 Shengnan Zhao
Shengnan Zhao Ling Li
Ling Li- Department of Pediatric Neurology, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, China
Streptococcus pneumoniae meningitis is a life-threatening bacterial infection of the central nervous system (CNS), and its unfavorable prognosis usually results from an intense inflammatory response. Recent studies have shown that brain-derived neurotrophic factor (BDNF) mediates anti-inflammatory and neuroprotective effects in CNS diseases; however, the distinct contribution of BDNF to pneumococcal meningitis (PM) remains unknown. In this study, we sought to investigate the effects of endogenous BDNF on the inflammatory response and brain damage in experimental PM. We used Camk2a-CreERT2 mice to delete Bdnf from the cerebral cortex and hippocampus, and meningitis was induced by intracisternal infection with S. pneumoniae. Clinical parameters were assessed during acute meningitis. At 24 h post-infection, histopathology, neutrophil granulocytes infiltration, and microglia/macrophage proliferation of brain tissues were evaluated. Additionally, cortical damage and hippocampal apoptosis were assessed using Nissl staining and terminal deoxynucleotidyl transferase dUTP-nick-end labeling (TUNEL), respectively. Pro-inflammatory cytokine levels were determined using real-time polymerase chain reaction (RT-PCR). Key molecules associated with the related signaling pathways were analyzed by RT-PCR and western blot. To investigate the role of microglia/macrophage in infected BDNF conditional knockout mice, GW2580 was used for microglia/macrophage depletion. Here, we, for the first time, found that BDNF conditional knockouts exhibited more profound clinical impairment, pathological severity, and neuron injury and enhanced microglia/macrophage proliferation than were observed in their littermate controls. Furthermore, the BDNF conditional knockouts showed an obviously increase in the expression of pro-inflammatory factors (Tnf-α, Il-1β, and Il-6). Mechanistically, loss of BDNF activated TLR2- and NOD2-mediated downstream nuclear factor kappa B (NF-κB) p65 and p38 mitogen-activated protein kinase (MAPK) pathways associated with S. pneumoniae infection. Furthermore, targeted depletion of microglia/macrophage population decreased the resistance of mice to PM with diminishing neuroinflammation in BDNF conditional knockouts. Our findings suggest that loss of BDNF may enhance the inflammatory response and contribute to brain injury during PM at least partially by modulating TLR2- and NOD2-mediated signaling pathways, thereby providing a potential therapeutic target for future interventions in bacterial meningitis pathologies.
Introduction
Bacterial meningitis (BM) is a serious infection of the central nervous system (CNS) characterized by inflammation of the meninges and subarachnoid space (1). The most common causative pathogen of BM is Streptococcus pneumoniae, which accounts for nearly half of total cases (2). Even with conjugate vaccines and timely antibiotic therapy, pneumococcal meningitis (PM) has a high mortality rate ranging from 20 to 51% in the worldwide (3). About 50% survivors may present with brain damage and neurological deficits, such as sensory-motor deficits, hearing loss, aphasia, epilepsy, and cognitive impairment (4). These neurofunctional consequences during BM correlate with neuronal apoptosis, particularly in the hippocampus dentate gyrus (DG) (5). It has become evident that the development and progression of PM and the extensive tissue damage that occurs during infection are not simply due to the infectious agent itself but are also largely dependent on the intense activation of the host immune response (6). Bacterial lysis such as peptidoglycan, cell wall fragments generated during antibiotics treatment is highly immunogenic and may mediate neuronal cell injury and death by promoting the production of pro-inflammatory factors such as tumor necrosis factor (TNF)-α and interleukin (IL)−1β that exacerbate neurodegenerative pathology (7, 8). Possible therapeutic approaches to block TNF-α and/or IL-1β release are investigated in animal models of BM (9, 10).
Although the exact mechanism underlying immune activation in PM remains largely unclear, recent studies have indicated that pattern-recognition receptors (PRRs) can induce the release of inflammatory cascades that initiate brain immune damage (6, 11, 12). Toll-like receptor 2 (TLR2) recognizes various bacterial compounds and is identified as a main PRR involved in the detection of microbial infection (13). The activation of TLR2 results in the recruitment of the downstream signaling molecule myeloid differentiation factor 88 (MyD88), which subsequently upregulates the production of pro-inflammatory mediators (14). More importantly, TLR2 was involved in the pathogenesis of brain injury, and enhanced inflammation and associated hippocampal apoptosis were found after administration of a TLR2 agonist and pneumococci in wildtype mice but not in TLR2 knockout mice (15). We previously showed that MyD88-deficient mice display a markedly diminished inflammatory reaction in PM (16). In addition, pathogen-associated molecular patterns (PAMPs) are detected intracellularly by cytosolic receptors via nucleotide-binding oligomerization domain 2 (NOD2) (17). More importantly, emerging studies suggest the pivotal role of NOD2 activation in macrophage recruitment, phagocytosis, and S. pneumoniae clearance, while NOD2 deficiency results in reduced inflammatory reactions after S. pneumoniae infection in vivo and in vitro (18, 19). The activation of both TLR2 and NOD2 trigger the production of inflammatory cytokines, mainly including tumor TNF-α, IL-1β, and IL-6, via stimulation of the nuclear translocation of nuclear factor kappa B (NF-κB) as well as mitogen-activated protein kinases (MAPKs) (20, 21). Mounting evidence has shown that phosphorylated p38 MAPK and NF-κB p65 are involved in the S. pneumoniae induced expression of inflammatory mediators in the brain (22, 23). This overwhelming inflammatory response could cause severe injury to the brain, thus resulting in the frequently unfavorable prognosis reported during the progression of PM (24, 25). Hence, new adjunctive therapies capable of reducing inflammation and brain damage are needed to improve outcomes in PM.
Brain-derived neurotrophic factor (BDNF) is mainly distributed in the cerebral cortex and hippocampus and is a key member of the neurotrophin family that has demonstrated an ability to promote neural survival, development, differentiation, and plasticity in the CNS (26). There is increasing evidence from animal experiments and clinical studies indicating that BDNF exerts neuroprotective effects on bacterial meningitis. For example, in our previous study, we reported that the level of BDNF increased following administration of dexamethasone and further reduced mortality and sequelae in PM (27). Similarly, the serum and cerebrospinal fluid (CSF) BDNF levels were also increased in pediatric patients with bacterial meningitis (28). Furthermore, pretreatment with exogenous BDNF could improve the neurogenesis of endogenous neural stem cells (NSCs) in the hippocampus in a rat model of PM (29). Conversely, Barichello et al. (30) found that downregulating BDNF in the hippocampus following the long-term phase of meningitis was associated with behavioral deficits in experimental PM. In addition to its neuroprotective effects, we previously found that administration of exogenous BDNF suppressed the expression of inflammatory mediators after S. pneumoniae meningitis (31). A recent study demonstrated that BDNF prevented ischemia-induced apoptosis and inflammation in vivo by suppressing the TLRs/MyD88 signaling pathway (32). Therefore, BDNF has the double function in that it reduces both neuroinflammation and neurological sequelae and might constitute a promising adjuvant treatment for PM. We hypothesized that the loss of BDNF might contribute to an uncontrolled inflammatory response and subsequent brain damage. The early neonatal lethality of BDNF homozygous null knockouts mice prevents their use in further studies (33). To circumvent the neurodevelopmental problems associated with BDNF absence, we crossed floxed Bdnf mice with the Camk2a-driven CreERT2 strain to selectively delete BDNF in the cerebral cortex and hippocampus (34, 35). In this study, we investigate whether endogenous BDNF modulates localized inflammation and subsequent brain damage in the infected brain. We further clarify whether TLR2- and NOD2-mediated signaling pathways are involved in these processes. We report here that BDNF knockout results in the augmented activation of the TLR2 and NOD2 downstream molecules NF-κB p65 and p38 MAPK to significantly enhance the inflammatory response and brain injury in a murine model of PM.
Materials and Methods
Transgenic Mice and Experimental Design
Camk2a-CreERT2 mice express tamoxifen-inducible modified Cre recombinase in the neural tissues (including the cortex, hippocampus, and other structures). Homozygous floxed Bdnf mice (Bdnf F/F; 021055, Jackson Laboratories) in a C57BL/6 background possess loxP sites flanking exon 5 in the Bdnf gene and are viable but obese and poor breeders. To produce forebrain-restricted BDNF mutant mice, BdnfF/+ mice were interbred with Camk2a-CreERT2 mice to obtain BdnfF/+; Camk2a-CreERT2 mice and these mice were mated to BdnfF/+mice to obtain Bdnf conditional knockout mice (Camk2a-CreERT2/BdnfF/F; Cre+/FF) as well as control mice (BdnfF/F; Cre-/FF). Genotyping of mice for BdnfF/F and Camk2a-CreERT2 was performed by standard PCR using the following primers: Camk2a-CreERT2, (forward) 5′ACCTGGATGCTGACGAAGGC3′ and (reverse) 5′CGGTTATTCAACTTGCACCA3′; and Bdnf, (forward) 5′ GGTCTGAAATTACAAGCAGATGG3' and (reverse) 5′TGTCCGTGGACGTTTACTTCT3′. PCR products were loaded on agarose gels to check for the presence of the Camk2a-CreERT2 transgene (306 bp), the wild type Bdnf allele (198 bp), the mutant allele (~250 bp), and the heterozygote allele (198 bp and ~250 bp). Mice were injected intraperitoneally with the exogenous estrogen-receptor ligand tamoxifen (Sigma-Aldrich, St. Louis, MO, USA, catalog #T-5648) once per day for 5 consecutive days; then, mice were given a 2-week break before treatment sessions. The Cre-/FF littermate controls received the same treatment in parallel. The 6–8-week-old Bdnf conditional knockout mice and controls in a C57BL/6 background were used in our experiments. All experiments used both sex of mice at ~50:50 ratio. Before establishing the animal model of PM, 6 Cre+/FF mice and 6 Cre-/FF mice were used to determine BDNF expression in cortex, hippocampus, and brainstem. Animals were divided into the following groups (n = 8 per group): (1) Cre-/FF mice injected intracisternally with normal saline (NS); (2) Cre-/FF mice injected intracisternally with an S. pneumoniae suspension; (3) Cre+/FF mice injected intracisternally with NS; (4) Cre+/FF mice injected intracisternally with an S. pneumoniae suspension. The animals were housed at 23°C with 60% humidity under a 12 h/12 h light/dark cycle with food and tap water available ad libitum. The animal experiments were approved by the Animal Ethical and Welfare Committee of Xinhua Hospital affiliated with Shanghai Jiaotong University School of Medicine.
Infecting Organisms
The standard serotype 3 strain S. pneumoniae (American Type Culture Collection, Manassas, VA, USA) was cultured on a sheep blood agar plate for 18 h followed by inoculation in VITAL AER broth overnight at 37°C in air with 5% CO2 to reach the logarithmic phase. The culture broth was centrifuged for 20 min at 5,000 g and resuspended twice in sterile saline to reach an approximate density of 1 × 104 colony forming units (cfu)/ml using a nephelometer (Bio-Merieux, Marcy-l'Étoile, France).
