 Fangying Lei1,2†
Fangying Lei1,2† Xueduan Liu1,2†
Xueduan Liu1,2† Haonan Huang1,2
Haonan Huang1,2 Shaodong Fu1,2
Shaodong Fu1,2 Kai Zou1,2Shuangfei Zhang1,2Li Zhou3
Kai Zou1,2Shuangfei Zhang1,2Li Zhou3 Jianguo Zeng3
Jianguo Zeng3 Hongwei Liu1,2Luhua Jiang1,2Bo Miao1,2Yili Liang1,2*
Hongwei Liu1,2Luhua Jiang1,2Bo Miao1,2Yili Liang1,2*- 1School of Minerals Processing and Bioengineering, Central South University, Changsha, China
- 2Key Laboratory of Biometallurgy, Ministry of Education, Changsha, China
- 3Hunan Key Laboratory of Traditional Chinese Veterinary Medicine, Hunan Agricultural University, Changsha, China
Endophytes are highly associated with plant growth and health. Exploring the variation of bacterial communities in different plant niches is essential for understanding microbe-plant interactions. In this study, high-throughput gene sequencing was used to analyze the composition and abundance of bacteria from the rhizospheric soil and different parts of the Macleaya cordata. The results indicated that the bacterial community structure varied widely among compartments. Bacterial diversity was observed to be the highest in the rhizospheric soil and the lowest in fruits. Proteobacteria, Actinobacteria, and Bacteroidetes were found as the dominant phyla. The genera Sphingomonas (∼47.77%) and Methylobacterium (∼45.25%) dominated in fruits and leaves, respectively. High-performance liquid chromatography (HPLC) was employed to measure the alkaloid content of different plant parts. Significant correlations were observed between endophytic bacteria and alkaloids. Especially, Sphingomonas showed a significant positive correlation with sanguinarine and chelerythrine. All four alkaloids were negatively correlated with the microbiota of stems. The predicted result of PICRUST2 revealed that the synthesis of plant alkaloids might lead to a higher abundance of endophytic microorganisms with genes related to alkaloid synthesis, further demonstrated the correlation between bacterial communities and alkaloids. This study provided the first insight into the bacterial community composition in different parts of Macleaya cordata and the correlation between the endophytic bacteria and alkaloids.
Introduction
Endophytes are widely present inside plants during part or all stages of their life cycle. They are able to survive in the root, stem, leaf, fruit, flower, and seed due to the specific colonization conditions provided by plant tissues and organs (Loaces et al., 2010; Bulgarelli et al., 2013; Wellner et al., 2013; Glassner et al., 2015). Plant microbial community composition is influenced by environmental factors, including climate, temperature, geographic location, vegetation density, and host genotype (Overbeek and Elsas, 2008; Meyer and Leveau, 2012; Ramond et al., 2013; Campisano et al., 2017; Yana et al., 2017; Ting et al., 2019; Sharaby et al., 2020). However, several studies have shown that plant compartment is the main driver of bacterial community composition, while the season, location, and plant species only play a minor role (Miller and Rudgers, 2014; Junker and Keller, 2015; Fonseca-García et al., 2016; Cregger et al., 2018). Soil microorganisms were partially the source of endophytic bacteria (Zarraonaindia et al., 2015; Mangeot-Peter et al., 2020). They migrated and colonized the area around the root of plants under the influence of the roots’ secretions (Tahtamouni et al., 2015). Rhizospheric microbial community composition was significantly influenced by the physical and chemical parameters in soil (Winston et al., 2017; Hartman et al., 2018; Ziyuan et al., 2020), which also affected the plant compartments in part (Mighell et al., 2019). However, Two studies on Arabidopsis showed that the endophytic microbial community composition in the Arabidopsis plants growing on four different soils was similar (Davide et al., 2012; Lundberg et al., 2012), indicating the existence of host mediated control mechanisms. Significant plant compartment effects were also observed in the microbiome of plants such as Populus, Cycas panzhihuaensis, and Stellera chamaejasme L (Jin et al., 2014; Cregger et al., 2018; Zheng and Gong, 2019).
Endophytes and plants can be considered as mutualistic symbiosis. They survived and evolved together (Hardoim et al., 2015; Vandenkoornhuyse et al., 2015). Endophytes are an essential part of the plant micro-ecosystem. They were thought to complement the host plant’s gene library and could regulate metabolism, enhance stress resistance, and transform host plants’ secondary metabolites (Zhou et al., 2015; Prasad et al., 2018; Cordovez et al., 2019; Víctor and Juan, 2019). Some endophytic bacteria were able to synthesize bioactive compounds such as saponins, terpenoids, and alkaloids, which are potential sources of antibacterial, anti-insect, anticancer, and other properties (Yu et al., 2010; Pimentel et al., 2011; Gutierrez et al., 2012). Considering the various effects of plant-associated bacteria on plants, recording the spatial variability of these bacterial communities is critical for further understanding of plant-microbe interactions and the potential value of endophytic bacteria.
Macleaya cordata, a perennial plant mainly distributes in China, Europe, and North America, has been considered as a traditional folk herbal medicine. Its chemical composition and biological activity were a matter of concern because of its detoxifying, analgesic, anti-inflammatory, antimicrobial, and antitumoral properties (Liu et al., 2013; Khadem et al., 2014; Li et al., 2017; Huang et al., 2018). Its extract has been used as a good alternative of antibiotics in feed additives for animal production, and has achieved European Food Safety Certification for its effectiveness in treating inflammation and regulating the intestinal flora of livestock and poultry. Modern pharmacological studies showed that isoquinoline alkaloids are the primary bioactive substances in Macleaya cordata. Some of them possess a prominent apoptotic effect on cancer cells. For instance, sanguinarine could inhibit cell invasion and the MMP-9 and COX-2 expression in TPA-induced breast cancer cells by inducing HO-1 expression (Park et al., 2014). The four main alkaloids in Macleaya cordata are sanguinarine, chelerythrine, allocrytopine, and protopine (Kosina et al., 2010). Significant differences in the accumulation of these four alkaloids were reported in the root, stem, leaf, and fruit of Macleaya cordata. Sanguinarine and chelerythrine were observed to accumulate mainly in the fruits, with allocrytopine and protopine being most abundant in roots and leaves, respectively. The alkaloids content in the stem was very low. Endophytic bacterial communities in different parts might be affected by this difference.
Bacterial communities across different niches covering below-ground and above-ground tissue-level microbial habitats (rhizospheric soil, root, stem, leaf, and fruit) were characterized in this study by using 16S rRNA gene-targeted Illumina MiSeq sequencing. The content of four main alkaloids was also detected to explore the relationship between alkaloids and endophytic bacteria. We hypothesized that: (i) Niche differentiation among different sample types influenced the composition and diversity of the Macleaya cordata associated microbial communities (ii) Alkaloids might contribute to the variation of bacterial communities in different niches. This experiment can support the subsequent search of functional microorganisms and guide the cultivation, protection, and increase of crucial metabolites and resource utilization of Macleaya cordata.
Materials and Methods
Study Location and Sampling Methods
Macleaya cordata used in this study were collected from an experimental cultivar trial in Hunan province at a site managed by the Hunan Agricultural University in July 2019. Mature and healthy roots, stems, leaves, fruits, and rhizospheric soil were selected from six individual plants on the experimental blocks. All six trees were growing in the same general area and experienced similar developing conditions. Samples were collected randomly and cleaned with distilled water, followed by cutting three pieces from each leaf (from the bottom, middle, and top) with five leaves from different branches. Roots, stems, and fruits were also collected from the same location from each tree (Roots and stems were cut into 2.5 cm long pieces using a sterilized scalpel). All samples were collected aseptically wearing bioclean gloves and put in marked aseptic bags. They were placed in dry ice foam boxes and taken back to the lab as soon as possible. The part for alkaloid content detection was frozen in liquid nitrogen immediately and stored in a −80°C freezer. The part for microbiological analysis would be surface sterilized firstly. Then samples were crushed and homogenized to powder using a sterilized mortar and pestle, frozen in liquid nitrogen, and stored at −80°C until DNA extraction. The surface disinfection process is as follows: The samples were rinsed in tap water, sterilized by 75% ethanol (1 min), washed with sterile water three times, soaked in 8% sodium hypochlorite (5 min), washed with sterilized water five times, and left to dry with sterile filter paper.
Preparation of Standard Alkaloid Solutions and Sample Solutions
All solvents used were of liquid chromatography (LC) grade. The sanguinarine and chelerythrine standards were purchased from Sigma (Shanghai, China), while protopine and allocryptopine standards were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Standards were dissolved in methanol and diluted to obtain a series of working solutions (5.12–409.52, 4.20–84.04, 8.61–550.75, 7.80–499.30 μg/ml, respectively) and establish the standard curves at 280 nm. 5.0 g of the powder (60 mesh) of each sample was placed in 250 ml corked conical flasks and extracted with 100 ml of methanol-1% HCL (50:50) in an ultrasonic bath (60 min, 35°C, 200 W, 40 KHz), the weight loss was compensated with methanol-1% HCL, and the supernatant was collected. The supernatant was filtered through a 0.22 μm membrane before HPLC analysis. Then a 20 μl aliquot of the solution was injected into the HPLC system. For accurate analysis of high content alkaloids, the sample solution was diluted to avoid a concentration that lies outside the linear range.
HPLC Analysis
Analysis was carried out using a Waters alliance 1,260 liquid chromatographic system (Milford, MA, United States). LC separations were accomplished on a 5 μm, 250 mm × 4.6 mm Ultimate XB C18 column (Welch Materials, Ellicott City, United States) at 35°C. Mobile phases consisted of (A) water containing Acetonitrile and (B) 1% phosphoric acid solution. Gradient elution program was as follows: 0–11 min 25% A, 11–27 min 25–60% A, 27–29 min 60–25% A, 29–35 min 25% A. The flow rate was set at 1 ml/min. The injection volume was 20 μl. A wavelength of 280 nm was selected for quantification. The limit of detection (LOD) was 1.35, 0.97, 0.89, 0.93 μg/ml for protopine, allocryptopine, sanguinarine, and chelerythrine.
