, 127). Moreover, in vitro studies showed that CD1c+ DCs isolated from healthy donors and prostate cancer patients are able to prime tumor-specific CD8+ T cells (108, 128).
In contrast to mDCs, pDCs are specialized in the detection and control of viral infections (110, 129). Viral infections are rapidly detected by pDCs via the engagement of TLR7 and/or TLR9 (116, 129). TLR triggering by viral agents leads to a rapid burst of type I interferons (IFNs) and induces cytotoxic functions in pDCs as well as natural killer (NK) cells (110, 130, 131). These outstanding antiviral activities make pDCs the key effector cells in early antiviral immunity (110). In a steady state, pDCs are characterized by low expression of MHC class II and co-stimulatory molecules (111). This phenotype is associated with tolerance induction and TH2 immunity, properties that are unfavorable for anti-cancer immunity (132). However, activation of pDCs leads to an upregulation of these proteins, turning pDCs into professional APCs that efficiently prime both, CD4+ and CD8+ T cells (108, 110, 131, 133). The strong release of type I IFN by pDCs leads to an IL-12 independent TH1 polarization characterized by strong secretion of IFN-γ and IL-10 (110, 134–136). Despite low antigen uptake and limited phagocytosis, pDCs isolated from blood, tonsils, and spleen were shown to efficiently cross-present antigens to CD8+ T cells (113, 120, 127, 137). Moreover, several studies reported that pDCs are able to prime potent melanoma-specific CD8+ T cells, which produce IFN-γ and are able to locate to melanoma lesions (108, 120, 138, 139). Finally, pre-clinical mouse models showed that pDCs are able to induce a tumor-specific T cell response in vivo, leading to control of tumor growth (138, 140).
Naturally Circulating DC-Based Immunotherapy
Due to the low occurrence of naturally circulating DCs in blood, conclusive clinical evidence on their usability for immunotherapy is lacking. In 2006, a small-scale study by Davis et al. reported on a vaccine that employed Flt3 ligand (Flt3L)-mobilized naturally circulating DCs (53). The treatment was safe and strong immune responses were detected in several patients. However, the purity of the employed DCs was generally low and, as it turned out, the administration of Flt3L induced the expansion of regulatory T cells in melanoma patients (53, 141).
Encouraged by the promising pre-clinical data, we initiated the first clinical trial on a cellular vaccine based on purified pDC in 2008 at RadboudUMC in the Netherlands (142). PDCs were isolated from leukapheresis products using MACS separation kits and cultured overnight in IL-3. On the next morning, pDCs were activated with a conventional Frühsommer-meningoencephalitis (FSME; English: tick-borne encephalitis) vaccine, which has the benefit of sustained secretion of T cell stimulatory cytokines due to natural triggering of TLRs (143). Subsequently, pDCs were loaded with TAA peptides, and injected intranodally.
Initial tests revealed only mild side effects of pDC vaccinations and the toxicity was even lower as compared to moDC vaccinations (142). Further, pDCs were able to activate the innate immune system, indicated by a systemic type I IFN signature. PDCs were also shown to efficiently migrate to efferent lymph nodes and FSME-specific adaptive immune responses were detected in 14 of 15 enrolled patients. The potent stimulatory capacities of pDCs were reflected in the cancer-specific immune response, as 7 of 15 patients showed increased frequencies of gp100-specific T cells after vaccination. Strikingly, TAA-specific T cell clones with high avidity could be identified after vaccination, indicating the induction of a strong functional response. Nevertheless, the overall magnitude of the induced melanoma-specific immune response appeared to be limited compared to previous moDC vaccination trials, as the total frequency of specific T cells in blood of pDC-vaccine patients was rather low (65, 93). Further analysis of skin-infiltrating lymphocytes obtained from delayed-type hypersensitivity reactions against tumor antigens – a sensitive assay to analyze functionality, migration, and specificity of anti-cancer T cells – showed positive responses in only 2 out of 15 tested patients (142, 144). Despite this, the overall survival of patients treated with pDCs was greatly increased in comparison to matched controls treated with standard chemotherapy. However, assumptions on clinical efficacy have to be taken with caution, as the study was primarily designed to assess the safety and toxicity of pDC-based immunotherapy.
Nevertheless, the prominent survival benefit of vaccinated patients is especially interesting in respect to the low frequency of TAA-specific T cells. Two explanations for this phenomenon are likely: (I) T cells induced by pDCs might be more potent and functional as compared to moDC primed T cells. This could be due to different cytokine secretion patterns, differential expression of co-stimulatory molecules, improved migratory capacities, or prolonged survival. (II) Alternatively, instead of – or in addition to – inducing T cell responses, the focus of pDC-mediated anti-cancer immunity might lie on the activation of NK cells and the innate immune system. Evidence for this comes from the lasting type I IFN signature induced in vaccinated patients (142). Strikingly, various studies report on pDC-dependent, IFN-α-mediated activation of natural DC subsets in arteriosclerosis, autoimmunity, and infections (145–147). Furthermore, it could be shown that type I IFNs are able to activate NK cells, induce IFN-γ secretion, and enhance cytotoxicity (148, 149). However, in comparison to subjects that underwent recombinant IFN-α therapy, patients vaccinated with pDCs showed longer overall survival indicating that the observed clinical benefits were not induced by type I IFNs alone (150–152). Interestingly, it was shown that contact-dependent interactions between pDCs and lymph node DCs greatly enhance Ag presentation and priming of anti-herpes simplex virus CTLs (153). The authors identified CD2–CD2L and CD40–CD40L as key mediators of this effect. PDCs can thus activate other DC subsets, for instance mDCs, to potentiate the immune response. However, this synergy not only acts in one direction: mDCs were shown to mature pDCs and enhance their Ag presentation capabilities during bacterial exposure (116, 154). Interestingly, in one scenario pDCs only act as APCs without instructing T cells with polarizing cytokines (116). Together, these results show that natural DCs of various subsets cooperate with each other to enhance the immune response and that the roles in this regulatory network are variable and depending on the stimulus. However, the studies also indicate a hierarchical organization within natural DC synergies, with one DC subset orchestrating and polarizing the immune response, and the other merely acting as “zombie” APC without instructive capabilities (116).