Mouse Model of S. pneumoniae Meningitis
The operation performed to induce S. pneumoniae meningitis has been previously described (16). Briefly, after all mice were anesthetized intraperitoneally with pentobarbital sodium (50 mg/kg), they were intracisternally injected with a 10 μl volume containing either 1 × 104 cfu/ml viable S. pneumoniae or sterile saline. Briefly, animals were immobilized on a stereotaxic device under anesthesia. A natural indentation of cisterna magna can usually be touched on the midline of craniocervical junction where the microinjector is most likely to enter the occipital hole. At 24 h post-infection, the mice were weighed, and the severity of the disease was evaluated by assigning a clinical score in a blinded manner. For the clinical score, no apparent behavioral abnormality was scored as 0, moderate lethargy (an apparent decrease in spontaneous activity) as 1, severe lethargy (rare spontaneous movements, but walking after stimulation by the investigator) as 2, unable to walk as 3, and dead as 4 (16, 36). After clinical evaluation, the animals were anesthetized and perfused through the left ventricle with 50 ml of pyrogen-free saline. Bacterial titers were determined in samples of cerebellar homogenates by plating serial dilutions on the sheep blood agar plates under 37°C and 5% CO2 environments overnight. The brains were segment into two hemispheres. The left hemispheres were fixed in 4% paraformaldehyde overnight at 4°C for histological analysis. The brain tissues separated from the right hemispheres were frozen immediately and stored at −80°C for proteins and RNA preparation.
In vivo Microglia/Macrophages Depletion
To evaluate the role of microglia/macrophages proliferation in the worsen outcomes caused by BDNF deficiency, GW2580 (Selleckchem, Houston, TX, USA, catalog #S8042), a selective inhibitor of the colony stimulating factor-1 (CSF-1) receptor kinase was used for microglia/macrophages depletion. GW2580 was dissolved in 0.5% hydroxypropyl methylcellulose and 0.1% Tween 80. Briefly, BDNF conditional knockout mice were divided into the following groups (n = 10 per group): (1) NS+0.5% hydroxypropyl methylcellulose/0.1% Tween 80; (2) PM+0.5% hydroxypropyl methylcellulose/0.1% Tween 80; (3) PM+GW2580; (4) NS+GW2580. PM was induced in BDNF conditional knockouts with 1 × 104 cfu/ml S. pneumoniae as described above. Mice were treated with GW2580 at 80 mg/kg by oral gavage 1 day prior infection and every 12 h after S. pneumoniae infection until the animals were sacrificed 24 h later.
Tissue Histopathological Evaluation
Paraffin-embedded brain tissues were sectioned coronally through the entire dentate gyrus using a microtome (4–6 μm thick). Brain inflammation and injury were analyzed by hematoxylin & eosin (H&E; Beyotime Biotechnology, Beijing, China, catalog #C0105), double-label immunofluorescence staining of Ly6G/IBA-1, and Nissl staining (Servicebio, Wuhan, China, catalog #G1036) according to the directions provided by the manufacturer. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) immunofluorescence staining was performed using an in-situ Cell Death Detection Kit (Roche, Basel, Switzerland, catalog #11684809910) to examine apoptosis-like cell death in the hippocampus DG. The numbers of TUNEL-stained nuclei and surviving neurons were counted blindly by two investigators.
Immunohistochemical Staining
Immunohistochemical staining was performed to quantify BDNF, Ly6G and IBA-1 in the hippocampus and cerebral cortex. Briefly, brain sections were incubated with primary antibodies, including rabbit polyclonal anti-BDNF (1:800, Servicebio, Wuhan, China, catalog #GB11559), rabbit polyclonal anti-Ly6G (1:500, Servicebio, Wuhan, China, catalog #GB11229) or goat polyclonal anti-IBA-1 (1:500, Abcam, Cambridge, UK, catalog #ab5076) antibodies, overnight at 4°C. After three washes in phosphate-buffered saline (PBS) for 5 min each, the sections were incubated with secondary antibodies, including goat anti-rabbit IgG and donkey anti-goat IgG (Abcam, Cambridge, UK, catalog #ab150077, #ab150129), for 50 min at room temperature in a dark room and then washed twice with PBS. Finally, the sections were incubated with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime Biotechnology, Beijing, China, catalog #C1002) for 10 min. Then, the sections were rinsed and visualized using a fluorescence microscope (Nikon, Japan).
RNA Extraction and Real-Time Polymerase Chain Reaction (RT-PCR)
The brain tissues, including the hippocampus and cerebral cortex, were lysed, and RNA was extracted using a Total RNA Kit (TaKaRa, Shiga, Japan, catalog #9767) according to the manufacturer's protocol. The RNA was reverse-transcribed into cDNA using a PrimeScript One Step RT-PCR kit (TaKaRa, Shiga, Japan, catalog #RR036A). Subsequently, real-time PCR analyses were performed using a SYBR Premix Dimmer Eraser kit (TaKaRa, Shiga, Japan, catalog #RR420A) to assess cDNA template amplification on an ABI7500 system (Applied Biosystems, Carlsbad, CA, USA). The gene-specific primer sequences used in this study are listed in Table 1. The results for targeted gene expression in each sample were normalized to β-actin expression, and the 2−ΔΔCt method was used to calculate the relative expression of messenger RNA (mRNA).
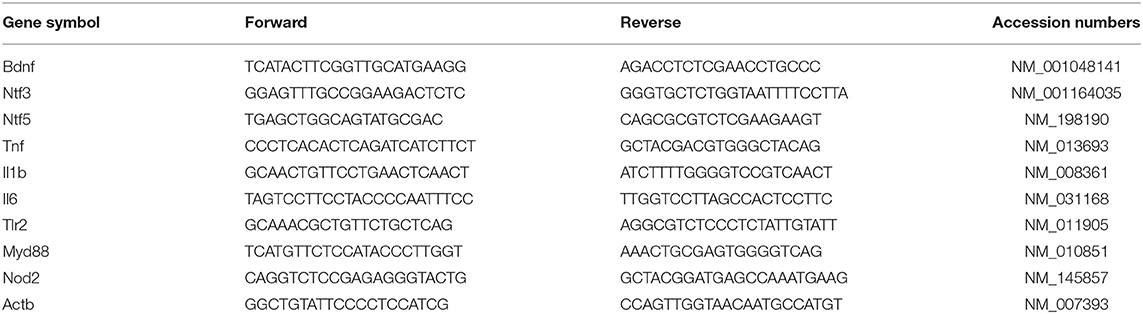
Table 1. The primers used in real-time PCR.
Western Blotting
Brain tissues (hippocampus and cortex) were sonicated (amplitude 38%, ultrasound 5 s, stop 3 s) in protein lysis buffer (Sigma-Aldrich, St. Louis, MO, USA, catalog #C3228) containing a Halt™ Protease Inhibitor Cocktail (Thermo Scientific, Waltham, MA, USA, catalog #87786) under ice water for five times and centrifuged at 12,000 g for 15 min at 4°C. The total protein concentration of each sample was determined using a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA, catalog #23227) according to the manufacturer's instructions. Protein samples (50 μg) were resolved by 10% sodium dodecyl sulfate polyacrylamide (SDS-PAGE) gels and then transferred to polyvinylidene fluoride (PVDF) membranes (0.4 μm; Millipore, Billerica, MA, USA, catalog #IPVH00010). The membranes were blocked with 5% bovine serum albumin (BSA; Sangon Biotech, Shanghai, China, catalog #C500626) for 2 h at room temperature and probed at 4°C overnight with antibodies against TLR2 (1:1000; Abcam, Cambridge, UK, catalog #ab213676), NOD2 (1:500; Santa Cruz Biotechnology, Dallas, TX, catalog #sc-56168), MyD88, p38 MAPK, phospho-p38, NF-κB p65, phospho-p65 or β-actin (1:1000; Cell Signaling Technologies, Danvers, MA, USA, catalog #4283S, #8690, #4511, #8242, #3033, #3700). The membranes were rinsed with TBST for 3 × 15 min and incubated with secondary antibodies for 1 h at room temperature. Reactive signals were detected by ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA, #32109) and ChemiDoc™ XRS+ System (Bio-Rad, USA) and quantitated using Image Lab software version 3.0.
Statistical Analysis
Differences between two groups were analyzed by two-tailed Student's t-tests. Multiple groups were compared using two-way analysis of variance (ANOVA; BDNF and meningitis) or one-way ANOVA followed by Tukey's post-hoc test. The data are expressed as the mean ± standard error of the mean (SEM). p < 0.05 was considered to indicate a significant difference. All graphs were generated using GraphPad Prism 5.0. Statistical analyses were performed using SPSS software version 17.0.
Results
Generation of Cortical and Hippocampal BDNF Knockout Mice
To generate BDNF knockout mice, we crossed floxed Bdnf mice with Camk2a-CreERT2 mice. Subsequently, standard PCR was used to analyze the genotypes of offspring. A floxed Bdnf band and a Camk2a-CreERT2 band defined BDNF conditional knockouts (Cre+/FF). In contrast, the Cre-/FF littermate controls exhibited only a floxed Bdnf band (Figure 1A). BDNF levels were compared in control and conditional null mutants by RT-PCR and showed that the Bdnf mRNA was reduced by ~70 and 71% in the cortex and hippocampus, respectively, after tamoxifen injection (Figure 1B). In accordance with these findings, an immunofluorescence study showed that there were fewer BDNF-immunoreactive neurons in sections of the hippocampal DG, CA1 and cortex taken from Cre+/FF mice than in their Cre-/FF littermate controls (Figure 1C). To confirm that the deletion is very targeted, we measured BDNF expression in brainstem by RT-PCR and immunofluorescence staining. The results showed that there were no differences between the genotypes in the expression levels of BDNF in the brainstem (Figure S1). BDNF, neurotrophin (NT) −3 and NT-4/5 function through a common tropomyosin-receptor kinase B (TrkB) receptor (37, 38). To test whether the levels of these neurotrophic factors were increased to compensate for BDNF deletion, we measured the gene expression levels of these factors after tamoxifen administration. We found that there were no differences between the genotypes in the expression levels of Ntf3 and Ntf5 in the cortex (Figure 1D) and the hippocampus (Figure 1E).
