 Maria Andrea Desbats
Maria Andrea Desbats Isabella Giacomini3
Isabella Giacomini3 Monica Montopoli
Monica Montopoli- 1Department of Medicine, University of Padova, Padova, Italy
- 2Veneto Institute of Molecular Medicine, Padova, Italy
- 3Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova, Italy
- 4U.O.C. Urology, Azienda Ospedaliera di Padova, Padova, Italy
Resistance of cancer cells to chemotherapy is the first cause of cancer-associated death. Thus, new strategies to deal with the evasion of drug response and to improve clinical outcomes are needed. Genetic and epigenetic mechanisms associated with uncontrolled cell growth result in metabolism reprogramming. Cancer cells enhance anabolic pathways and acquire the ability to use different carbon sources besides glucose. An oxygen and nutrient-poor tumor microenvironment determines metabolic interactions among normal cells, cancer cells and the immune system giving rise to metabolically heterogeneous tumors which will partially respond to metabolic therapy. Here we go into the best-known cancer metabolic profiles and discuss several studies that reported tumors sensitization to chemotherapy by modulating metabolic pathways. Uncovering metabolic dependencies across different chemotherapy treatments could help to rationalize the use of metabolic modulators to overcome therapy resistance.
Metabolic Reprogramming
The metabolic program between non-proliferating and proliferating cells is different. Non-proliferating cells rely mostly on catabolic reactions while proliferating cells must balance catabolic and anabolic reactions required to sustain enhanced cellular growth (1–3). In normally proliferating cells most ATP from glucose is obtained by glycolisis, tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OxPhos), while nucleotides, aminoacids, and lipids are provided by intermediate metabolites of these pathways; such as acetyl-CoA for fatty acids, glycolytic intermediates for non-essential aminoacids, and ribose for nucleotides. Tumor cells are characterized by metabolic hallmarks similar to highly proliferating normal cells but, in addition, they develop a high plasticity to metabolic rewiring to sustain enhanced cellular growth in changing microenvironmental conditions (4).
Back in the 1920's, Otto Warburg observed that many tumors depended on glycolysis as the sole source of ATP; even in the presence of oxygen (aerobic glycolysis) (5). Accordingly, the rate of glucose entry to cancer cells was found 20-to-30- fold higher than in normal cells (6), and glucose transporters and key glycolytic enzymes were heavily upregulated (7). Cancer cells under hypoxia induce pyruvate dehydrogenase kinase (PDK) that inactivates pyruvate dehydrogenase (PDH) (8). Thus, most glucose-derived pyruvate does not enter the TCA cycle and is converted in lactate by the action of lactate dehydrogenase (LDH) (9). This is because most tumors produce great quantities of lactate, which is very striking, since glycolysis produces only 2 ATP molecules for each glucose, while oxidative phosphorylation between 30 and 32 ATPs.
Later it became clear that in cancer cells glucose is consumed mainly to supply glycolitic intermediates for anabolic pathways. Glucose-6-phosphate can be oxidized by glucose-6-phosphate dehydrogenase (G6PD) to produce reduced nicotinamide adenine dinucleotide phosphate (NADPH) and ribose-5-phosphate (R5P) through the pentose phosphate pathway (PPP). NADPH and R5P are required for nucleotide synthesis, but also to sustain biosynthetic reactions and to maintain the redox capacity of the cell (1). Moreover, 3-phosphoglycerate could serve as a precursor for serine and glycine metabolism through the one-carbon cycle (10). Pyruvate instead that can be converted into alanine by alanine aminotransferase (ALT) (11). In turn, these aminoacids can be metabolized for nucleotide synthesis, DNA methylation, glutathione production and NADPH generation (12). Interestingly, several PPP enzymes and 3-phosphoglycerate dehydrogenase (PHGDH) were found upregulated in some cancer (13–16).
Unlike originally thought, aerobic glycolysis in cancer cells is not a sign of defective oxidative phosphorylation. Instead, high rates of glycolysis inhibit mitochondrial respiration, a phenomenon termed the “Crabtree effect” (17). Indeed, mitochondria function is essential for cancer cell proliferation (18). Mitochondrial redox homeostasis is crucial for maintaining cellular aspartate levels critical for nucleotide synthesis (19). Indeed, aspartate was shown essential for in vivo tumor growth (20).
Of great significance, cancer cells require TCA cycle intermediates for biosynthetic pathways and NADPH production (21). The TCA cycle generates citrate that can be exported to the cytosol through the mitochondrial tricarboxylate carrier (SLC25A1) to be converted into acetyl-CoA and oxaloacetate by ATP citrate lyase (ACLY). (22). Acetyl-CoA can either be employed for fatty acid and cholesterol synthesis (to support membrane biogenesis) or used for protein acetylation reactions, which regulate nuclear transcription as well as cytoplasmic processes like autophagy (23). The TCA cycle also provides metabolic precursors for the synthesis of non-essential amino acids, such as aspartate and asparagine from oxaloacetate, or proline, arginine and glutamate from α-ketoglutarate. To cope with the continuous efflux of intermediates cancer cells replenish the TCA cycle by increasing or developing the ability to use various carbon sources; including glutamine, acetate, lactate, serine, and glycine (24–27). In particular, tumor cells consume great quantities of aminoacids.
Glutamine is the major contributor of TCA intermediates in many cancer cell lines (28). Glutamine is transported into the cell through plasma membrane transporters, like SLC1A5 (ASCT2) and SLC7A5 (29) and converted into glutamate by glutaminase (GLS). Then glutamate is transformed into α-ketoglutarate, by either glutamate dehydrogenase (GDH) or transaminases; and α-ketoglutarate enters the TCA cycle to maintain the production of citrate. Glutamine can also be directly converted into citrate by reductive carboxylation. The reductive carboxylation of α-ketoglutarate by the inverse reaction of isocitrate dehydrogenase (IDH) generates citrate (30). Glutamine reductive carboxylation is particularly important in tumor cells under hypoxic conditions or when mitochondrial respiration is impaired (31). Moreover, GLS and GDH are upregulated in a wide variety of tumors and its inhibition has been shown to diminish tumorigenesis (32, 33).
Another contributor of TCA intermediates is lactate. Some cancer cells can use lactate produced by aerobic glycolysis as a source of energy. More than 50% of the total TCA cycle intermediates in breast cancer cells under glucose deprivation derived from lactate (34). Moreover, overexpression of lactate transporters (MCTs) is a common finding in some cancers (35).
Enhanced glycolisis and glutamine metabolism in cancer cells support the increase of de novo fatty acids synthesis (36). Fast-proliferating cancer cells use fatty acids and cholesterol for biosynthesis of cell membranes, cell signaling and secondary messengers (37), as well as for lipid catabolism through fatty acid β-oxidation (FAO) during nutrient deprivation (38). In some cancers such us prostate cancer and lymphoma, lipid-dependent metabolism becomes essential for energy production (39). In physiological conditions, lipid synthesis is restricted to specialized tissues, such as the liver and adipose tissues. Normal cells uptake lipids from the bloodstream, while cancer cells could obtain lipids and lipoproteins exogenously or by de novo synthesis (38). A wide variety of tumors have increased expression of crucial lipogenic enzymes such us ACLY, acetyl-CoA-carboxylase (ACC), fatty acid synthase (FASN) (38, 40, 41); as well as present an increase in the transcriptional activities of the sterol regulatory element-binding proteins (SREBPs) (42, 43). The upregulation of lipogenic enzymes seems required for tumor progression (40). Interesstingly, some cancer cells harbor adipocyte characteristics like storing excess lipids in lipid droplets (LD) (44). LD are intracellular storage organelles of neutral lipids mainly found in adipose tissue, but observed in several cell types and tissues (45, 46). LDs are dynamic, and their accumulation seem to confer survival advantages to cancer cells (47). Drugs that specifically target LD formation are thought to hold greater therapeutic potential compared with general lipid biosynthesis inhibitors (48, 49).
Enhanced glycolisis, glutamine metabolism and fatty acids synthesis are features shared by many cancer cell lines. However, the metabolic phenotype of the tumor in vivo is highly heterogeneous, resulting from the combination of intrinsic (genetic and epigenetic changes, tissue of origin, state of differentiation) and extrinsic (oxygen and nutrient availability, metabolic interactions within the tumor microenvironment) factors (50).
Role of Oncogenes and Tumor Suppressor Genes in Metabolism Reprogramming
One of the intrinsic factors that determine the tumor metabolic phenotype is the activation of oncogenes or deactivation of tumor suppressor genes which result in a metabolic rewiring (51). Tumor metabolism is distinct in tumors harboring different oncogenic alterations. Oncogenes such as RAS, MYC, or PI3K, favor glycolysis over oxidative phosphorylation; whereas tumor suppressors such as p53, PTEN, Von Hippel–Lindau (VHL), or liver kinase B1 (LKB1) have the opposite effect (52).
In particular, MYC expression could activate the pentose phosphate pathway, purine/pyrimidine synthesis and fatty acid oxidation under chemotherapy and radiotherapy (53). MYC directly regulates several glycolytic enzymes such as glucose transporter 1 (GLUT1), hexokinase 2 (HK2), phosphofructokinase muscle type (PFKM), and enolase 1 (ENO1); as well as lactate dehydrogenase A (LDHA) (54). Moreover, MYC expression increases glutamine uptake and glutaminolysis (55, 56) by inducing the expression of glutamine transporters SLC1A5 and SLC7A5 and by repressing the transcription of microRNA-23a/b which targets glutaminase 1 (GLS1) (56, 57).
p53 can directly or indirectly influence the expression of genes involved in glucose, OxPhos and lipid metabolism, among other pathways (58). p53 inhibits glycolysis dowregulating GLUTs and the glycolytic enzyme phosphoglycerate mutase (PGAM) (59, 60). p53 also induce the expression of TIGAR (TP53-induced glycolysis and apoptosis regulator), which indirectly inhibits phosphofructose kinase 1 (PFK1) diverting glycolytic intermediates into the PPP (61). p53 decreases fatty acid synthesis by also inhibiting the PPP. p53 directly binds and inhibits G6PD leading to decreased production of NADPH (14). Moreover, p53 directly repress the expression of SREBP-1 which regulates the expression of fatty acid synthesis enzymes (62). On the other hand p53 enhances fatty acid oxidation. p53 induces two important enzymes involved in fatty acid oxidation, Lipin 1 and carnitine palmitoyltransferase (CPT1C) (63, 64). p53 was also reported to transcriptionally induce malonyl-CoA decarboxylase (MCD), which catalyzes the conversion of malonyl-CoA to acetyl-CoA, to promote fatty acid oxidation and prevent lipid accumulation (65). p53 enhances mitochondrial OxPhos by inducing the expression of the cytochrome c oxidase (COX, complex IV) assembly factor SCO2 (66) or the expression of AIF; which maintains the integrity of mitochondrial NADH:ubiquinone oxidoreductase (complex I) (67). In addition, p53 promotes OxPhos by repressing the transcription of pyruvate dehydrogenase kinase 2 (PDK2), which inhibits PDH (68). PDK2 repression activates PDH reducing the conversion of pyruvate to lactate and increasing the conversion of pyruvate to acetyl-CoA (68).
RAS can influence the glycolytic metabolism through the PI3K-mTOR pathway, or by upregulating glucose flux through hexosamine biosynthesis pathway (HBP) or the PPP (53). In addition, mutant KRAS activated lipogenesis through induction of FAS (69). In a Kras-driven mutant model of spontaneous lung tumorigenesis the uptake and utilization of branched-chain amino acids (BCAAs) such as leucine and valine, were increased, as well as the expression of the enzymes responsible for their catabolism (70).
PTEN decreases glycolysis and promotes oxidative phosphorylation. MEFs from PTEN tg mice present high levels of peroxisome proliferator-activated receptor g coactivator-1α (PGC-1α), increase the number of mitochondria, increment the levels of oxygen consumption and ATP production, and diminish lactate secretion. Moreover, PTEN decreases the levels of pyruvate kinase isozyme M2 (PKM2) and 6- phosphofructo-1-kinase/fructose-2,6-biphosphatase isoform 3 (PFKFB3); and elicits the inhibition of the pro-tumorigenic glutaminase GLS1 thus contributing to the cancer-protection (71).
Of note, most studies on the role of oncogenes/tumor suppressor genes in metabolic reprogramming were performed using cell models with single genetic modifications. It's hard to translate this findings to the tumor in vivo which harbors many genetic defects, and whose metabolic profile will depend on their combination.
Metabolic Heterogeneity in Tumors
Based on the metabolic strategies prioritized by several solid cancers Lehuede et al. (72) proposed a classification of cancer-specific metabolic phenotypes in glycolytic and oxidative tumors. While lung, liver, colorectal cancers, and leukemias rely mostly on glycolysis; lymphomas, melanomas, and glioblastomas behave as oxidative tumors (72). However, there is not a uniform metabolic phenotype across tumors with a similar genetic defect in different organs or genetically different tumors in the same organ (73). A large study of metabolic features in 180 patient-derived melanoma xenografts excluded a general metabolomic signature (74).
Cancer cells reside in poor oxygen and nutrition environments and hence attempt to reprogram the preexisting tissue metabolism for survival (75). The fact that some regions of the tumor could have more access to oxygen or various carbon sources than others (73) explains why tumor cells are metabolically heterogeneous. Intra-tumoral metabolic heterogeneity is maintained through coupled metabolic interactions between distinct cell populations coexisting in the tumor microenvironment. Stromal and tumor cells can compete or alternatively form symbiotic relationships where the metabolic products of a population become a nutrient of another (76).
Tumor cells can promote a “Reverse Warburg effect” in neighboring Cancer-associated fibroblasts (CAFs) (77). CAFs are stromal cells which often dominate the tumor microenvironment. Reactive oxygen species (ROS) produced by cancer cells activates HIF-1α and NFkB in CAFs inducing the production and release of energy-rich metabolites as lactate. Cancer cells could in turn take up lactate to fuel mitochondria respiration for energy production and anabolic metabolism (78, 79). This metabolic symbiosis may be controlled by the differential expression of lactate monocarboxylate transporters MCT1 and MCT4. Lactate is released from CAFs by MCT4 and taken up by MCT1 in cancer cells. Indeed, lactate transporters inhibition reduces lactate uptake, induces a switch to glycolysis, and blocks metabolic symbiosis and tumor progression (80). Interestingly, higher levels of MCT1 confer a higher metastatic potential to melanoma cells as metastasizing cells depend on MCT1 to manage oxidative stress (81).
Cancer-associated adipocytes (CAAs) are adipocytes infiltrated into the tumor tissue (82). They provide carbon sources, growth factors, and cytokines affecting tumor growth, metastasis, and drug responses (83). CAAs frequently release fibroblast growth factors like (FGFs), leptin, adiponectin, IL-1β, IL-6, TNF-α, CCL2, and CCL5; while cancer cells produce signaling molecules to trigger adipocyte lipolysis (84). In the presence of CAAs some cancer cells can acquire exogenous free fatty acids (FFAs) released by CAAs through the cell surface fatty acid translocase CD36 and switch their metabolic program from glycolysis to FAO (38). In vitro, ovarian cancer cells induce white adipocytes lipolysis, fatty acids uptake and FAO (85).