Strikingly, mouse experiments demonstrated that injection of a mixture of ex vivo activated and antigen-loaded mDCs and pDCs induces a superior immune response against tumors (155). Moreover, therapeutic efficiency, as assessed by overall survival and tumor burden, was greatly improved when mice received simultaneous injections of both subsets compared to injections of one subset alone (155). The observed synergistic effect was mainly based on enhanced antigen presentation by mDCs induced by contact-dependent interactions with pDCs. These observations might explain why patients in our pDC vaccination trial showed significantly increased overall survival despite low frequencies of vaccination-specific CTLs (142, 155). Injected pDCs might have activated mDCs present at the site of injection leading to the induction of a TH1 and CTL response. As the in situ activated mDCs then would present naturally processed melanoma antigens expressed at the site of the tumor, the subsequently induced anti-cancer immune response would not be fully detectable when examining the vaccine-specific T cell response only.
Subsequent to the pDC-based vaccine, we conducted a phase I trial vaccinating metastatic melanoma patients with ex vivo activated and antigen-loaded autologous blood CD1c+ mDCs. Preliminary results confirm the safety and feasibility of mDC-based vaccines and could identify clinical responses in a number of patients (manuscript in preparation). Considering the results of these studies and the synergistic effects of pDCs and mDCs observed in mice and in in vitro models, the next step would be to initialize a human vaccination trial using a cocktail of activated and antigen-loaded mDCs and pDCs. Once injected in, e.g., the lymph node, these natural DC subsets might synergize and potentiate the T cell response.
Importantly, before clinical trials can exploit the synergy between mDCs and pDCs a number of questions need to be addressed: first: what ratio of mDCs and pDCs should be chosen and should one DC subset dominate the immune response? How should both DC subsets be activated in vitro? How does the simultaneous secretion of two different T cell polarizing cytokines (IFN-α by pDCs, IL-12 by mDCs) influence naive T cell priming? And what impact does this have on other immune cells? In addition, does the synergy between mDCs and pDCs also help to induce tumor-specific antibodies by B-cells? Does it increase the anti-cancer activity of the innate immune system?
In vitro studies and pre-clinical mouse models suggest answers to some of these questions. Mouse models, for instance, indicate that activated pDCs need to be cocultured with immature mDCs to induce maximal expression of IL-12 as well as co-stimulatory molecules CD40, CD80, and CD86 (Table 1) (155). This was cell–cell contact-dependent and also crucial for the induction of a superior CD8+ T cell response. Secretion of IFN-α by pDCs did not influence the secretion of IL-12 by mDCs, indicating that mDCs retain their strong TH1 polarizing capacities when administered together with pDCs. In vitro studies on human DCs, however, are not as conclusive and report on both, impaired and increased production of IL-12 by mDCs when cultured in IFN-α supplemented media (156–159). The induction of CD8+ T cells, however, seems to be augmented by the combined effect of IFN-α and IL-12 as comprehensive and lasting immune responses including effector and memory T cells could only be detected when T cells were cocultured with both cytokines (160, 161).
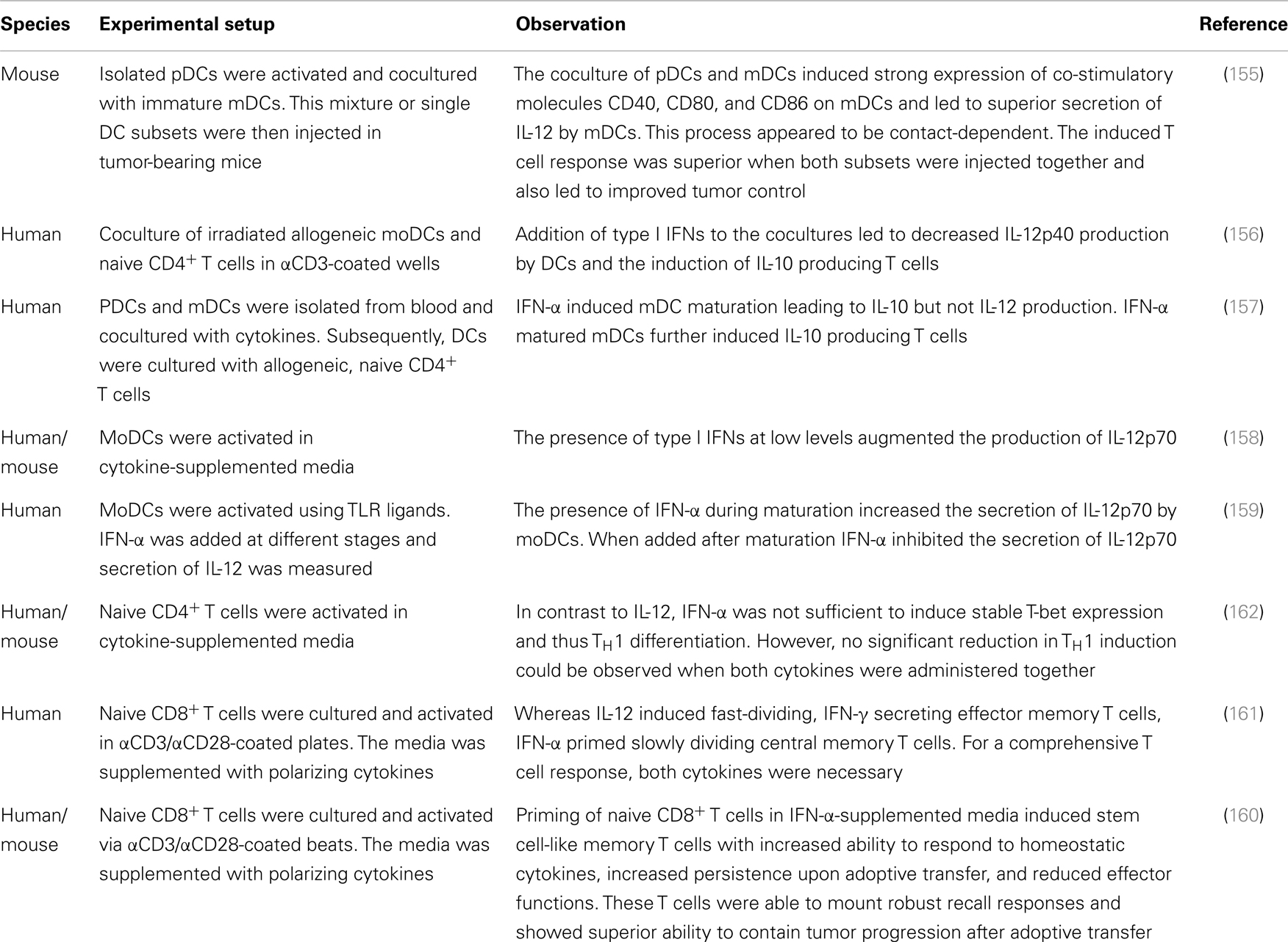
Table 1. Controversial effect of IL-12 and IFN-α on immune activation and T cell priming.