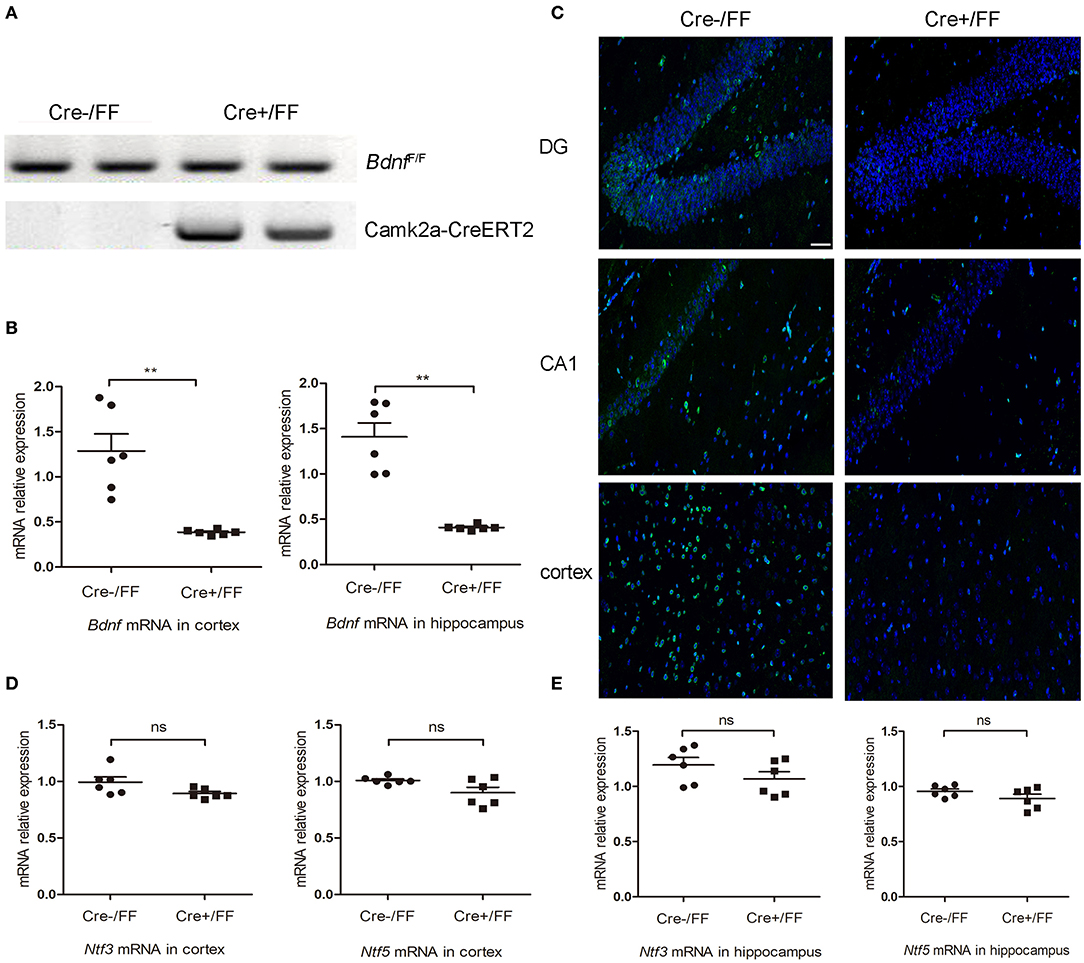
Figure 1. Selective deletion of BDNF in the cerebral cortex and hippocampus. (A) To produce forebrain-restricted BDNF mutant mice, BdnfF/+ mice were interbred with Camk2a-CreERT2 mice. Standard PCR was used to detect the genotypes of Camk2a-CreERT2/BdnfF/F mice and BdnfF/F mice. Mice exhibited the floxed Bdnf band and the Camk2a-CreERT2 band were defined as Cre+/FF mice; mice only exhibited the floxed Bdnf band were defined as Cre-/FF littermate controls. (B) Mice were injected intraperitoneally with tamoxifen once per day for 5 consecutive days followed by a 2-week break. RT-PCR was used to compare the expression level of the Bdnf mRNA with that of actin in Cre+/FF mice and Cre-/FF littermate controls in the cortex and hippocampus (n = 6 mice per group). (C) Immunocytochemical staining for BDNF (green) and DAPI (blue) was performed in sections of the hippocampal CA1 and DG and the cortex areas in tissues obtained from Cre+/FF mice and Cre-/FF littermate control mice. The BDNF expression was reduced in the cortex and hippocampus in the Cre+/FF mice. (D,E) RT-PCR analysis of the expression of Ntf3 and Ntf5 mRNA in the cortex and hippocampus in mice with different genotypes. There were no differences between the genotypes in the expression levels of Ntf3 and Ntf5 in the cortex and hippocampus (n = 6 mice per group). ns: not significant (p > 0.05); **p < 0.01. Mean comparisons between groups were performed by two-tailed Student's t-tests. Error bars indicate SEM. The data shown are from three independent experiments. Scale bar: 50 μm.
Conditional BDNF Deletion in Mice Aggravated the Clinical Severity of PM
As shown in Table 2, animals infected with S. pneumoniae showed positive bacterial cultures from cerebellar homogenates, whereas no pneumococci grew from cerebellar homogenates obtained from saline inoculated animals. Moreover, the infected Cre+/FF mice showed higher bacterial titers as compared with those from the infected Cre-/FF mice. Approximately 18 h after S. pneumoniae challenge, all infected mice began to display symptoms associated with PM, including weight loss, a lag in response speed and coma. Control animals injected with pyrogen-free saline showed a normal health status. Of the 8 infected animals in each PM group, one (1/8) animal died in the Cre-/FF group and two (2/8) animals died in the Cre+/FF group at 24 h (Table 2). Weight loss was significantly more pronounced in the Cre+/FF mice than in their Cre-/FF littermate controls (Table 2). In addition, clinical scores for disease severity showed markedly more severity in infected Cre+/FF mice than in control mice (Table 2).

Table 2. Bacterial titer, weight loss, clinical score, and mortality in different groups (mean ± standard error).
Conditional BDNF Deletion in Mice Aggravated the Pathological Severity of PM
When histopathology was examined at 24 h post-infection, large numbers of inflammatory cells were found to have infiltrated the subarachnoid space, and subarachnoidal and cortical hemorrhage was observed. Additionally, a significant increase in the number of cells infiltrated with inflammatory factors was observed in the brains of infected Cre+/FF mice than in their Cre-/FF littermate controls (Figure 2A). This finding was consistent with our previous study, which indicated that exogenous BDNF pretreatment alleviated pathological severity after pneumococcal infection (31). From the H&E stained slides, we observed that most exudative cells were infiltrated neutrophil granulocytes, which is a characteristic feature of acute bacterial meningitis. We performed double-label immunofluorescence staining of Ly6G+ neutrophil granulocytes and IBA-1+ microglia/macrophage and found massive infiltration of Ly6G+ neutrophil granulocytes into the subarachnoid space from the circulation. Moreover, BDNF conditional knockouts exhibited enhanced neutrophil granulocytes infiltration than were observed in their littermate controls at 24 h post-infection (Figure 2B).
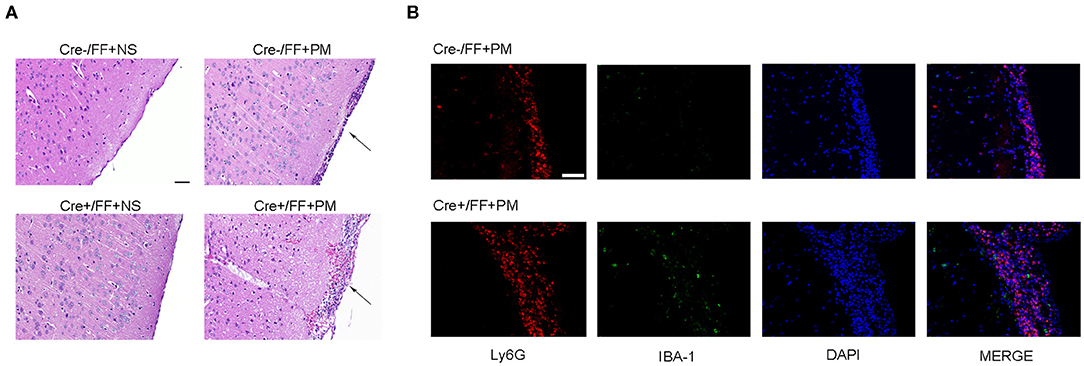
Figure 2. BDNF conditional knockout aggravated the pathological severity of PM. Mice were intracisternally injected with a 10 μl volume containing either 104 cfu/ml S. pneumoniae or sterile saline. At 24 h post-infection, all survivals were sacrificed to harvest the brain. (A) H&E staining of coronal brain sections obtained in each group. Uninfected mice showed no inflammatory infiltrates, whereas all infected mice showed infiltration of inflammatory exudate in the subarachnoid space. Cre+/FF mice had a more aggravated pathological severity than was observed in their Cre-/FF littermate control mice. (B) Double-label immunofluorescence staining of Ly6G+ neutrophil granulocytes (red) and IBA-1+ microglia/macrophages (green). All infected mice showed massive infiltration of Ly6G+ neutrophil granulocytes into the subarachnoid space from the circulation in the acute phage of infection. Cre+/FF mice exhibited enhanced neutrophil granulocytes infiltration than were observed in their littermate controls. Scale bar: 50 μm.
Conditional BDNF Deletion in Mice Increased Neuronal Injury and Hippocampal Apoptosis Associated With PM
In recent years, studies have indicated that a wide spectrum of brain injuries are associated with PM, including focal necrosis of cortical neurons and apoptotic neuronal cell death in the DG, which are regularly detected in animals with PM (39). Therefore, we analyzed hippocampal apoptosis and neuronal injury in the cortex using TUNEL and Nissl staining, respectively. Our results indicated that cortical neuron loss was more remarkable in infected Cre+/FF mice than in control mice and that the knockouts also had more severely incomplete neuronal structures at 24 h post-infection (Figure 3A). There were significantly fewer surviving neurons in the infected Cre+/FF mice than in the Cre-/FF littermate controls (Figure 3B). Furthermore, S. pneumoniae infection caused obvious apoptosis in the hippocampal DG (Figure 3C), and the number of TUNEL-positive cells was significantly higher in infected Cre+/FF mice than in infected control mice (Figure 3D).
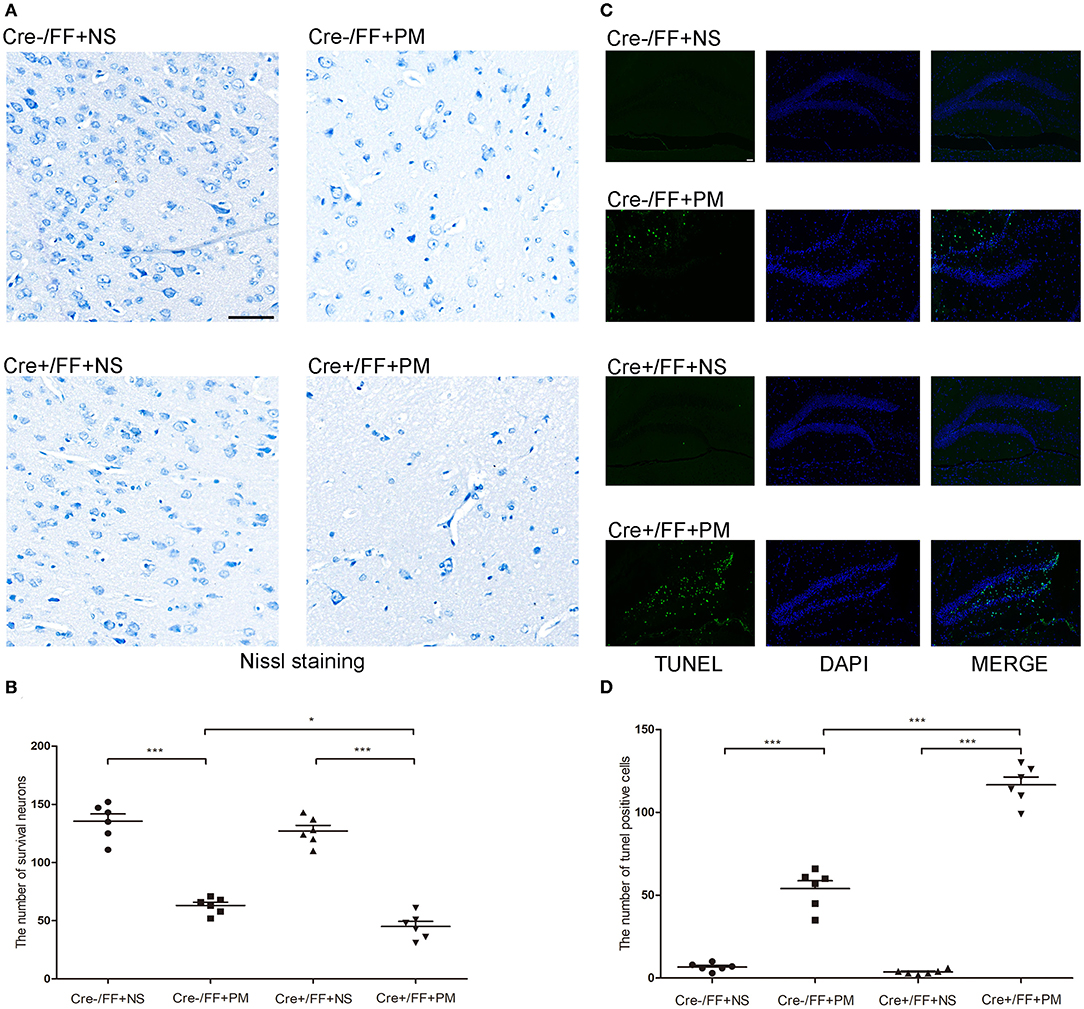
Figure 3. BDNF conditional knockout increased the neuronal injury and hippocampal apoptosis associated with PM. Mice were intracisternally injected with a 10 μl volume containing either 104 cfu/ml S. pneumoniae or sterile saline. At 24 h post-infection, all survivals were sacrificed to harvest the brain. (A) The number of surviving neurons was assessed in the cerebral cortex by Nissl staining in all groups at 24 h post-infection. In infected Cre+/FF mice group, the neuron loss is more severe than in Cre-/FF mice. (B) Quantitative analysis of the Nissl results (n = 6 mice per group). (C) TUNEL staining was used to assess apoptotic neurons in the hippocampal DG. There are more TUNEL-positive cells in infected Cre+/FF mice than in infected control mice. (D) Quantitative analysis of the TUNEL results (n = 6 mice per group). *p < 0.05; ***p < 0.001. Mean comparisons between groups were performed by two-way ANOVA followed by Tukey's post-hoc test. Error bars indicate SEM. The data shown are from three independent experiments. Scale bar: 50 μm.