Tumor metabolism also modulates the activity of tumor-associated immune populations. Activated T cells and cancer cells share some metabolic similarities (86) giving rise to a competition for nutrients which could impair the immune function (87). Naive CD4 T cells use mitochondrial OxPhos as a primary energy source, but upon activation they increase the expression of GLUT1 and shift to aerobic glycolysis (88). Also TAMs Tumor Associated Macrophages (TAMs) M1 rely on glycolysis to meet increased energetic demands (89). Moreover, increased lactate levels due to enhanced tumoral glycolisis can lead to diminished antitumour immunity. Lactate inhibits FIP200, leading to defective autophagy and increased apoptosis of naive T cells (90). Lactate can also suppress NK and dendritic cell function but does not affect regulatory T (Treg) cell function (91). Lactate could also induce the conversion of M1 to M2 pro-tumoral macrophages (92). CD8+ T cells and NK cells, are also sensitive to glutamine, serine, glycine, leucine, isoleucine and valine aminoacid restriction (93, 94). Moreover, limited availability of extracellular glutamine shifted the balance from Th1 to Treg cells (95).
Some cancer cells harbor an “hybrid glycolysis/ OxPhos phenotype” which allows them to use both glycolysis and the byproducts from glycolysis by OxPhos for energy production and biomass synthesis (96). Metabolic plasticity may be specifically associated with metastasis and therapy-resistance because a hybrid metabolism could maintain low ROS levels which induce a moderate stress response and the appearance of mutations that further stimulate tumorigenesis and metastasis (97). Dual inhibition of glycolysis (by 2-Deoxy-d-glucose, 2-DG) and OxPhos (by metformin) has been shown to effectively repress tumor growth and metastasis across multiple preclinical cancer models (98). Thus, a combination of glycolytic and OxPhos inhibitors could effectively eliminate the tumor survival potential of hybrid cells (99).
Understanding the factors that influence tumor heterogeneity is fundamental for the development of therapies that could act modulating tumor metabolism. Up to now tumor heterogeneity and toxicity issues has limited the success of most clinical trials targeting metabolic pathways (Figure 1).
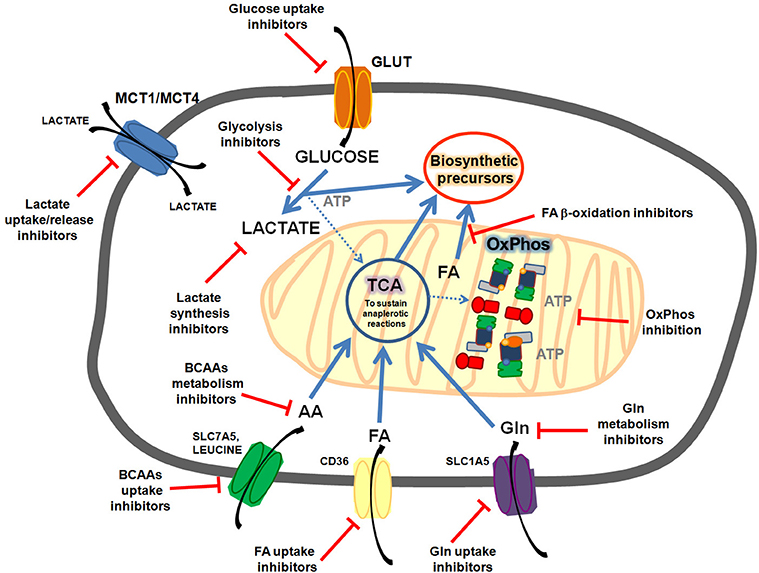
Figure 1. Common metabolic features targeted in cancer cells. Cancer cells could present enhanced glycolisis and lactate release, enhanced FA (fatty acids) synthesis, uptake and oxidation; enhanced OxPhos activity, enhanced glutamine uptake and metabolism, enhanced branched amino acids (BCAAs) uptake and oxidation, etc. Targeting these pathways could sensitize cancer cells to chemotherapy.
Targeting Cancer Metabolism to Overcome Drug Resistance
Recently, metabolic reprogramming has been shown to play a role in the response of cancer cells to widely-used first-line chemotherapeutics (100).
Chemotherapeutic drugs target a differential feature of cancer cells that help them to actively proliferate. The main types of chemotherapy agents used in the clinics are: alkylating agents and platinants (damage DNA), such as cisplatin (101); cytotoxic antibiotics (bind DNA to prevent DNA and/or RNA synthesis); inhibitors of topoisomerase (damage DNA), such as daunorubicin, doxorubicin, irinotecan and etoposide; antimetabolites (interfere with intermediary metabolism of proliferating cells), such as gemcitabine; anti-microtubule agents (target microtubules and associated proteins required in cell division), such as paclitaxel and docetaxel (102); hormonal agents (inhibit hormone synthesis or function as hormone receptor agonist/antagonist) (103), such as tamoxifen or enzalutamide; and immunotherapy (target cancer cells that express a specific antigen or boost the natural ability of T cells to fight cancer), such as trastuzumab.
Tumor recurrence results from the ability of specific tumor subpopulations to resist treatment and expand. Resistance constitutes a lack of response to drug-induced tumor growth inhibition and it may be inherent to a subpopulation of cancer cells or can be acquired as a consequence of drug exposure. Chemoresistance is caused through genetic mutations in various proteins involved in cellular mechanisms such as cell cycle, apoptosis and cell adhesion (104). Reported chemoresistance mechanisms include: altered drug membrane transport, mutation, increased expression of drug targets, decreased drug activation, increased drug degradation due to altered expression of drug-metabolizing enzymes, drug inactivation due to conjugation with glutathione, altered drug subcellular redistribution, drug interactions, enhanced DNA repair, overexpression of anti-apoptotic genes, inactivation of apoptotic gene products, among others (105).
In the last decades, several studies have demonstrated that metabolic reprogramming plays an important role in the onset of chemotherapy resistance (106). Mostly due to the fact that chemotherapy agents used in the clinics cause a compensatory metabolic reprogramming in cancer cells (Figure 2). Thus, implementation of combinatorial therapies with chemotherapeutic drugs and metabolic modulators (Table 1) might provide a way to overcome therapy resistance.
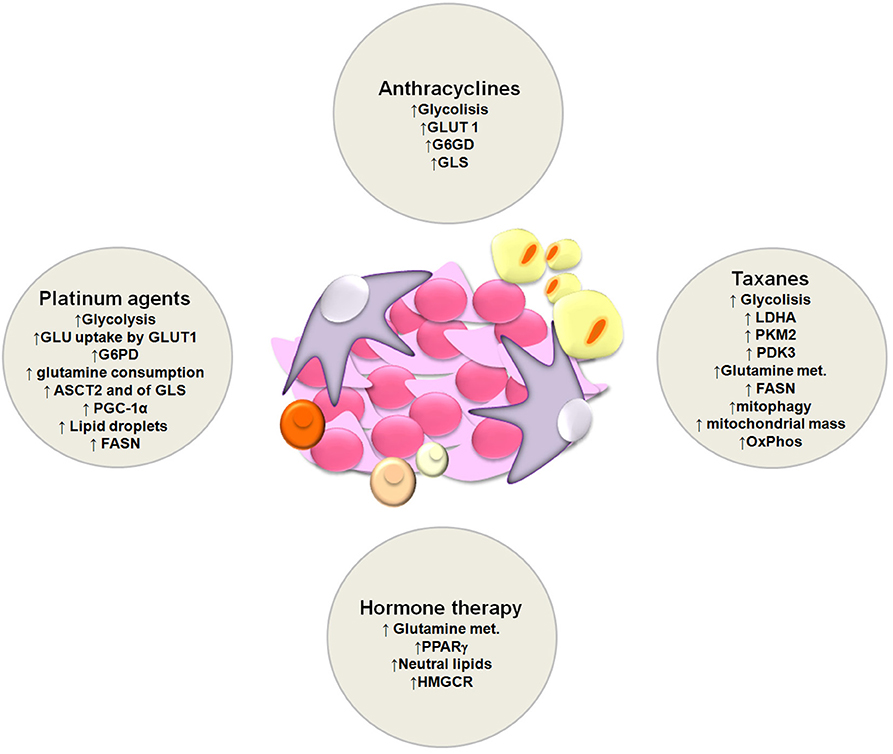
Figure 2. Schematic representation of metabolic alterations involved in the onset of resistance to platinum agents, anthracyclines, taxanes and hormone therapy.
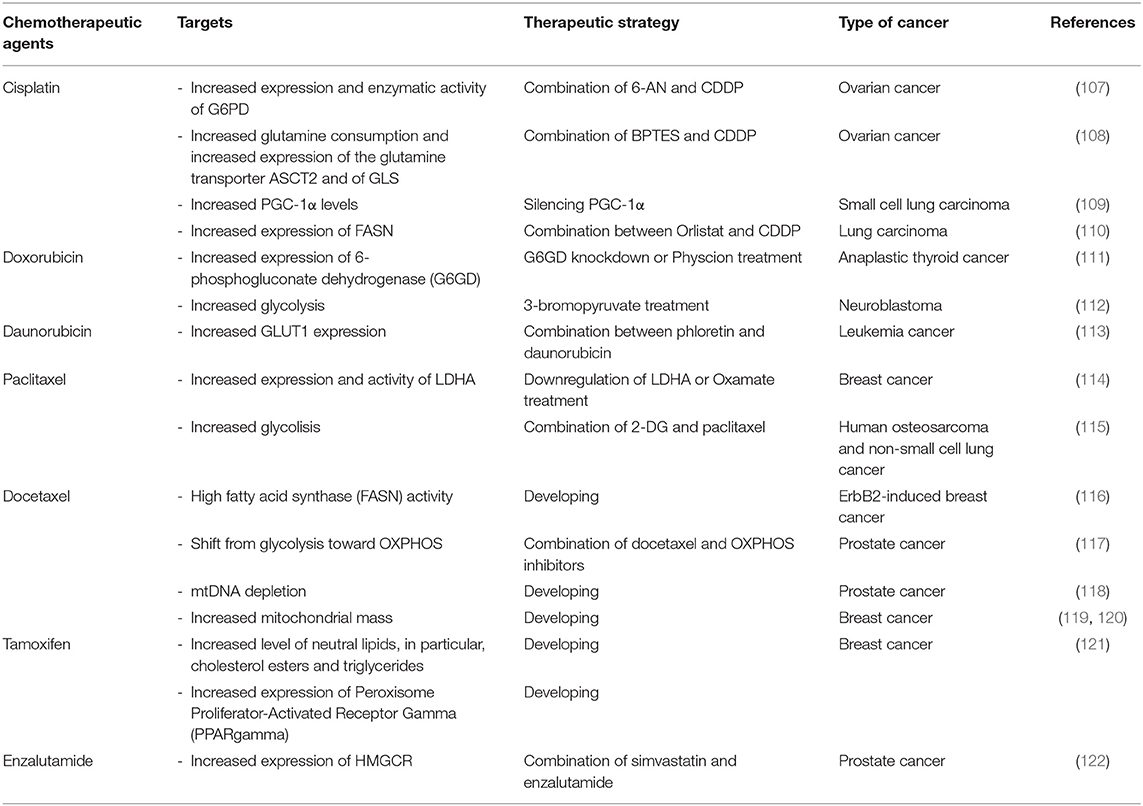
Table 1. Overview of promising combination therapy of chemotherapeutic agents and metabolic modulators.
Targeting Glucose Metabolism
As we already mentioned above, intensive aerobic glycolysis generates the accumulation of lactate that results in acidosis and promotes tumor progression and metastasis by inducing immunosuppression, vascularization, aggressive proliferation, migration, invasion and therapy resistance (123, 124). It has been demonstrated that enhanced glucose uptake and improved aerobic glycolysis are capable to induce the intrinsic or acquired resistance to chemotherapy in several tumor cells such as breast (125), or ovarian (107). Several key glycolytic enzymes and glucose transporters inhibitors are currently in preclinical or clinical development to counteract resistance to chemotherapeutic drugs (100, 107, 126–129).
Some reports proposed that aerobic glycolysis is an important pathway for colorectal cancer (CRC) development. In fact, the overexpression of the immune checkpoint protein B7-H3 in CRC cells enhanced glucose consumption and lactate release by HK2 expression, while B7-H3 knockdown had the opposite effect. Moreover, it is known that the depletion of HK2 expression or HK2 inhibition blocked aerobic glycolysis and CRC chemo-resistance (130). Recent studies reported that human colorectal adenocarcinoma doxorubicin-resistant cells (LoVo DOX) presents over expression of GLUT1. Thus, the treatment with silybin (a modulator of GLUTs) resulted synergic with the chemotherapeutic agents and it was able to overcome doxorubicin resistance (131).
Several key glycolytic enzymes, comprising HK2, PFK, and PKM2, are highly expressed in ovarian cancer cells and were implicated in anti-apoptotic and cell survival processes which correlate with chemo-resistance. These enzymes are controlled by oncogenes (e.g., Akt, mTOR) and tumor suppressors (e.g., p53) that may drive deregulated metabolism and ovarian cancer development (132). A few publications reported that ovarian cell lines with high glycolysis rate also presented high OxPhos activity, showing that most ovarian tumor cell lines prefer a highly glycolytic metabolic phenotype (133). Several inhibitors of glycolysis, such as 2-DG, 3-bromopyruvate (3-BrPA) or lonidamide (LND), have been studied in recent years. The combined treatment between metformin and 2-DG decreased the cellular growth of ovarian cancer cells (134). Moreover, 2-DG was able to sensitize cisplatin (CDDP)-resistant and radio-resistant cervical CaSki cell lines (135). 3-BrPA (pyruvate analog) is an inhibitor of HK2 and an alkylating agent. LND is another inhibitor of HK2, that enhanced CDDP activity in ovarian cancer cells (136). LND in combination with the chemotherapeutic agents, CDDP, and paclitaxel, presented a good activity and tolerability (137). The PPP is another pathway involved in ovarian drug resistance. It was demonstrated that ovarian CDDP-resistant cells (C13) showed increased glucose uptake, the up-regulation of the glucose transporter GLUT1 and increased expression and activity of G6PD, in comparison to CDDP-sensitive clones (2008). A combination of 6-nicotinamide (6-AN) (the G6PD inhibitor) and CDDP leads to a resensitization of CDDP-resistant cells (107). Moreover, since another ovarian cisplatin-resistant cancer cell line, IGROV PT, presented a higher expression of G6PD, the same combination has been loaded in liposomes and tested. The results showed a resensitization of resistant cells to cisplatin (138).