Although many studies report synergistic effects of IFN-α and IL-12 on T cell priming and immune activation, it is hard to predict how these and other factors integrate in the complex microenvironment found in neoplastic lesions of melanoma patients. Following initial clinical trials focusing on safety and feasibility, future studies thus need to explore the interactions between DC subsets in patients and improve various vaccination parameters.
Concluding Remarks
Although randomized clinical trials are needed to further prove the clinical efficacy of vaccination with natural blood DCs, DC therapy has major advantages over treatment with FDA-approved checkpoint inhibitors like ipilimumab, as DC therapy with natural DC is less costly and associated with only very mild side effects. Before anti-cancer therapy with natural DCs can be implemented as standard therapy for melanoma, some issues still need to be overcome. First, DC vaccination, in particular DC vaccination with natural DCs, is currently performed only in a limited number of medical centers. However, the isolation technique with magnetic beads is FDA-approved for stem cell isolation and common practice, thus enabling robust standardization for use in multiple centers in the future. In addition, as it is not feasible yet to perform mRNA electroporation on these rare cells, antigen loading still depends on HLA-binding tumor-peptides, thus excluding patients that do not have the matching HLA-phenotype. Efforts are made to enable peptide-loading for a broader HLA-repertoire, including MHC class II epitopes, to induce broader immune responses and enable inclusion of more patients.
As the field of moDC vaccinations appears to have reached some level of maturity, naturally circulating DC-based vaccinations are just at the beginning of their clinical development. However, the lessons learned from moDC-based vaccination trials will surely contribute to accelerate the development of mDC/pDC-based vaccines, hopefully leading to highly efficient DC-based immunotherapies and benefits for an increasing number of cancer patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Dutch Cancer Society (KUN2010-4722, KUN2009-4402), The Netherlands Organization for Scientific Research (NWO-95103002 and NWO-95100106), the Swedish Research Council, and a Radboud University Medical Center PhD grant. Carl G. Figdor received the NWO Spinoza award and an ERC Adv grant.
Abbreviations
APC, antigen-presenting cell; CTL, cytotoxic T lymphocyte; DC, dendritic cell; FSME, Frühsommer-meningoencephalitis; HLA, human leukocyte antigen; i.d., intradermal; IFN, interferon; IL, interleukin; i.v., intravenous; KLH, keyhole limpet hemocyanin; mDC, myeloid dendritic cell; MHC, major histocompatibility complex; moDC, monocyte-derived dendritic cell; NK cell, natural killer cell; pDC, plasmacytoid dendritic cell; PRR, pathogen recognition receptor; TAA, tumor-associated antigen; TLR, toll-like receptor.
References
1. Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol (2012) 23(Suppl 8):viii10–4. doi: 10.1093/annonc/mds257
2. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Web Page]. Lyon: International Agency for Research on Cancer (2012).
3. Dennis LK. Analysis of the melanoma epidemic, both apparent and real: data from the 1973 through 1994 surveillance, epidemiology, and end results program registry. Arch Dermatol (1999) 135(3):275–80. doi:10.1001/archderm.135.3.275
4. de Vries E, Coebergh JW. Melanoma incidence has risen in Europe. BMJ (2005) 331(7518):698. doi:10.1136/bmj.331.7518.698
5. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol (2009) 27(36):6199–206. doi:10.1200/JCO.2009.23.4799
6. Garbe C, Peris K, Hauschild A, Saiag P, Middleton M, Spatz A, et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline – update 2012. Eur J Cancer (2012) 48(15):2375–90. doi:10.1016/j.ejca.2012.06.013
7. Ko JM, Fisher DE. A new era: melanoma genetics and therapeutics. J Pathol (2011) 223(2):241–50. doi:10.1002/path.2804
8. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364(26):2507–16. doi:10.1056/NEJMoa1103782
9. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med (2012) 366(8):707–14. doi:10.1056/NEJMoa1112302
10. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science (2013) 342(6165):1432–3. doi:10.1126/science.342.6165.1432
11. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas: with a report of ten original cases. Am J Med Sci (1893) 105:487–511. doi:10.1097/00000441-189305000-00001
12. Anichini A, Fossati G, Parmiani G. Clonal analysis of cytotoxic T-lymphocyte response to autologous human metastatic melanoma. Int J Cancer (1985) 35(5):683–9. doi:10.1002/ijc.2910350518
13. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (1991) 254(5038):1643–7. doi:10.1126/science.1840703
14. Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today (1997) 18(4):175–82. doi:10.1016/S0167-5699(97)84664-6
15. Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med (2000) 6(9):1018–23. doi:10.1038/79526
16. Bioley G, Jandus C, Tuyaerts S, Rimoldi D, Kwok WW, Speiser DE, et al. Melan-A/MART-1-specific CD4 T cells in melanoma patients: identification of new epitopes and ex vivo visualization of specific T cells by MHC class II tetramers. J Immunol (2006) 177(10):6769–79.