Conditional BDNF Deletion in Mice Increased Microglia/macrophage Proliferation Associated With PM
IBA-1 staining was performed to evaluate the local status of inflammation in the CNS during PM. As shown in Figure 4, microglia/macrophage were detected in all infected animals after S. pneumoniae infection. Moreover, infected Cre+/FF mice showed significantly enhanced IBA-1 fluorescence intensity in the cortex (Figure 4A) and hippocampus CA1 (Figure 4C) than was observed in the infected controls, and the number of IBA-1 positive cells was significantly higher in infected Cre+/FF mice than in infected Cre-/FF littermate controls (Figures 4B,D).
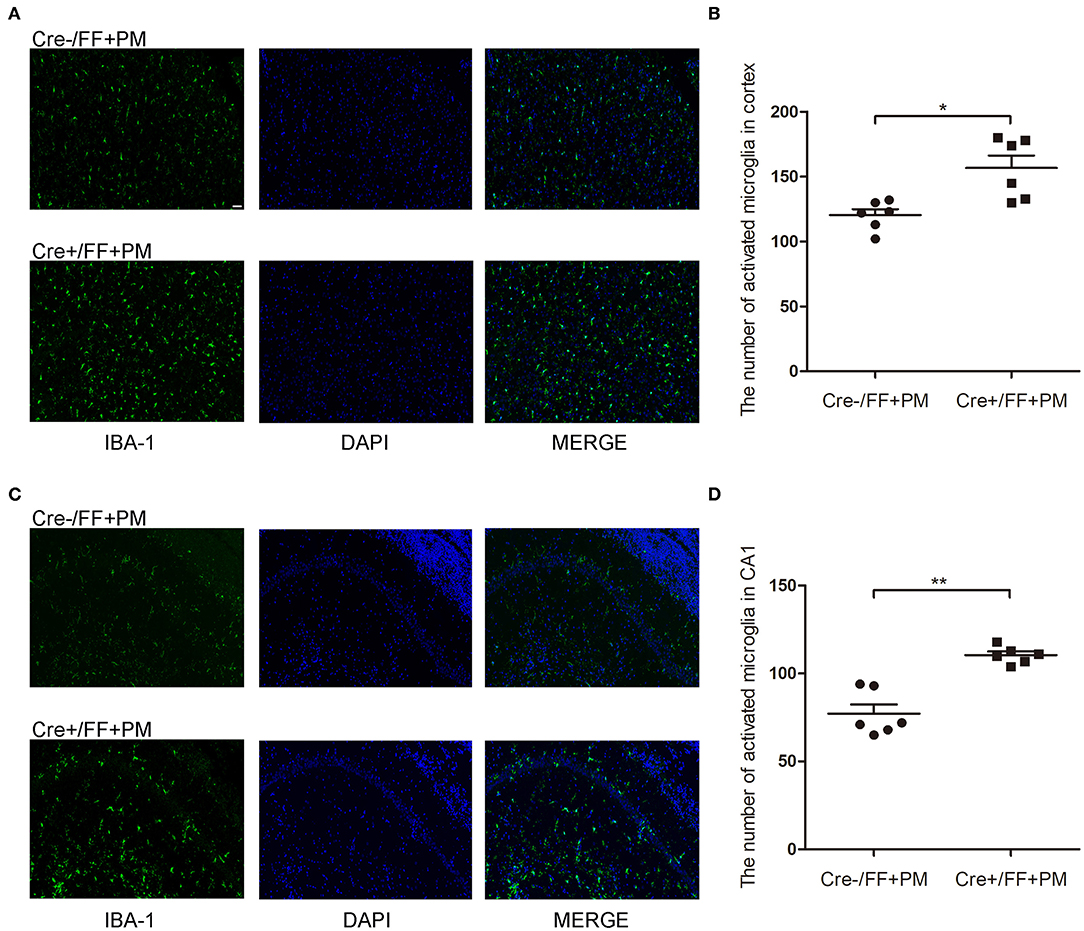
Figure 4. BDNF conditional knockout increased the microglia/macrophage proliferation associated with PM. Mice were intracisternally injected with a 10 μl volume containing either 104 cfu/ml S. pneumoniae or sterile saline. At 24 h post-infection, all survivals were sacrificed to harvest the brain. (A,C) Immunofluorescent staining showing IBA-1 (green) positive cells counterstained with DAPI (blue) in the cortex and in the CA1 hippocampal region. All infected animals showed microglia/macrophage proliferation and the infected Cre+/FF mice showed significantly enhanced IBA-1 fluorescence intensity than was observed in the infected controls. (B,D) Quantitative analysis of IBA-1-positive cell numbers in the cortex and in the CA1 region of the hippocampus (n = 6 mice per group). *p < 0.05; **p < 0.01. Mean comparisons between groups were performed by two-tailed Student's t-tests. Error bars indicate SEM. The data shown are from three independent experiments. Scale bar: 50 μm.
Conditional BDNF Deletion in Mice Upregulated Inflammatory Factor Expression Associated With PM in Brain Tissues
At 24 h post-infection, brains (hippocampus and cerebral cortex) were harvested to examine the expression of proinflammatory factors (Tnf-α, Il-1β, and Il-6) by RT-PCR. The results of RT-PCR showed that the mRNA levels of all the analyzed inflammatory factors were significantly higher after infection. Moreover, the levels of inflammatory factors were also higher in Cre+/FF mice in the cortex (Figure 5A) and the hippocampus (Figure 5B) relative to the results observed in infected control mice. These data indicated that BDNF deletion was significantly associated with the expression of pro-inflammatory factors following exposure to pneumococcal infection.
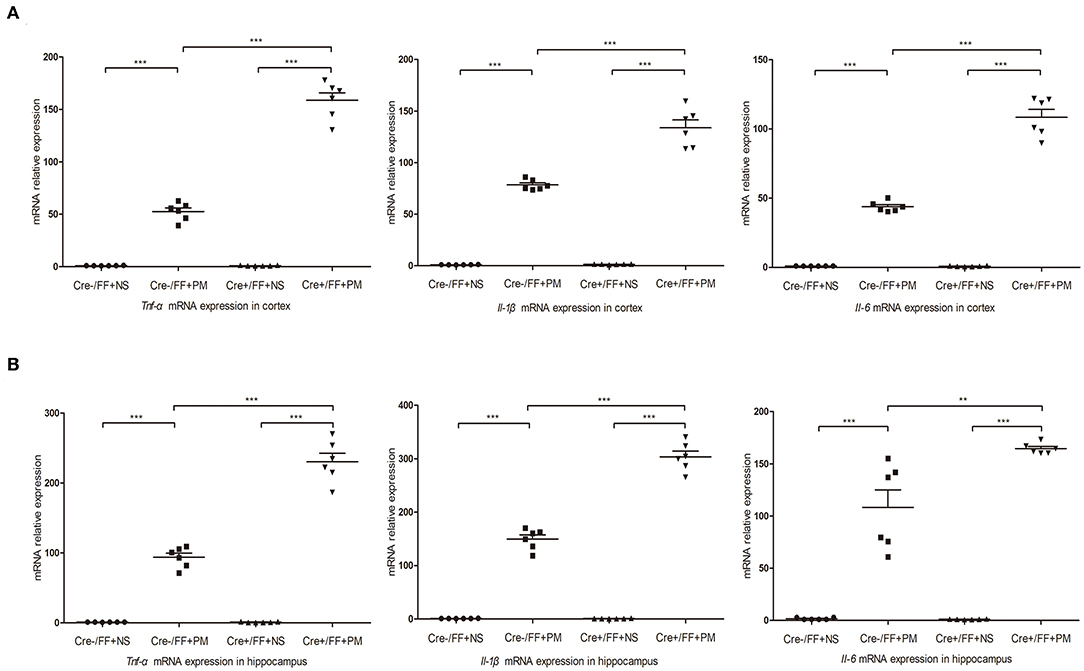
Figure 5. BDNF conditional knockout upregulated pro-inflammatory cytokine expression in PM. Mice were intracisternally injected with a 10 μl volume containing either 104 cfu/ml S. pneumoniae or sterile saline. At 24 h post-infection, all survivals were sacrificed to harvest the brain. (A,B) The levels of pro-inflammatory cytokines (Tnf-α, Il-1β, and Il-6) were measured using RT-PCR in both the cortex and hippocampus at 24 h post-infection. In cortex and hippocampus homogenates from infected Cre+/FF mice, a significant increased expression of Tnf-α, Il-1β, and Il-6 was detected than infected control mice (n = 6 mice per group). **p < 0.01; ***p < 0.001. Mean comparisons between groups were performed by two-way ANOVA followed by Tukey's post-hoc test. Error bars indicate SEM. The data shown are from three independent experiments.