DNA Extraction and PCR Amplification of the Bacterial 16S rRNA
Total plant DNA and soil microbial DNA were extracted using DNeasy® Plant Mini Kit (250) (Qiagen, Inc., Duesseldorf, Germany) and ALFA-SEQ Advanced Soil DNA Kit (Guangdong Magigene Biotechnology Co., Ltd.), respectively. All steps were performed strictly according to the manufacturer’s protocols. The concentration and purity of DNA were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, United States). 1% (w/v) TAE-agarose gel stained with EB was also used to detect the extracted DNA’s quality. The V5 and V6 hypervariable regions of the bacteria 16S rRNA genes were amplified with the primers 799F (5′-AAC MGG ATT AGA TAC CCK G-3′); 1115R (5′-AGG GTT GCG CTC GTT G-3′). The extracted DNA was amplified using a thermocycler polymerase chain reaction (PCR) system with the following process: denaturation for 1 min at 94°C; 35 cycles of 20 s at 94°C, annealing for 25 s at 57°C, elongation for 45 s at 68°C; and a final extension step for 10 min at 68°C. The PCR was performed with reaction volumes of 50 μl containing 25 μl 2 × Taq Master Mix plus (Vazyme), 1.5 μl each primer (10 μM), 2 μl template DNA (100 ng), 20 μl PCR water. The PCR products were assessed by 1.5% (w/v) TAE-agarose gel stained with EB and further purified using the Gel Extraction Kit D2500 (OMEGA bio-tek, Norcross, Georgia, United States). High-throughput paired-end sequencing of the purified amplicons was conducted on the Illumina Miseq platform (Miseq PE250).
Sequencing Data Processing
Gene sequencing data of raw 16S rRNA were imported to the QIIME2 platform to generate the OTU table. The cutadapt program was used to remove the forward and reverse primers. The paired-end fastq files were merged, denoised, chimeras removed using the dada2 program. Meanwhile, they were clustered to operational taxonomic units (OTUs) at 97% similarity. Clean data were classified using BLAST with the Greengenes reference databases. Contaminant sequences unclassified at the domain (bacteria/archaea), mitochondria, and chloroplasts were filtered during the taxa filter-table process. All sequences produced from Illumina sequencing had been uploaded to the sequence read archive (SRA) of the NCBI database. The accession number of all samples is PRJNA714300.
Statistical Analysis
Alpha-diversity indexes, including Shannon index and Pielou evenness, and Non-metric Multidimensional Scaling (NMDS) for assessing the beta-diversity differences in community composition were calculated on Institute for Galaxy | Denglab pipeline1. Dissimilarity tests of OTUs and predicted function genes (by PICRUSt2) were computed using MRPP, ANOSIM, and PERMANOVA with Bray-Curtis distance matrices to identify whether the habitat had a significant effect on community composition and the abundance of functional genes. Statistical analyses were conducted using Minitab 19.0 software (Minitab Inc., Pennsylvania State University, Pennsylvania, United States). One-way ANOVA analyses, followed by Tukey’s test, were carried out to identify the gene abundance difference. Correlation between bacterial community and four alkaloids was carried out based on the Spearman correlation coefficients and Canonical Correspondence Analysis (CCA). PICRUSt2 was employed to predict each sample’s function genes based on the 16S rRNA sequencing data, and the KEGG database was used to match the selected reference OTU.
Results
Analysis of Sequencing Data
A total of 4,298,014 raw reads were obtained among 30 samples. After quality control and contaminant sequences removal, 1,218,950 high-quality paired 16S rRNA sequences with an average read length of 300 bp remained, which can be clustered into 6,866 OTUs (Supplementary Table 1). CPM standardization was performed on all samples before analysis of microbial communities. Rarefaction curves were constructed for each sample showing the number of observed OTUs. As expected, endophytic microorganisms had lower observed OTUs than rhizospheric soil bacterial communities (Supplementary Figure 1). Rarefaction curves evaluating the OTU richness per sample generally approached saturation, indicating that the sequencing depth was adequate.
Diversity of the Microbial Communities Associated With Macleaya cordata
Bacterial diversity was observed to be highest in soil, while the situation in plants was relatively complex, with bacterial diversity showing no significant differences in roots, stems and leaves, but a significant decrease in fruits. The highest evenness of bacterial communities was found in the stems among plant parts (Figure 1). NMDS revealed strong clustering of bacterial communities based on plant compartments (Figure 2A). Specifically, Coordinate1 (x-axis) separated the rhizospheric soil, roots, and above-ground (stems, leaves, fruit) samples. Rhizospheric soil exhibited a significant difference in community composition comparing with the other four plant compartments. However, the bacterial communities in the fruit and leaf were highly similar and separated from other endosphere samples in Coordinate2 (y-axis). Dissimilarity tests based on Bray-Curtis distance, including MRPP, ANOSIM, and PERMANOVA, also showed that the bacterial community composition of these five groups was significantly different (P < 0.05) (Table 1).
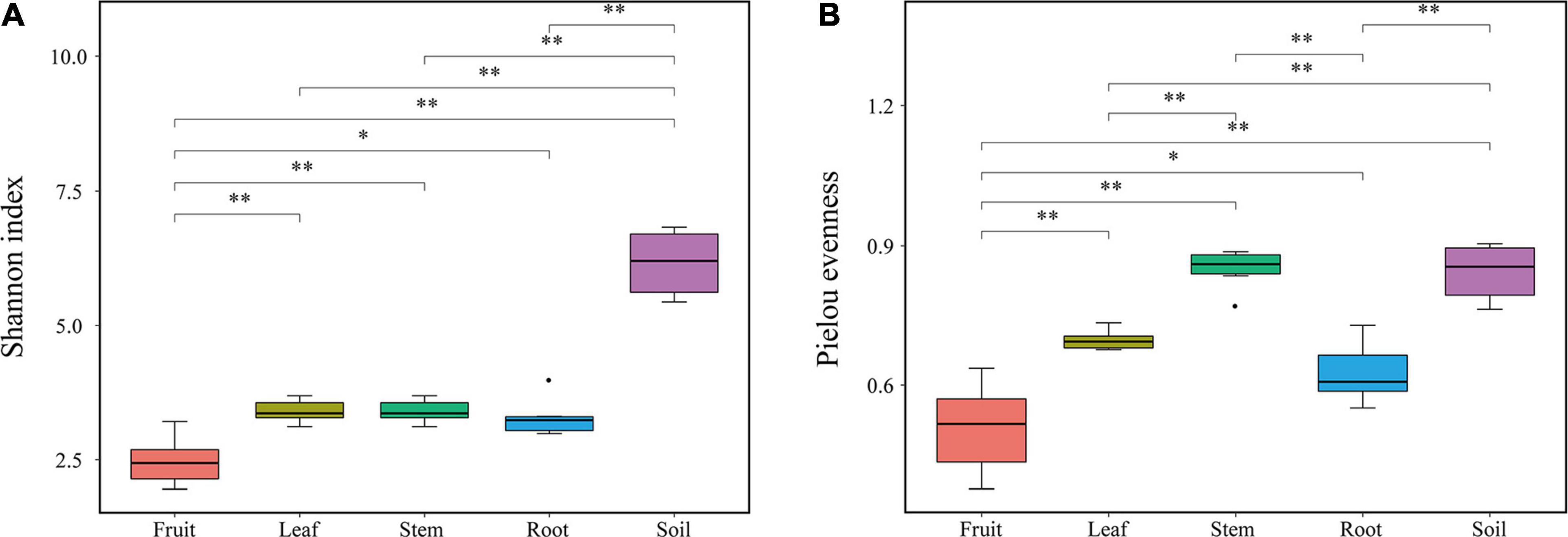
Figure 1. Diversity indexes of endophytic bacterial communities. (A) Shannon index (B) Pielou’s evenness.