17. Flamand V, Sornasse T, Thielemans K, Demanet C, Bakkus M, Bazin H, et al. Murine dendritic cells pulsed in vitro with tumor antigen induce tumor resistance in vivo. Eur J Immunol (1994) 24(3):605–10. doi:10.1002/eji.1830240317
18. Clemente CG, Mihm MG, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer (1996) 77(7):1303–10. doi:10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5
19. Haanen JB, Baars A, Gomez R, Weder P, Smits M, de Gruijl TD, et al. Melanoma-specific tumor-infiltrating lymphocytes but not circulating melanoma-specific T cells may predict survival in resected advanced-stage melanoma patients. Cancer Immunol Immunother (2006) 55(4):451–8. doi:10.1007/s00262-005-0018-5
20. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
21. Engell-Noerregaard L, Hansen TH, Andersen MH, Thor Straten P, Svane IM. Review of clinical studies on dendritic cell-based vaccination of patients with malignant melanoma: assessment of correlation between clinical response and vaccine parameters. Cancer Immunol Immunother (2009) 58(1):1–14. doi:10.1007/s00262-008-0568-4
22. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi:10.1016/1074-7613(95)90125-6
23. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270(5238):985–8. doi:10.1126/science.270.5238.985
24. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi:10.1126/science.271.5256.1734
25. Sutmuller RP, van Duivenvoorde LM, van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med (2001) 194(6):823–32. doi:10.1084/jem.194.6.823
26. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
27. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
28. Pardoll DM. Immunology beats cancer: a blueprint for successful translation. Nat Immunol (2012) 13(12):1129–32. doi:10.1038/ni.2392
29. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi:10.1038/nm0902-1039c
30. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi:10.1056/NEJMoa1104621
31. Steinman RM, Gutchinov B, Witmer MD, Nussenzweig MC. Dendritic cells are the principal stimulators of the primary mixed leukocyte reaction in mice. J Exp Med (1983) 157(2):613–27. doi:10.1084/jem.157.2.613
32. Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol (2009) 10(12):1237–44. doi:10.1038/ni.1822
33. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol (2000) 18(1):767–811. doi:10.1146/annurev.immunol.18.1.767
34. Young JW, Steinman RM. Accessory cell requirements for the mixed-leukocyte reaction and polyclonal mitogens, as studied with a new technique for enriching blood dendritic cells. Cell Immunol (1988) 111(1):167–82. doi:10.1016/0008-8749(88)90061-5
35. Markowicz S, Engleman EG. Granulocyte-macrophage colony-stimulating factor promotes differentiation and survival of human peripheral blood dendritic cells in vitro. J Clin Invest (1990) 85(3):955–61. doi:10.1172/JCI114525
36. Mehta-Damani A, Markowicz S, Engleman EG. Generation of antigen-specific CD8+ CTLs from naive precursors. J Immunol (1994) 153(3):996–1003.
37. Takamizawa M, Fagnoni F, Mehta-Damani A, Rivas A, Engleman EG. Cellular and molecular basis of human gamma delta T cell activation. Role of accessory molecules in alloactivation. J Clin Invest (1995) 95(1):296–303. doi:10.1172/JCI117654
38. Inaba K, Metlay JP, Crowley MT, Steinman RM. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J Exp Med (1990) 172(2):631–40. doi:10.1084/jem.172.2.631
39. Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med (1996) 2(1):52–8. doi:10.1038/nm0196-52
40. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med (1994) 179(4):1109–18. doi:10.1084/jem.179.4.1109
41. Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med (1998) 4(3):328–32. doi:10.1038/nm0398-328
42. De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res (2003) 63(1):12–7.
43. Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med (1995) 182(2):389–400. doi:10.1084/jem.182.2.389
44. Jiang WP, Swiggard WJ, Heufler C, Peng M, Mirza A, Steinman RM, et al. The receptor Dec-205 expressed by dendritic cells and thymic epithelial-cells is involved in antigen-processing. Nature (1995) 375(6527):151–5. doi:10.1038/375151a0
45. Engering AJ, Cella M, Fluitsma D, Brockhaus M, Hoefsmit EC, Lanzavecchia A, et al. The mannose receptor functions as a high capacity and broad specificity antigen receptor in human dendritic cells. Eur J Immunol (1997) 27(9):2417–25. doi:10.1002/eji.1830270941
46. Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol (1998) 28(9):2760–9. doi:10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N
47. D’Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, Vecchi A, et al. Uncoupling of inflammatory chemokine receptors by IL-10: generation of functional decoys. Nat Immunol (2000) 1(5):387–91. doi:10.1038/80819
48. Saeki H, Moore AM, Brown MJ, Hwang ST. Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol (1999) 162(5):2472–5.
49. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med (2001) 193(2):233–8. doi:10.1084/jem.193.2.233
50. Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol (1997) 27(12):3135–42. doi:10.1002/eji.1830271209
51. Schreibelt G, Benitez-Ribas D, Schuurhuis D, Lambeck AJ, van Hout-Kuijer M, Schaft N, et al. Commonly used prophylactic vaccines as an alternative for synthetically produced TLR ligands to mature monocyte-derived dendritic cells. Blood (2010) 116(4):564–74. doi:10.1182/blood-2009-11-251884
52. Palucka AK, Ueno H, Connolly J, Kerneis-Norvell F, Blanck JP, Johnston DA, et al. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J Immunother (2006) 29(5):545–57. doi:10.1097/01.cji.0000211309.90621.8b
53. Davis ID, Chen Q, Morris L, Quirk J, Stanley M, Tavarnesi ML, et al. Blood dendritic cells generated with Flt3 ligand and CD40 ligand prime CD8+ T cells efficiently in cancer patients. J Immunother (2006) 29(5):499–511. doi:10.1097/01.cji.0000211299.29632.8c
54. de Vries IJM, Lesterhuis WJ, Scharenborg NM, Engelen LPH, Ruiter DJ, Gerritsen MJP, et al. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin Cancer Res (2003) 9(14):5091–100.
55. Jonuleit H, Giesecke-Tuettenberg A, Tuting T, Schuler-Thurner B, Stuge TB, Paragnik L, et al. A comparison of two types of dendritic cell as adjuvants for the induction of melanoma-specific T-cell responses in humans following intranodal injection. Int J Cancer (2001) 93(2):243–51. doi:10.1002/ijc.1323
56. McIlroy D, Gregoire M. Optimizing dendritic cell-based anticancer immunotherapy: maturation state does have clinical impact. Cancer Immunol Immunother (2003) 52(10):583–91. doi:10.1007/s00262-003-0414-7
57. Schuler-Thurner B, Schultz ES, Berger TG, Weinlich G, Ebner S, Woerl P, et al. Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med (2002) 195(10):1279–88. doi:10.1084/jem.20012100
58. Schuler-Thurner B, Dieckmann D, Keikavoussi P, Bender A, Maczek C, Jonuleit H, et al. Mage-3 and influenza-matrix peptide-specific cytotoxic T cells are inducible in terminal stage HLA-A2.1+ melanoma patients by mature monocyte-derived dendritic cells. J Immunol (2000) 165(6):3492–6.
59. Thurner B, Haendle I, Roder C, Dieckmann D, Keikavoussi P, Jonuleit H, et al. Vaccination with Mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med (1999) 190(11):1669–78. doi:10.1084/jem.190.11.1669
60. Mullins DW, Sheasley SL, Ream RM, Bullock TN, Fu YX, Engelhard VH. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med (2003) 198(7):1023–34. doi:10.1084/jem.20021348
61. Dudda JC, Simon JC, Martin S. Dendritic cell immunization route determines CD8+ T cell trafficking to inflamed skin: role for tissue microenvironment and dendritic cells in establishment of T cell-homing subsets. J Immunol (2004) 172(2):857–63.
62. Fong L, Brockstedt D, Benike C, Wu L, Engleman EG. Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol (2001) 166(6):4254–9.
63. Schuurhuis DH, Verdijk P, Schreibelt G, Aarntzen EH, Scharenborg N, de Boer A, et al. In situ expression of tumor antigens by messenger RNA-electroporated dendritic cells in lymph nodes of melanoma patients. Cancer Res (2009) 69(7):2927–34. doi:10.1158/0008-5472.CAN-08-3920
64. Aarntzen EH, Srinivas M, Bonetto F, Cruz LJ, Verdijk P, Schreibelt G, et al. Targeting of 111In-labeled dendritic cell human vaccines improved by reducing number of cells. Clin Cancer Res (2013) 19(6):1525–33. doi:10.1158/1078-0432.CCR-12-1879
65. Lesterhuis WJ, de Vries IJ, Schreibelt G, Lambeck AJ, Aarntzen EH, Jacobs JF, et al. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific T cells in advanced melanoma patients. Clin Cancer Res (2011) 17(17):5725–35. doi:10.1158/1078-0432.CCR-11-1261
66. Kyte JA, Mu L, Aamdal S, Kvalheim G, Dueland S, Hauser M, et al. Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther (2006) 13(10):905–18. doi:10.1038/sj.cgt.7700961
68. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature (1998) 393(6684):474–8. doi:10.1038/30989
69. Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature (1998) 393(6684):478–80. doi:10.1038/30996
70. Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature (1998) 393(6684):480–3. doi:10.1038/31002
71. Baxevanis CN, Voutsas IF, Tsitsilonis OE, Gritzapis AD, Sotiriadou R, Papamichail M. Tumor-specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J Immunol (2000) 164(7):3902–12.
72. Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature (2003) 421(6925):852–6. doi:10.1038/nature01441
73. Faiola B, Doyle C, Gilboa E, Nair S. Influence of CD4 T cells and the source of major histocompatibility complex class II-restricted peptides on cytotoxic T-cell priming by dendritic cells. Immunology (2002) 106(1):122–3.
74. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med (2000) 192(11):1553–62. doi:10.1084/jem.192.11.1553
75. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi:10.1084/jem.192.11.1545
76. Lorvik KB, Haabeth OA, Clancy T, Bogen B, Corthay A. Molecular profiling of tumor-specific T1 cells activated in vivo. Oncoimmunology (2013) 2(5):e24383. doi:10.4161/onci.24383
77. Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature (2013) 494(7437):361–5. doi:10.1038/nature11824
78. D’Alessandro G, Zardawi I, Grace J, McCarthy WH, Hersey P. Immunohistological evaluation of MHC class I and II antigen expression on nevi and melanoma: relation to biology of melanoma. Pathology (1987) 19(4):339–46. doi:10.3109/00313028709103880
79. Aarntzen EH, De Vries IJ, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, et al. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res (2013) 73(1):19–29. doi:10.1158/0008-5472.CAN-12-1127
80. Duran-Aniotz C, Segal G, Salazar L, Pereda C, Falcon C, Tempio F, et al. The immunological response and post-treatment survival of DC-vaccinated melanoma patients are associated with increased Th1/Th17 and reduced Th3 cytokine responses. Cancer Immunol Immunother (2013) 62(4):761–72. doi:10.1007/s00262-012-1377-3
81. Shimizu K, Thomas EK, Giedlin M, Mule JJ. Enhancement of tumor lysate- and peptide-pulsed dendritic cell-based vaccines by the addition of foreign helper protein. Cancer Res (2001) 61(6):2618–24.
82. Kierstead LS, Ranieri E, Olson W, Brusic V, Sidney J, Sette A, et al. gp100/pmel17 and tyrosinase encode multiple epitopes recognized by Th1-type CD4+T cells. Br J Cancer (2001) 85(11):1738–45. doi:10.1054/bjoc.2001.2160
83. Cochlovius B, Stassar M, Christ O, Raddrizzani L, Hammer J, Mytilineos I, et al. In vitro and in vivo induction of a Th cell response toward peptides of the melanoma-associated glycoprotein 100 protein selected by the TEPITOPE program. J Immunol (2000) 165(8):4731–41.