The increment in glycolysis is a common characteristic of drug-resistant breast cancer cells independent of the chemotherapeutic treatment, but this augmented activity is regulated in different ways in several resistant breast tumors. It has been demonstrated that triple-negative breast cancer (TNBC) and HER2 positive breast cancer possess higher rate of glycolytic activity than estrogen receptor-positive (ER+) breast cancer cells (139–141). In TNBC, it was shown that EGF pathways are activator of the first step in glycolysis (142) and that MYC modulates this metabolic phenotype by inhibiting the expression of the thioredoxin-interacting protein (143). The different expression of GLUT isoforms in breast cancer correlates with tumor cell differentiation, pathological grade, and prognosis. Invasive breast cancer, HER-2 positive, and TNBC, mostly present with a higher glycolysis rate due to the highest expression of GLUT (139). The most invasive breast cancer type, TNBC, has the highest GLUT-1 expression (139). Moreover, overexpression of ErbB2 increased the expression of LDHA; promoting glycolysis and breast tumor growth (144). Increased glycolytic rate and a higher sensitivity toward inhibition of glycolysis were demonstrated in lapatinib-resistant BT474 breast cancer cells by a multi-omics approach (125). Curiously, the increase glycolytic activity in BT474 cells was not resulting from an overexpression of glycolytic enzymes, but merely from modifications in the phosphorylation state of glycolytic enzymes, demonstrating that post-translational changes alone can modulate glycolysis. In trastuzumab-resistant ErbB2-positive breast cancer cells, the improved glycolytic rate is regulated by heat shock factor 1 and LDHA and inhibition of glycolysis with 2-DG and the LDH inhibitor oxamate by-pass trastuzumab resistance (145). Finally, in paclitaxel-resistant breast cancer cells, synergistic effects on inducing apoptosis were shown in LDHA downregulated cells or with oxamate (a pyruvate analog that inhibits the conversion of pyruvate to lactate) association (146). LDHA expression and activity are higher in taxol-resistant breast cancer cells. Downregulation of LDHA or oxamate treatment resensitizes taxol-resistant cells to taxol (146).
Differently to other tumor cell types that showed a higher rate of glucose consumption early in the modification process, prostate cancer (PCa) cells shift to the Warburg effect only in the metastatic stage, excluding the possibility to use advanced diagnostic procedures like standard FDG-PET scan for the detection of cancer in the early stages (147, 148). Glucose transporters have not been contemplated in PCa evolution because glucose metabolism in the prostate gland is different than in other organs. However, the relevance of GLUTs transporters has been lately proposed since the importance of increasing nutrients uptake, comprising glucose is clearly confirmed in PCa. In PCa androgens induce glucose uptake, upregulation of GLUT transporters and increased the AMPK pathway (149, 150). Glycolysis varies between androgen-sensitive and insensitive cells and it has been demonstrated that more aggressive tumors showed a higher glucose dependence (151). Indeed, prostate cancer cells switch to aerobic glycolysis only in the metastatic stage (147, 148). Even if the metabolic mechanism that supports prostate cancer metastasis has not been elucidated, in androgen-sensitive cells LNCaP and VCaP, androgen signaling induces both glycolysis and OxPhos (152). An augmented activity of key glycolytic enzymes by androgens has been established. HK2 phosphorylation is prompt by androgens via PKA signaling, while PFKFB2 is induced by direct binding of androgen receptor (AR) to PFKFB2 promoter. Activation of PFKFB2 produces a constitutive activation of 6-phosphofructo-2-kinase 2 (PFK2), which is entailed in the second irreversible reaction of the glycolytic pathway (150).
Combining different glycolytic inhibitors with chemotherapeutic agents could be a strategy to overcome drug resistance. To increase the anti-tumor activity 2-DG was used in vitro and in vivo in combination with inhibitors of lysosomal permeabilization (153). Moreover, the combined treatment between 2-DG and fenofibrate (PPARα agonist) caused a synergic effect in cancer cell growth (154). The combination of 2-DG and paclitaxel in mouse xenografts models of human osteosarcoma and non-small cell lung cancer resulted in a significant reduction in tumor growth (155). 3-BrPa use in vivo conditions resulted in anti-tumor activity after a single injection in a rabbit VX2 hepatoma model (156). Moreover, cells treated with 3-BrPa enhanced doxorubicin-resistant cells response to the drug (112). Dichloroacetate (DCA), a PDK1 inhibitor, was frequently used in combination with different chemotherapeutics agents (157). The combined treatment of DCA with paclitaxel was able to sensitize NSCLC resistant cells (158). Moreover, the combined treatment of DCA and CDDP was able to decrease tumor growth in advanced bladder cancer (159). The silencing of PKM2 in lung cancer cells enhanced the efficacy of docetaxel (160). Another group reported that PDK3 knockdown inhibited hypoxia-induced glycolysis and increased the susceptibility of cancer cells to paclitaxel (161). Cao et al. demonstrated that leukemia daunorubicin-resistant cells show increased expression of GLUT1 and that the combined treatment between daunorubicin and phloretin, an inhibitor of glucose transporter sensitizes K562/Dox cells (113). Doxorubicin-resistant cell lines from anaplastic thyroid cancer presented a high expression of G6GD (an enzyme of PPP). G6GD knockdown or the anthraquinone physcion decreased G6GD activity and resensitized doxorubicin-resistant cells (111).
Targeting Glutamine Metabolism
Tumor cells increase glutamine metabolism to preserve the citric acid cycle, especially given the loss of the entry from pyruvate, in order to adapt to the modifications in the glycolytic pathway (162). Also, glutamine metabolism contributes to cancer cell chemoresistance. Recent studies demonstrated that the use of small molecules, of which several are in clinical trials, to inhibit key enzymes in glutaminolysis pathways is effective in slowing the proliferation of cancer cells (163–168).
Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) was recognized to be the first allosteric inhibitor of GLS1 (169). It has demonstrated to reduce the proliferation in several cancer cell types in vitro and in xenograft models. Unfortunately, its poor metabolic stability and low solubility have discouraged its clinical development (169). For this reason, new inhibitors have been developed, such as CB-839 that is a more potent inhibitor of GLS1 than BPTES (170). CB-839 reduced proliferation of mouse HCC cells at very low concentration, while BPTES, at the same concentration, had no activity (171). CB-839 is ongoing in phase 1 clinical trial for the treatment of various cancer types [Study of the Glutaminase Inhibitor CB-839 in Solid Tumors https://clinicaltrials.gov/ct2/show/NCT02071862]. Glutamine analogs, such as azaserine, acivicin, and 6-diazo-5-oxo-L-norleucine (DON), are one strategy to disrupt glutamine metabolic pathways They form covalent bonds with Ser286 in the active site of GLS1 (166). These compounds have demonstrated to block the proliferation of a variety of cancers and have shown their efficacy in some clinical trials (163). Unfortunately, the main problem related to the clinical use of azaserine, acivicin, and DON is their lower selectivities toward GLS1, since they inhibit other glutamine-dependent enzymes. Then, compound 968 was identified as an allosteric inhibitor of GLS1; and was shown to have cytotoxic effects in lymphoma, breast cancer, glioblastoma, and lung cancer (172–176).
It has been demonstrated that high expression of GLS can promote glutamine-independent growth and resistance to therapies that limit glutamine metabolism (177, 178). Thus, other pharmacological approaches to target glutamine metabolism include the use of glutamine synthetase or GLUD (Glutamate dehydrogenase) inhibitors (179).
Analysis in vitro demonstrated that a high glutamine flux protected MCF7 cells from tamoxifen-induced apoptosis (180). Indeed, a higher content of glutamate was correlated with breast cancer outcomes in patients (181). Metabolomic analysis of 270 breast tumor samples and 97 normal breast samples showed that breast tumor cells had a higher glutamate-to-glutamine ratio than normal cells (182). Another study showed that highly invasive and drug-resistant breast cancer cells presented increased glutamine metabolism, increased glutamate-to-glutamine ratio, and a higher glutaminase expression compared to non-invasive breast cancer cells (172). Moreover, deprivation of glutamine or BPTES treatment in combination with CDDP or etoposide enhanced chemotherapy cytotoxicity on breast cancer HCC1937 cells (183). Anti-proliferative effects of 1,4-di(5-amino-1,3,4-thiadiazol-2-yl)butane compound, GLS1 inhibitor, on human breast tumor lines are similar to BPTES or CB-839 (184). Co-treatment of CB-839 and everolimus interrupts the growth of these endocrine-resistant xenografts (185).
GLS1 and GLS2 inhibitors or BPTES co-administered with doxorubicin demonstrated a synergistic activity decreasing proliferation of the human pancreas adenocarcinoma ascites metastasis (AsPC-1) cells (186). Disruption of glutamine metabolic pathways improved the efficacy of gemcitabine treatment (nucleoside analog that works by blocking DNA replication) in pancreatic cancer (187).
Some studies have revealed that some invasive ovarian tumor cells improve the use of glutamine to fuel TCA cycle (188). Yuan et al. demonstrated that compound 968 is able to block cell proliferation and sensitize paclitaxel in ovarian cancer (189). Moreover, it has been demonstrated that ovarian cancer CDDP-resistant cells present increased glutamine consumption and increased expression of the glutamine transporter ASCT2 and GLS. Thus, the combined treatment of BPTES and CDDP resensitized CDDP-resistant cells (108). Another molecule is epigallocatechin gallate (EGCG), which is a GLUD inhibitor GLUD. This compound combined with CDDP had a synergic effect on A2780(cisR) ovarian cancer cells becoming a strategy to overcome cisplatin resistance (190).
mTOR inhibitors-resistant glioblastoma cells have a compensatory increase in glutamine metabolism, suggesting that combined inhibition of GLS1 and mTOR could potentially overcome resistance (191).
Targeting Lipid Metabolism
Lipid metabolism is another important player in the development of chemoresistance. The interest in therapeutic strategies directed to block lipid synthesis, lipid uptake, intracellular lipolysis/lipid utilization, and lipid storage is growing (192).
Among the enzyme that regulates lipid metabolism, FASN is an important one and it correlates with poor prognosis in various types of cancer and also interferes with drug efficacy (193). Moreover, FASN overexpression induces resistance to antitumoral drugs such as adriamycin and mitoxantrone in breast cancer cells (194), gemcitabine-resistant pancreatic cells (195), cisplatin-resistant ovarian cancer cells (110), and radiotherapy resistant head and neck squamous cell carcinomas (196).
Inhibitor compounds targeting lipogenic enzymes (such as FASN, ACLY, and ACC) have been studied and their anticancer activity has been demonstrated in several preclinical models (197–199). Besides the promising data, serious side effects of these compounds have led to their clinical development exclusion. Various FASN inhibitors, such as Cerulenin, C75, orlistat, C93, C247, and GSK837149A, have been identified (200). The inhibition of FASN demonstrated to represent an excellent target, when used in in vitro, in xenograft and genetically induced mouse model analysis (200). Inhibitors of FASN induced cancer cells death directly or sensitized them to chemotherapic drugs, such as 5-fluorouracil and trastuzumab (201–204).
It has been reported, by genomic profiling, that CPT1 and fatty acid input into an oxidative pathway are decreased in four aggressive cancer cells, including melanoma, breast, ovarian, and prostate malignancies, respect to their non-aggressive counterparts (205). Recent studies reported a negative correlation between FASN inhibition and the consequent effect on body weight and food intake. In fact, a worsen eating that leads to weight loss was observed in mice treated with cerulenin and C75 and the cause seemed to be related to the CPT-1 inhibition in the hypothalamus (206–208).
The SPHK1 sphingosine Kinase 1 isozyme has been largely studied and its several functions in tumor development have been demonstrated, while the SPHK2 has not been as well-studied (209–213). Several studies in vitro and in vivo (only preclinical) demonstrated that ABC294640, the SPHK2-specific inhibitor, is able to inhibit proliferation of cancer cells or tumors more effectively or similarly than agents targeting SPHK1, in several tumor models, including ovarian (214), multiple myeloma (215), lung (216), kidney (217), breast (218), prostate (219), and pancreatic cancers (220).
Liver X receptor (LXR) is a crucial transcriptional regulator of cholesterol homeostasis and SR9243 is an LXR inverse agonist. SR9243 is able to kill selectively cancer cells because it inhibits the Warburg effect and lipogenesis and so the inhibition leads to the formation of an environment not favorable to cancer cells (221).
Recently, several studies have shown that dysregulated sphingolipid metabolism is a key contributor to the progression and resistance of ovarian cancer. By RNA-seq, Dobbin and colleagues revealed transcriptional variants between matched pairs of carboplatin and paclitaxel-treated vs. control patient-derived xenograft (PDX) models of ovarian cancer. In particular, they identified that S1P signaling is modified pathways following chemotherapy treatment (222). Sphingolipid metabolizing enzymes strictly related in modulating the ceramide-sphingosine-S1P rheostat play a key role in cell proliferation and have been directly correlated with drug resistance in ovarian cancer (223, 224). Specifically, increased expression of ceramide transport protein (CERT), SPHK1, SPHK2, and glucosylceramide synthase (GCS) have been correlated with resistance to paclitaxel, doxorubicin, and N-(4-hydroxylphenyl) retinamide (fenretinide) chemotherapies and cytotoxicity (225–230). The sphingolipid-mediated sphingosine-1-phosphate (S1P) pathway may represent a promising new pharmacological target to counteract the chemoresistance in ovarian cancer cells. Few therapeutic compounds specifically target S1P pathway proteins, but this pathway can modify the response of several chemotherapeutic treatments, including docetaxel, doxorubicin, and cyclophosphamide (231–234). Several approaches have been studied for modulating sphingolipid metabolism, and some of them consist in the use of combined treatment between ceramide analogs and chemotherapeutic agents (235–237). Treatments that showed activity in resistance ovarian cancer models include the use of synthetic ceramide analogs, inhibitors of SPHK, neutralization of secreted S1P, and S1PR antagonists. For example, the combined treatment of C6-ceramide with paclitaxel showed a synergic effect in suppressing cell proliferation and migration of CAOV3 ovarian cancer cells (238, 239). Moreover, drug delivery systems seem to be useful. In fact, a resensitization to paclitaxel of taxane-resistant SKOV3.TR ovarian cancer cells have been shown with the combination of paclitaxel with C6-ceramide-encapsulated in poly(ethylene oxide)-modified poly(epsilon-caprolactone) (PEO-PCL) nanoparticles (235). Kelly M. and colleagues demonstrated that the combined treatment of tamoxifen with the Sphingosine kinase 1 (SK1) inhibitor FTY720 blocks proliferation of both ERα-positive and ERα-negative drug-resistant cell lines and an ERα-positive PDX model of ovarian tumor (240). The multiple mechanisms of action of tamoxifen and its relatively high therapeutic index provide a strong rationale for combining tamoxifen with FTY720, as a strategy for treating ovarian tumors and circumventing drug resistance (226, 241–243).