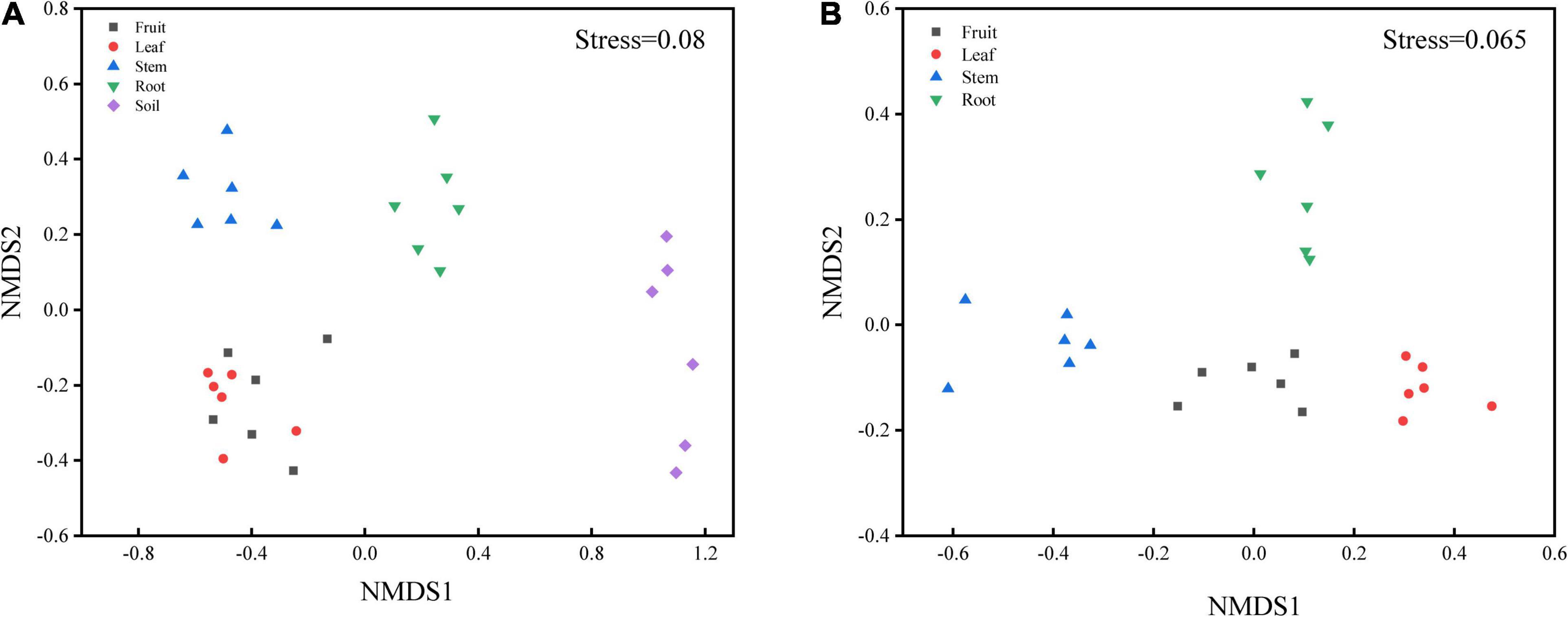
Figure 2. Non-metric multidimensional scaling (NMDS) carried out with (A) OTU table based on Bray-Curtis distance (S = 0.08) and (B) predicted function gene table based on Bray-Curtis distance (S = 0.065).
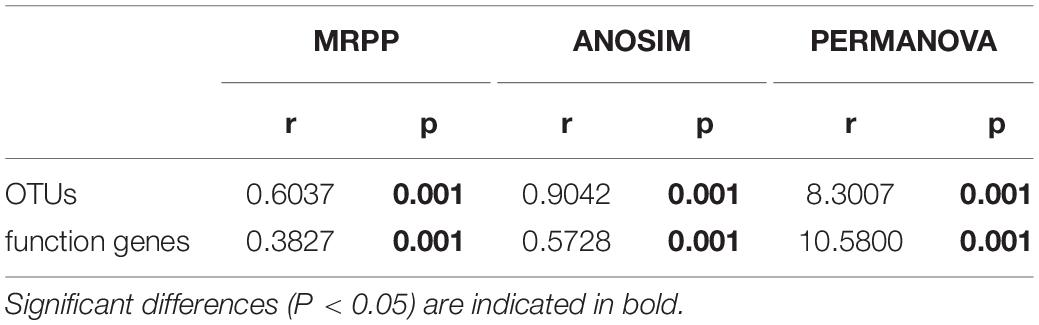
Table 1. Dissimilarity tests of OTUs and predicted function genes (by PICRUSt) based on Bray-Curtis distance.
To better understand the OTUs distribution within different niches, the number of OTUs shared by all compartments and OTUs uniquely identified in each sample type were calculated (Supplementary Figure 2). The bacterial communities in the rhizospheric soil had the highest number of OTUs, followed by roots. 302, 155, 236, 245, and 5,391 bacterial OTUs were uniquely identified for root, stem, leaf, fruit, and rhizospheric soil, respectively, with 19 OTUs shared among the five sample groups. A high overlap (208) of OTUs from rhizospheric soil and root was observed.
The Bacterial Composition Differences Across Habitat at Phylum Level and Genera Level
At the phylum level, the bacterial community composition showed high variabilities among different compartments (Figure 3A). Twenty-five dominant bacterial phyla (≥0.1% relative abundance) were detected across this study. Proteobacteria phyla was the dominant group in all niches (soil, root, stem, leaf, fruit), representing 28.61–54.84%, 37.76–88.36%, 60.43–79.27%, 69.53–88.93%, and 90.30–97.10% of all bacteria, respectively. Alphaproteobacteria accounted for the highest proportion in leaves. The phylum Thermi showed a significantly high abundance in the above-ground part, almost absent in the underground part. Firmicutes and Tenericutes were detected in stems but rare in other parts. The highest abundance of Actinobacteria was found in the roots, followed by the rhizospheric soil, with lower abundance in the above-ground parts. Bacteroidetes had greater abundance in soils and roots than in the above-ground parts whereas TM7 had a greater abundance in root and stem habitats than other habitats. Bacterial community composition was different between rhizospheric soil and plant compartments. Acidobacteria, Bacteroidetes, Gemmatimonadetes, Chloroflexi, Verrucomicrobia, AD3, and Nitrospirae were relatively abundant in the soil compared to other compartments. However, some of them were also detected in endosphere samples in very low abundance.
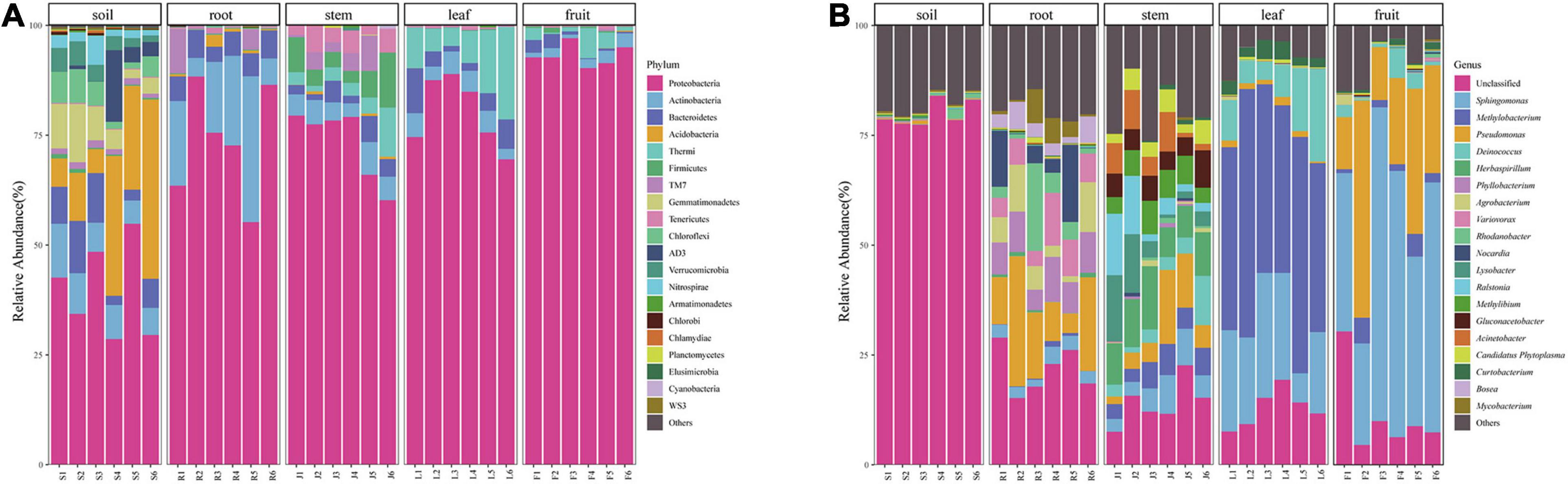
Figure 3. Bacterial community composition of different niches (A) bacterial community at the Phylum level and (B) bacterial community at the genera level.
At the genera level, 334 bacteria were identified from all sequences. Dominant bacteria differed among five compartments. Only the top 20 genera were shown in Figure 3B. Sphingomonas (47.77%) and Pseudomonas (25.09%) were abundant in the fruit, whereas Methylibium (45.25%) and Deinococcus (10.20%) were mainly distributed in the leaves. Pseudomonas (10.09%), Nocardia (11.42%), and Rhodanobacter (8.82%) were the dominant genera in the roots, and their abundance did not differ much. The microorganisms in stems were relatively uniform. The dominant genera in fruit, leaves, and roots were also abundant in stems. Besides, some microorganisms with low abundance in other endophytic tissues like Herbaspirillum (9.79%), Lysobacter (6.30%), and Ralstonia (6.03%) were also present in relatively high abundance in stems. Most bacterial genera detected in soil could not be identified.
Alkaloids Content of Macleaya cordata
The contents of four kinds of alkaloids in each tissue (fruit, leaf, stem, and root) of the six mature plants were calculated according to the standard curve regression equation (Supplementary Table 2). Alkaloid content showed a significant difference among organs (Figure 4A). The highest content of sanguinarine and chelerythrine was observed in fruit (4.04 and 1.65 mg/g). They were low in roots and leaves and almost zero in the stem. The accumulation of alkaloids in leaves and roots was mainly composed of protopine and allocrytopine. Roots contained the highest allocrytopine (11.67 mg/g) than other organs, while the leaf’s protopine content (12.81 mg/g) was the highest. The accumulation of four kinds of alkaloids in the stem was much lower than that in the other three organs.