84. Osen W, Soltek S, Song M, Leuchs B, Steitz J, Tuting T, et al. Screening of human tumor antigens for CD4 T cell epitopes by combination of HLA-transgenic mice, recombinant adenovirus and antigen peptide libraries. PLoS One (2010) 5(11):e14137. doi:10.1371/journal.pone.0014137
85. Schultz ES, Schuler-Thurner B, Stroobant V, Jenne L, Berger TG, Thielemanns K, et al. Functional analysis of tumor-specific Th cell responses detected in melanoma patients after dendritic cell-based immunotherapy. J Immunol (2004) 172(2):1304–10.
86. Tuyaerts S, Michiels A, Corthals J, Bonehill A, Heirman C, de Greef C, et al. Induction of influenza matrix protein 1 and MelanA-specific T lymphocytes in vitro using mRNA-electroporated dendritic cells. Cancer Gene Ther (2003) 10(9):696–706. doi:10.1038/sj.cgt.7700622
87. Markovic SN, Dietz AB, Greiner CW, Maas ML, Butler GW, Padley DJ, et al. Preparing clinical-grade myeloid dendritic cells by electroporation-mediated transfection of in vitro amplified tumor-derived mRNA and safety testing in stage IV malignant melanoma. J Transl Med (2006) 4:35. doi:10.1186/1479-5876-4-35
88. Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, et al. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol (2004) 172(11):6649–57.
89. Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol (2008) 180(1):309–18.
90. Bonehill A, Van Nuffel AM, Corthals J, Tuyaerts S, Heirman C, Francois V, et al. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res (2009) 15(10):3366–75. doi:10.1158/1078-0432.CCR-08-2982
91. Van Nuffel AM, Benteyn D, Wilgenhof S, Pierret L, Corthals J, Heirman C, et al. Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol Ther (2012) 20(5):1063–74. doi:10.1038/mt.2012.11
92. Kyte JA, Gaudernack G. Immuno-gene therapy of cancer with tumour-mRNA transfected dendritic cells. Cancer Immunol Immunother (2006) 55(11):1432–42. doi:10.1007/s00262-006-0161-7
93. Aarntzen EH, Schreibelt G, Bol K, Lesterhuis WJ, Croockewit AJ, de Wilt JH, et al. Vaccination with mRNA-electroporated dendritic cells induces robust tumor antigen-specific CD4+ and CD8+ T cells responses in stage III and IV melanoma patients. Clin Cancer Res (2012) 18(19):5460–70. doi:10.1158/1078-0432.CCR-11-3368
94. Van Nuffel AM, Benteyn D, Wilgenhof S, Corthals J, Heirman C, Neyns B, et al. Intravenous and intradermal TriMix-dendritic cell therapy results in a broad T-cell response and durable tumor response in a chemorefractory stage IV-M1c melanoma patient. Cancer Immunol Immunother (2012) 61(7):1033–43. doi:10.1007/s00262-011-1176-2
95. Wilgenhof S, Van Nuffel AM, Corthals J, Heirman C, Tuyaerts S, Benteyn D, et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother (2011) 34(5):448–56. doi:10.1097/CJI.0b013e31821dcb31
96. Bakker AB, van der Burg SH, Huijbens RJ, Drijfhout JW, Melief CJ, Adema GJ, et al. Analogues of CTL epitopes with improved MHC class-I binding capacity elicit anti-melanoma CTL recognizing the wild-type epitope. Int J Cancer (1997) 70(3):302–9. doi:10.1002/(SICI)1097-0215(19970127)70:3<302::AID-IJC10>3.0.CO;2-H
97. Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol (1996) 157(6):2539–48.
98. Lesterhuis WJ, Schreibelt G, Scharenborg NM, Brouwer HM, Gerritsen MJ, Croockewit S, et al. Wild-type and modified gp100 peptide-pulsed dendritic cell vaccination of advanced melanoma patients can lead to long-term clinical responses independent of the peptide used. Cancer Immunol Immunother (2011) 60(2):249–60. doi:10.1007/s00262-010-0942-x
99. Stuge TB, Holmes SP, Saharan S, Tuettenberg A, Roederer M, Weber JS, et al. Diversity and recognition efficiency of T cell responses to cancer. PLoS Med (2004) 1(2):e28. doi:10.1371/journal.pmed.0010028
100. Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB, et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol (2006) 17(4):563–70. doi:10.1093/annonc/mdj138
101. Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy – revisited. Nat Rev Drug Discov (2011) 10(8):591–600. doi:10.1038/nrd3500
102. Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, et al. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol (2004) 5(11):1143–8. doi:10.1038/ni1129
103. Ridolfi R, Riccobon A, Galassi R, Giorgetti G, Petrini M, Fiammenghi L, et al. Evaluation of in vivo labelled dendritic cell migration in cancer patients. J Transl Med (2004) 2(1):27. doi:10.1186/1479-5876-2-27
104. Kalinski P, Schuitemaker JH, Hilkens CM, Wierenga EA, Kapsenberg ML. Final maturation of dendritic cells is associated with impaired responsiveness to IFN-gamma and to bacterial IL-12 inducers: decreased ability of mature dendritic cells to produce IL-12 during the interaction with Th cells. J Immunol (1999) 162(6):3231–6.
105. Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol (2000) 1(4):311–6. doi:10.1038/79758
106. Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol (2006) 24(19):3089–94. doi:10.1200/JCO.2005.04.5252
107. Malarkey MA, Witten CM. Approval Letter – Provenge [Web Page]. Silver Spring, MD: U.S. Food and Drug Administration (2012).
108. Tel J, Schreibelt G, Sittig SP, Mathan TS, Buschow SI, Cruz LJ, et al. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood (2013) 121(3):459–67. doi:10.1182/blood-2012-06-435644
109. Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, et al. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J Immunol (2000) 165(11):6037–46.
110. Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell (2001) 106(3):259–62. doi:10.1016/S0092-8674(01)00456-1
111. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31(1):563–604. doi:10.1146/annurev-immunol-020711-074950
112. MacDonald KP, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DN. Characterization of human blood dendritic cell subsets. Blood (2002) 100(13):4512–20. doi:10.1182/blood-2001-11-0097
113. Mittag D, Proietto AI, Loudovaris T, Mannering SI, Vremec D, Shortman K, et al. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J Immunol (2011) 186(11):6207–17. doi:10.4049/jimmunol.1002632
114. Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol (2007) 81(3):584–92. doi:10.1189/jlb.0806510
115. Skrzeczynska-Moncznik J, Bzowska M, Loseke S, Grage-Griebenow E, Zembala M, Pryjma J. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand J Immunol (2008) 67(2):152–9. doi:10.1111/j.1365-3083.2007.02051.x
116. Piccioli D, Sammicheli C, Tavarini S, Nuti S, Frigimelica E, Manetti AG, et al. Human plasmacytoid dendritic cells are unresponsive to bacterial stimulation and require a novel type of cooperation with myeloid dendritic cells for maturation. Blood (2009) 113(18):4232–9. doi:10.1182/blood-2008-10-186890
117. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med (2010) 207(6):1247–60. doi:10.1084/jem.20092140
118. Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med (2010) 207(6):1273–81. doi:10.1084/jem.20100348
119. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207(6):1261–71. doi:10.1084/jem.20092618
120. Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med (2013) 210(5):1035–47. doi:10.1084/jem.20121103
121. Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature (2009) 458(7240):899–903. doi:10.1038/nature07750
122. Zhang JG, Czabotar PE, Policheni AN, Caminschi I, Wan SS, Kitsoulis S, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity (2012) 36(4):646–57. doi:10.1016/j.immuni.2012.03.009
123. Hemont C, Neel A, Heslan M, Braudeau C, Josien R. Human blood mDC subsets exhibit distinct TLR repertoire and responsiveness. J Leukoc Biol (2013) 93(4):599–609. doi:10.1189/jlb.0912452
124. Piccioli D, Tavarini S, Borgogni E, Steri V, Nuti S, Sammicheli C, et al. Functional specialization of human circulating CD16 and CD1c myeloid dendritic-cell subsets. Blood (2007) 109(12):5371–9. doi:10.1182/blood-2006-08-038422
125. Schreibelt G, Tel J, Sliepen KH, Benitez-Ribas D, Figdor CG, Adema GJ, et al. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol Immunother (2010) 59(10):1573–82. doi:10.1007/s00262-010-0833-1
126. Nizzoli G, Krietsch J, Weick A, Steinfelder S, Facciotti F, Gruarin P, et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood (2013) 122(6):932–42. doi:10.1182/blood-2013-04-495424
127. Hoeffel G, Ripoche AC, Matheoud D, Nascimbeni M, Escriou N, Lebon P, et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity (2007) 27(3):481–92. doi:10.1016/j.immuni.2007.07.021
128. Wilkinson R, Kassianos AJ, Swindle P, Hart DN, Radford KJ. Numerical and functional assessment of blood dendritic cells in prostate cancer patients. Prostate (2006) 66(2):180–92. doi:10.1002/pros.20333
129. Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells – virus experts of innate immunity. Semin Immunol (2005) 17(4):253–61. doi:10.1016/j.smim.2005.05.008
130. Mathan TS, Figdor CG, Buschow SI. Human plasmacytoid dendritic cells: from molecules to intercellular communication network. Front Immunol (2013) 4:372. doi:10.3389/fimmu.2013.00372
131. Tel J, Smits EL, Anguille S, Joshi RN, Figdor CG, de Vries IJ. Human plasmacytoid dendritic cells are equipped with antigen-presenting and tumoricidal capacities. Blood (2012) 120(19):3936–44. doi:10.1182/blood-2012-06-435941
132. Kadowaki N, Antonenko S, Lau JY, Liu YJ. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med (2000) 192(2):219–26. doi:10.1084/jem.192.2.219
133. Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, et al. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood (2003) 101(9):3520–6. doi:10.1182/blood-2002-10-3063
134. Ito T, Yang M, Wang YH, Lande R, Gregorio J, Perng OA, et al. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med (2007) 204(1):105–15. doi:10.1084/jem.20061660
135. Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol (2000) 1(4):305–10. doi:10.1038/79747
136. Farkas L, Kvale EO, Johansen FE, Jahnsen FL, Lund-Johansen F. Plasmacytoid dendritic cells activate allergen-specific TH2 memory cells: modulation by CpG oligodeoxynucleotides. J Allergy Clin Immunol (2004) 114(2):436–43. doi:10.1016/j.jaci.2004.04.035
137. Di Pucchio T, Chatterjee B, Smed-Sorensen A, Clayton S, Palazzo A, Montes M, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol (2008) 9(5):551–7. doi:10.1038/ni.1602
138. Salio M, Palmowski MJ, Atzberger A, Hermans IF, Cerundolo V. CpG-matured murine plasmacytoid dendritic cells are capable of in vivo priming of functional CD8 T cell responses to endogenous but not exogenous antigens. J Exp Med (2004) 199(4):567–79. doi:10.1084/jem.20031059
139. Tel J, Anguille S, Waterborg CE, Smits EL, Figdor CG, de Vries IJ. Tumoricidal activity of human dendritic cells. Trends Immunol (2014) 35(1):38–46. doi:10.1016/j.it.2013.10.007
140. Schlecht G, Garcia S, Escriou N, Freitas AA, Leclerc C, Dadaglio G. Murine plasmacytoid dendritic cells induce effector/memory CD8+ T-cell responses in vivo after viral stimulation. Blood (2004) 104(6):1808–15. doi:10.1182/blood-2004-02-0426