It has been demonstrated that tamoxifen-resistant breast cells, T-47D, present an increased level of neutral lipids, in particular, cholesterol esters and triglycerides, and increased expression of Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) (121). CDDP-resistant human ovarian cancer cell lines shift their metabolism toward a lipogenic phenotype and accumulate lipid droplets (244). Moreover, CDDP-resistant lung cells have an increased expression of FASN and that inhibiting FASN could decrease the metastatic potential of CDDP-resistant cells (245). It has been demonstrated that a combination of orlistat, an inhibitor of FASN and cisplatin, in vivo, causes a delay in tumor growth (110). A high fatty acid synthase (FASN) activity is also involved in ErbB2-induced breast cancer chemoresistance to docetaxel (116). It has been demonstrated that prostate-resistant cells, C4-2R and MR49F (enzalutamide-resistant cells) respect to C4-2 and LNCaP have increased expression of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR). They showed that the combined treatment between simvastatin and enzalutamide sensitizes resistant cells in vitro. Moreover, tests in vivo in xenografts mice demonstrate a decrease in tumor cell proliferation (122).
Inhibitors of CPT1, such as etomoxir or ranolazine, have demonstrated promising results in different types of tumors. In fact, the combined treatment of etomoxir and orlistat is able to inhibit cell proliferation in LnCaP and VCaP prostate cancer cells (246). Moreover, the treatment of human leukemia cells with etomoxir or ranolazine can induce apoptosis cell death (115).
Targeting Mitochondria Metabolism
During cancer cells' adaptation to an hypoxic microenvironment, mitochondria have been demonstrated to be fundamental during solid tumor metastasis and in chemoresistance (247–249). Targeting mitochondrial-dependent metabolism to overcome drug resistance is an area of intense research. The increase of antioxidant pathways that help cancer cells to neutralize mitochondrial ROS is a common strategy adopted by some tumors to become drug-resistant (250). Moreover, mitochondria could promote therapy resistance by reducing the mitochondrial permeability transition (MPT) and inducing apoptosis resistance (251).
Mitochondria also appear responsible for the accumulation of oncometabolites such as fumarate, succinate, and 2-hydroxyglutarate (2-HG). Indeed, Succinate dehydrogenase complex iron-sulfur subunit B (SDHB), fumarate hydratase (FH), isocitrate dehydrogenase [NADP(+)] 1, cytosolic (IDH1), isocitrate dehydrogenase [NADP(+)] 2 and mitochondrial (IDH2) may be affected by germline or somatic mutations in a variety of human tumors (252). Fumarate, succinate and 2-HG accumulation is sufficient to drive malignant transformation and thus behave like bona fide oncometabolite (253). These oncometabolites share the capacity to inhibit α-ketoglutarate-dependent enzymes that control gene expression at the epigenetic level, such as Jumonji domain (JMJ) histone lysine demethylases (254) as well as ten-eleven translocation (TET) dioxygenases (255), resulting in the expression of a potentially oncogenic transcriptional program associated with a block in terminal differentiation (256).
Dysregulation of mitophagy (removing of abnormal mitochondria) contributes to neoplastic progression and drug resistance in various tumors (257). Enhanced mitophagy can protect cancer cells during chemotherapy and radiotherapy preventing apoptosis (258). On the other hand, excessive mitochondrial clearance may induce metabolic disorders and cell death (259). Therefore, mitophagy likely plays a dual role in cancer drug resistance (260). Mitophagy inhibition enhances the sensitivity of breast cancer cells to classical paclitaxel (114).
mtDNA has an essential role on tumorigenesis and chemoresistance. mtDNA pathogenic point mutations and changes in copy number, were shown to induce cancer progression (261) and have been associated with cancer development to a more malignant phenotype with poor prognosis in vivo (262–264). However, most mtDNA mutations are neutral missense mutations present in homoplasmy (265), suggesting that severe mutations are negatively selected. Indeed, mtDNA mutations per se are not able to induce carcinogenesis (266) but some mtDNA polymorphisms correlated with tumor development (267–270).
In particular, mutations in mitochondria encoded Complex I (CI) subunits could affect tumor progression depending on their mutational load and its detrimental activity (271). Based on these observations, Gasparre et al. introduced the concept of oncojanus: severe CI assembly mutations can promote tumorigenesis below a threshold level; but above that level they behave as antitumorigenic due to CI assembly defects. On the other side, non-disassembling mild mtDNA CI mutations could stimulate tumor proliferation and metastases. The oncojanus function of CI subunits was described for both mitochondrial and nuclear encoded CI subunits (272, 273). CI disruption inhibits OxPhos, promote NADH accumulation, inhibition of α-ketoglutarate dehydrogenase and increase the α-Ketoglutarate (KG)/succinate ratio. The α-Ketoglutarate (KG)/succinate imbalance activates prolyl-hydroxylases (PDH) enzymes responsible for the hydroxylation and degradation of HIF-1α even in hypoxic conditions (271, 274). Of note, genetical and pharmacological targeting of CI activity in osteosarcoma and colorectal cancer cell models successfully converted a carcinoma into a benign low-proliferating and non-invasive oncocytic tumor (273).
Moreover, the oncojanus effect was also observed in ovarian cancer after chemotherapy (275). A missense mtDNA point mutation in the MTND4 subunit of CI appeared after carboplatin treatment and generated a mild energetic defect allowing paclitaxel chemoresistance. When mutated MTND4 arrived to a certain threshold CI activity was impaired turning cancer cells into an oncocytic phenotype. Later it was demonstrated that the accumulation of deleterious mtDNA mutations induced by carboplatin in ovarian cancer cell lines conferred paclitaxel resistance through the reduction of filamentous tubulin (276). The treatment of A549 non-small cell lung cancer cells with CDDP induced an homoplasmic shift of a non-synonymous mutation in the CI protein MTND2 resulting in chemoresistance to cisplatin; which was correlated with the upregulation of the nuclear PGC-1α and PGC-1β and increased mitochondrial biogenesis (277).
mtDNA depletion in cancer cells under drug treatment promotes invasion and metastasis, induces expression of epithelial-to-mesenchymal (EMT) proteins (278) and activates pro-survival and antiapoptotic pathways (279, 280). Although the detailed molecular mechanism remains to be determined, several studies have demonstrated that reduced mtDNA content promotes activation of a mitochondria-to-nucleus signaling leading to increased expression of anti-apoptotic genes, including Bcl-2, and activation of pro-survival enzymes, such as Akt (280), that likely play a role in conferring resistance to apoptosis induced by drug treatment. mtDNA depletion in androgen-dependent LNCaP prostate cancer cells resulted in the loss of androgen dependence and increased resistance to paclitaxel (118, 119).
Horizontal transfer of mtDNA to cancer cells with a low respiratory function was correlated with recovery of respiration and increased tumor-initiating efficacy (281). mtDNA exchange through intercellular bridges or exosomes (extracellular vesicles implicated in cell-cell communication and transmission of disease states) could induce drug resistance by promoting a cancer stem cell (CSC) phenotype (282) Interestingly, exosomes containing mtDNA and mitochondrial proteins involved in mitochondrial fusion and biogenesis were found in the serum of prostate cancer patients as well as in the tumor itself (283). MSCs also protected AML leukemia cells from chemotherapeutic cytotoxicity by transferring them functional mitochondria. These effects occur together with mitochondrial fragmentation controlled by ERK-mediated Drp1 phosphorylation. Thus, disruption of leukemia cells/stromal interactions and targeting mitochondrial dynamics may provide a novel strategy that could be combined with conventional chemotherapeutic agents for leukemia treatment (284). It was demonstrated that the co-culture of leukemia cells with mesenchymal stem cells (MSCs) increased the expression of uncoupling protein 2 (UCP2) in leukemia cells; uncoupling oxidative phosphorylation and decreasing ROS production (285, 286). Recent studies have demonstrated the protective role of mitochondrial metabolism on cancer cells exposed to chemotherapy cytotoxicity. In acute myeloid leukemia (AML) adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) signaling promoted glucose uptake and a shift to glycolysis decreasing intracellular ROS (287).
Phenformin is a biguanide similar to metformin that targets complex I of mitochondria. It was identified to be more potent in decreasing cell growth in non-small cell lung cancer, but unfortunately drug-resistance emerged (288). It has been hypothezed that the development of resistance is dependent on functional LKB1-AMPK signaling, which improves a switch in their metabolism to bypass inhibitory effects of phenformin. Since the serious side effects of the drug, it was withdrawned from the market. Besides this, new studies on phenformin have been conducted (288).
PGC-1α is a transcription co-activator that regulates mitochondria biogenesis and it is involved in energy metabolism. It has been reported that CDDP treatment increases PGC-1α levels in a small cell lung carcinoma cell line and PGC-1α silencing sensitizes cells to this drug (109). Increased mitochondrial mass confers stem-like properties to breast cancer cell lines MDA-MB-231 and MCF7 and enables their resistance to paclitaxel (117). PGC-1α induction may also cause chemoresistance by activating a metabolic shift to bypass ATP request, as shown for cells treated with inhibitors of BRAF (289). This is also confirmed for 5-FU resistance, which increased PGC-1α expression and so modified cellular cancer metabolism to modulate the energetic stress induced by treatment (290–292).
Numerous emerging studies demonstrated a correlation between mitochondrial dynamics and cell survival (293–298). The co-culture of leukemia cells with MSCs also altered the mitochondrial dynamics of leukemia cells due to an increase of the activating phosphorylation of Dynamin-1-like protein (Drp1) at S616. Drp1 is a GTPase that regulates mitochondrial fission. Leukemia cells overexpressing wild-type Drp1 or Drp1 S616E presented fragmented mitochondria, reduced mitochondrial ROS levels, increased glycolysis, and improved drug resistance (299–301). Drp1 S616 phosphorylation through the stimulation of mitochondria fission and glycolysis seemed required to RAS-induced transformation (302). Moreover, inhibition of Drp1 activity caused mitochondrial fusion and impeded tumor growth (303). The cell cycle inhibitor Cytarabine is a normally chemotherapeutic therapy for leukemias and lymphomas, with reduced clinical implications due to the development of resistance (304, 305). Among the probable mechanisms, Cytarabine treatment improved the increase of a chemoresistant leukemic stem cell population with high FAO/OxPhos activity. In light of this, the FAO inhibitor etomoxir blocks oxygen consumption in acute myeloid leukemia cells and sensitized cells to cytarabine (306).
Recent evidence suggests that chemoresistant ovarian cancer has an increase in OxPhos dependence. Improved OxPhos in ovarian cancer cells increase IL-6 production (307) which facilitates tumor cell survival and proliferation (308), changing efficacy to chemotherapy, and reduce progression-free survival of ovarian cancer patients (309). Ovarian cancer cell migration was shown to be sustained by pyruvate, involving the mitochondrial activity during metastasis (310). Other studies have revealed that some invasive ovarian tumor cells improve the use of glutamine to fuel the TCA cycle (188). CD44+CD117+ ovarian tumor stem cells derived from epithelial ovarian cancer patients exhibited both high glucose uptake and a high OxPhos phenotype, which was correlated with their amplified capacity to live under a glucose-free context (311). On the other hand, CD44+MyD88+ cancer stem cells had a mainly glycolytic phenotype and suggested that therapy with glycolytic inhibitors could be favorable to increase patient's survival (312). Mouse ovarian cancer-initiating cells (putative cancer stem cells) harbor a highly flexible metabolic phenotype, whereby they could use either glycolysis or OxPhos under stress (313). It was proposed that most ovarian tumor cells may use either glycolysis or OXPHOS and that such plasticity increased their “cellular fitness” (310, 314, 315). The shift from glycolysis to OxPhos has also been showed upon MYC/KRAS or MYC/ERBB2 removal in breast cancer cells (316, 317), and also in glioma cells because of the acquired resistance to phosphoinositide-3-kinase (PI3K) (318). Moreover, PI3K resistance in breast cancer cells is related to the shift from glucose to lactate (34). Inhibitors of mitochondrial respiration become therapeutic strategies in ovarian cancer cells because of their dependence on OxPhos. In fact, cancer-selective inhibition of the electron transport chain (ETC) could kill ovarian cancer cells directly without affecting normal cells. The complex I inhibitor BAY 87-2243 has been studied in a Phase 1 study (NCT01297530) but results were not showed (Clinicaltrials.gov). Several strategies targeting mitochondria CIII complex, such as the thiol modifier β-phenylethylisothiocyanates (PEITC), have been developed. In particular, PEITC can enhance the ROS level, decreasing OxPhos and, consequently, causing prostate cancer cell death by apoptosis (319). Moreover, the same compound combined with metformin demonstrated cytotoxicity in human ovarian cancer cells (320). Also, treatment with ABT-737, the inhibitor of Bcl-2 proteins (321) and the FAO inhibitor perhexiline also was capable of sensitized CDDP-resistant ovarian cancer cells (322) by modulating mitochondrial metabolism. The main drugs to target mitochondria or disrupt OxPhos are antibiotic or anti-parasitic agents. Among these, azithromycin and doxycycline target mitochondrial protein synthesis, while salinomycin targets mitochondrial K+/H+ exchange (323). Azithromycin, combined with CDDP and paclitaxel, is able to reduce side effects and to enhance patients' survival (324). Doxycycline is able to inhibit cellular growth of ovarian cells SKOV3 and SKOV3/DPP and to sensitize resistant cells to CDDP (325). Salinomycin is another antibiotic able to inhibit cell growth, especially on cancer cells compared to normal epithelial cells (326).
Resistance to 5-FU has been associated to aumgmented mitochondrial mass and activity, increase ETC enzymes expression and higher level of oxygen consumption (290, 327). So, due to their OxPhos-dependency, resistant cells were showed to be sensitive to Complex I inhibition by metformin (327). OxPhos involvement to 5-FU resistance was correlated to the development of stemness-related phenotype, stricly linking CSCs to mitochondrial metabolism as described previously (328, 329). According, the activation of mitochondrial FAO is able to promote stemness in gastric cancer cells and consequently there is chemoresistance to 5-FU induced by tumor-associated mesenchymal stem cells. In fact, treatment with the FAO inhibitor etomoxir was capable to partially reduce FU resistance (330).
BRAFV600E is a mutation found in stage IIIc or stage IV melanoma. Chapman and co-workers demonstrated that its inhibition with vemurafenib leads to the shift to OxPhos and the switch is useful to treat resistant melanoma cells (331). Elesclomol sodium (STA-4783) is a compound targeting ROS in tumor cells. Its mechanism is strictly related to inhibition of electron transport flux and so increase ROS generation causing oxidative stress in both malignant and healthy cells. However, as cancer cells have already higher ROS production, this drug will be capable to cause cytotoxicity selectively in malignant cells, resulting in activation of apoptosis in melanoma cancer cells (332). STA-4783 alone and in association with paclitaxel revealed promising results in phase I/II clinical studies in patients with refractory solid tumors (333, 334), but unfortunately serious side effects lead to stop phase III study in melanoma patients (335).