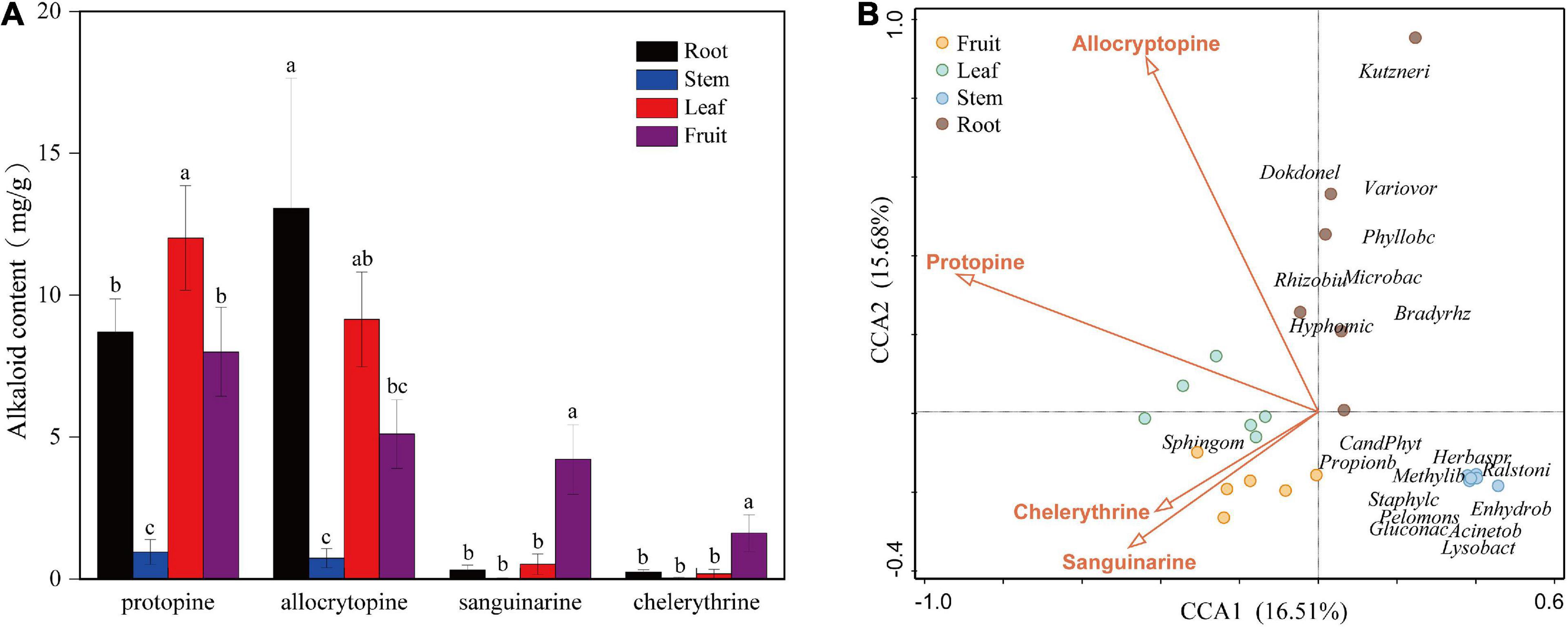
Figure 4. (A) Contents of four kinds of alkaloids in different organs (fruit, leaf, stem, and root) of Macleaya cordata. (B) Canonical Correspondence Analysis (CCA) of the top 20 bacterial genera (with significant correlation coefficients) and four critical alkaloids in Macleaya cordata.
Correlation Between the Endophytic Bacterial Abundances and Four Alkaloids
The Spearman correlation analysis was performed to explore the relationship between the endophytic bacteria at the phylum and genus level (relative abundance > 0.01%) and alkaloids to quantify the environmental influence (Tables 2, 3). Tenericutes showed a significant (P < 0.05) and negative correlation with all four alkaloids. Actinobacteria was significantly (P < 0.05) negatively correlated with sanguinarine and chelerythrine. However, most of the genera with higher abundance in this phylum showed a significant (P < 0.05) and positive correlation with allocryptopine. The Firmicutes, β- proteobacteria, and γ-proteobacteria were significantly (P < 0.05) negatively correlated with protopine and allocryptopine. Only the α-proteobacteria was significantly (P < 0.05) and positively correlated with protopine, allocryptopine, and sanguinarine. At the genera level, most endophytic bacteria were significantly (P < 0.05) correlated with alkaloids. Sphingomonas showed a significant (P < 0.05) positive correlation with sanguinarine and chelerythrine, while Herbaspirillum, Methylibium, and Pelomonas showed a significant (P < 0.01) and negative correlation with them. Canonical Correspondence Analysis (CCA) of the top 20 bacterial genera (with significant correlation coefficients) and four critical alkaloids in Macleaya cordata was also conducted (Figure 4B). The results were consistent with the above analysis. The sanguinarine and chelerythrine were more associated with the fruit bacterial communities, whereas protopine and allocryptopine had greater effects on leaf and root bacterial communities, respectively. However, all four alkaloids were negatively correlated with the microbiota of the stem.
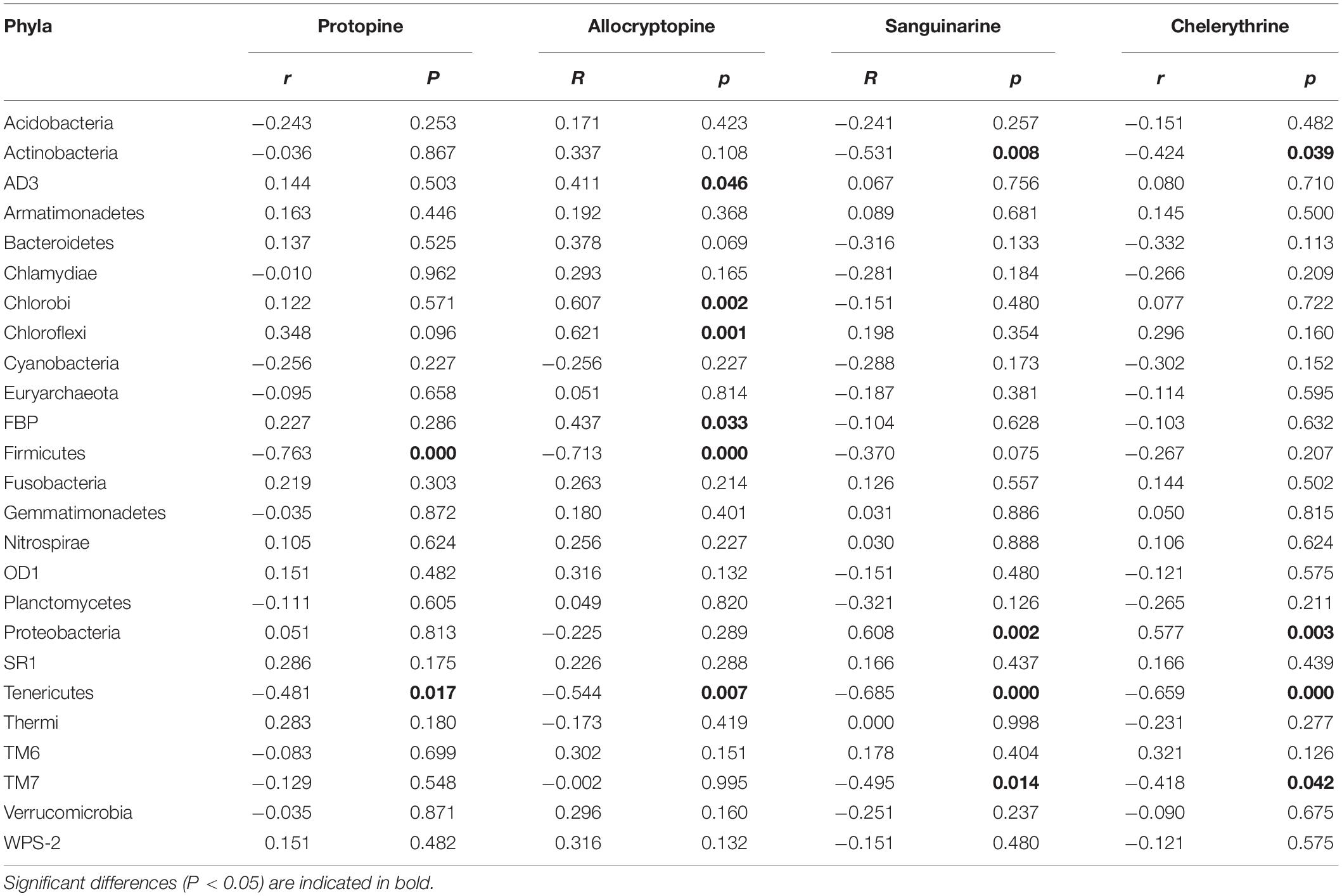
Table 2. Spearman Correlation coefficients showing the relationships between endophytic bacterial phyla and four alkaloids.