141. Klein O, Ebert LM, Zanker D, Woods K, Tan BS, Fucikova J, et al. Flt3 ligand expands CD4+ FoxP3+ regulatory T cells in human subjects. Eur J Immunol (2013) 43(2):533–9. doi:10.1002/eji.201242603
142. Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res (2013) 73(3):1063–75. doi:10.1158/0008-5472.CAN-12-2583
143. de Vries IJ, Tel J, Benitez-Ribas D, Torensma R, Figdor CG. Prophylactic vaccines mimic synthetic CpG oligonucleotides in their ability to modulate immune responses. Mol Immunol (2011) 48(6–7):810–7. doi:10.1016/j.molimm.2010.12.022
144. Aarntzen EH, Bol K, Schreibelt G, Jacobs JF, Lesterhuis WJ, Van Rossum MM, et al. Skin-test infiltrating lymphocytes early predict clinical outcome of dendritic cell-based vaccination in metastatic melanoma. Cancer Res (2012) 72(23):6102–10. doi:10.1158/0008-5472.CAN-12-2479
145. Niessner A, Shin MS, Pryshchep O, Goronzy JJ, Chaikof EL, Weyand CM. Synergistic proinflammatory effects of the antiviral cytokine interferon-alpha and toll-like receptor 4 ligands in the atherosclerotic plaque. Circulation (2007) 116(18):2043–52. doi:10.1161/CIRCULATIONAHA.107.697789
146. Kramer M, Schulte BM, Eleveld-Trancikova D, van Hout-Kuijer M, Toonen LW, Tel J, et al. Cross-talk between human dendritic cell subsets influences expression of RNA sensors and inhibits picornavirus infection. J Innate Immun (2010) 2(4):360–70. doi:10.1159/000300568
147. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science (2001) 294(5546):1540–3. doi:10.1126/science.1064890
148. Romagnani C, Della Chiesa M, Kohler S, Moewes B, Radbruch A, Moretta L, et al. Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T helper cells and CD4+ CD25hi T regulatory cells. Eur J Immunol (2005) 35(8):2452–8. doi:10.1002/eji.200526069
149. Benlahrech A, Donaghy H, Rozis G, Goodier M, Klavinskis L, Gotch F, et al. Human NK cell up-regulation of CD69, HLA-DR, interferon gamma secretion and cytotoxic activity by plasmacytoid dendritic cells is regulated through overlapping but different pathways. Sensors (Basel) (2009) 9(1):386–403. doi:10.3390/s90100386
150. Kirkwood JM, Ernstoff MS. Role of interferons in the therapy of melanoma. J Invest Dermatol (1990) 95(6 Suppl):180S–4S. doi:10.1111/1523-1747.ep12875497
151. Creagan ET, Ahmann DL, Green SJ, Long HJ, Rubin J, Schutt AJ, et al. Phase II study of recombinant leukocyte A interferon (rIFN-alpha A) in disseminated malignant melanoma. Cancer (1984) 54(12):2844–9. doi:10.1002/1097-0142(19841215)54:12<2844::AID-CNCR2820541205>3.0.CO;2-Q
152. Legha SS, Papadopoulos NE, Plager C, Ring S, Chawla SP, Evans LM, et al. Clinical evaluation of recombinant interferon alfa-2a (roferon-A) in metastatic melanoma using two different schedules. J Clin Oncol (1987) 5(8):1240–6.
153. Yoneyama H, Matsuno K, Toda E, Nishiwaki T, Matsuo N, Nakano A, et al. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J Exp Med (2005) 202(3):425–35. doi:10.1084/jem.20041961
154. Lozza L, Farinacci M, Fae K, Bechtle M, Staber M, Dorhoi A, et al. Crosstalk between human DC subsets promotes antibacterial activity and CD8+ T-cell stimulation in response to bacille Calmette-Guerin. Eur J Immunol (2014) 44(1):80–92. doi:10.1002/eji.201343797
155. Lou Y, Liu C, Kim GJ, Liu YJ, Hwu P, Wang G. Plasmacytoid dendritic cells synergize with myeloid dendritic cells in the induction of antigen-specific antitumor immune responses. J Immunol (2007) 178(3):1534–41.
156. McRae BL, Semnani RT, Hayes MP, van Seventer GA. Type I IFNs inhibit human dendritic cell IL-12 production and Th1 cell development. J Immunol (1998) 160(9):4298–304.
157. Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol (2001) 166(5):2961–9.
158. Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med (2005) 201(9):1435–46. doi:10.1084/jem.20041964
159. Heystek HC, den Drijver B, Kapsenberg ML, van Lier RA, de Jong EC. Type I IFNs differentially modulate IL-12p70 production by human dendritic cells depending on the maturation status of the cells and counteract IFN-gamma-mediated signaling. Clin Immunol (2003) 107(3):170–7. doi:10.1016/S1521-6616(03)00060-3
160. Hervas-Stubbs S, Mancheno U, Riezu-Boj JI, Larraga A, Ochoa MC, Alignani D, et al. CD8 T cell priming in the presence of IFN-alpha renders CTLs with improved responsiveness to homeostatic cytokines and recall antigens: important traits for adoptive T cell therapy. J Immunol (2012) 189(7):3299–310. doi:10.4049/jimmunol.1102495
161. Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood (2009) 113(22):5516–25. doi:10.1182/blood-2008-11-188458
Keywords: dendritic cell vaccination, immunotherapy, naturally circulating dendritic cells, melanoma, monocyte-derived dendritic cells, plasmacytoid dendritic cells, myeloid dendritic cells
Citation: Wimmers F, Schreibelt G, Sköld AE, Figdor CG and De Vries IJM (2014) Paradigm shift in dendritic cell-based immunotherapy: from in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front. Immunol. 5:165. doi: 10.3389/fimmu.2014.00165
Received: 03 February 2014; Paper pending published: 01 March 2014;
Accepted: 28 March 2014; Published online: 11 April 2014.
Edited by:
Marianne Boes, University Medical Centre Utrecht, NetherlandsCopyright: © 2014 Wimmers, Schreibelt, Sköld, Figdor and De Vries. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: I. Jolanda M. De Vries, Department of Tumor Immunology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Geert Grooteplein 26, P.O. Box 9101, Nijmegen 6500 HB, Netherlands e-mail:ai5kZXZyaWVzQG5jbWxzLnJ1Lm5s