Lee and coworkers demonstrated that an enhanced mitochondrial oxidative phosphorylation characterizes irinotecan-resistant NSCLC cells. They tested a combined treatment between gossypol (a molecule that inhibits aldehyde dehydrogenase) and phenformin (a molecule that inhibits mitochondrial complex I), concluding that the combination leads to sensitization of irinotecan-resistant NSCLC cells (336). Another drug targeting mitochondria is apogossypol, which is semisynthetic. It has been demonstrated to be cytotoxic in murine B cells (337). Metformin, a drug usually used for the treatment of type 2 diabetes, has demonstrated anti-cancer properties. In fact, the combined treatment of metformin and paclitaxel showed anticancer activity in vivo and was able to arrest the cell cycle in vitro in human breast MCF-7 and human lung A459 cancer cells (338). PC3 prostate cancer cells docetaxel-resistant shift their metabolism from glycolysis toward OXPHOS and this is linked to EMT phenotype. The combination of chemotherapy and OXPHOS inhibitors limited docetaxel-associated drug resistance and progression toward metastasis (120).
In short, targeting glycolysis, PPP, OxPhos, and fatty acid synthesis and oxidation could enhance chemotherapy and radiation responsiveness and overcome therapy resistance. Importantly, therapy-resistant tumors present different metabolic phenotypes related to non-treated tumors, thus it's needed a better understanding of the new dependencies and vulnerabilities across different chemotherapy treatments in different tumors to reduce toxicity and to increase the efficacy of chemotherapeutic drugs.
Concluding Remarks
Metabolic deregulation is an established hallmark of cancer, thus the elucidation of novel therapy combinations based on new tumor-specific metabolic liabilities after chemotherapy will be essential to the development rational metabolic therapeutic strategies to overcome drug resistance.
Author Contributions
MD, IG, TP-G, and MM wrote the manuscript.
Funding
This work was supported by AIRC Inverstigator Grant 2018_22030 and MIUR PRIN 2017_2017237P5X.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell. (2008) 134:703–7. doi: 10.1016/j.cell.2008.08.021
2. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
3. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
4. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
5. Warburg O, Posener K, Negelein E. Über den stoffwechsel der carcinomzelle. Biochem Z. (1924) 152:309–344.
6. Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. (2009) 121:29–40. doi: 10.1016/j.pharmthera.2008.09.005
7. Sreedhar A, Zhao Y. Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomed Rep J. (2018) 8:3–10. doi: 10.3892/br.2017.1022
8. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
9. Fantin VR, St.-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. (2006) 9:425–34. doi: 10.1016/j.ccr.2006.04.023
10. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. (2016) 16:650–62. doi: 10.1038/nrc.2016.81
11. Felig P. Amino acid metabolism in man. Annu Rev Biochem. (1975) 44:933–55. doi: 10.1146/annurev.bi.44.070175.004441
12. Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. (2016) 73:377–92. doi: 10.1007/s00018-015-2070-4
13. Li D, Zhu Y, Tang Q, Lu H, Li H, Yang Y. et al. A new G6PD knockdown tumor-cell line with reduced proliferation and increased susceptibility to oxidative stress. Cancer Biother Radiopharm. (2009) 24:81–90. doi: 10.1089/cbr.2008.0494
14. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. (2011) 13:310–16. doi: 10.1038/ncb2172
15. Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology. (2015) 149:1587–98.e11. doi: 10.1053/j.gastro.2015.07.050
16. Zogg CK. Phosphoglycerate dehydrogenase: potential therapeutic target and putative metabolic oncogene. J Oncol. (2014) 2014:524101. doi: 10.1155/2014/524101
17. Crabtree HG. Observations on the carbohydrate metabolism of tumours. Biochem J. (1929) 23:536–45. doi: 10.1042/bj0230536
18. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. (2007) 11:37–51. doi: 10.1016/j.ccr.2006.10.020
19. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. (2015) 162:552–63. doi: 10.1016/j.cell.2015.07.017
20. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N. et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. (2016) 24:716–27. doi: 10.1016/j.cmet.2016.09.006
21. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. (2007) 104:19345–50. doi: 10.1073/pnas.0709747104
22. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. (2015) 21:805–21. doi: 10.1016/j.cmet.2015.05.014
23. Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. (2014) 53:710–25. doi: 10.1016/j.molcel.2014.01.016
24. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. (2012) 336:1040–4. doi: 10.1126/science.1218595
25. Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS ONE. (2013) 8:e75154. doi: 10.1371/journal.pone.0075154
26. Maddocks OD, Labuschagne CF, Adams PD, Vousden KH. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol Cell. (2016) 61:210–21. doi: 10.1016/j.molcel.2015.12.014
27. Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. (2014) 159:1603–14. doi: 10.1016/j.cell.2014.11.025
28. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. (2017) 3:169–80. doi: 10.1016/j.trecan.2017.01.005
29. Bhutia YD, Ganapathy V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim Biophys Acta. (2016) 1863:2531–9. doi: 10.1016/j.bbamcr.2015.12.017
30. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. (2011) 481:380–4. doi: 10.1038/nature10602
31. Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, et al. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun. (2013) 4:2236. doi: 10.1038/ncomms3236
32. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. (2017) 36:1302–15. doi: 10.15252/embj.201696151
33. Yielding KL, Tomkins GM. An effect of L-leucine and other essential amino acids on the structure and activity of glutamic dehydrogenase. Proc Natl Acad Sci USA. (1961) 47:983–9. doi: 10.1073/pnas.47.7.983
34. Park S, Chang CY, Safi R, Liu X, Baldi R, Jasper JS, et al. ERRa-regulated lactate metabolism contributes to resistance to targeted therapies in breast cancer. Cell Rep. (2016) 15:323–35. doi: 10.1016/j.celrep.2016.03.026
35. Pinheiro C, Morais-Santos F, Baltazar F, Longatto-Filho A, Garcia EA, Scapulatempo-Neto C, et al. Lactate transporters and vascular factors in HPV-induced squamous cell carcinoma of the uterine cervix. BMC Cancer. (2014) 14:1–12. doi: 10.1186/1471-2407-14-751
36. Kuhajda FP. Fatty acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. (2000) 16: 202–8. doi: 10.1016/S0899-9007(99)00266-X
37. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. (2006) 9:358–65. doi: 10.1097/01.mco.0000232894.28674.30
38. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. (2016) 5:e189. doi: 10.1038/oncsis.2015.49
39. Liu Y, Zuckier LS, Ghesani NV. Dominant uptake of fatty acid over glucose by prostate cells: a potential new diagnostic and therapeutic approach. Anticancer Res. (2010) 30:369–74. doi: 10.1097/00003072-200505000-00038
40. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
41. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. (2012) 72:3709–14. doi: 10.1158/0008-5472.CAN-11-4112
42. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. (2004) 64:2212–21. doi: 10.1158/0008-5472.CAN-2148-2
43. Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal. (2009) 2:ra82. doi: 10.1126/scisignal.2000446
44. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. (2017) 541:41–5. doi: 10.1038/nature20791
45. Tirinato L, Pagliari F, Limongi T, Marini M, Falqui A, Seco J, et al. An overview of lipid droplets in cancer and cancer stem cells. Stem Cells Int. (2017) 2017:1656053. doi: 10.1155/2017/1656053
46. Apffel CA, Baker JR. Lipid droplets in the cytoplasm of malignant cells. Cancer. (1964) 17:176–84. doi: 10.1002/1097-0142(196402)17:2<176::aid-cncr2820170207>3.0.co;2-2
47. Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res. (2012) 10:133–42. doi: 10.1158/1541-7786.MCR-11-0206
48. Shyu P, Wong XFA, Crasta K, Thibault G. Dropping in on lipid droplets: insights into cellular stress and cancer. Biosci Rep. (2018) 38:BSR20180764. doi: 10.1042/BSR20180764
49. Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. (2019) 11:e10698. doi: 10.15252/emmm.201910698
50. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. (2017). 168:657–669. doi: 10.1016/j.cell.2016.12.039
51. Tarrado-Castellarnau M, de Atauri P, Cascante M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget. (2016) 7:62726–53. doi: 10.18632/oncotarget.10911
52. Iurlaro R, León-Annicchiarico CL, Muñoz-Pinedo C. Regulation of cancer metabolism by oncogenes and tumor suppressors. Methods Enzymol. (2014) 542:59–80. doi: 10.1016/B978-0-12-416618-9.00003-0
53. Neugent ML, Goodwin J, Sankaranarayanan I, Yetkin CE, Hsieh MH, Kim JW. A new perspective on the heterogeneity of cancer glycolysis. Biomol Ther. (2018) 26:10–18. doi: 10.4062/biomolther.2017.210
54. Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. (2006) 16:253–264. doi: 10.1016/j.semcancer.2006.07.014
55. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. (2007) 178:93–105. doi: 10.1083/jcb.200703099
56. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
57. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. (2009) 458:762–5. doi: 10.1038/nature07823
58. Puzio-Kuter AM. The role of p53 in metabolic regulation. Genes Cancer. (2011) 2:385–91. doi: 10.1177/1947601911409738
59. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. (2004) 64:2627–33. doi: 10.1158/0008-5472.CAN-03-0846
60. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. (2005) 65:177–85.
61. Kruiswijk F, Labuschagne CF, Vousden KH. P53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. (2015) 16:393–405. doi: 10.1038/nrm4007
62. Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, et al. p53 Activation in adipocytes of obese mice. J Biol Chem. (2003) 278:25395–400. doi: 10.1074/jbc.M302364200
63. Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. (2011) 44:491–501. doi: 10.1016/j.molcel.2011.08.038
64. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. (2013) 20:659–68. doi: 10.1038/cdd.2012.168
65. Liu Y, He Y, Jin A, Tikunov AP, Zhou L, Tollini LA, et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating mcd and promoting fatty acid oxidation. Proc Natl Acad Sci USA. (2014) 111:E2414–22. doi: 10.1073/pnas.1315605111
66. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. (2006) 312:1650–3. doi: 10.1126/science.1126863
67. Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, et al. Regulation of AIF expression by p53. Cell Death Differ. (2006) 13:2140–9. doi: 10.1038/sj.cdd.4401965
68. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. (2012) 72:560–7. doi: 10.1158/0008-5472.CAN-11-1215
69. Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci USA. (2017) 114:4300–5. doi: 10.1073/pnas.1617709114
70. Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. (2016) 353:1161–5. doi: 10.1126/science.aaf5171
71. Ortega-Molina A, Serrano M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab. (2013) 24:184–9. doi: 10.1016/j.tem.2012.11.002
72. Lehuédé C, Dupuy F, Rabinovitch R, Jones RG, Siegel PM. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. (2016) 76:5201–8. doi: 10.1158/0008-5472.CAN-16-0266
73. Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol. (2017) 48:47–53. doi: 10.1016/j.ceb.2017.05.006
74. Shi X, Tasdogan A, Huang F, Hu Z, Morrison SJ, DeBerardinis RJ. The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci Adv. (2017) 3:5268. doi: 10.1126/sciadv.aao5268
75. Hu J, Locasale JW, Bielas JH, O'Sullivan J, Sheahan K, Cantley LC, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. (2013) 31:522–9. doi: 10.1038/nbt.2530
76. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. (2017) 27:863–75. doi: 10.1016/j.tcb.2017.06.003
77. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
78. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. (2010) 9:3506–14. doi: 10.4161/cc.9.17.12731
79. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016) 16:582–98. doi: 10.1038/nrc.2016.73
80. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. (2008) 118:3930–42. doi: 10.1172/JCI36843
81. Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature. (2019) 577:115–20. doi: 10.1038/s41586-019-1847-2
82. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. (2011) 71:2455–65. doi: 10.1158/0008-5472.CAN-10-3323
83. Bochet L, Meulle A, Imbert S, Salles B, Valet P, Muller C. Cancer-associated adipocytes promote breast tumor radioresistance. Biochem Biophys Res Commun. (2011) 411:102–6. doi: 10.1016/j.bbrc.2011.06.101
84. Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. (2013) 1831:1533–41. doi: 10.1016/j.bbalip.2013.02.010
85. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
86. Andrejeva G, Rathmell JC. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. (2017) 26:49–70. doi: 10.1016/j.cmet.2017.06.004
87. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression, Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
88. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
89. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. (2012) 15:813–26. doi: 10.1016/j.cmet.2012.04.023
90. Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S, et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naïve T cell apoptosis and affects tumor immunity. Sci Immunol. (2017) 2:4631. doi: 10.1126/sciimmunol.aan4631
91. O'Sullivan D, Sanin DE, Pearce EJ, Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol. (2019) 19:324–35. doi: 10.1038/s41577-019-0140-9
92. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
93. Ren W, Liu G, Yin J, Tan B, Wu G, Bazer FW, et al. Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis. (2017) 8:e2757. doi: 10.1038/cddis.2017.207
94. Loftus RM, Assmann N, Kedia-Mehta N, O'Brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. (2018) 9:2341. doi: 10.1038/s41467-018-04719-2
95. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal. (2015) 8:ra97. doi: 10.1126/scisignal.aab2610
96. Jia D, Park JH, Jung KH, Levine H, Kaipparettu BA. Elucidating the metabolic plasticity of cancer: mitochondrial reprogramming and hybrid metabolic states. Cells. (2018) 7:21. doi: 10.3390/cells7030021
97. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. (2015) 22:590–605. doi: 10.1016/j.cmet.2015.08.015
98. Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. (2011) 10:2350–62. doi: 10.1158/1535-7163.MCT-11-0497
99. Jia D, Lu M, Jung KH, Park JH Yu L, Onuchic JN, et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci USA. (2019) 116:3909–18. doi: 10.1073/pnas.1816391116
100. Zaal EA, Berkers CR. The influence of metabolism on drug response in cancer. Front Oncol. (2018) 8:500. doi: 10.3389/fonc.2018.00500
101. Aldossary S. A. Review on pharmacology of cisplatin: clinical use, toxicity and mechanism of resistance of cisplatin. Biomed Pharmacol J. (2019) 12:7–15. doi: 10.13005/bpj/1608
102. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. (2004) 4:253–65. doi: 10.1038/nrc1317
103. Beretta GL, Zaffaroni N. Androgen Receptor-Directed Molecular Conjugates for Targeting Prostate Cancer. Front Chem. (2019) 7:369. doi: 10.3389/fchem.2019.00369
104. Alimbetov D, Askarova S, Umbayev B, Davis T, Kipling D. Pharmacological targeting of cell cycle, apoptotic and cell adhesion signaling pathways implicated in chemoresistance of cancer cells. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19061690
105. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. (2017) 7:339–48. doi: 10.15171/apb.2017.041
106. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
107. Catanzaro D, Gaude E, Orso G, Giordano C, Guzzo G, Rasola A, et al. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget. (2015) 6:30102–14. doi: 10.18632/oncotarget.4945
108. Hudson CD, Savadelis A, Nagaraj AB, Joseph P, Avril S, DiFeo A, et al. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget. (2016) 7:41637–49. doi: 10.18632/oncotarget.9317
109. Cruz-Bermúdez A, Laza-Briviesca R, Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S, et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS inhibition. Free Radic Biol Med. (2019) 135:167–81. doi: 10.1016/j.freeradbiomed.2019.03.009
110. Papaevangelou E, Almeida GS, Box C, DeSouza NM, Chung YL. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int J Cancer. (2018) 143:992–1002. doi: 10.1002/ijc.31392
111. Ma L, Cheng Q. Inhibiting 6-phosphogluconate dehydrogenase reverses doxorubicin resistance in anaplastic thyroid cancer via inhibiting NADPH-dependent metabolic reprogramming. Biochem Biophys Res Commun. (2018) 04:498:912–7. doi: 10.1016/j.bbrc.2018.03.079
112. Bean JF, Qiu YY Yu S, Clark S, Chu F, Madonna MB. Glycolysis inhibition and its effect in doxorubicin resistance in neuroblastoma. J Pediatr Surg. (2014) 49:981–4. doi: 10.1016/j.jpedsurg.2014.01.037
113. Cao X, Fang L, Gibbs S, Huang Y, Dai Z, Wen P, et al. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother Pharmacol. (2007) 59:495–505. doi: 10.1007/s00280-006-0291-9
114. Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL, et al. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. (2015) 11:1259–79. doi: 10.1080/15548627.2015.1056970
115. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. (2010) 120:142–56. doi: 10.1172/JCI38942
116. Menendez JA, Lupu R, Colomer R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of HER−2/ neu -overexpressing human breast cancer cells to docetaxel (taxotere). Breast Cancer Res Treat. (2004) 84:183–95. doi: 10.1023/B:BREA.0000018409.59448.60
117. Farnie G, Sotgia F, Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. (2015) 6:30472–86. doi: 10.18632/oncotarget.5401
118. Higuchi M, Kudo T, Suzuki S, Evans TT, Sasaki R, Wada Y, et al. Mitochondrial DNA determines androgen dependence in prostate cancer cell lines. Oncogene. (2006) 25:1437–45. doi: 10.1038/sj.onc.1209190
119. Moro L, Arbini AA, Marra E, Greco M. Mitochondrial DNA depletion reduces PARP-1 levels and promotes progression of the neoplastic phenotype in prostate carcinoma. Cell Oncol. (2008) 30:307–22. doi: 10.3233/clo-2008-0427
120. Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget. (2016) 7:61890–904. doi: 10.18632/oncotarget.11301
121. Hultsch S, Kankainen M, Paavolainen L, Kovanen RM, Ikonen E, Kangaspeska S, et al. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer. (2018) 18:850. doi: 10.1186/s12885-018-4757-z
122. Kong Y, Cheng L, Mao F, Zhang Z, Zhang Y, Farah E, et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J Biol Chem. (2018) 293:14328–41. doi: 10.1074/jbc.RA118.004442
123. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol Aug. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
124. Polet F, Feron O. Endothelial cell metabolism and tumour angiogenesis: glucose and glutamine as essential fuels and lactate as the driving force. J Intern Med. (2013) 273:156–65. doi: 10.1111/joim.12016
125. Ruprecht B, Zaal EA, Zecha J, Wu W, Berkers CR, Kuster B, et al. Lapatinib resistance in breast cancer cells is accompanied by phosphorylation-mediated reprogramming of glycolysis. Cancer Res. (2017) 77:1842–53. doi: 10.1158/0008-5472.CAN-16-2976
126. Li SS, Ma J, Wong AS. T. Chemoresistance in ovarian cancer: exploiting cancer stem cell metabolism. J Gynecol Oncol. (2018) 03:29:e32. doi: 10.3802/jgo.2018.29.e32
127. Li X, Zhong Y, Lu J, Axcrona K, Eide L, Syljuåsen RG, et al. MtDNA depleted PC3 cells exhibit Warburg effect and cancer stem cell features. Oncotarget. (2016) 7:40297–313. doi: 10.18632/oncotarget.9610
128. Sottnik JL, Lori JC, Rose BJ, Thamm DH. Glycolysis inhibition by 2-deoxy-D-glucose reverts the metastatic phenotype in vitro and in vivo. Clin Exp Metastasis. (2011) 28:865–75. doi: 10.1007/s10585-011-9417-5
129. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. (2013) 12:152. doi: 10.1186/1476-4598-12-152
130. Shi T, Ma Y, Cao L, Zhan S, Xu Y, Fu F, et al. B7-H3 promotes aerobic glycolysis and chemoresistance in colorectal cancer cells by regulating HK2. Cell Death Dis. (2019) 10:308. doi: 10.1038/s41419-019-1549-6
131. Catanzaro D, Gabbia D, Cocetta V, Biagi M, Ragazzi E, Montopoli M, et al. (2018). Silybin counteracts doxorubicin resistance by inhibiting GLUT1 expression. Fitoterapia. 124:42–8. doi: 10.1016/j.fitote.2017.10.007
132. Han CY, Patten DA, Richardson RB, Harper ME, Tsang BK. Tumor metabolism regulating chemosensitivity in ovarian cancer. Genes cancer. (2018) 9:155–75. doi: 10.18632/genesandcancer.176.
133. Fabian C, Koetz L, Favaro E, Indraccolo S, Mueller-Klieser W, Sattler UG. Protein profiles in human ovarian cancer cell lines correspond to their metabolic activity and to metabolic profiles of respective tumor xenografts. FEBS J. (2012) 279:882–91. doi: 10.1111/j.1742-4658.2012.08479.x
134. Zhu J, Zheng Y, Zhang H, Sun H. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am J Transl Res. (2016) 8:4812–21.
135. Rashmi R, Huang X, Floberg JM, Elhammali AE, McCormick ML, Patti GJ, et al. Radioresistant cervical cancers are sensitive to inhibition of glycolysis and redox metabolism. Cancer Res. (2018) 78:1392–403. doi: 10.1158/0008-5472.CAN-17-2367
136. Silvestrini R, Zaffaroni N, Villa R, Orlandi L, Costa A. Enhancement of cisplatin activity by lonidamine in human ovarian cancer cells. Int J Cancer. (1992) 52:813–7. doi: 10.1002/ijc.2910520524
137. De Lena M, Lorusso V, Latorre A, Fanizza G, Gargano G, Caporusso L. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur J Cancer. (2001) 37:364–8. doi: 10.1016/S0959-8049(00)00400-7
138. Catanzaro D, Nicolosi S, Cocetta V, Salvalaio M, Pagetta A, Ragazzi E, et al. Cisplatin liposome and 6-amino nicotinamide combination to overcome drug resistance in ovarian cancer cells. Oncotarget. (2018) 9:16847–60. doi: 10.18632/oncotarget.24708
139. Choi J, Jung WH, Koo JS. Metabolism-related proteins are differentially expressed according to the molecular subtype of invasive breast cancer defined by surrogate immunohistochemistry. Pathobiology. (2013) 80:41–52. doi: 10.1159/000339513
140. Pelicano H, Zhang W, Liu J, Hammoudi N, Dai J, Xu RH, et al. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Res. (2014) 16:434. doi: 10.1186/s13058-014-0434-6
141. Lanning NJ, Castle JP, Singh SJ, Leon AN, Tovar EA, Sanghera A, et al. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metabol. (2017) 5:6. doi: 10.1186/s40170-017-0168-x
142. Lim SO, Li CW, Xia W, Lee HH, Chang SS, Shen J, et al. EGFR signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. (2016) 76:1284–96. doi: 10.1158/0008-5472.CAN-15-2478
143. Shen A, Wang L, Huang M, Sun J, Chen Y, Shen YY, et al. c-Myc alterations confer therapeutic response and acquired resistance to c-Met inhibitors in MET-addicted cancers. Cancer Res. (2015) 75:4548–59. doi: 10.1158/0008-5472.CAN-14-2743
144. Zhao YH, Zhou M, Liu H, Ding Y, Khong HT Yu D, et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. (2009) 28:3689–701. doi: 10.1038/onc.2009.229
145. Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP, Wilson GL, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. (2011) 71:4585–97. doi: 10.1158/0008-5472.CAN-11-0127
146. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, et al. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer. (2010) 9:33. doi: 10.1186/1476-4598-9-33
147. Schöder H, Larson SM. Positron emission tomography for prostate, bladder, and renal cancer. Semin Nucl Med. (2004) 34:274–92. doi: 10.1053/j.semnuclmed.2004.06.004
148. Testa C, Pultrone C, Manners DN, Schiavina R, Lodi R. Metabolic imaging in prostate cancer: where we are. Front Oncol. (2016) 6:225. doi: 10.3389/fonc.2016.00225
149. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. (2011) 30:2719–33. doi: 10.1038/emboj.2011.158
150. Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ, KIM J-W, et al. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 2 in prostate cancer cells. Biochem J. (2011) 433:225–33. doi: 10.1042/BJ20101104
151. Vaz CV, Alves MG, Marques R, Moreira PI, Oliveira PF, Maia CJ, et al. Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int J Biochem Cell Biol. (2012) 44:2077–84. doi: 10.1016/j.biocel.2012.08.013
152. Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene. (2014) 33:5251–61. doi: 10.1038/onc.2013.463
153. Kosic M, Arsikin-Csordas K, Paunovic V, Firestone RA, Ristic B, Mircic A, et al. Synergistic anticancer action of lysosomal membrane permeabilization and glycolysis inhibition. J Biol Chem. (2016) 291:22936–48. doi: 10.1074/jbc.M116.752113
154. Liu H, Kurtoglu M, León-Annicchiarico CL, Munoz-Pinedo C, Barredo J, Leclerc G, et al. Combining 2-deoxy-D-glucose with fenofibrate leads to tumor cell death mediated by simultaneous induction of energy and ER stress. Oncotarget. (2016) 7:36461–73. doi: 10.18632/oncotarget.9263
155. Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, et al. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. (2004) 64:31–4. doi: 10.1158/0008-5472.CAN-03-3294
156. Jae HJ, Chung JW, Park HS, Lee MJ, Lee KC, Kim HC, et al. The antitumor effect and hepatotoxicity of a hexokinase II inhibitor 3-bromopyruvate: in vivo investigation of intraarterial administration in a rabbit VX2 hepatoma model. Korean J Radiol. (2009) 10:596–603. doi: 10.3348/kjr.2009.10.6.596
157. Tataranni T, Piccoli C. Dichloroacetate (DCA) and Cancer: an overview towards clinical applications. Oxid Med Cell Longev. (2019) 2019:1–14. doi: 10.1155/2019/8201079
158. Sun H, Zhu A, Zhou X, Wang F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget. (2017) 8:52642–50. doi: 10.18632/oncotarget.16991
159. Woolbright BL, Choudhary D, Mikhalyuk A, Trammel C, Shanmugam S, Abbott E, et al. The role of pyruvate dehydrogenase kinase-4 (PDK4) in bladder cancer and chemoresistance. Mol Cancer Ther. (2018) 17:2004–12. doi: 10.1158/1535-7163.MCT-18-0063
160. Shi HS Li D, Zhang J, Wang YS, Yang L, Zhang HL, et al. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci. (2010) 101:1447–53. doi: 10.1111/j.1349-7006.2010.01562.x
161. Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. (2008) 283:28106–14. doi: 10.1074/jbc.M803508200
162. Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget. (2010) 1:734–40. doi: 10.18632/oncotarget.208
163. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. (2013) 123:3678–84. doi: 10.1172/JCI69600
164. Mondesir J, Willekens C, Touat M, de Botton S. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Med. (2016) 7:171–80. doi: 10.2147/JBM.S70716
165. Cho YS, Levell JR, Liu G, Caferro T, Sutton J, Shafer CM, et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med Chem Lett. (2017) 8:1116–21. doi: 10.1021/acsmedchemlett.7b00342
166. Shapiro RA, Clark VM, Curthoys NP. Inactivation of rat renal phosphate-dependent glutaminase with 6-diazo-5-oxo-L-norleucine. Evidence for interaction at the glutamine binding site. J Biol Chem. (1979) 254:2835–8.
167. Gatzemeier U, Cavalli F, Häussinger K, Kaukel E, Koschel G, Martinelli G, et al. Phase III trial with and without lonidamine in non-small cell lung cancer. Semin Oncol. (1991) 18(Suppl.4):42–8.
168. Di Cosimo S, Ferretti G, Papaldo P, Carlini P, Fabi A, Cognetti F. Lonidamine: efficacy and safety in clinical trials for the treatment of solid tumors. Drugs Today. (2003) 39:157–74. doi: 10.1358/dot.2003.39.3.799451
169. Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1, 2, 4-thiadiazol-2-yl) ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. (2012) 55:10551–63. doi: 10.1021/jm301191p
170. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) 13:890–901. doi: 10.1158/1535-7163.MCT-13-0870
171. Xiang Y, Stine ZE, Xia J, Lu Y, O'Connor RS, Altman BJ, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest. (2015) 125:2293–306. doi: 10.1172/JCI75836
172. Simpson NE, Tryndyak VP, Beland FA, Pogribny IP. An in vitro investigation of metabolically sensitive biomarkers in breast cancer progression. Breast Cancer Res Treat. (2012) 133:959–68. doi: 10.1007/s10549-011-1871-x
173. Katt WP, Ramachandran S, Erickson JW, Cerione RA. Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther. (2012) 11:1269–78. doi: 10.1158/1535-7163.MCT-11-0942
174. Simpson NE, Tryndyak VP, Pogribna M, Beland FA, Pogribny IP. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics. (2012) 7:1413–20. doi: 10.4161/epi.22713
175. Kahlert UD, Cheng M, Koch K, Marchionni L, Fan X, Raabe EH, et al. Alterations in cellular metabolome after pharmacological inhibition of N otch in glioblastoma cells. Int J Cancer. (2016) 138:1246–55. doi: 10.1002/ijc.29873
176. Xie C, Jin J, Bao X, Zhan WH, Han TY, Gan M, et al. Inhibition of mitochondrial glutaminase activity reverses acquired erlotinib resistance in non-small cell lung cancer. Oncotarget. (2016) 7:610–21. doi: 10.18632/oncotarget.6311
177. Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H, et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol. (2016) 10:73–84. doi: 10.1016/j.molonc.2015.08.003
178. Hernandez-Davies JE, Tran TQ, Reid MA, Rosales KR, Lowman XH, Pan M, et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J Transl Med. (2015) 13:210. doi: 10.1186/s12967-015-0581-2
179. Choi YK, Park KG. Targeting glutamine metabolism for cancer treatment. Biomol Ther. (2018) 26:19–28. doi: 10.4062/biomolther.2017.178
180. Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance. Cancer Biol Ther. (2011) 12:1085–97. doi: 10.4161/cbt.12.12.18671
181. Asiago VM, Alvarado LZ, Shanaiah N, Gowda GN, Owusu-Sarfo K, Ballas RA, et al. Early detection of recurrent breast cancer using metabolite profiling. Cancer Res. (2010) 70:8309–18. doi: 10.1158/0008-5472.CAN-10-1319
182. Budczies J, Pfitzner BM, Györffy B, Winzer KJ, Radke C, Dietel M, et al. Glutamate enrichment as new diagnostic opportunity in breast cancer. Int J Cancer. (2015) 136:1619–28. doi: 10.1002/ijc.29152
183. Chen L, Cui H, Fang J, Deng H, Kuang P, Guo H, et al. Glutamine deprivation plus BPTES alters etoposide- and cisplatin-induced apoptosis in triple negative breast cancer cells. Oncotarget. (2016) 7:54691–701. doi: 10.18632/oncotarget.10579
184. Zimmermann SC, Wolf EF, Luu A, Thomas AG, Stathis M, Poore B, et al. Allosteric glutaminase inhibitors based on a 1, 4-di (5-amino-1, 3, 4-thiadiazol-2-yl) butane scaffold. ACS Med Chem Lett. (2016) 7:520–24. doi: 10.1021/acsmedchemlett.6b00060
185. Demas DM, Demo S, Fallah Y, Clarke R, Nephew KP, Althouse S, et al. Glutamine metabolism drives growth in advanced hormone receptor positive breast cancer. Front Oncol. (2019) 9:686. doi: 10.3389/fonc.2019.00686
186. Abdel-Magid AF. Glutaminase GLS1 inhibitors as potential cancer treatment. ACS Med Chem Lett. (2016) 7:207–8. doi: 10.1021/acsmedchemlett.6b00016
187. Chen R, Lai LA, Sullivan Y, Wong M, Wang L, Riddell J, et al. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci Rep. (2017) 7:7950. doi: 10.1038/s41598-017-08436-6
188. Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol. (2014) 10:728. doi: 10.1002/msb.20134892
189. Yuan L, Sheng X, Clark LH, Zhang L, Guo H, Jones HM, et al. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am J Transl Res. (2016) 8:4265–77.
190. Mazumder MEH, Beale P, Chan C, Yu JQ, Huq F. Epigallocatechin gallate acts synergistically in combination with cisplatin and designed trans-palladiums in ovarian cancer cells. Anticancer Res. (2012) 32:4851–60.
191. Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. (2015) 125:1591–602. doi: 10.1172/JCI78239
192. Diao XY, Lin T. Progress in therapeutic strategies based on cancer lipid metabolism. Thorac Cancer. (2019) 10:1741–3. doi: 10.1111/1759-7714.13146
193. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. (2013) 13:227–32. doi: 10.1038/nrc3483
194. Liu H, Liu Y, Zhang JT. A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Ther. (2008) 7:263–70. doi: 10.1158/1535-7163.MCT-07-0445
195. Yang Y, Liu H, Li Z, Zhao Z, Yip-Schneider M, Fan Q, et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int J Biochem Mol Biol. (2011) 2:89−98.
196. Mims J, Bansal N, Bharadwaj MS, Chen X, Molina AJ, Tsang AW, et al. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat Res. (2015) 183:291–304. doi: 10.1667/RR13828.1
197. Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. (2010) 6:551–62. doi: 10.2217/fon.10.11
198. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. (2005) 8:311–21. doi: 10.1016/j.ccr.2005.09.008
199. Li S, Qiu L, Wu B, Shen H, Zhu J, Zhou L, et al. TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Mol Med Rep. (2013) 8:373–8. doi: 10.3892/mmr.2013.1505
200. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. (2010) 10:267–77. doi: 10.1038/nrc2817
201. Li JN, Gorospe M, Chrest FJ, Kumaravel TS, Evans MK, Han WF, et al. Pharmacological inhibition of fatty acid synthase activity produces both cytostatic and cytotoxic effects modulated by p53. Cancer Res. (2001) 61:1493–9.
202. Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, et al. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. (2000) 60:213–8.
203. Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol. (2007) 31:769–76. doi: 10.3892/ijo.31.4.769
204. Vazquez-Martin A, Ropero S, Brunet J, Colomer R, Menendez JA. Inhibition of Fatty Acid Synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol Rep. (2007) 18:973–80. doi: 10.3892/or.18.4.973
205. Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. (2010) 70:8117–26. doi: 10.1158/0008-5472.CAN-09-3871
206. Kato H, Nishitoh H. Stress responses from the endoplasmic reticulum in cancer. Front Oncol. (2015) 5:93. doi: 10.3389/fonc.2015.00093
207. Mylonis I, Sembongi H, Befani C, Liakos P, Siniossoglou S, Simos G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J Cell Sci. (2012) 125:3485–93. doi: 10.1242/jcs.106682
208. Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. (2013) 110:4628–33. doi: 10.1073/pnas.1217611110
209. Loizides-Mangold U. On the future of mass-spectrometry-based lipidomics. FEBS J. (2013) 280:2817–29. doi: 10.1111/febs.12202
210. Le TT, Yue S, Cheng JX. Shedding new light on lipid biology with coherent anti-Stokes Raman scattering microscopy. J Lipid Res. (2010) 51:3091–102. doi: 10.1194/jlr.R008730
211. Dessi S, Batetta B, Pulisci D, Spano O, Anchisi C, Tessitore L, et al. Cholesterol content in tumor tissues is inversely associated with high-density lipoprotein cholesterol in serum in patients with gastrointestinal cancer. Cancer. (1994) 73:253–8. doi: 10.1002/1097-0142(19940115)73:2<253::aid-cncr2820730204>3.0.co;2-f
212. Grosse PY, Bressolle F, Pinguet F. Antiproliferative effect of methyl-betacyclodextrin in vitro and in human tumour xenografted athymic nude mice. Br J Cancer. (1998) 78:1165–9. doi: 10.1038/bjc.1998.648
213. Mohammad N, Malvi P, Meena AS, Singh SV, Chaube B, Vannuruswamy G, et al. Cholesterol depletion by methyl-beta-cyclodextrin augments tamoxifen induced cell death by enhancing its uptake in melanoma. Mol Cancer. (2014) 13:204. doi: 10.1186/1476-4598-13-204
214. White MD, Chan L, Antoon JW, Beckman BS. Targeting ovarian cancer and chemoresistance through selective inhibition of sphingosine kinase-2 with ABC294640. Anticancer Res. (2013) 33:3573–9.
215. Venkata JK, An N, Stuart R, Costa LJ, Cai H, Coker W, et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood. (2014) 124:1915–25. doi: 10.1182/blood-2014-03-559385
216. Guan S, Liu YY, Yan T, Zhou J. Inhibition of ceramide glucosylation sensitizes lung cancer cells to ABC294640, a first-in-class small molecule SphK2 inhibitor. Biochem Biophys Res Commun. (2016) 476:230–6. doi: 10.1016/j.bbrc.2016.05.102
217. Gao P, Smith CD. Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol Cancer Res. (2011) 9:1509–19. doi: 10.1158/1541-7786.MCR-11-0336
218. Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, et al. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. (2010) 151:5124–35. doi: 10.1210/en.2010-0420
219. Schrecengost RS, Keller SN, Schiewer MJ, Knudsen KE, Smith CD. Downregulation of critical oncogenes by the selective SK2 inhibitor ABC294640 hinders prostate cancer progression. Mol Cancer Res. (2015) 13:1591–601. doi: 10.1158/1541-7786.MCR-14-0626
220. Lewis CS, Voelkel-Johnson C, Smith CD. Suppression of c-Myc and RRM2 expression in pancreatic cancer cells by the sphingosine kinase-2 inhibitor ABC294640. Oncotarget. (2016) 7:60181–92. doi: 10.18632/oncotarget.11112
221. Flaveny CA, Griffett K, El-Gendy B-D, Kazantzis M, Sengupta M, Amelio AL, et al. Broad anti-tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer cell. (2015) 28:42–56. doi: 10.1016/j.ccell.2015.05.007
222. Dobbin ZC, Katre AA, Steg AD, Erickson BK, Shah MM, Alvarez RD, et al. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget. (2014) 5:8750–64. doi: 10.18632/oncotarget.2373
223. Liu Y-Y, Hill RA, Li Y-T. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv Cancer Res. (2013) 117:59–89. doi: 10.1016/B978-0-12-394274-6.00003-0
224. Lee AJ, Roylance R, Sander J, Gorman P, Endesfelder D, Kschischo M, et al. CERT depletion predicts chemotherapy benefit and mediates cytotoxic and polyploid-specific cancer cell death through autophagy induction. J Pathol. (2012) 226:482–94. doi: 10.1002/path.2998
225. Lee JW Ryu JY, Yoon G, Jeon HK, Cho YJ, Choi JJ, et al. Sphingosine kinase 1 as a potential therapeutic target in epithelial ovarian cancer. Int J Cancer. (2015) 137:221–9. doi: 10.1002/ijc.29362
226. Devalapally H, Duan Z, Seiden MV, Amiji MM. Modulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticles. Clin Cancer Res. (2008) 14:3193–203. doi: 10.1158/1078-0432.CCR-07-4973
227. Prinetti A, Basso L, Appierto V, Villani MG, Valsecchi M, Loberto N, et al. Altered sphingolipid metabolism in N-(4-hydroxyphenyl)-retinamide-resistant A2780 human ovarian carcinoma cells. J Biol Chem. (2003) 278:5574–83. doi: 10.1074/jbc.M207269200
228. Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell. (2007) 11:498–512. doi: 10.1016/j.ccr.2007.04.011
229. Kolesnick R, Altieri D, Fuks Z. A CERTain role for ceramide in taxane-induced cell death. Cancer Cell. (2007) 11:473–5. doi: 10.1016/j.ccr.2007.05.003
230. Liu YY, Patwardhan GA, Bhinge K, Gupta V, Gu X, Jazwinski SM. Suppression of glucosylceramide synthase restores p53-dependent apoptosis in mutant p53 cancer cells. Cancer Res. (2011) 71:2276–85. doi: 10.1158/0008-5472.CAN-10-3107
231. Ponnusamy S, Meyers-Needham M, Senkal CE, Saddoughi SA, Sentelle D, Selvam SP, et al. Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. (2010) 6:1603–24. doi: 10.2217/fon.10.116
232. Pyne NJ, Tonelli F, Lim KG, Long JS, Edwards J, Pyne S. Sphingosine 1-phosphate signalling in cancer. Biochem Soc Trans. (2012) 40:94–100. doi: 10.1042/BST20110602
233. Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. (2004) 4:604–16. doi: 10.1038/nrc1411
234. Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. (2010) 10:489–503. doi: 10.1038/nrc2875
235. Kitai Y, Ariyama H, Kono N, Oikawa D, Iwawaki T, Arai H. Membrane lipid saturation activates IRE1alpha without inducing clustering. Genes Cells. (2013) 18:798–809. doi: 10.1111/gtc.12074
236. Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem. (2010) 285:22027–35. doi: 10.1074/jbc.M110.126870
237. Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. (2013) 1:3. doi: 10.1186/2049-3002-1-3
238. Williams KJ, Argus JP, Zhu Y, Wilks MQ, Marbois BN, York AG, et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. (2013) 73:2850–62. doi: 10.1158/0008-5472.CAN-13-0382-T
239. Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. (2015) 126:1925–9. doi: 10.1182/blood-2014-12-617498
240. Kreitzburg KM, van Waardenburg RCAM, Yoon KJ. Sphingolipid metabolism and drug resistance in ovarian cancer. Cancer Drug Resist. (2018) 1:181–97. doi: 10.20517/cdr.2018.06
241. Morad SA, Levin JC, Shanmugavelandy SS, Kester M, Fabrias G, Bedia C, et al. Ceramide-antiestrogen nanoliposomal combinations–novel impact of hormonal therapy in hormone-insensitive breast cancer. Mol Cancer Ther. (2012) 11:2352–61. doi: 10.1158/1535-7163.MCT-12-0594
242. Markman M, Bookman MA. Second-line treatment of ovarian cancer. Oncologist. (2000) 5:26–35. doi: 10.1634/theoncologist.5-1-26
243. Morad SA, Cabot MC. Tamoxifen regulation of sphingolipid metabolism–therapeutic implications. Biochim Biophys Acta. (2015) 1851:1134–45. doi: 10.1016/j.bbalip.2015.05.001
244. Montopoli M, Bellanda M, Lonardoni F, Ragazzi E, Dorigo P, Froldi G, et al. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr Cancer Drug Targets. (2011) 11:226–35. doi: 10.2174/156800911794328501
245. Yang L, Zhang F, Wang X, Tsai Y, Chuang KH, Keng PC, et al. A FASN-TGF-β1-FASN regulatory loop contributes to high EMT/metastatic potential of cisplatin-resistant non-small cell lung cancer. Oncotarget. (2016) 7:55543–54. doi: 10.18632/oncotarget.10837
246. Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther. (2014) 13:2361–71. doi: 10.1158/1535-7163.MCT-14-0183
247. Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. (2017) 1858:686–99. doi: 10.1016/j.bbabio.2017.01.012
248. Guerra F, Guaragnella N, Arbini AA, Bucci C, Giannattasio S, Moro L. Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front Oncol. (2017) 7:295. doi: 10.3389/fonc.2017.00295
249. Altieri DC. Mitochondrial dynamics and metastasis. Cell Mol Life Sci. (2019) 76:827–35. doi: 10.1007/s00018-018-2961-2
250. Cocetta V, Ragazzi E, Montopoli M. Mitochondrial involvement in cisplatin resistance. Int J Mol Sci. (2019) 20:3384. doi: 10.3390/ijms20143384
251. Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. (2016) 26:655–67. doi: 10.1016/j.tcb.2016.04.006
252. Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. (2012) 18:5562–71. doi: 10.1158/1078-0432.CCR-12-1773
253. Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. (2014) 2:10. doi: 10.1186/2049-3002-2-10
254. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. (2012) 26:1326–38. doi: 10.1101/gad.191056.112
255. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
256. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. (2012) 483:474–8. doi: 10.1038/nature10860
257. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J. (2015) 34:856–80. doi: 10.15252/embj.201490784
258. Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in cancer: a tale of adaptation. Cells. (2019) 8:493. doi: 10.3390/cells8050493
259. Um JH, Yun J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. (2017) 50:299–307. doi: 10.5483/BMBRep.2017.50.6.056
260. Yan C, Li TS. Dual role of mitophagy in cancer drug resistance. Anticancer Res. (2018) 38:617–21. doi: 10.21873/anticanres.12266
261. Cook CC, Higuchi M. The awakening of an advanced malignant cancer: an insult to the mitochondrial genome. Biochim Biophys Acta. (2012) 1820:652–62. doi: 10.1016/j.bbagen.2011.08.017
262. Cui H, Huang P, Wang Z, Zhang Y, Zhang Z, Xu W, et al. Association of decreased mitochondrial DNA content with the progression of colorectal cancer. BMC Cancer. (2013) 13:110. doi: 10.1186/1471-2407-13-110
263. Koochekpour S, Marlowe T, Singh KK, Attwood K, Chandra D. Reduced mitochondrial DNA content associates with poor prognosis of prostate cancer in African American men. PLoS ONE. (2013) 8:e74688. doi: 10.1371/journal.pone.0074688
264. Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, Lee LM, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer. (2006) 45:629–38. doi: 10.1002/gcc.20326
265. Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik-Zainal S, Ramakrishna M, et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. (2014) 3:e02935. doi: 10.7554/eLife.02935.028
266. Maybury BD. Mitochondrial DNA damage is uncommon in cancer but can promote aggressive behaviour. Anticancer Res. (2013) 33:3543–52.
267. Kloss-Brandstätter A, Schäfer G, Erhart G, Hüttenhofer A, Coassin S, Seifarth C, et al. Somatic mutations throughout the entire mitochondrial genome are associated with elevated PSA levels in prostate cancer patients. Am J Hum Genet. (2010) 87:802–12. doi: 10.1016/j.ajhg.2010.11.001
268. Bai R K, Leal SM, Covarrubias D, Liu A, Wong LJC. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. (2007) 67:4687–94. doi: 10.1158/0008-5472.CAN-06-3554
269. Canter JA, Kallianpur AR, Parl FF; Millikan RC. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res. (2005) 65:8028–33. doi: 10.1158/0008-5472.CAN-05-1428
270. Czarnecka AM, Krawczyk T, Zdrozny M, Lubinski J, Arnold RS, Kukwa W, et al. Mitochondrial NADH-dehydrogenase subunit 3 (ND3) polymorphism (A10398G) and sporadic breast cancer in Poland. Breast Cancer Res Treat. (2010) 121:511–8. doi: 10.1007/s10549-009-0358-5
271. Gasparre G, Kurelac I, Capristo M, Iommarini L, Ghelli A, Ceccarelli C, et al. A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene MTND1, an oncojanus function. Cancer Res. (2011) 71:6220–9. doi: 10.1158/0008-5472.CAN-11-1042
272. Su CY, Chang YC, Yang CJ, Huang MS, Hsiao M. The opposite prognostic effect of NDUFS1 and NDUFS8 in lung cancer reflects the oncojanus role of mitochondrial complex I. Sci. Rep. (2016) 6:31357. doi: 10.1038/srep31357
273. Kurelac I, Iommarini L, Vatrinet R, Amato LB, De Luise M, Leone G, et al. Inducing cancer indolence by targeting mitochondrial Complex I is potentiated by blocking macrophage-mediated adaptive responses. Nat Commun. (2019) 10:903. doi: 10.1038/s41467-019-08839-1
274. Porcelli AM, Ghelli A, Ceccarelli C, Lang M, Cenacchi G, Capristo M, et al. The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum Mol Genet. (2010) 19:1019–32. doi: 10.1093/hmg/ddp566
275. Guerra F, Perrone AM, Kurelac I, Santini D, Ceccarelli C, Cricca M, et al. Mitochondrial DNA mutation in serous ovarian cancer: implications for mitochondria-coded genes in chemoresistance. J. Clin Oncol. (2012) 30:e373–8. doi: 10.1200/JCO.2012.43.5933
276. Girolimetti G, Guerra F, Iommarini L, Kurelac I, Vergara D, Maffia M, et al. Platinum-induced mitochondrial DNA mutations confer lower sensitivity to paclitaxel by impairing tubulin cytoskeletal organization. Hum Mol Genet. (2017) 26:2961–74. doi: 10.1093/hmg/ddx186
277. Yao Z, Jones AW, Fassone E, Sweeney MG, Lebiedzinska M, Suski JM, et al. PGC-1beta mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene. (2013) 32:2592–600. doi: 10.1038/onc.2012.259
278. Cook CC, Kim A, Terao S, Gotoh A, Higuchi M. Consumption of oxygen: a mitochondrial-generated progression signal of advanced cancer Cell Death Dis. (2012) 3:e258. doi: 10.1038/cddis.2011.141
279. Arbini AA, Guerra F, Greco M, Marra E, Gandee L, Xiao G, et al. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis. (2013) 2:e82. doi: 10.1038/oncsis.2013.45
280. Srinivasan S, Guha M, Avadhani NG. Mitochondrial respiratory defects promote the Warburg effect and cancer progression. Mol Cell Oncol. (2016) 3:e1085120. doi: 10.1080/23723556.2015.1085120
281. Cho YM, Kim JH, Kim M, Park SJ, Koh SH, Ahn HS, et al. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE. (2012) 7:e32778. doi: 10.1371/journal.pone.0032778
282. Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by exosomes. Mol Cancer. (2019) 18:58. doi: 10.1186/s12943-019-0970-x
283. Philley JV, Kannan A, Qin W, Sauter ER, Ikebe M, Hertweck KL, et al. Complex-I alteration and enhanced mitochondrial fusion are associated with prostate cancer progression. J Cell Physiol. (2016) 231:1364–74. doi: 10.1002/jcp.25240
284. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood. (2016) 128:253–64. doi: 10.1182/blood-2015-07-655860
285. Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M. The Warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. (2008) 68:5198–205. doi: 10.1158/0008-5472.CAN-08-0555
286. Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. (2009) 69:2163–6. doi: 10.1158/0008-5472.CAN-08-3722
287. Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell. (2015) 17:585–96. doi: 10.1016/j.stem.2015.08.019
288. Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. (2013) 23:143–58. doi: 10.1016/j.ccr.2012.12.008
289. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. (2013) 23:302–15. doi: 10.1016/j.ccr.2013.02.003
290. Vellinga TT, Borovski T, Boer VCJ, de Fatrai S, van Schelven S, Trumpi K, et al. SIRT1/PGC1α-dependent increase in oxidative phosphorylation supports chemotherapy resistance of colon cancer. Clin Cancer Res. (2015) 21:2870–9. doi: 10.1158/1078-0432.CCR-14-2290
291. Yun CW, Han YS, Lee SH. PGC-1α controls mitochondrial biogenesis in drug-resistant colorectal cancer cells by regulating endoplasmic reticulum stress. Int J Mol Sci. (2019) 20:1707. doi: 10.3390/ijms20071707
292. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. (2007) 26:1324–37. doi: 10.1038/sj.onc.1210220
293. Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. (2016) 529:528–31. doi: 10.1038/nature16500
294. Kasahara A, Cipolat S, Chen Y, Dorn GW 2nd, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science. (2013) 342:734–7. doi: 10.1126/science.1241359
295. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. (2009) 11:958–66. doi: 10.1038/ncb1907
296. Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. (2009) 186:805–16. doi: 10.1083/jcb.200903065
297. Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, et al. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. (2012) 197:535–51. doi: 10.1083/jcb.201110034
298. Prieto J, Leon M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, et al. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. (2016) 7:11124. doi: 10.1038/ncomms11124
299. Cai J, Wang J, Huang Y, Wu H, Xia T, Xiao J, et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell death & disease. (2016) 7:e2459. doi: 10.1038/cddis.2016.370
300. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. (2007) 8:939–44. doi: 10.1038/sj.embor.7401062
301. Karbowski M, Youle R. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. (2003) 10:870–80. doi: 10.1038/sj.cdd.4401260
302. Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. (2015) 57:521–36. doi: 10.1016/j.molcel.2015.01.003
303. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. (2015) 57:537–51. doi: 10.1016/j.molcel.2015.01.002
304. Freiburghaus C, Emruli VK, Johansson A, Olsson R, Ek F, Geisler CH, et al. Resistance to cytarabine in mantle cell lymphoma is mediated by down-regulation of deoxycytidine kinase at the protein level. Blood. (2016) 128:1769. doi: 10.1182/blood.V128.22.1769.1769
305. Lokody I. Drug resistance: overcoming resistance in acute myeloid leukaemia treatment. Nat Rev Cancer. (2014) 14:452–3. doi: 10.1038/nrc3776
306. Farge T, Saland E, Toni FD, Aroua N, Hosseini M, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. (2017) 7:716–35. doi: 10.1158/2159-8290.CD-16-0441
307. Matassa DS, Amoroso MR, Lu H, Avolio R, Arzeni D, Procaccini C, et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. (2016) 23:1542–54. doi: 10.1038/cdd.2016.39
308. Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. (2005) 41:2502–12. doi: 10.1016/j.ejca.2005.08.016
309. Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front Oncol. (2013) 3:256. doi: 10.3389/fonc.2013.00256
310. Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for naplerosis, mitochondrial function, and migration. Am J Physiol Endocrinol Metab. (2012) 303:E1036–52. doi: 10.1152/ajpendo.00151.2012
311. Pastò A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget. (2014) 5:4305–19. doi: 10.18632/oncotarget.2010
312. Alvero AB, Montagna MK, Sumi NJ, Joo WD, Graham E, Mor G. Multiple blocks in the engagement of oxidative phosphorylation in putative ovarian cancer stem cells: implication for maintenance therapy with glycolysis inhibitors. Oncotarget. (2014) 5:8703–15. doi: 10.18632/oncotarget.2367
313. Anderson AS, Roberts PC, Frisard MI, Hulver MW, Schmelz EM. Ovarian tumor-initiating cells display a flexible metabolism. Exp Cell Res. (2014) 328:44–57. doi: 10.1016/j.yexcr.2014.08.028
314. Dier U, Shin DH, Hemachandra LP, Uusitalo LM, Hempel N. Bioenergetic analysis of ovarian cancer cell lines: profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS ONE. (2014) 9:e98479. doi: 10.1371/journal.pone.0098479
315. Dar S, Chhina J, Mert I, Chitale D, Buekers T, Kaur H, et al. Bioenergetic adaptations in chemoresistant ovarian cancer cells. Sci Rep. (2017) 7:8760. doi: 10.1038/s41598-017-09206-0
316. Havas KM, Milchevskaya V, Radic K, Alladin A, Kafkia E, Garcia M, et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J Clin Invest. (2017) 127:2091–105. doi: 10.1172/JCI89914
317. Porporato PE, Filigheddu N, Bravo-San Pedro JM, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. (2018) 28:265–80. doi: 10.1038/cr.2017.155
318. Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst. (2015) 107:dju502. doi: 10.1093/jnci/dju502
319. Xiao D, Powolny AA, Moura MB, Kelley EE, Bommareddy A, Kim SH, et al. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem. (2010) 285:26558–69. doi: 10.1074/jbc.M109.063255
320. Chan DK, Miskimins WK. Metformin and phenethyl isothiocyanate combined treatment in vitro is cytotoxic to ovarian cancer cultures. J Ovarian Res. (2012) 5:19. doi: 10.1186/1757-2215-5-19
321. Xu Y, Gao W, Zhang Y, Wu S, Liu Y, Deng X, et al. ABT737 reverses cisplatin resistance by targeting glucose metabolism of human ovarian cancer cells. Int J Oncol. (2018) 53:1055–68. doi: 10.3892/ijo.2018.4476
322. Zhu J, Wu G, Song L, Cao L, Tan Z, Tang M, et al. NKX2–8 deletion-induced reprogramming of fatty acid metabolism confers chemoresistance in epithelial ovarian cancer. EBioMedicine. (2019) 43:238–52. doi: 10.1016/j.ebiom.2019.04.041
323. Emmings E, Mullany S, Chang Z, Landen CN Jr, Linder S, Bazzaro M. Targeting mitochondria for treatment of chemoresistant ovarian cancer. Int J Mol Sci. (2019) 20:229. doi: 10.3390/ijms20010229
324. Chu DJ, Yao DE, Zhuang YF, Hong Y, Zhu XC, Fang ZR, et al. Azithromycin enhances the favorable results of paclitaxel and cisplatin in patients with advanced non-small cell lung cancer. Genet Mol Res. (2014) 13:2796–805. doi: 10.4238/2014.April.14.8
325. Wu W, Yu LH, Ma B, Xu MJ. The inhibitory effect of doxycycline on cisplatin-sensitive and -resistant epithelial ovarian cancer. PLoS ONE. (2014) 9:e89841. doi: 10.1371/journal.pone.0089841
326. Kaplan F, Teksen F. Apoptotic effects of salinomycin on human ovarian cancer cell line (OVCAR-3). Tumor Biol. (2016) 37:3897–903. doi: 10.1007/s13277-015-4212-6
327. Denise C, Paoli P, Calvani M, Taddei ML, Giannoni E, Kopetz S, et al. 5-Fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget. (2015) 6:41706–21. doi: 10.18632/oncotarget.5991
328. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. (2016) 114:1305–12. doi: 10.1038/bjc.2016.152
329. Jagust P, de Luxán-Delgado B, Parejo-Alonso B, Sancho P. Metabolism-based therapeutic strategies targeting cancer stem cells. Front Pharmacol. (2019) 10:203. doi: 10.3389/fphar.2019.00203
330. He W, Liang B, Wang C, Li S, Zhao Y, Huang Q, et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene. (2019) 38:4637–54. doi: 10.1038/s41388-019-0747-0
331. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. (2011) 364:2507–16. doi: 10.1056/NEJMoa1103782
332. Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang CY, Zhang M, et al. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. (2008) 7:2319–27. doi: 10.1158/1535-7163.MCT-08-0298
333. Berkenblit A, Eder JP, Ryan DP, Seiden MV, Tatsuta N, Sherman ML, et al. Phase i clinical trial of STA-4783 in combination with paclitaxel in patients with refractory solid tumors. Clin Cancer Res. (2007) 13:584–90. doi: 10.1158/1078-0432.CCR-06-0964
334. O'Day S, Gonzalez R, Lawson D, Weber R, Hutchins L, Anderson C, et al. Phase II, randomized, controlled, double-blinded trial of weekly elesclomol plus paclitaxel versus paclitaxel alone for stage IV metastatic melanoma. J Clin Oncol. (2009) 27:5452–8. doi: 10.1200/JCO.2008.17.1579
335. Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. (2010) 9:447–64. doi: 10.1038/nrd3137
336. Lee S, Lee JS, Seo J, Lee SH, Kang JH, Song J, et al. Targeting mitochondrial oxidative phosphorylation abrogated irinotecan resistance in NSCLC. Sci Rep. (2018) 8:15707. doi: 10.1038/s41598-018-33667-6
337. Kitada S, Kress CL, Krajewska M, Jia L, Pellecchia M, Reed JC. Bcl-2 antagonist apogossypol (NSC736630) displays single-agent activity in Bcl-2–transgenic mice and has superior efficacy with less toxicity compared with gossypol (NSC19048). Blood. (2008) 111:3211–9. doi: 10.1182/blood-2007-09-113647
Keywords: cancer, metabolic reprogramming, TCA cycle, Warburg effect, metabolic vulnerabilities, chemoresistance
Citation: Desbats MA, Giacomini I, Prayer-Galetti T and Montopoli M (2020) Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 10:281. doi: 10.3389/fonc.2020.00281
Received: 31 October 2019; Accepted: 18 February 2020;
Published: 06 March 2020.
Edited by:
Paolo E. Porporato, University of Turin, ItalyReviewed by:
Elisa Giannoni, University of Florence, ItalyFlora Guerra, University of Salento, Italy
Copyright © 2020 Desbats, Giacomini, Prayer-Galetti and Montopoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica Montopoli, bW9uaWNhLm1vbnRvcG9saUB1bmlwZC5pdA==