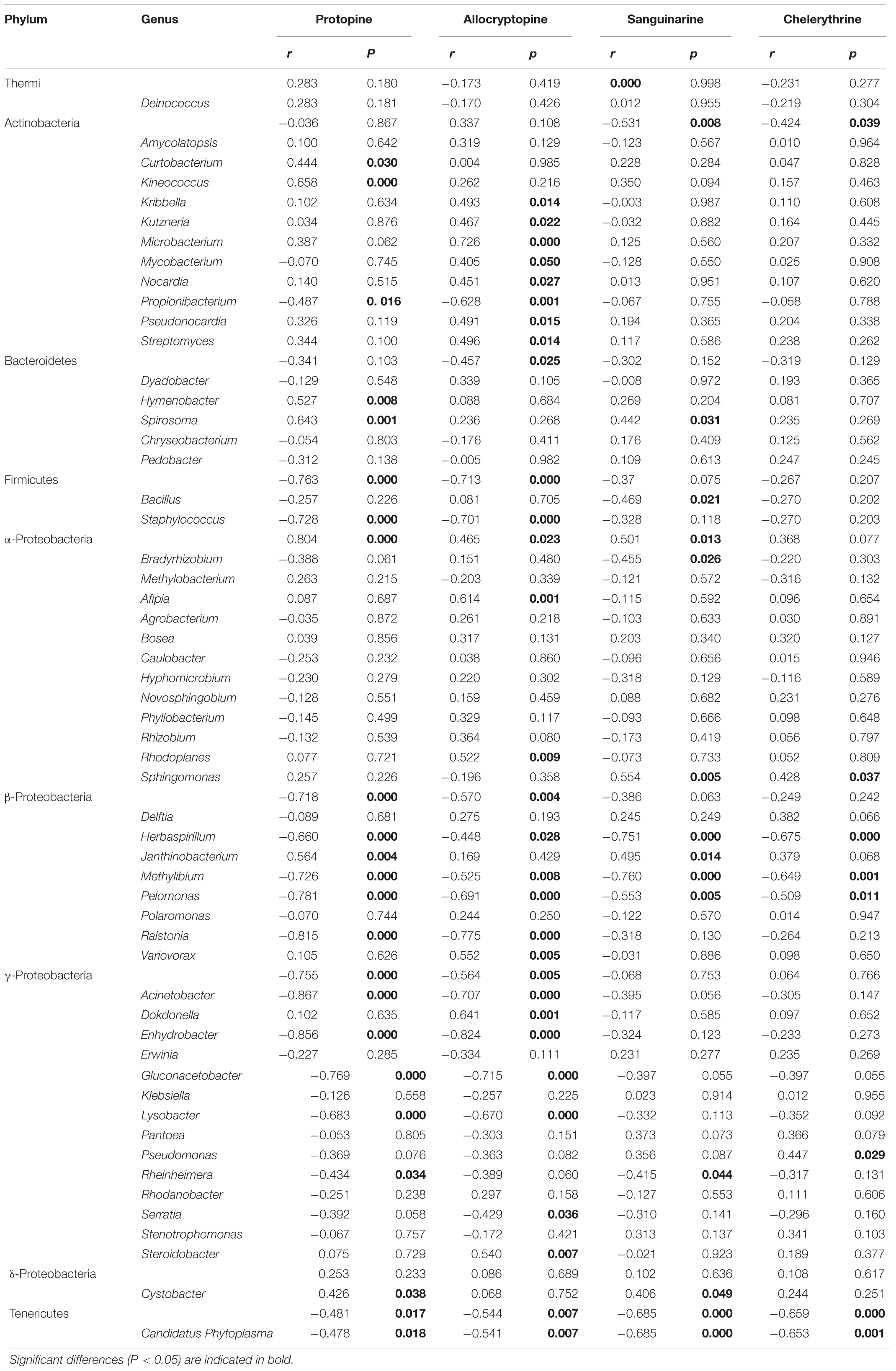
Table 3. Spearman Correlation coefficients showing the relationships between endophytic bacterial genera (relative abundance > 0.01%) and four alkaloids.
Functional Predictions by PICRUSt2
PICRUSt2 was applied as an exploratory predictive tool for functional annotation analysis. 7,185 KEGG Orthology groups (KOs) were predicted in Macleaya cordata endophytic communities. NMDS showed that these KOs were strongly clustered and separated from each other in different niches (Figure 2B). Dissimilarity tests of the relative abundance of KOs based on Bray-Curtis distance were also employed, including MRPP, ANOSIM, and PERMANOVA (Table 1). These results revealed that the relative abundance of KOs was significantly different among the root, stem, leaf, and fruit samples (P < 0.05).
Forty-two gene families were observed at Hierarchical Level 1 (KOs in KEGG). The majority of the KOs belonged to Genetic Information Processing, Environmental Information Processing, and Metabolism. As shown in Figure 5A, the frequencies of categories associated with membrane transport gradually increased from 4.57% in fruit to 5.91% in roots. In comparison, the frequencies of categories associated with signal transduction decreased from 4.79% in fruit to 3.23% in roots. The abundance of KOs related to the biosynthesis of other secondary metabolites was the lowest in the stem bacterial community and the highest in the root. To further identify microbiota function differences in the biosynthesis of secondary metabolites, 17 related KEGG functional categories at the second hierarchy level were analyzed (Figure 5B). In comparison between groups with different niches, eight pathways (e.g., Streptomycin biosynthesis, Caffeine metabolism, and Cytochrome P450) were significantly more abundant in the root, while the Staurosporine biosynthesis pathway was significantly more abundant in fruit.
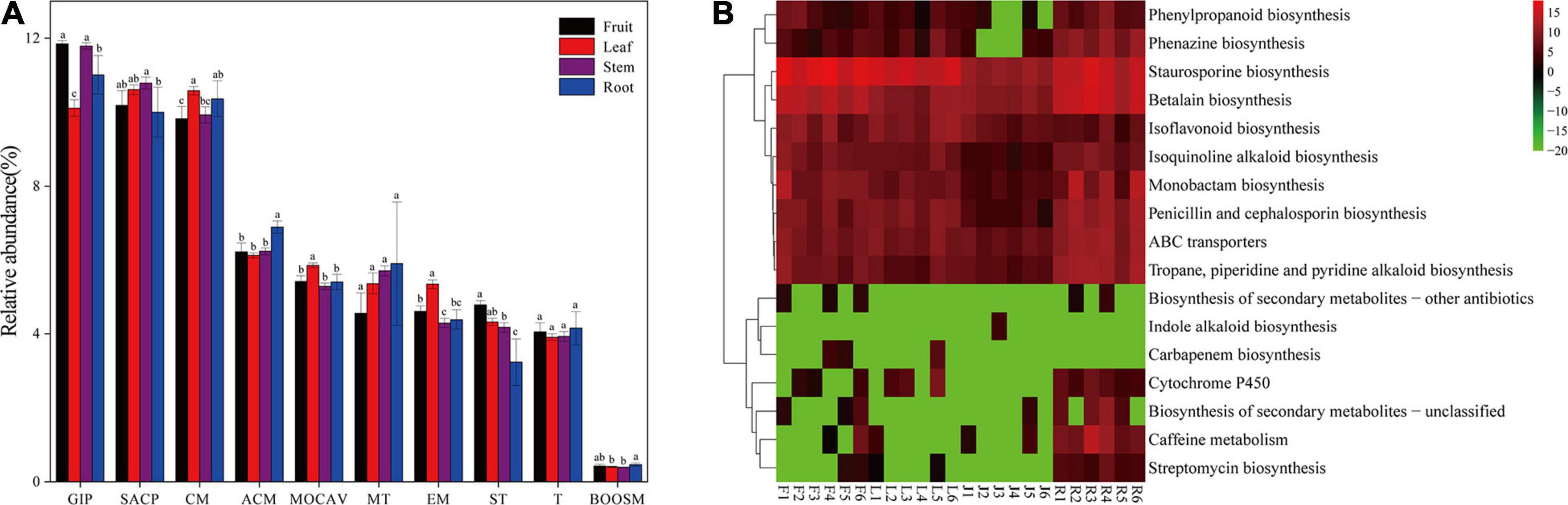
Figure 5. (A) Gene functional predictions of endophytic microbiota between different plant compartments. (GIP: genetic information processing; SACP: signaling and cellular processes; CM: Carbohydrate metabolism; ACM: Amino acid metabolism; MOCAV: Metabolism of cofactors and vitamins; MT: Membrane transport; EM: Energy metabolism; ST: Signal transduction; T: Translation; BOOSM: Biosynthesis of other secondary metabolites). (B) Comparisons of the seventeen gene pathways involved in the biosynthesis of secondary metabolites in the bacterial microbiota. Values of each functional gene (row) were log2 transformed. The second level of the KEGG pathway related to the biosynthesis of secondary metabolites was shown in the heatmap (S, Soil; R, Root; J, Stem; L: leaf; F: fruit).
Discussion
The Composition and Diversity of Bacterial Communities Differ in Niches
Bacterial community composition was significantly (P < 0.01) different among plant compartments (Table 1). The NMDS diagram (Figure 2A) also showed that the separation in the bacterial community structure between different plant compartments at the OTU level, indicating differences in the microbes in different niches. Although soil microorganisms were a source of plant endophytes, the migration of soil microorganisms to the root was still mediated by the host (Davide et al., 2012; Lundberg et al., 2012). For above-ground plant tissues, endophytes also derived from horizontal dispersal in the atmosphere and vertical transmission through seeds (Maignien et al., 2014; Fonseca-García et al., 2016), which resulted in bacterial differences between above-ground organs and soil. Similar results have also been reported in maize and wheat (Xiong et al., 2020), poplar (Beckers et al., 2017), Arabidopsis thaliana (Davide et al., 2012), and other plant species (Edwards et al., 2015; Lu et al., 2020).
As demonstrated by rarefaction curves and the alpha diversity indices, there were significant differences in species diversity between rhizospheric soil and plant compartments of Macleaya cordata. As an ideal habitat for various microorganisms, the bacterial diversity of rhizospheric soil was significantly higher than roots and above-ground niches. This finding is consistent with the general view of microbial colonization (Beckers et al., 2017). No significant difference was observed in the Shannon diversity of bacterial communities in roots, stems, and leaves, while the bacterial diversity in fruits was the lowest. The bacterial diversity within the plant compartments was low, probably because plant tissues were highly variable and complex. Bacterial colonization is limited by the host’s immune system, nutritional conditions, and metabolites (Compant et al., 2010). The highest evenness of bacterial communities was found in the stem among plant parts. It should be noticed that there was no significant difference in bacterial diversity between stems and roots, while many studies have determined that bacterial diversity was highest in roots (Santana et al., 2016; Wang et al., 2019). The variation of endophytic bacterial communities in different compartments was primarily driven by tissue-specific filtering mechanisms within the host (Bonito et al., 2014; Chao et al., 2020). Under the host-mediated control, only a limited number of microorganisms could maintain a symbiotic lifestyle with the host. This pressure sequentially increased from the soil to the plant compartments (Chao et al., 2020; Pasquale et al., 2020), which might be responsible for the lowest diversity of bacterial communities in fruits of Macleaya cordata.