Conditional BDNF Deletion in Mice Upregulated TLR2 and NOD2 Expression Associated With PM
To investigate whether TLR2 and NOD2 are involved in the inflammatory response to S. pneumoniae infection in BDNF knockout mice, we measured the expression levels of NOD2 and TLR2 and the downstream molecules MyD88 in the brain tissues of mice challenged with S. pneumoniae. Compared with non-infected mice, mice submitted to S. pneumoniae stimulation expressed higher levels of Tlr2 and Myd88 mRNA at 24 h after infection in both the cortex (Figures 6A,B) and the hippocampus (Figures 6C,D). Additionally, the bacteria-induced upregulation of Tlr2 and Myd88 was markedly stronger in Cre+/FF mice than in control mice. Western blot and grayscale analyses were used to evaluate total TLR2 and MyD88 protein levels in the cortex (Figures 6E,F) and hippocampus (Figures 6G,H), and the results agreed with the findings for the mRNA levels of these markers as measured by RT-PCR. Similarly, there was a remarkable increase in the expression of the Nod2 mRNA in response to pneumococcal stimulation in the cortex (Figure 7A) and hippocampus (Figure 7B), and Cre+/FF mice showed clearly higher expression levels of Nod2 than were observed in control mice. Western blot and grayscale analyses were used to evaluate NOD2 protein levels in the cortex (Figure 7C) and hippocampus (Figure 7D), and the results agreed with the findings for the mRNA levels of this marker. These data indicated that TLR2 and NOD2 signaling are involved in the inflammatory response observed in Cre+/FF mice after pneumococcal infection.
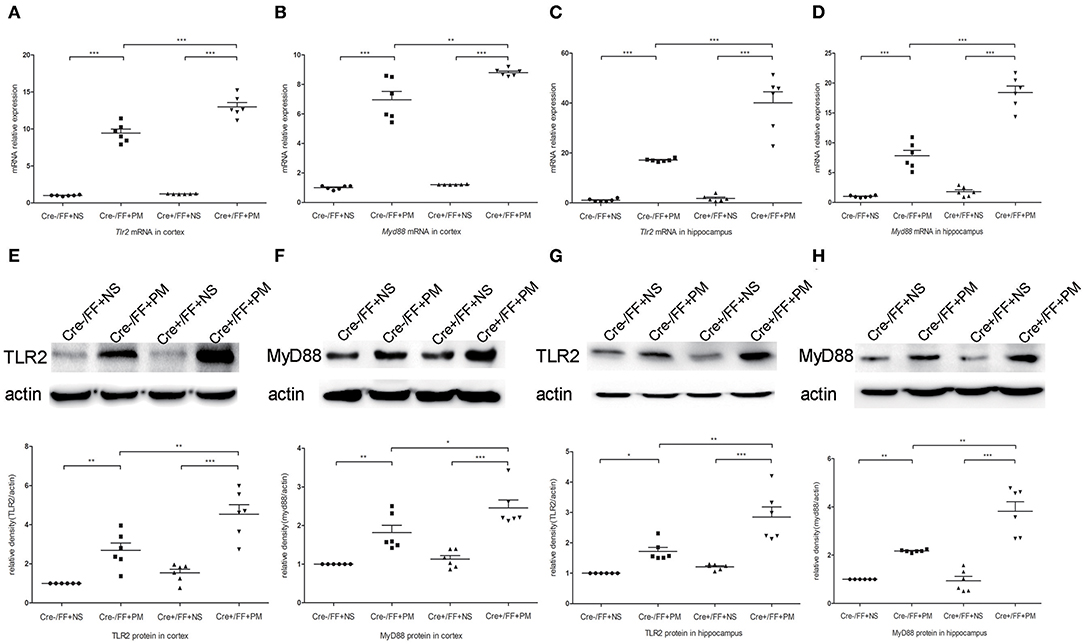
Figure 6. BDNF conditional knockout activated the TLR2 and MyD88 expression associated with PM. Mice were intracisternally injected with a 10 μl volume containing either 104 cfu/ml S. pneumoniae or sterile saline. At 24 h post-infection, all survivals were sacrificed to harvest the brain. (A–D) The mRNA levels of Tlr2 and Myd88 mRNA were measured in the cortex and hippocampus by RT-PCR at 24 h post-infection (n = 6 mice per group). (E–H) The relevant protein expression in cortical and hippocampal tissue lysates and a density analysis of TLR2, MyD88, and β-actin expression in each group (n = 6 mice per group). BDNF conditional knockout increased levels of TLR2 and MyD88 in the cortex and hippocampus associated with PM. *p < 0.05; **p < 0.01; ***p < 0.001. Mean comparisons between groups were performed by two-way ANOVA followed by Tukey's post-hoc test. Error bars indicate SEM. The data shown are from three independent experiments.
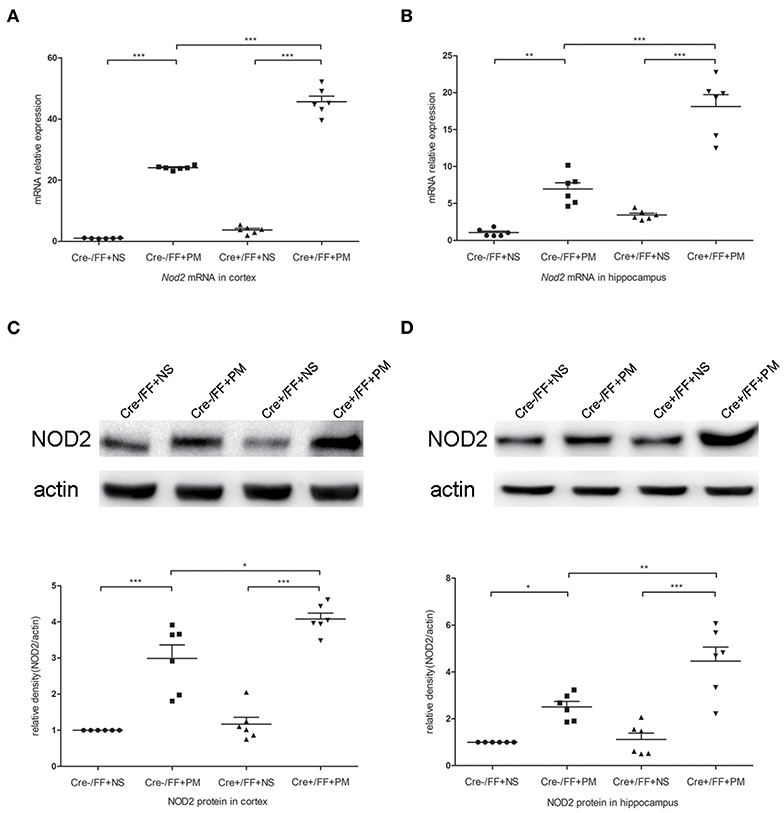
Figure 7. BDNF conditional knockout activated the NOD2 expression associated with PM. Mice were intracisternally injected with a 10 μl volume containing either 104 cfu/ml S. pneumoniae or sterile saline. At 24 h post-infection, all survivals were sacrificed to harvest the brain. (A,B) The mRNA expression levels of Nod2 in the cortex and hippocampus were assessed by RT-PCR at 24 h post-infection (n = 6 mice per group). (C,D) The relevant protein expression levels in cortical and hippocampal tissue lysates and a density analysis of NOD2 and β-actin expression were evaluated in each group (n = 6 mice per group). BDNF conditional knockout increased levels of NOD2 in the cortex and hippocampus associated with PM. *p < 0.05; **p < 0.01; ***p < 0.001. Mean comparisons between groups were performed by two-way ANOVA followed by Tukey's post-hoc test. Error bars indicate SEM. The data shown are from three independent experiments.
Conditional BDNF Deletion in Mice Further Activated NF-κB p65 and p38 MAPK in PM
We next investigated the influence of BDNF deletion on S. pneumoniae-induced activation of NF-κB p65 and p38 MAPK, which are two common downstream factors in both the TLR2 and the NOD2 signaling pathway (40, 41). Our data showed that S. pneumoniae infection activated the NF-κB pathway, as demonstrated by the substantially enhanced phosphorylation of p65 in the cortex (Figure 8A) and hippocampus (Figure 8B). The similar activation of the p38 MAPK pathway was also observed in all infected mice and was associated with the markedly enhanced phosphorylation of p38 in the cortex (Figure 8C) and hippocampus (Figure 8D). Moreover, the S. pneumoniae-induced phosphorylation of p65 and p38 was substantially stronger in the cortex and hippocampus of Cre+/FF mice exposed to S. pneumoniae infection than in their Cre-/FF littermate controls. These data indicated that BDNF deletion resulted in the augmented activation of both the NF-κB p65 and the p38 MAPK pathway in brain tissues during PM.
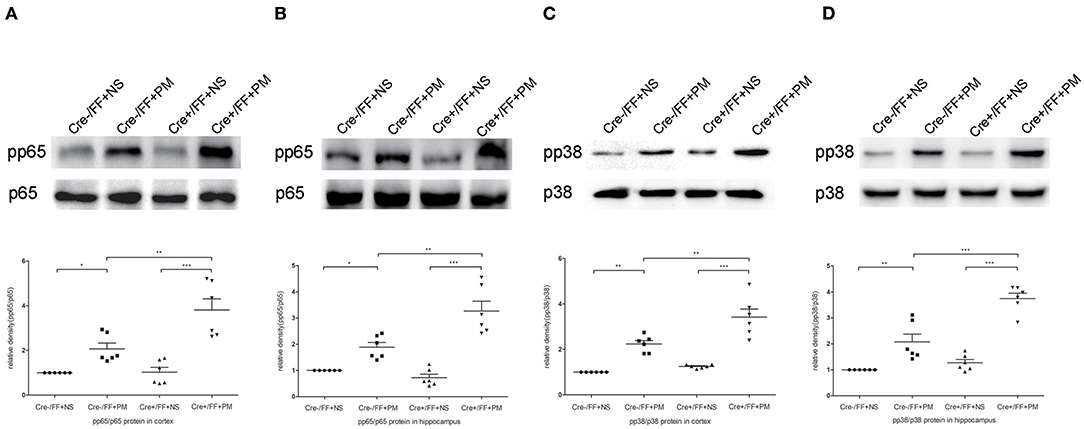
Figure 8. BDNF conditional knockout activated the NF-κB p65 and p38 MAPK signaling pathways in brain tissues. Mice were intracisternally injected with a 10 μl volume containing either 104 cfu/ml S. pneumoniae or sterile saline. At 24 h post-infection, all survivals were sacrificed to harvest the brain. (A–D) The relevant protein expression of p65, pp65, p38, and pp38 in the hippocampus and cortex were evaluated in each group. The graphs indicated the expression ratios of pp65/p65 in the cortex and hippocampus as well as those of pp38/p38 (n = 6 mice per group). BDNF conditional knockout increased levels of phosphorylated p65 and p38 associated with PM. *p < 0.05; **p < 0.01; ***p < 0.001. Mean comparisons between groups were performed by two-way ANOVA followed by Tukey's post-hoc test. Error bars indicate SEM. The data shown are from three independent experiments.
Microglia/macrophage Depletion Attenuated Neuroinflammation but Aggravated Clinical Impairment in PM
To assess the effect of GW2580 on the microglia/macrophage population in BDNF conditional knockout mice, we determined the expression of IBA-1 by immunofluorescence staining at 24 h post-infection. The increase in IBA-1-positive cell numbers after infection was significantly decreased with GW2580 treatment in cortex and hippocampus (Figure S2). The depletion of microglia/macrophages led to an increase of bacterial loads in the cerebellar homogenates and these higher bacterial titers resulted in more severe clinical symptoms and higher mortality (Table 3). However, we observed a decrease of leukocytes invasion in the subarachnoid space after S. pneumoniae infection in PM+GW2580 group compared with PM control group (Figure 9A). At 24 h post-infection, the mRNA expression of pro-inflammatory cytokines (i.e., Tnf-α, Il-1β, and Il-6) in both the cerebral cortex and hippocampus homogenates was evaluated by RT-PCR. Our results showed that S. pneumoniae infection led to massive pro-inflammatory cytokine release in BDNF conditional knockout mice. In contrast, the expression of Tnf-α, Il-1β, and Il-6 was significantly attenuated in microglia/macrophage depletion mice, reflecting impaired immune activation (Figures 9B,C).