Niche Preference Exists for Bacteria of Macleaya cordata
We identified several prokaryotic taxa (>0.1%), including microbiota members belonging to Proteobacteria, Actinobacteria, Acidobacteria, Bacteroidetes, TM7, Firmicutes. Bacterial diversity varied among plant-associated habitats, and the dominant phylum found in each habitat was highly comparable to other plant hosts in each habitat. Many studies have reported that plants’ bacterial microbiota is generally dominated by three major phyla (Proteobacteria, Actinobacteria, and Bacteroidetes) in both above- and below-ground tissues (Tahtamouni et al., 2015; Wang et al., 2020). This is also consistent with our study. Proteobacteria was the most abundant in the above-ground compartment, while Actinobacteria was widely distributed in the below-ground. Many microorganisms were detected in rhizospheric soil but hardly found in other niches, such as Acidobacteria, Bacteroidetes, and Gemmatimonadetes, which were often discovered in the soil in other studies (Cregger et al., 2018; Wang et al., 2020). Bacteria in the stem were affected by both the above-ground parts (leaf and fruit) and the below-ground part (root) and dominated by Proteobacteria with the enrichment of Actinobacteria, Bacilli, Mollicutes, and TM7. However, some of the microorganisms did not spread from the stem to the leaf and fruit. This might be caused by nonuniform colonization of different compartments, the microbial source difference, or other environmental factors.
As shown in Figure 3B, only a few bacterial genera were dominant in fruit and leaves. Sphingomonas and Pseudomonas were found to be the dominant bacterial genera in the fruit, and Methylobacterium, Sphingomonas, and Deinococcus were detected as predominant groups in the endophytic communities of the Macleaya cordata leaf. Different studies have reported that these genera represented a substantial part of various plant species’ endophytic microbiota (Mano and Morisaki, 2008; Delmotte et al., 2009). In the previous study, Sphingomonas played an essential role in plant stress tolerance, plant growth promotion, and biodegradation of polycyclic aromatic hydrocarbon (Wilkes et al., 1996; Halo et al., 2015; Asaf et al., 2020). Microbial colonization is related to its ability to adapt to the host’s internal environment and its utilization of substrates. Leaves are more often exposed to the vagaries of the environment, including nutrient stress, desiccation, and ultraviolet radiation, providing a special habitat for microorganisms (Hunter et al., 2010). Methylobacterium, often isolated from the leaf surface and interior, could specifically colonize the plant by profiting from methanol released by the plant (Galbally and Kirstine, 2002) and has been reported to be drought and radiation-resistant (Yoshida et al., 2017; Jorge et al., 2019; Kim et al., 2019). Therefore, Methylobacterium were able to successfully colonize the leave extensively (Delmotte et al., 2009). Cystobacter was found only in the fruits and leaves, and Erwinia was found only in above-ground tissues. Colonization of these two genera might be due to horizontal transmission (Frank et al., 2017).
Bacterial communities of different ecological niches were assembled under the effect of environmental filtering, ecological drift, and dispersal limitations (Nemergut et al., 2013; Moroenyane et al., 2021). However, there is still a lack of understanding of these processes. Plants are exposed to diverse and highly variable environmental factors, physiological structure (thickness and shape), chemical properties (nutrients contents, water, and secondary metabolites) of each compartment drive the differences in bacterial communities to some extent (Delmotte et al., 2009; Hunter et al., 2010; Arturo et al., 2012). In a study on soybean, the secondary metabolites (ethylamine and betaine) were considered as a robust environmental filter for bacterial communities (Shintaro et al., 2019). Furthermore, the bacterial communities in Stevia rebaudiana and Coptis teeta, were also demonstrated to be significantly correlated with secondary metabolites (Yu et al., 2015; Liu et al., 2020). On the other hand, the functional capacity of bacterial species is key to their recruitment by hosts, and Burke et al. (2011) proposed that bacterial community assembly is associated with function rather than the taxonomy.
The Alkaloids May Contribute to the Variation of the Endophytic Bacterial Communities
Protopine and allocrytopine are considered as precursors of sanguinarine and chelerythrine, respectively, and they are abundant in the root. In a previous study on the dynamics of the four alkaloids of Macleaya cordata, it was found that after entering the mature fruiting season, the content of protopine in the fruit decreased significantly, while the content of sanguinarine and chelerythrine increased rapidly. This suggested that protopine and allocrytopine were transported into the fruit and converted into sanguinarine and chelerythrine. The transcriptome, proteome, and metabolism data in a previous study conducted by Jianguo Zeng et al. (2013) revealed that the root of Macleaya cordata is the primary organ for the biosynthesis of isoquinoline alkaloids. In this study, significant differences were found in alkaloids accumulation in various organs. The content of allocrytopine in the root, leaf, and fruit showed a decreasing trend. Besides, the highest accumulation of protopine was found in leaves, followed by roots and fruit. The sanguinarine and chelerythrine in fruit were much higher than those in other tissues. The accumulation of all four alkaloids in stems was low. Some common endophytes, such as Firmicutes, have been reported to be abundant in the above-ground part, whereas in this study, it was only abundant in the stem. It can be speculated that this may explain to some extent the higher microbial diversity in stems since these four alkaloids have antibacterial effects (Beuria et al., 2005; Li et al., 2009; Razan et al., 2014; Yu et al., 2014). The CCA analysis also showed that all four alkaloids were negatively correlated with the microbiota of the stem of Macleaya cordata. The correlation tests between alkaloids and microorganisms also showed that most endophytic bacteria were significantly (P < 0.05) correlated with alkaloids.
Endophytes and host plants are closely related, and they adapt to each other and coevolve. Studies have shown that genes and abilities that evolve in one lineage are usually acquired steadily by another lineage (Papke and Gogarten, 2012). Direct gene transfer between species has occurred in all major taxa and seems to occur more frequently in prokaryotes (Moran, 2007). This leads to the possibility that microorganisms could respond to environmental toxins by selecting specific gene sequences that give them a competitive advantage over other organisms. It can be hypothesized that the microbes colonized in the plant might be affected by alkaloids produced by the host. Through the analysis of the functional annotation in Hierarchical level 2, endophytic bacteria in roots was found to contribute most to the gene abundance of the Cytochrome P450, ABC transporters, and secondary metabolite synthesis pathway including the isoquinoline alkaloid synthesis pathway. This was consistent with Jianguo Zeng’s reports that all enzymes for protopine and allocrytopine biosynthesis were highly expressed in the host root. Cytochrome P450 is considered as a large family of enzymes involved in many important metabolic pathways, and many enzymes related to the biosynthesis of benzylisoquinoline alkaloids belong to the P450 family (Zeng et al., 2013). Simultaneously, the ABC transporters are involved in transporting secondary metabolites such as alkaloids (Shitan and Yazaki, 2007). The number of genes annotated to the Staurosporine biosynthesis pathway was significantly higher in fruits than that in other parts, which may be due to the high accumulation of sanguinarine in the fruit. Staurosporine, an alkaloid with a diindole chemical structure, has been reported to partially block the accumulation of sanguinarine induced by a fungal activator (Peter et al., 1996). Since the primary function of stems is transport and there is almost no alkaloid stored in the stem, this may contribute to the low gene abundance of the microbiota genes related to the synthesis of secondary metabolites in the stem.
Conclusion
This study provided the first insight into the bacterial communities of different plant tissues and rhizospheric soil in Macleaya cordata. There were significant differences in bacterial communities among different ecological niches under the influence of plants’ vertical stratification structure. A strong correlation between the endophytic bacteria and the alkaloids was found by the Spearman correlation analysis. The predicted results of PICRUST2 further demonstrated that alkaloids might contribute to the variation of bacterial communities in different niches. All in all, this experiment can support the subsequent search of functional microorganisms and guide the cultivation, protection, and increase of crucial metabolites and resource utilization of Macleaya cordata.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
FL, XL, and YL conceived and designed the work. FL, HH, and SF performed the experiments. FL and XL wrote and revised the paper. KZ and SZ helped with the sample collection. FL and XL contributed equally to this study. All authors read and approved the final manuscript.
Funding
This work was supported by the following grants: Chinese National Science and Technology Support Program (2013BAC09B00), National Natural Science Foundation of China (31570113), National Key R&D Program of China (2017YFD0501500), and Graduate Research and Innovation Project (1053320170629) in Central South University, China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.681210/full#supplementary-material
Footnotes
References
Arturo, S.-A., Yumi, O., Fernandes, G. W., Ball, R. A., and Gamon, J. (2012). Relationships between endophyte diversity and leaf optical properties. Trees Struct. Funct. 26, 291–299. doi: 10.1007/s00468-011-0591-5
Asaf, S., Numan, M., Khan, A. L., and Al-Harrasi, A. (2020). Sphingomonas: from diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 40, 138–152. doi: 10.1080/07388551.2019.1709793
Beckers, B., Op De Beeck, M., Weyens, N., Boerjan, W., and Vangronsveld, J. (2017). Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 5:25.
Beuria, T. K., Santra, M. K., and Panda, D. (2005). Sanguinarine blocks cytokinesis in bacteria by inhibiting FtsZ assembly and bundling. Biochemistry 44, 16584–16593. doi: 10.1021/bi050767%2B
Bonito, G., Reynolds, H., Robeson, M. S. II, Nelson, J., Hodkinson, B. P., Tuskan, G., et al. (2014). Plant host and soil origin influence fungal and bacterial assemblages in the roots of woody plants. Mol. Ecol. 23, 3356–3370. doi: 10.1111/mec.12821
Bulgarelli, D., Schlaeppi, K., Spaepen, S., van Themaat, E., and Schulze-Lefert, P. (2013). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
Burke, C., Steinberg, P., Rusch, D., Kjelleberg, S., and Thomas, T. (2011). Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. U S A 108, 14288–14293. doi: 10.1073/pnas.1101591108
Campisano, A., Albanese, D., Yousaf, S., Pancher, M., Donati, C., and Pertot, I. (2017). Temperature drives the assembly of endophytic communities’ seasonal succession. Environ. Microbiol. 19, 3353–3364. doi: 10.1111/1462-2920.13843
Chao, X., YongGuan, Z., JunTao, W., Brajesh, S., LiLi, H., JuPei, S., et al. (2020). Host selection shapes crop microbiome assembly and network complexity. New Phytol. 229, 1091–1104.
Compant, S., Clément, C., and Sessitsch, A. (2010). Plant growth-promoting bacteria in the rhizo- and endosphere of plants: their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 42, 669–678. doi: 10.1016/j.soilbio.2009.11.024
Cordovez, V., Dini-Andreote, F., Carrión, V. J., and Raaijmakers, J. M. (2019). Ecology and evolution of plant microbiomes. Annu. Rev. Microbiol. 73, 69–88. doi: 10.1146/annurev-micro-090817-062524
Cregger, M. A., Veach, A. M., Yang, Z. K., Crouch, M. J., Vilgalys, R., Tuskan, G. A., et al. (2018). The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6:31.
Davide, B., Matthia, R., and Klaus, S. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Delmotte, N., Knief, C., Chaffron, S., Innerebner, G., Roschitzki, B., Schlapbach, R., et al. (2009). Community proteogenomics reveals insights into the physiology of Phyllosphere bacteria. Proc. Natl. Acad. Sci. U S A 106, 16428–16433. doi: 10.1073/pnas.0905240106
Edwards, J., Johnson, C., Santos-Medellin, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U S A 112, E911–E920.
Fonseca-García, C., Coleman-Derr, D., Garrido, E., Visel, A., Tringe, S. G., and Partida-Martínez, L. P. (2016). The Cacti Microbiome: interplay between habitat-filtering and host-specificity. Front. Microbiol. 7:150. doi: 10.3389/fmicb.2016.00150
Frank, A. C., Saldierna Guzmán, J. P., and Shay, J. E. (2017). Transmission of bacterial endophytes. Microorganisms 5:70. doi: 10.3390/microorganisms5040070
Galbally, I. E., and Kirstine, W. (2002). The production of methanol by flowering plants and the global cycle of methanol. J. Atmospheric chem. 43, 195–229.
Glassner, H., Zchori-Fein, E., Compant, S., Sessitsch, A., Katzir, N., Portnoy, V., et al. (2015). Characterization of endophytic bacteria from cucurbit fruits with potential benefits to agriculture in melons (Cucumis meloL.). FEMS Microbiol. Ecol. 91:fiv074. doi: 10.1093/femsec/fiv074
Gutierrez, R. M. P., Gonzalez, A. M. N., and Ramirez, A. M. (2012). Compounds derived from endophytes : a Review of phytochemistry and. Curr. Med. Chem. 19, 2992–3030. doi: 10.2174/092986712800672111
Halo, B. A., Khan, A. L., Waqas, M., Al-Harrasi, A., Hussain, J., Ali, L., et al. (2015). Endophytic bacteria (Sphingomonassp. LK11) and gibberellin can improveSolanum lycopersicumgrowth and oxidative stress under salinity. J. Plant Interact. 10, 117–125. doi: 10.1080/17429145.2015.1033659
Hardoim, P. R., van Overbeek, L. S., Berg, G., Pirttila, A. M., Compant, S., Campisano, A., et al. (2015). The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 79, 293–320. doi: 10.1128/mmbr.00050-14
Hartman, K., van der Heijden, M. G. A., Wittwer, R. A., Banerjee, S., Walser, J.-C., and Schlaeppi, K. (2018). Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 6:14.
Huang, P., Zhang, Y., Xiao, K., Jiang, F., Wang, H., Tang, D., et al. (2018). The chicken gut metagenome and the modulatory effects of plant-derived benzylisoquinoline alkaloids. Microbiome 6:211.
Hunter, P. J., Hand, P., Pink, D., Whipps, J. M., and Bending, G. D. (2010). Both leaf properties and microbe-microbe interactions influence within-species variation in bacterial population diversity and structure in the lettuce (Lactuca Species) phyllosphere. Appl. Environ. Microbiol. 76, 8117–8125. doi: 10.1128/aem.01321-10
Jin, H., Yang, X.-Y., Yan, Z.-Q., Liu, Q., Li, X.-Z., Chen, J.-X., et al. (2014). Characterization of rhizosphere and endophytic bacterial communities from leaves, stems and roots of medicinal Stellera chamaejasme L. Syst. Appl. Microbiol. 37, 376–385. doi: 10.1016/j.syapm.2014.05.001
Jorge, G. L., Kisiala, A., Morrison, E., Aoki, M., Nogueira, A. P. O., and Emery, R. J. N. (2019). Endosymbiotic Methylobacterium oryzae mitigates the impact of limited water availability in lentil (Lens culinaris Medik.) by increasing plant cytokinin levels. Environ. Exper. Bot. 162, 525–540. doi: 10.1016/j.envexpbot.2019.03.028
Junker, R. R., and Keller, A. (2015). Microhabitat heterogeneity across leaves and flower organs promotes bacterial diversity. FEMS Microbiol. Ecol. 91:fiv097. doi: 10.1093/femsec/fiv097
Khadem, A., Soler, L., Everaert, N., and Niewold, T. A. (2014). Growth promotion in broilers by both oxytetracycline and Macleaya cordata extract is based on their anti-inflammatory properties. Br. J. Nutr. 112, 1110–1118. doi: 10.1017/s0007114514001871
Kim, J., Chhetri, G., Kim, I., Kim, H., Kim, M. K., and Seo, T. (2019). Methylobacterium terrae sp. nov., a radiation-resistant bacterium isolated from gamma ray-irradiated soil. J. Microbiol. 57, 959–966. doi: 10.1007/s12275-019-9007-9
Kosina, P., Gregorova, J., Gruz, J., Vacek, J., Kolar, M., Vogel, M., et al. (2010). Phytochemical and antimicrobial characterization of Macleaya cordata herb. Fitoterapia 81, 1006–1012. doi: 10.1016/j.fitote.2010.06.020
Li, H., Wang, J., Zhao, J., Lu, S., Wang, J., and Jiang, W. (2009). Isoquinoline Alkaloids from Macleaya cordata active against plant microbial pathogens. Nat. Product Commun. 4, 1557–1560.
Li, L., YanChun, L., JiaLu, H., XiuBin, L., ZhiXing, Q., JianGuo, Z., et al. (2017). Medicinal plants of the genus Macleaya (Macleaya cordata, Macleaya microcarpa): a review of their phytochemistry, pharmacology, and toxicology. Phytother. Res. 32, 19–48. doi: 10.1002/ptr.5952
Liu, M., Lin, Y.-L., Chen, X.-R., Liao, C.-C., and Poo, W.-K. (2013). In vitro assessment of Macleaya cordata crude extract bioactivity and anticancer properties in normal and cancerous human lung cells. Exper. Toxicol. Pathol. 65, 775–787. doi: 10.1016/j.etp.2012.11.004
Liu, T.-H., Zhang, X.-M., Tian, S.-Z., Chen, L.-G., and Yuan, J.-L. (2020). Bioinformatics analysis of endophytic bacteria related to berberine in the Chinese medicinal plant Coptis teeta Wall. 3 Biotech 10:96.
Loaces, I., Ferrando, L., and Fernández Scavino, A. (2010). Dynamics, diversity and function of endophytic siderophore-producing bacteria in rice. Microb. Ecol. 61, 606–618. doi: 10.1007/s00248-010-9780-9
Lu, Y., Zhang, E., Hong, M., Yin, X., Cai, H., Yuan, L., et al. (2020). Analysis of endophytic and rhizosphere bacterial diversity and function in the endangered plant Paeonia ludlowii. Arch. Microbiol. 202, 1717–1728. doi: 10.1007/s00203-020-01882-3
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Jase, G., Stephanie, M., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–94. doi: 10.1038/nature11237
Maignien, L., DeForce, E. A., Chafee, M. E., Eren, A. M., and Simmons, S. L. (2014). Ecological succession and stochastic variation in the assembly of Arabidopsis thaliana phyllosphere communities. mBio 5:e00682–13.
Mangeot-Peter, L., Tschaplinski, T. J., Engle, N. L., Veneault-Fourrey, C., Martin, F., and Deveau, A. (2020). Impacts of soil microbiome variations on root colonization by fungi and bacteria and on the Metabolome of Populus tremula × alba. Phytobiomes J. 4, 142–155. doi: 10.1094/pbiomes-08-19-0042-r
Mano, H., and Morisaki, H. (2008). Endophytic bacteria in the rice plant. Microb. Environ. 23, 109–117.
Meyer, K. M., and Leveau, J. H. J. (2012). Microbiology of the phyllosphere: a playground for testing ecological concepts. Oecologia 168, 621–629. doi: 10.1007/s00442-011-2138-2
Mighell, K., Saltonstall, K., Turner, B. L., Espinosa-Tasón, J., and Bael, S. A. V. (2019). Abiotic and biotic drivers of endosymbiont community assembly in Jatropha curcas. Ecosphere 10:e02941. doi: 10.1002/ecs2.2941
Miller, T. E. X., and Rudgers, J. A. (2014). Niche differentiation in the dynamics of host-symbiont interactions: symbiont prevalence as a coexistence problem. Am. Nat. 183, 506–518. doi: 10.1086/675394
Moran, N. A. (2007). Symbiosis as an adaptive process and source of phenotypic complexity. PNAS 104, 8627–8633. doi: 10.1073/pnas.0611659104
Moroenyane, I., Mendes, L., Tremblay, J., Tripathi, B., and Yergeau, É (2021). Plant compartments and developmental stages modulate the balance between Niche-Based and neutral processes in Soybean Microbiome. Microbial. Ecol. ∗∗vol&pg.,
Nemergut, D. R., Schmidt, S. K., Fukami, T., O’Neill, S. P., Bilinski, T. M., Stanish, L. F., et al. (2013). Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 77, 342–356.