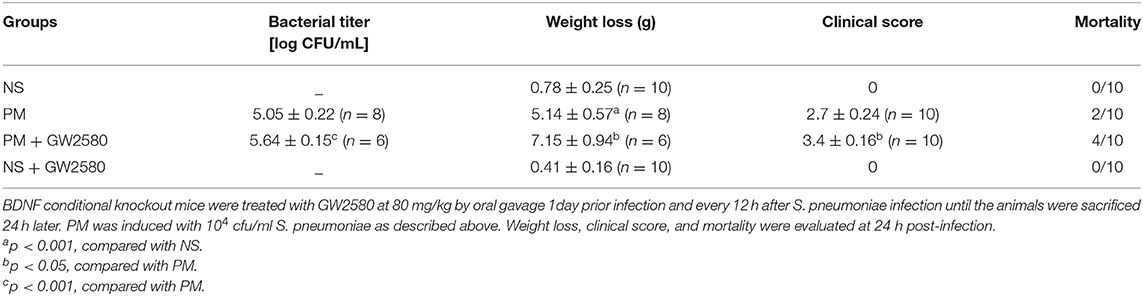
Table 3. Bacterial titer, weight loss, clinical score, and mortality in different groups (mean ± standard error).
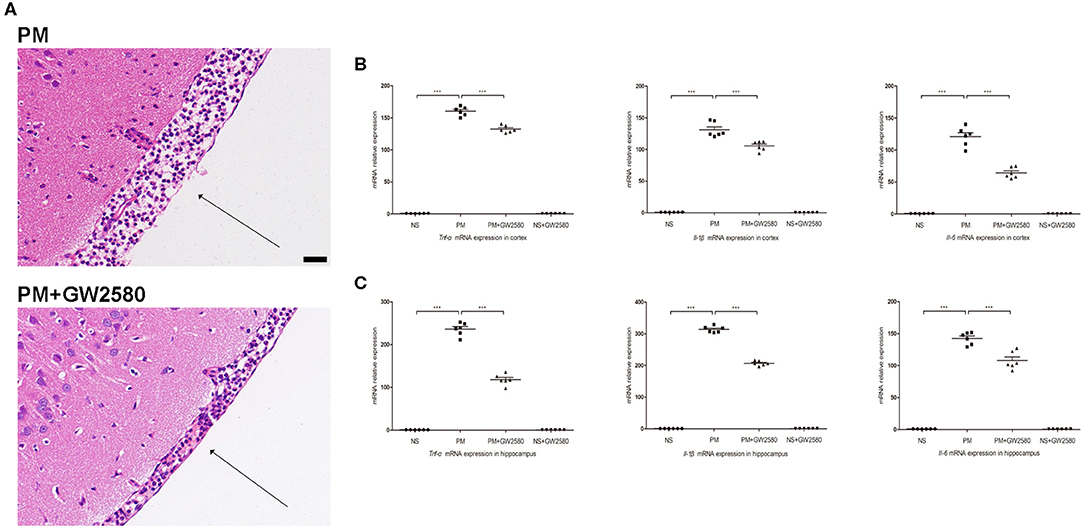
Figure 9. Macrophage/microglia depletion attenuated neuroinflammation in PM. BDNF conditional knockout mice were treated with GW2580 at 80 mg/kg by oral gavage 1 day prior infection and every 12 h after S. pneumoniae infection until the animals were sacrificed 24 h later. PM was induced with 104 cfu/ml S. pneumoniae as described above. (A) H&E staining of coronal brain sections obtained in infected groups. All infected mice showed infiltration of inflammatory exudate in the subarachnoid space. PM+GW2580 mice had a decrease of leukocyte invasion in the subarachnoid space than was observed in PM control mice. (B,C) The levels of pro-inflammatory cytokines (Tnf-α, Il-1β, and Il-6) were measured using RT-PCR in both the cortex and hippocampus at 24 h post-infection. In cortex and hippocampus homogenates from PM+GW2580 mice, a significant decreased expression of Tnf-α, Il-1β, and Il-6 was detected than PM control mice (n = 6 mice per group). ***p < 0.001. Mean comparisons between groups were performed by one-way ANOVA followed by Tukey's post-hoc test. Error bars indicate SEM. The data shown are from three independent experiments. Scale bar: 50 μm.
Discussion
In this study, we conditionally deleted the Bdnf gene from the cortex and hippocampus in mice to determine the contribution of this marker to S. pneumoniae infection processing. The results of this study demonstrated that conditional BDNF knockout aggravated the clinical and pathological severity observed after S. pneumoniae infection and intensified the inflammatory response, neuron injury and hippocampal apoptosis that are correlated with S. pneumoniae meningitis. Additionally, we found that BDNF deletion enhanced neurological impairment at least partially by modulating the TLR2 and NOD2 signaling pathways. Targeted depletion of microglia/macrophage population attenuated neuroinflammation during PM, however, it failed to reverse the poor outcomes of infected BDNF conditional knockout mice. Collectively, our results suggested a novel regulatory mechanism by which the inflammatory response was mediated by endogenous BDNF in PM, and these results increased our understanding of the pathogenesis of this disease.
Tissue-specific gene knockout can yield remarkable information about the relative therapeutic targets in a disease, and time-specific gene ablation surmounts potential physiological compensatory mechanisms that can cover the true phenotypic contribution of a gene (42, 43). Camk2a is a calcium-activated serine/threonine kinase that is abundantly present in the forebrain (including the cortex, hippocampus, and other structures), but is mostly absent from mid-hind brain regions. It is widely used to drive a tamoxifen-inducible CreERT2 recombinase construct that permits tissue-specific gene deletion in adult animals (44, 45). BDNF has a wide array of functions in the brain and is expressed at the highest levels in the hippocampus and cerebral cortex (46, 47). Here, to identify the role of endogenous BDNF in the inflammatory reaction and brain injury during PM, we crossed floxed Bdnf mice with the Camk2a-driven CreERT2 strain to delete Bdnf . RT-PCR was used to assess the effects of conditional Bdnf deletion, and the results showed that tamoxifen injection reduced the mRNA expression level of Bdnf by ~70% in the cortex and hippocampus. The remaining ~30% of Bdnf mRNA may be produced by cells that did not undergo gene recombination (34). Although BDNF, NT-3 and NT-4/5 all activate the TrkB receptor, there were no differences in the expression levels of Ntf3 or Ntf5 between Cre+/FF mice and their Cre-/FF littermate controls. These results showed that Ntf3 and Ntf5 expression levels were not upregulated to compensate for the absence of BDNF after Bdnf gene deletion, in line with previous findings (34).
Despite the fact that significant progress has been made toward treatments for PM, the mortality and neurological sequelae rates associated with PM remain high (48, 49). The brain damage caused by S. pneumoniae infection is correlated with an enhanced inflammatory response in the CNS, especially after antibiotic treatment, which results in large amounts of bacterial components being released and recognized by brain-resident immune cells (14, 50). Subsequently, bacterial lysis stimulates the release of pro-inflammatory cytokines, such as TNF-α and IL-1β in the CNS, eventually resulting in mitochondrial dysfunction, neuronal apoptosis, and brain damage (51). In recent decades, therapeutic targets have popularly targeted the regulation of the inflammatory response and eliminating side effects correlated with the excessive activation of the immune system. Although numerous therapeutic adjunctive approaches have been investigated to decrease the harmful effects of the inflammatory response in vitro and in animal models treated with corticosteroids, IL-1β receptor antagonists, or TNF-α inhibitors, the clinical efficacy of these treatments remains unsatisfactory (24, 49, 52). Recent studies have indicated that BDNF exerts neuroprotective effects in diverse CNS-related diseases, and this may provide a promising adjunctive therapy for acute BM. Our recent study verified that BDNF administration decreased the inflammatory response and hippocampal apoptosis by modulating immune-related intracellular signaling pathways in rats submitted to PM (31). In addition, we demonstrated that BDNF was obviously upregulated after S. pneumoniae inoculation for 24 h but then declined with time (53). Moreover, the expression of BDNF in brains was further reduced after infected animals were treated with antibiotics but was increased when the animals were treated with an antibiotic plus dexamethasone (53, 54). These findings suggested that decreased BDNF levels were associated with the aggravation of infection-induced intracranial complications; however, direct evidence for the involvement of endogenous BDNF in S. pneumoniae meningitis is lacking.
In our animal model, the intracisternal injection with 104 cfu/ml of standard serotype 3 S. pneumoniae induced pathological manifestations that resemble the human situation: microglial activation, neutrophil granulocytes recruitment, and indicated neuronal injury in the cortex and hippocampus (55). Although, the unphysiological mode of bacterial infection is a theoretical disadvantage, the directly injection of bacteria into CSF is easy to perform and ensures all bacterial fluid distributed to the intracranial compartments. In contrast, the bacteremia-derived meningitis model may result in rapid bacterial clearance from the bloodstream not necessarily leading to BM (data not shown).
In the present study, we showed that the loss of BDNF decreased the resistance of mice to S. pneumoniae meningitis and was associated with more weight loss and worse clinical features. There was a tendency for infected Cre+/FF mice to have a lower survival rate than their littermate controls. We suggest that more animals and the complete removal of the Bdnf gene will be required to further analyze this difference in mortality.
BDNF deletion resulted in increased microglia/macrophage proliferation and neutrophil infiltration of subarachnoid space during S. pneumoniae infection than was observed in infected control mice. Activated microglia/macrophage can produce large amounts of cytokines and neurotoxic mediators, leading to neuronal cell injury and even the perpetuation of neuronal damage (56). These results are in accordance with the finding that overwhelmingly higher amounts of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) are produced and released by immune cells in Cre+/FF mice than in their infected Cre-/FF littermate controls. Microglia activation is widely regarded as a double-edged sword possessing both protective and destructive effects. In our present study, targeted depletion of microglia/macrophage cell population decreased the resistance of mice to PM with diminishing neuroinflammation in BDNF conditional knockout mice. Once sensing the invasion of S. pneumoniae, microglia transform into activated phenotypes, and proliferate at the site of injury (57). The activated microglial cells release cytokines or chemokines that induce the infiltration of circulating immune cells during infection. Infiltrated leukocytes support bacterial clearance but also contribute to neuronal damage (58). The severity of clinical symptoms during PM depends on both the bacterial load and the host immune response. In the BDNF conditional knockout mice, the overgrowth of bacterial caused by microglia/macrophage depletion may be more detrimental for the outcomes of PM than the dismissed host immune response. Although our study found that BDND deficiency led to enhanced microglia/macrophage proliferation after infection, it may also result in other effects (e.g., increased bacterial tiers, neutrophil infiltration, activation of inflammatory pathway, and reduced neuroprotective effect) that contributes to the worsen outcomes in our animal model.
We found that the number of surviving neurons in the cortex was significantly lower and the number of apoptotic bodies was higher in the DG of infected Cre+/FF mice than in infected control mice. Significant brain damage arises from the mechanisms involved in neuronal apoptosis, especially in the hippocampus during PM, and autopsies performed in cases of PM have revealed that neuronal apoptosis occurs in the DG in this disease (5). Although a growing body of evidence has shown that BDNF participates in regulating intracellular signaling molecules to inhibit the expression of pro-inflammatory cytokines during infection, the exact mechanism by which BNDF exerts its effects in S. pneumoniae infection remains unclear (59).
We found BDNF deletion resulted in significantly higher post-S. pneumoniae infection levels of TLR2/MyD88 and NOD2 expression than were observed in infected control mice. TLR2 and NOD2 act as critical PRRs by detecting bacterial molecules. Upon activation of these receptors, they induce the activation of NF-κB and MAPK pathways, which eventually results in the transcription of the targeted inflammatory genes and production of proinflammatory cytokines (39, 60). Previous research has shown that in vivo, the selective activation of TLR2 led to inflammatory changes and high leukocyte concentrations in the CNS and neuronal apoptosis in the hippocampal DG. Of note, the TLR2-deficient mice submitted to S. pneumoniae showed lower levels of CSF pleocytosis and less excess neuronal damage than were found in controls (15). Similarly, MyD88-decient mice showed an obviously compromised inflammatory response, including reduced leucocyte infiltration and lower levels of cytokine release during S. pneumoniae infection (61). Moreover, the increase in NOD2 expression observed after S. pneumoniae infection further promotes inflammatory cell infiltration and blood-brain barrier (BBB) permeability within the CNS, leading to cerebral injury (12, 62). In the present study, we further demonstrated that BDNF deletion resulted in a strong increase in the S. pneumoniae-induced phosphorylation of NF-κB p65 and the phosphorylation of p38 MAPK than were observed in control mice. NF-κB p65 is a widely expressed transcription factor that is a strong inducer of many proinflammatory genes that are associated with the pathogenesis of PM. Early studies revealed that in PM, the pharmacological inhibition of NF-κB p65 obviously reduced the host inflammatory response and attenuated CNS complications, such as BBB disruption and cerebrovascular failure (63). Moreover, increased p38 MAPK activation was observed after stimulation with pneumolysin, and the inhibition of p38 resulted in a strong improvement in cell survival (64). Importantly, p38 MAPK is a critical regulator of inflammatory cytokine production, and some p38 MAPK-blocking drugs that reduce IL-1 and TNF-α expression have been successfully used in the treatment of inflammatory diseases (65, 66). Here, we provide the first evidence showing that BDNF deletion resulted in a strong increase in the S. pneumoniae-induced phosphorylation of NF-κB p65 and phosphorylation of p38 MAPK through TLR2/MyD88 and NOD2 downstream signaling, lead to the excessive secretion of pro-inflammatory cytokines, thereby exacerbating brain inflammation and further injury (Figure 10).
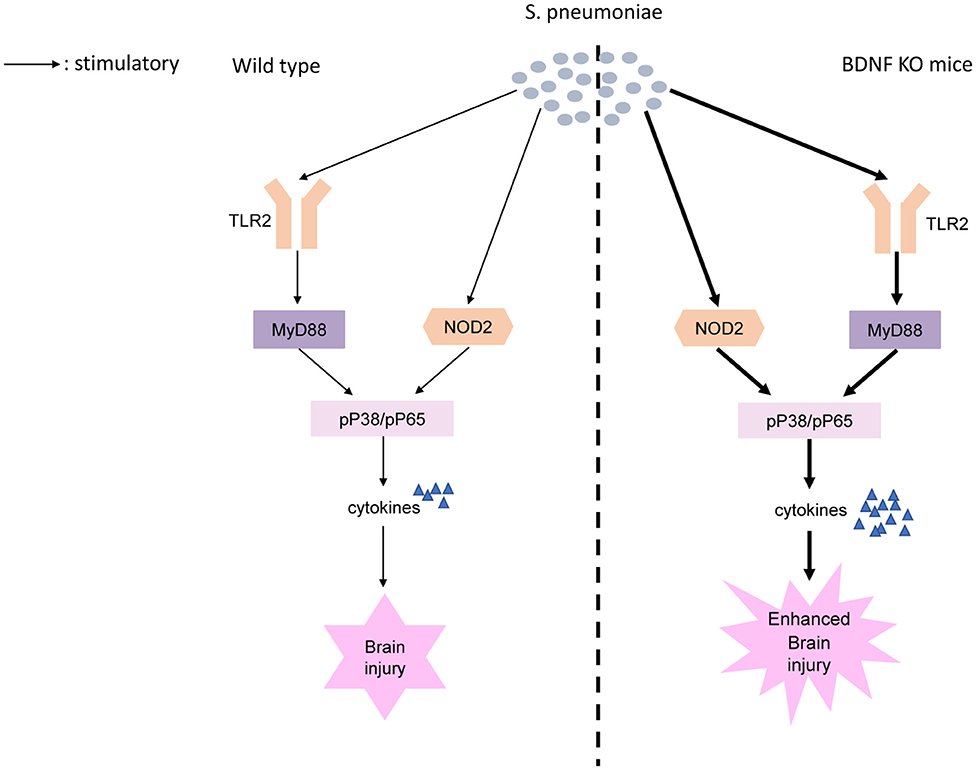
Figure 10. Proposed model of BDNF knockout exacerbates neuroinflammation and brain injury in experimental pneumococcal meningitis. S. pneumoniae is recognized by PRRs including TLR2 and NOD2. BDNF deletion results in a strong increase in the S. pneumoniae-induced phosphorylation of NF-κB p65 and phosphorylation of p38 MAPK through TLR2/MyD88 and NOD2 downstream signaling. Increased phosphorylation of NF-κB p65 and phosphorylation of p38 MAPK, as two of the major factors responsible for the transcription of the targeted inflammatory genes, lead to the excessive secretion of pro-inflammatory cytokines, thereby exacerbating brain inflammation and further injury.
The role of BDNF may change over time as the infection evolves. There is also strong evidence to suggest important roles of BDNF in regulating neurogenesis, survival, plasticity, and neuronal differentiation (67, 68). Our previous studies showed that BDNF adjunctive treatment with antibiotics for 7 d could rescue cortical and hippocampal neurons in a rat model of pneumococcal meningitis (69). Moreover, administration of exogenous BDNF for 7–28 days might increase the proliferation and differentiation of NSCs in the hippocampus after bacterial meningitis (29). Our current study only evaluated the acute phase of the disease, and in subsequent studies, the role of endogenous BDNF at different time points during infection process should be investigated.
Conclusion
In summary, the results of our study indicate that conditional Bdnf deletion exacerbates brain damage and augments the innate immunity-associated inflammatory response, likely by activating the TLR2- and NOD2-mediated downstream signaling pathways during Streptococcus pneumoniae meningitis; therefore, BDNF may be a promising candidate therapeutic target in BM.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal experiments were approved by the Animal Ethical and Welfare Committee of Xinhua Hospital (approval ID: 2014041).
Author Contributions
SZ performed research, analyzed data, and wrote the manuscript. ZZ and DX helped with the animal experiment. YW helped with the molecular biological experiments. LL provided the necessary guidance on the performance of all the experiment. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 81571180 and 81271337).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01357/full#supplementary-material
Abbreviations
CNS, Central nervous system; BDNF, Brain-derived neurotrophic factor; PM, Pneumococcal meningitis; BM, Bacterial meningitis; PRRs, Pattern-recognition receptors; TLR2, Toll-like receptor 2; MyD88, Molecule myeloid differentiation factor 88; NOD2, Nucleotide-binding oligomerization domain 2; TNF, Tumor necrosis factor; IL, Interleukin; NF-κB, Nuclear factor kappa B; MAPKs, Mitogen-activated protein kinases; NSCs, Endogenous neural stem cells; NS, Normal saline; TUNEL, terminal deoxynucleotidyl transferase dUTP-nick-end labeling; DG, Dentate gyrus; NT-3, Neurotrophin-3; NT-4, Neurotrophin-4; TrkB, Tropomyosin-receptor kinase B; BBB, Blood-brain barrier; PAMPs, Pathogen-associated molecular patterns.
References
1. van de Beek D, de Gans J, Spanjaard L, Weisfelt M, Reitsma JB, Vermeulen M. Clinical features and prognostic factors in adults with bacterial meningitis. N Engl J Med. (2004) 351:1849–59. doi: 10.1056/NEJMoa040845
2. Castelblanco RL, Lee M, Hasbun R. Epidemiology of bacterial meningitis in the USA from 1997 to 2010: a population-based observational study. Lancet Infect Dis. (2014) 14:813–9. doi: 10.1016/S1473-3099(14)70805-9
3. Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev. (2010) 23:467–92. doi: 10.1128/CMR.00070-09
4. Lucas MJ, Brouwer MC, van de Beek D. Neurological sequelae of bacterial meningitis. J Infect. (2016) 73:18–27. doi: 10.1016/j.jinf.2016.04.009
5. Nau R, Soto A, Bruck W. Apoptosis of neurons in the dentate gyrus in humans suffering from bacterial meningitis. J Neuropathol Exp Neurol. (1999) 58:265–74. doi: 10.1097/00005072-199903000-00006
6. Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, et al. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. (2003) 170:438–44. doi: 10.4049/jimmunol.170.1.438
7. Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. (2013) 2013:480739. doi: 10.1155/2013/480739
8. Scheld WM, Dacey RG, Winn HR, Welsh JE, Jane JA, Sande MA. Cerebrospinal fluid outflow resistance in rabbits with experimental meningitis. Alterations with penicillin and methylprednisolone. J Clin Invest. (1980) 66:243–53. doi: 10.1172/JCI109850
9. Peterson PK, Hu S, Sheng WS, Kravitz FH, Molitor TW, Chatterjee D, et al. Thalidomide inhibits tumor necrosis factor-alpha production by lipopolysaccharide- and lipoarabinomannan-stimulated human microglial cells. J Infect Dis. (1995) 172:1137–40. doi: 10.1093/infdis/172.4.1137
10. Koedel U, Winkler F, Angele B, Fontana A, Flavell RA, Pfister HW. Role of Caspase-1 in experimental pneumococcal meningitis: evidence from pharmacologic Caspase inhibition and Caspase-1-deficient mice. Ann Neurol. (2002) 51:319–29. doi: 10.1002/ana.10103
11. Chen X, Meng X, Foley NM, Shi X, Liu M, Chai Y, et al. Activation of the TLR2-mediated downstream signaling pathways NF-kappaB and MAPK is responsible for B7-H3-augmented inflammatory response during S. pneumoniae infection. J Neuroimmunol. (2017) 310:82–90. doi: 10.1016/j.jneuroim.2017.07.002
12. Wang Y, Liu X, Liu Q. NOD2 Expression in Streptococcus pneumoniae meningitis and its influence on the blood-brain barrier. Can J Infect Dis Med Microbiol. (2018) 2018:7292084. doi: 10.1155/2018/7292084
13. Schröder NW, Morath S, Alexander C, Hamann L, Hartung T, Zähringer U, et al. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem. (2003) 278:15587–94. doi: 10.1074/jbc.M212829200
14. Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev. (2011) 24:557–91. doi: 10.1128/CMR.00008-11
15. Hoffmann O, Braun JS, Becker D, Halle A, Freyer D, Dagand E, et al. TLR2 mediates neuroinflammation and neuronal damage. J Immunol. (2007) 178:6476–81. doi: 10.4049/jimmunol.178.10.6476
16. Xu D, Lian D, Zhang Z, Liu Y, Sun J, Li L. Brain-derived neurotrophic factor is regulated via MyD88/NF-κB signaling in experimental Streptococcus pneumoniae meningitis. Sci Rep. (2017) 7:3545. doi: 10.1038/s41598-017-03861-z
17. Liu X, Han Q, Leng J. Analysis of nucleotide-binding oligomerization domain proteins in a murine model of pneumococcal meningitis. BMC Infect Dis. (2014) 14:648. doi: 10.1186/s12879-014-0648-3
18. Davis KM, Nakamura S, Weiser JN. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest. (2011) 121:3666–76. doi: 10.1172/JCI57761
19. Liu X, Chauhan VS, Young AB, Marriott I. NOD2 mediates inflammatory responses of primary murine glia to Streptococcus pneumoniae. Glia. (2010) 58:839–47. doi: 10.1002/glia.20968
20. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. (2007) 449:819–26. doi: 10.1038/nature06246
21. Shaw MH, Reimer T, Kim YG, Nuñez G. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol. (2008) 20:377–82. doi: 10.1016/j.coi.2008.06.001
22. Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. (2003) 300:1524–5. doi: 10.1126/science.1085536
23. Chen X, Li Y, Blankson S, Liu M, Huang D, Redmond HP, et al. B7-H3 Augments inflammatory responses and exacerbates brain damage via amplifying NF-κB p65 and MAPK p38 activation during experimental pneumococcal meningitis. PLoS ONE. (2017) 12:e0171146. doi: 10.1371/journal.pone.0171146
24. Kastenbauer S, Pfister HW. Pneumococcal meningitis in adults: spectrum of complications and prognostic factors in a series of 87 cases. Brain. (2003) 126:1015–25. doi: 10.1093/brain/awg113
25. Klein M, Koedel U, Pfister HW. Oxidative stress in pneumococcal meningitis: a future target for adjunctive therapy? Prog Neurobiol. (2006) 80:269–80. doi: 10.1016/j.pneurobio.2006.11.008
26. Horch HW, Krüttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. (1999) 23:353–64. doi: 10.1016/S0896-6273(00)80785-0
27. Li L, Shui QX, Zhao ZY, Zhu XD, Bao WQ. Dynamic changes of TrkB gene expression in Streptococcus pneumoniae meningitis after treatment with antibiotics and dexamethasone. World J Pediatr. (2011) 7:250–6. doi: 10.1007/s12519-011-0285-5
28. Morichi S, Kashiwagi Y, Takekuma K, Hoshika A, Kawashima H. Expressions of brain-derived neurotrophic factor (BDNF) in cerebrospinal fluid and plasma of children with meningitis and encephalitis/encephalopathy. Int J Neurosci. (2013) 123:17–23. doi: 10.3109/00207454.2012.721829
29. Lian D, He D, Wu J, Liu Y, Zhu M, Sun J, et al. Exogenous BDNF increases neurogenesis in the hippocampus in experimental Streptococcus pneumoniae meningitis. J Neuroimmunol. (2016) 294:46–55. doi: 10.1016/j.jneuroim.2016.03.014
30. Barichello T, Simões LR, Generoso JS, Sharin VS, Souza LB, Jornada LK, et al. Depression-like adult behaviors may be a long-term result of experimental pneumococcal meningitis in wistar rats infants. Neurochem Res. (2016) 41:2771–8. doi: 10.1007/s11064-016-1992-z
31. Xu D, Lian D, Wu J, Liu Y, Zhu M, Sun J, et al. Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J Neuroinflammation. (2017) 14:156. doi: 10.1186/s12974-017-0930-6
32. Lu H, Liu X, Zhang N, Zhu X, Liang H, Sun L, et al. Neuroprotective effects of brain-derived neurotrophic factor and noggin-modified bone mesenchymal stem cells in focal cerebral ischemia in rats. J Stroke Cerebrovasc Dis. (2016) 25:410–8. doi: 10.1016/j.jstrokecerebrovasdis.2015.10.013
33. Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. (1994) 368:147–50. doi: 10.1038/368147a0
34. Meng L, Ohman-Gault L, Ma L, Krimm RF. Taste bud-derived BDNF is required to maintain normal amounts of innervation to adult taste buds. eNeuro. (2015) 2:ENEURO.0097–15.2015. doi: 10.1523/ENEURO.0097-15.2015
35. Sikandar S, Minett MS, Millet Q, Santana-Varela S, Lau J, Wood JN, et al. Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain. (2018) 141:1028–39. doi: 10.1093/brain/awy009
36. Kuhn PL, Wrathall JR. A mouse model of graded contusive spinal cord injury. J Neurotrauma. (1998) 15:125–40. doi: 10.1089/neu.1998.15.125
37. Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. (1994) 25:1386–403. doi: 10.1002/neu.480251107
38. Fariñas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A. Characterization of neurotrophin and Trk receptor functions in developing sensory ganglia: direct NT-3 activation of TrkB neurons in vivo. Neuron. (1998) 21:325–34. doi: 10.1016/S0896-6273(00)80542-5
39. Koedel U, Scheld WM, Pfister HW. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis. (2002) 2:721–36. doi: 10.1016/S1473-3099(02)00450-4
40. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
41. Yao Q. Nucleotide-binding oligomerization domain containing 2: structure, function, and diseases. Semin Arthritis Rheum. (2013) 43:125–30. doi: 10.1016/j.semarthrit.2012.12.005
42. Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. (1997) 237:752–7. doi: 10.1006/bbrc.1997.7124
43. Metzger D, Chambon P. Site- and time-specific gene targeting in the mouse. Methods. (2001) 24:71–80. doi: 10.1006/meth.2001.1159
44. Cao M, Wu JI. Camk2a-Cre-mediated conditional deletion of chromatin remodeler Brg1 causes perinatal hydrocephalus. Neurosci Lett. (2015) 597:71–6. doi: 10.1016/j.neulet.2015.04.041
45. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. (2010) 13:133–40. doi: 10.1038/nn.2467
46. Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. (1997) 17:2295–313. doi: 10.1523/JNEUROSCI.17-07-02295.1997
47. Edelmann E, Lessmann V, Brigadski T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology. (2014) 76:610–27. doi: 10.1016/j.neuropharm.2013.05.043
48. Brouwer MC, Heckenberg SG, de Gans J, Spanjaard L, Reitsma JB, van de Beek D. Nationwide implementation of adjunctive dexamethasone therapy for pneumococcal meningitis. Neurology. (2010) 75:1533–9. doi: 10.1212/WNL.0b013e3181f96297
49. Weisfelt M, van de Beek D, Spanjaard L, Reitsma JB, de Gans J. Clinical features, complications, and outcome in adults with pneumococcal meningitis: a prospective case series. Lancet Neurol. (2006) 5:123–9. doi: 10.1016/S1474-4422(05)70288-X
50. Gerber J, Nau R. Mechanisms of injury in bacterial meningitis. Curr Opin Neurol. (2010) 23:312–8. doi: 10.1097/WCO.0b013e32833950dd
51. Barichello T, Generoso JS, Collodel A, Moreira AP, Almeida SM. Pathophysiology of acute meningitis caused by Streptococcus pneumoniae and adjunctive therapy approaches. Arq Neuropsiquiatr. (2012) 70:366–72. doi: 10.1590/S0004-282X2012000500011
52. Cooper DD, Seupaul RA. Is adjunctive dexamethasone beneficial in patients with bacterial meningitis? Ann Emerg Med. (2012) 59:225–6. doi: 10.1016/j.annemergmed.2011.10.014
53. Li L, Shui QX, Zhao ZY. Regulation of brain-derived neurotrophic factor (BDNF) expression following antibiotic treatment of experimental bacterial meningitis. J Child Neurol. (2003) 18:828–34. doi: 10.1177/088307380301801202
54. Blaser C, Wittwer M, Grandgirard D, Leib SL. Adjunctive dexamethasone affects the expression of genes related to inflammation, neurogenesis and apoptosis in infant rat pneumococcal meningitis. PLoS ONE. (2011) 6:e17840. doi: 10.1371/journal.pone.0017840
55. Berman PH, Banker BQ. Neonatal meningitis. A clinical and pathological study of 29 cases. Pediatrics. (1966) 38:6–24.
56. Marques CP, Cheeran MC, Palmquist JM, Hu S, Lokensgard JR. Microglia are the major cellular source of inducible nitric oxide synthase during experimental herpes encephalitis. J Neurovirol. (2008) 14:229–38. doi: 10.1080/13550280802093927
57. Mariani MM, Kielian T. Microglia in infectious diseases of the central nervous system. J Neuroimmune Pharmacol. (2009) 4:448–61. doi: 10.1007/s11481-009-9170-6
58. Nau R, Bruck W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. (2002) 25:38–45. doi: 10.1016/S0166-2236(00)02024-5
59. Takeda K, Tokunaga N, Aida Y, Kajiya M, Ouhara K, Sasaki S, et al. Brain-Derived neurotrophic factor inhibits peptidoglycan-induced inflammatory cytokine expression in human dental pulp cells. Inflammation. (2017) 40:240–7. doi: 10.1007/s10753-016-0474-4
60. Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol. (2012) 14:460–6. doi: 10.1111/j.1462-5822.2011.01746.x
61. Koedel U, Rupprecht T, Angele B, Heesemann J, Wagner H, Pfister HW, et al. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain. (2004) 127:1437–45. doi: 10.1093/brain/awh171
62. Zheng Y, Shang F, An L, Zhao H, Liu X. NOD2-RIP2 contributes to the inflammatory responses of mice in vivo to Streptococcus pneumoniae. Neurosci Lett. (2018) 671:43–9. doi: 10.1016/j.neulet.2018.01.057
63. Koedel U, Bayerlein I, Paul R, Sporer B, Pfister HW. Pharmacologic interference with NF-kappaB activation attenuates central nervous system complications in experimental Pneumococcal meningitis. J Infect Dis. (2000) 182:1437–45. doi: 10.1086/315877
64. Stringaris AK, Geisenhainer J, Bergmann F, Balshüsemann C, Lee U, Zysk G, et al. Neurotoxicity of pneumolysin, a major pneumococcal virulence factor, involves calcium influx and depends on activation of p38 mitogen-activated protein kinase. Neurobiol Dis. (2002) 11:355–68. doi: 10.1006/nbdi.2002.0561
65. English JM, Cobb MH. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol Sci. (2002) 23:40–5. doi: 10.1016/S0165-6147(00)01865-4
66. Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J Clin Pharmacol. (2003) 43:406–13. doi: 10.1177/0091270002250615
67. Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin Sci (Lond). (2006) 110:167–73. doi: 10.1042/CS20050163
68. Leal G, Comprido D, Duarte CB. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology. (2013) 76:639–56. doi: 10.1016/j.neuropharm.2013.04.005
Keywords: Streptococcus pneumoniae meningitis, brain-derived neurotrophic factor, microglia/macrophage, neuroinflammation, brain injury
Citation: Zhao S, Zhang Z, Xu D, Wang Y and Li L (2020) Selective Loss of Brain-Derived Neurotrophic Factor Exacerbates Brain Injury by Enhancing Neuroinflammation in Experimental Streptococcus pneumoniae Meningitis. Front. Immunol. 11:1357. doi: 10.3389/fimmu.2020.01357
Received: 09 March 2020; Accepted: 27 May 2020;
Published: 26 June 2020.
Edited by:
Sermin Genc, Dokuz Eylul University, TurkeyReviewed by:
Pankaj Baral, Harvard Medical School, United StatesLuiz Eduardo Baggio Savio, Federal University of Rio de Janeiro, Brazil
Copyright © 2020 Zhao, Zhang, Xu, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Li, bGlsaW5nQHhpbmh1YW1lZC5jb20uY24=