Overbeek, L. V., and Elsas, J. D. V. (2008). Effects of plant genotype and growth stage on the structure of bacterial communities associated with potato (Solanum tuberosum L.). FEMS Microbiol. Ecol. 64, 283–296. doi: 10.1111/j.1574-6941.2008.00469.x
Papke, R. T., and Gogarten, J. P. (2012). Ecology. How bacterial lineages emerge. Science 336, 45–46. doi: 10.1126/science.1219241
Park, S. Y., Jin, M. L., Kim, Y. H., Lee, S.-J., and Park, G. (2014). Sanguinarine inhibits invasiveness and the MMP-9 and COX-2 expression in TPA-induced breast cancer cells by inducing HO-1 expression. Oncol. Rep. 31, 497–504. doi: 10.3892/or.2013.2843
Pasquale, A., Sylvia, S., Silvia, P., and Massimiliano, C. (2020). Diversity and structure of the endophytic bacterial communities associated with Three Terrestrial Orchid Species as revealed by 16S rRNA Gene metabarcoding. Front. Microbiol. 11:604964. doi: 10.3389/fmicb.2020.604964
Peter, J. F., Alison, G. J., Julie, P., and Vincenzo, D. L. (1996). Uncoupled defense gene expression and antimicrobial alkaloid accumulation in elicited opium poppy CeII cuitures. Plant Physiol. 111, 687–697. doi: 10.1104/pp.111.3.687
Pimentel, M. R., Molina, G., Dionisio, A. P., Marostica Junior, M. R., and Pastore, G. M. (2011). The use of endophytes to obtain bioactive compounds and their application in biotransformation process. Biotechnol. Res. Int. 2011:576286.
Prasad, V. S. S. K., Davide, G., and Emilio, S. (2018). Plant growth promoting and biocontrol activity of Streptomyces spp. as Endophytes. Int. J. Mol. Sci. 19:952. doi: 10.3390/ijms19040952
Ramond, J.-B., Tshabuse, F., Bopda, C. W., Cowan, D. A., and Tuffin, M. I. (2013). Evidence of variability in the structure and recruitment of rhizospheric and endophytic bacterial communities associated with arable sweet sorghum (Sorghum bicolor (L) Moench). Plant and Soil 372, 265–278. doi: 10.1007/s11104-013-1737-6
Razan, H., Jurgen, R., and Michael, W. (2014). Synergistic antimicrobial activity of combinations of sanguinarine and EDTA with vancomycin against multidrug resistant bacteria. Drug Metabol. Lett. 8:10.
Santana, R. S. M., Fernandes, G. W., Ávila, M. P., Reis, M. P., Araújo, F. M. G. D., Salim, A. C. M., et al. (2016). Endophytic microbiota associated with the root tips and leaves of Baccharis dracunculifolia. Brazilian Arch. Biol. Technol. 59:e16160287.
Sharaby, Y., Rodríguez-Martínez, S., Lalzar, M., Halpern, M., and Izhaki, I. (2020). Geographic partitioning or environmental selection: what governs the global distribution of bacterial communities inhabiting floral nectar? Sci. Total Environ. 749:142305. doi: 10.1016/j.scitotenv.2020.142305
Shintaro, H., Masatoshi, M., and Kiwamu, M. (2019). Growth stage-dependent bacterial communities in soybean plant tissues: Methylorubrum Transiently dominated in the flowering stage of the soybean shoot. Microb. Environ. 34, 446–450. doi: 10.1264/jsme2.me19067
Shitan, N., and Yazaki, K. (2007). Accumulation and membrane transport of plant alkaloids. Curr. Pharm. Biotechnol. 8, 244–252. doi: 10.2174/138920107781387429
Tahtamouni, M. E., Khresat, S., Lucero, M., Sigala, J., and Unc, A. (2015). Diversity of endophytes across the soil-plant continuum for Atriplex spp. in arid environments. J. Arid Land 8, 241–253. doi: 10.1007/s40333-015-0061-9
Ting, O., Wei-Fang, X., Fei, W., Gary, S., Ze-Yang, Z., Zhong-Huai, X., et al. (2019). A Microbiome study reveals seasonal variation in endophytic bacteria among different mulberry cultivars. Comput. Struct. Biotechnol. J. 17, 1091–1100. doi: 10.1016/j.csbj.2019.07.018
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
Víctor, J. C., and Juan, P.-J. (2019). Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 366, 606–612. doi: 10.1126/science.aaw9285
Wang, Y., Wang, C., Gu, Y., Wang, P., Song, W., Ma, J., et al. (2020). The variability of bacterial communities in both the endosphere and ectosphere of different niches in Chinese chives (Allium tuberosum). PLoS One 15:e0227671. doi: 10.1371/journal.pone.0227671
Wang, Y., Zhang, W., Zhang, B., Huang, Q., Huang, R., and Su, X. (2019). Endophytic communities of transgenic poplar were determined by the environment and niche rather than by transgenic events. Front. Microbiol. 10:588. doi: 10.3389/fmicb.2019.00588
Wellner, S., Lodders, N., Glaeser, S. P., and Kampfer, P. (2013). Methylobacterium trifolii sp. nov. and Methylobacterium thuringiense sp. nov., methanol-utilizing, pink-pigmented bacteria isolated from leaf surfaces. Int. J. Syst. Evol. Microbiol. 63, 2690–2699. doi: 10.1099/ijs.0.047787-0
Wilkes, H., Wittich, R., Timmis, K. N., Fortnagel, P., and Francke, W. (1996). Degradation of Chlorinated Dibenzofurans and Dibenzo-p-Dioxins by Sphingomonas sp. Strain RW1. Appl. Environ. Microbiol. 62, 367–371. doi: 10.1128/aem.62.2.367-371.1996
Winston, M. E., Hampton-Marcell, J., Zarraonaindia, I., Owens, S. M., Moreau, C. S., Gilbert, J. A., et al. (2017). Understanding cultivar-specificity and soil determinants of the cannabis microbiome. PLoS One 9:e99641. doi: 10.1371/journal.pone.0099641
Xiong, C., Yong-Guan, Z., Jun-Tao, W., Singh, B., Li-Li, H., Shen, J. P., et al. (2020). Host selection shapes crop microbiome assembly and network complexity. New Phytol. 229, 1091–1104. doi: 10.1111/nph.16890
Yana, A.-G., Ido, I., and Malka, H. (2017). From microhabitat of floral nectar up to Biogeographic Scale: novel insights on neutral and niche bacterial assemblies. Microbial. Ecol. 74, 128–139. doi: 10.1007/s00248-017-0935-9
Yoshida, S., Hiradate, S., Koitabashi, M., Kamo, T., and Tsushima, S. (2017). Phyllosphere Methylobacterium bacteria contain UVA-absorbing compounds. J. Photochem. Photobiol. B 167, 168–175. doi: 10.1016/j.jphotobiol.2016.12.019
Yu, H., Zhang, L., Li, L., Zheng, C., Guo, L., Li, W., et al. (2010). Recent developments and future prospects of antimicrobial metabolites produced by endophytes. Microbiol. Res. 165, 437–449. doi: 10.1016/j.micres.2009.11.009
Yu, X., Gao, X., Zhu, Z., Cao, Y., Zhang, Q., Tu, P., et al. (2014). Alkaloids from the tribe Bocconieae (Papaveraceae): a chemical and biological review. Molecules 19, 13042–13060. doi: 10.3390/molecules190913042
Yu, X., Yang, J., Wang, E., Li, B., and Yuan, H. (2015). Effects of growth stage and fulvic acid on the diversity and dynamics of endophytic bacterial community in Stevia rebaudiana Bertoni leaves. Front. Microbiol. 6:867. doi: 10.3389/fmicb.2015.00867
Zarraonaindia, I., Owens, S. M., Weisenhorn, P., West, K., Hampton-Marcell, J., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. mBio 6:e002527–14.
Zeng, J., Liu, Y., Liu, W., and Liu, X. (2013). Integration of transcriptome, proteome and metabolism data reveals the alkaloids biosynthesis in macleaya cordata and macleaya microcarpa. PLoS One 8:e53409. doi: 10.1371/journal.pone.0053409
Zheng, Y., and Gong, X. (2019). Niche differentiation rather than biogeography shapes the diversity and composition of microbiome of Cycas panzhihuaensis. Microbiome 7:152.
Zhou, J.-Y., Yuan, J., Li, X., Ning, Y.-F., and Dai, C.-C. (2015). Endophytic bacterium-triggered reactive oxygen species directly increase oxygenous sesquiterpenoid content and diversity in Atractylodes lancea. Appl. Environ. Microbiol. 82, 1577–1585. doi: 10.1128/aem.03434-15
Keywords: Macleaya cordata, 16S rRNA, bacterial community structure, niche differentiation, alkaloids
Citation: Lei F, Liu X, Huang H, Fu S, Zou K, Zhang S, Zhou L, Zeng J, Liu H, Jiang L, Miao B and Liang Y (2021) The Macleaya cordata Symbiont: Revealing the Effects of Plant Niches and Alkaloids on the Bacterial Community. Front. Microbiol. 12:681210. doi: 10.3389/fmicb.2021.681210
Received: 16 March 2021; Accepted: 07 May 2021;
Published: 09 June 2021.
Edited by:
Rodrigo Pulgar Tejo, University of Chile, ChileCopyright © 2021 Lei, Liu, Huang, Fu, Zou, Zhang, Zhou, Zeng, Liu, Jiang, Miao and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yili Liang, bGlhbmd5aWxpNkBjc3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship