 Katja Eloranta1
Katja Eloranta1 Stefano Cairo2
Stefano Cairo2 Emmi Liljeström1Tea Soini1,3
Emmi Liljeström1Tea Soini1,3 Antti Kyrönlahti1Jean-Gabriel Judde2
Antti Kyrönlahti1Jean-Gabriel Judde2 David B. Wilson4,5Markku Heikinheimo1,4†
David B. Wilson4,5Markku Heikinheimo1,4† Marjut Pihlajoki1*†
Marjut Pihlajoki1*†- 1Pediatric Research Center, Children's Hospital, Helsinki University Hospital, University of Helsinki, Helsinki, Finland
- 2Xentech, Evry, France
- 3Department of Medicine, Center for Infectious Medicine, Karolinska Institutet, Stockholm, Sweden
- 4Department of Pediatrics, Washington University School of Medicine, St. Louis Children's Hospital, St. Louis, MO, United States
- 5Department of Developmental Biology, Washington University School of Medicine, St. Louis, MO, United States
Background: Hepatoblastoma (HB) is the most common pediatric liver malignancy. Despite advances in chemotherapeutic regimens and surgical techniques, the survival of patients with advanced HB remains poor, underscoring the need for new therapeutic approaches. Chloroquine (CQ), a drug used to treat malaria and rheumatologic diseases, has been shown to inhibit the growth and survival of various cancer types. We examined the antineoplastic activity of CQ in cell models of aggressive HB.
Methods: Seven human HB cell models, all derived from chemoresistant tumors, were cultured as spheroids in the presence of relevant concentrations of CQ. Morphology, viability, and induction of apoptosis were assessed after 48 and 96 h of CQ treatment. Metabolomic analysis and RT-qPCR based Death Pathway Finder array were used to elucidate the molecular mechanisms underlying the CQ effect in a 2-dimensional cell culture format. Quantitative western blotting was performed to validate findings at the protein level.
Results: CQ had a significant dose and time dependent effect on HB cell viability both in spheroids and in 2-dimensional cell cultures. Following CQ treatment HB spheroids exhibited increased caspase 3/7 activity indicating the induction of apoptotic cell death. Metabolomic profiling demonstrated significant decreases in the concentrations of NAD+ and aspartate in CQ treated cells. In further investigations, oxidation of NAD+ decreased as consequence of CQ treatment and NAD+/NADH balance shifted toward NADH. Aspartate supplementation rescued cells from CQ induced cell death. Additionally, downregulated expression of PARP1 and PARP2 was observed.
Conclusions: CQ treatment inhibits cell survival in cell models of aggressive HB, presumably by perturbing NAD+ levels, impairing aspartate bioavailability, and inhibiting PARP expression. CQ thus holds potential as a new agent in the management of HB.
Introduction
Hepatoblastoma (HB) is a malignant liver neoplasm that usually occurs in children younger than 4-years (1). It is the most common primary hepatic malignancy in the pediatric population with an annual incidence of 1.2–1.5/1,000,000 (2). Risk factors for HB include low birth weight, pre-maturity, and certain congenital disorders such as Beckwith-Wiedemann syndrome and familial adenomatous polyposis (FAP) (3–5). Complete tumor resection is paramount for optimal outcome, but majority of HBs are not surgically treatable at the time of diagnosis (6–8). Platinum-derivatives, cisplatin, and carboplatin, combined with doxorubicin form the backbone of neoadjuvant and adjuvant chemotherapy in HB treatment (9). Nonetheless, severe adverse effects limit the use of these agents, and chemoresistance is a hallmark of aggressive HB (10–12). Thus, new and less toxic therapeutic modalities would be desirable to further improve the results of HB management.
Chloroquine (CQ), a drug discovered in 1934, has been used broadly in the treatment of malaria, rheumatoid arthritis, and systemic lupus erythematosus (13–16). CQ also has documented antineoplastic activity in various solid cancer types including hepatocellular carcinoma, non-small cell lung cancer, glioblastoma multiforme, bladder cancer, pancreatic adenocarcinoma, and colon cancer (17–23). In addition to its efficacy as monotherapy, CQ has shown potential to sensitize cancer cells to conventional therapy after primary treatment has failed (24–28). Currently, there are 21 clinical trials evaluating efficacy and safety of CQ in oncology practice (Supplementary Table 1).
Multiple mechanisms underlying the tumor suppressive activity of CQ have been described. Autophagy is a process linked to metastasis and activation of chemoresistance in a context of aggressive cancer, and its inhibition is a widely recognized mechanism of CQ action (29–32). In vitro studies have shown that the transforming growth factor beta (TGF-β), hedgehog, and p53 signaling pathways are affected by CQ treatment (33–37). Other biological processes impacted by CQ administration include G2/M cell cycle arrest, increased apoptosis, altered inflammatory responses, and tumor vessel normalization (38–42).
Previous studies have shown that autophagy promotes survival of immortalized HB cells and tumor growth in vivo, suggesting that therapeutic interventions disturbing autophagic flux may hold potential in HB management (43, 44). In this study, we demonstrate the efficacy of CQ in 3-dimensional (3D) cell models of HB established from patients whose response to the first line treatments was suboptimal. Further, we shed new light on the molecular mechanisms of CQ action in HB cells.
Materials and Methods
Cell Cultures and Treatments
Immortalized human HB cell line HUH6 was purchased from Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan) and maintained as described earlier (45). Recently established human HB cell lines HB-284, HB-282, HB-303, HB-243, HB-295, and HB-279 were obtained through collaboration with XenTech (Evry, France) and cultured in Advanced DMEM/F12 (Gibco, Waltham, MA, USA) supplemented with 8% fetal bovine serum, 2 μM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin sulfate, and 20 μM rock kinase inhibitor Y-27632 (SelleckChem, Houston, TX, USA). Cultures were maintained at 37°C in a humified incubator containing 5% CO2.
Cells were treated with CQ diphosphate (#ab142116; Abcam, Cambridge, MA, USA) dissolved in sterile water as a 10 mM stock solution. Further dilutions were prepared in adequate cell culture medium. Cell culture medium without CQ served as a control treatment. For all the experiments fresh culture medium with or without CQ was replaced daily.
3D Spheroid Cultures
Cells were seeded at density of 3,000 cells/well to low attachment CellCarrier spheroid ULA 96-well-plates (PerkinElmer, Waltham, MA, USA). After 72 h incubation, spheroids were established and treatment with CQ or control medium was initiated. Images were captured at treatment timepoints of 0, 48, and 96 h with Eclipse TS100 microscope supplemented with DS-Fi1 digital imaging system (Nikon, Tokyo, Japan).
3D Viability Measurements
Viability of spheroids was assessed with ATPlite™ 3D monitoring system (PerkinElmer) as described in the manufacturer's instructions at treatment timepoints of 48 and 96 h. Luminescence was measured with EnSpire Multimode Plate Reader (PerkinElmer).
Apoptosis Assay
Caspase 3/7 Glo assay (Promega, Madison, WI, USA) was used to measure apoptosis in 3D cultures as instructed. Luminescence was recorded with EnSpire Multimode Plate Reader (PerkinElmer).
Clonogenic Cell Survival Assay
HUH6, HB-284, and HB-243 cells were seeded at low density in 12-well-plates and cultured for 4 or 14 days after attachment in absence of CQ or with CQ in concentrations 1, 5, or 10 μM. Cells were fixed with 4% paraformaldehyde, permeabilized with 100% methanol, and subsequently stained with crystal violet. Images were captured with Bio-Rad ChemiDoc XRS+ Imaging System (Bio-Rad, Hercules, CA, USA). Colony number was determined with ColonyArea Plugin in ImageJ Software (46).
Metabolomic Profiling
HUH6 cells were treated with control medium or 5 μM CQ for 96 h. Next, 1.5 × 106 cells/sample were collected, and cell pellets were snap-frozen in liquid nitrogen for analysis. Acquity UPLC-MS/MS system and XEVO TQ-S Triple Quadrupole LC/MS (Waters Corporation, Milford, MA, USA) were used to analyze the samples. Normalized concentrations of 100 metabolites were analyzed with web-based software MetaboAnalyst 4.0 (http://www.metaboanalyst.ca) (47). Detailed information of sample processing, reagents, and full protocol are given elsewhere (48).
NAD/NADH Assay
NAD/NADH ratio was measured with NAD/NADH assay kit purchased from Abcam. Briefly, HUH6 cells were cultured with or without 5 μM CQ for 96 h. Cells were lysed and processed following manufacturer's instructions as described (49). The reaction was allowed to develop for 1.5 h, and then absorbance was measured at 450 nm with Multiskan FC microplate reader (Thermo Fisher Scientific, Vantaa, Finland). NAD/NADH ratio was calculated using normalized concentrations by equation ([NADtotal—NADH])/[NADH].
Aspartate Rescue Experiment
Cells were treated with 5 μM CQ or 5 μM CQ supplemented with 10 mM L-aspartic acid (Sigma Aldrich, St. Louis, MO, USA) for 96 h. The impact of 10 mM L-aspartic acid alone on growth was also monitored. Cell viability was assessed by ATPLite Luminescence assay system using Enspire Multimode Plate Reader (both from PerkinElmer).
RNA Extraction and RT-qPCR
Total RNA was extracted from HUH6 cells treated with 5 μM CQ or control media for 96 h utilizing RNAeasy Mini Kit (QIAGEN, Valencia, CA, USA) as instructed. Reverse transcription was performed with Reverse transcriptase Core kit (Eurogentec, Seraing, Belgium). RT2 Profiler Cell Death Pathway Finder qPCR array (QIAGEN) was performed as described in the manufacturer's instructions. Geometric mean of B2M, HPRT1, and GAPDH expression served as a reference.
Protein Extraction and Western Blotting
Proteins were extracted utilizing NucleoSpin RNA/Protein extraction kit (Macherey-Nagel, Düren, Germany). Ten micrograms of protein was separated by electrophoresis using Mini-Protean TGX Stain-Free Gels (Bio-Rad). Proteins were transferred onto polyvinyl fluoride membrane and non-specific binding was blocked with 5% non-fat milk in 0.1% Tris-buffered Tween saline buffer. Membranes were incubated with following primary antibodies at +4°C for overnight: anti-human PARP1 rabbit IgG in dilution 1:1,500 (#9532; Cell Signaling Technology, Danvers, MA, USA) and anti-human PARP2 rabbit IgG in dilution 1:1,000 (#NBP2-47337; Novus Biologicals, Littleton, CO, USA). Next, goat anti-rabbit IgG secondary antibody (#111-035-144 in dilution 1:10,000; Jackson ImmunoResearch, West Grove, PA, USA) incubation was performed at room temperature for 1 h. Protein bands were detected utilizing Enhanced Chemiluminescence detection kit (Amersham ECL reagent; GE Healthcare, Barrington, IL) and analyzed with Image Lab Software 6.0 (Bio-rad). Band intensity was normalized to total protein amount in each lane.
Immunofluorescence Staining
HUH6 cells (200,000/well) were grown with or without 5 μM of CQ for 96 h on 2-well-chamber slides pre-coated with Matrigel (Corning, Corning, NY, USA). Subsequently, cells were fixed and permeabilized with ice-cold 100% methanol (5 min, room temperature). Unspecific binding was blocked with UltraVision Protein Block (Thermo Fisher Scientific). Next, cells were incubated with primary antibody at room temperature for 1 h (#9532 human anti-rabbit PARP1 at 1:800 dilution, Cell Signaling Technologies). Secondary antibody incubation was performed with goat anti-rabbit IgG (H+L) AlexaFluor 647 (1 h, room temperature) at 1:500 dilution (A32733, Invitrogen, Carlsbad, CA, USA). Images were captured with Zeiss Axio Imager M2 (objective: EC Plan Neofluar 40 × /0.75 Ph 2 M27) (Carl-Zeiss, Oberkochen, Germany).
Statistical Analyses
For viability assays, apoptosis measurements, and RNA and protein quantifications statistical analyses were performed using JMP Software (JMP Pro; version 14.1.0, SAS Institute Inc.). All the experiments were carried out at least in triplicate. Statistical significance was assessed using Student's t-test or one-way ANOVA depending the experimental design. *P < 0.05 was considered as a statistically significant and **p < 0.01 as a statistically highly significant. Metabolomic profiling analyses were completed using MetaboAnalyst 4.0. software (47).
Results
HB Spheroids Treated With CQ Show Decreased ATP Availability and Increased Cell Death
Since traditional 2D cell cultures do not accurately represent the architecture and interactions of solid tumors, we assayed the effect of CQ treatment using 3D HB spheroid models. Six of the cell lines used in this study were established from aggressive HB tumors (Clinical details in Table 1) thus representing a situation of an unfavorable treatment outcome for first line therapy (50). The seventh cell line, HUH6, is a gold standard in HB research. Spheroids were treated with three different concentrations of CQ in an acceptable range (51). Spheroid morphology was monitored after 48 and 96 h of treatment by capturing brightfield images with inverted phase contrast microscope. CQ treatment triggered a time and dose-dependent increase in the necrotic non-viable zone and loss of proliferative edge in all 7 spheroid models studied (Figures 1A–C, Supplementary Figures 1A–D). Characteristics of proliferating, quiescent, and necrotic spheroid morphology are shown in Figure 1D. Next, the viability of HB spheroids was quantified by ATP measurements. After 48 h of CQ treatment a statistically significant decrease in viability was observed in two out of seven models (Figures 1B,C). By 96 h, six out of seven models showed a significant decrease in viability (Figures 1A–C, Supplementary Figures 1A,B,D). Since increased apoptotic cell death has been reported following CQ treatment (40, 52), we next measured activation of caspase 3/7 in HB cell spheroids. After 48 h, all 7 models showed a significant increase in apoptosis with 10 μM CQ compared to control (Figures 1A–C, Supplementary Figures 1A–D). An even more enhanced caspase 3/7 activation was detected in four out of seven cell models (Figures 1A–C, Supplementary Figure 1A) after 96 h, and three out of these four proceeded toward apoptotic cell death at a lower CQ concentration (5 μM).
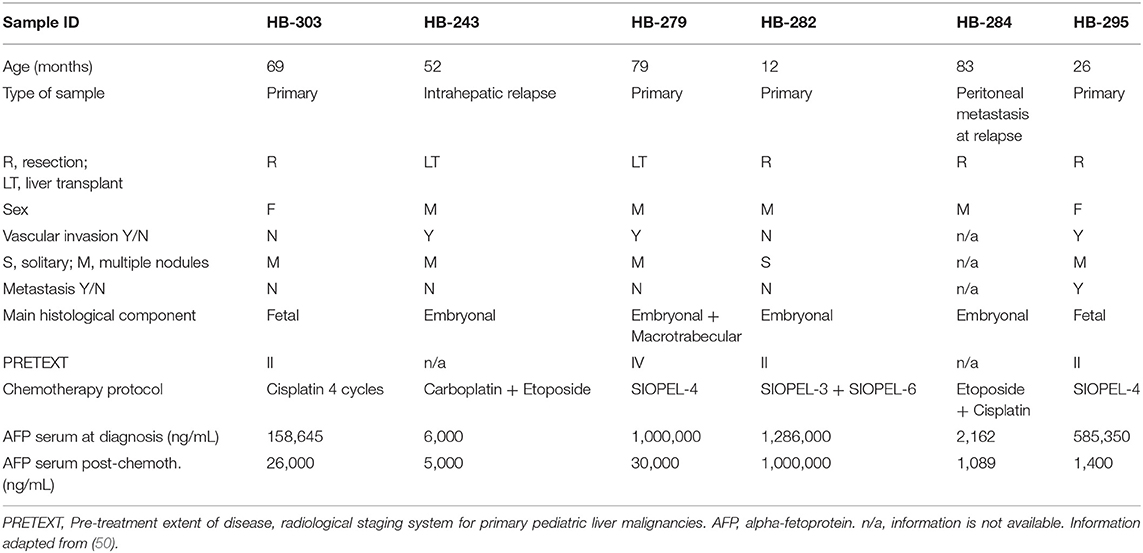
Table 1. Clinical information of HB patients.
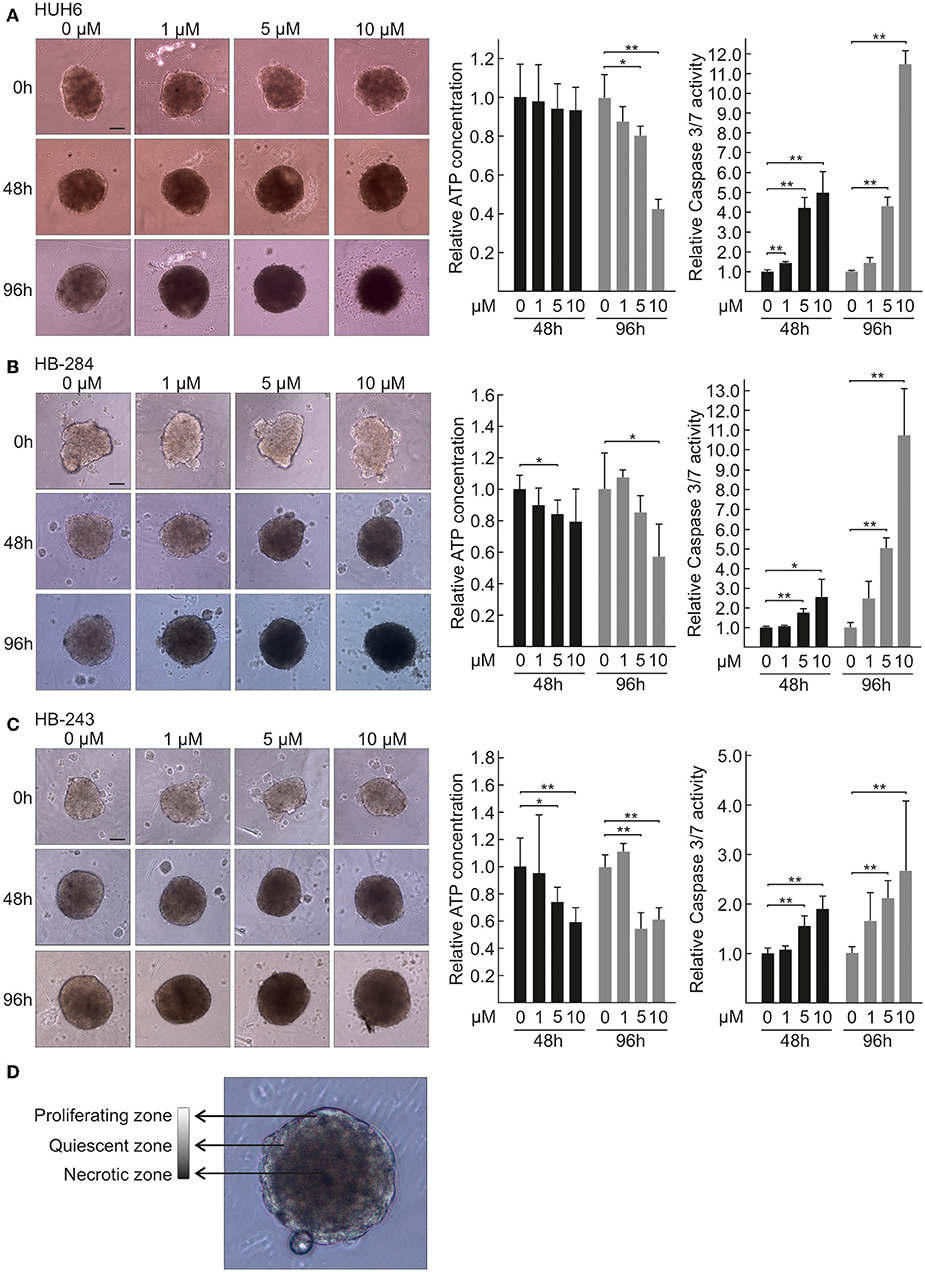
Figure 1. Morphology, viability, and caspase 3/7 activation of HB spheroids treated with CQ. Morphology of HUH6 (A), HB-284 (B), and HB-243 (C) derived spheroids treated with control medium (0 μM) or CQ at concentrations of 1, 5, and 10 μM (left panel). Relative ATP concentration (middle panel) and relative caspase 3/7 activation (right panel) in HUH6 (A), HB-284 (B), HB-243 (C) derived spheroids after 48 and 96 h CQ treatment. CQ concentrations; 1, 5, and 10 μM. *P < 0.05. **P < 0.01. Statistical significance was assessed with one-way ANOVA. Bar plots are presented as relative values of mean ± RSD (N = 3). Characteristics of proliferating, quiescent, and necrotic spheroid morphology (D). Pictures were captured at initiation of treatment (0 h) and after 48 h and 96 h of CQ administration. Magnification 10×, scale bar = 10 μm.
CQ Treatment Impairs Viability in HB 2D Cell Cultures
Due to low cell density, spheroids are not suitable for many high throughput applications. To circumvent this limitation, we validated our 3D spheroid findings in HB cells cultured in a 2D format. Clonogenic potential was measured after short term (4 days) and long term (14 days) CQ administration. A statistically highly significant decrease in clone formation was evident already after 4 days of treatment at concentrations of 5 and 10 μM (Figures 2A–C), and long-term exposure had the same effect on cell viability (Figures 2A–C).
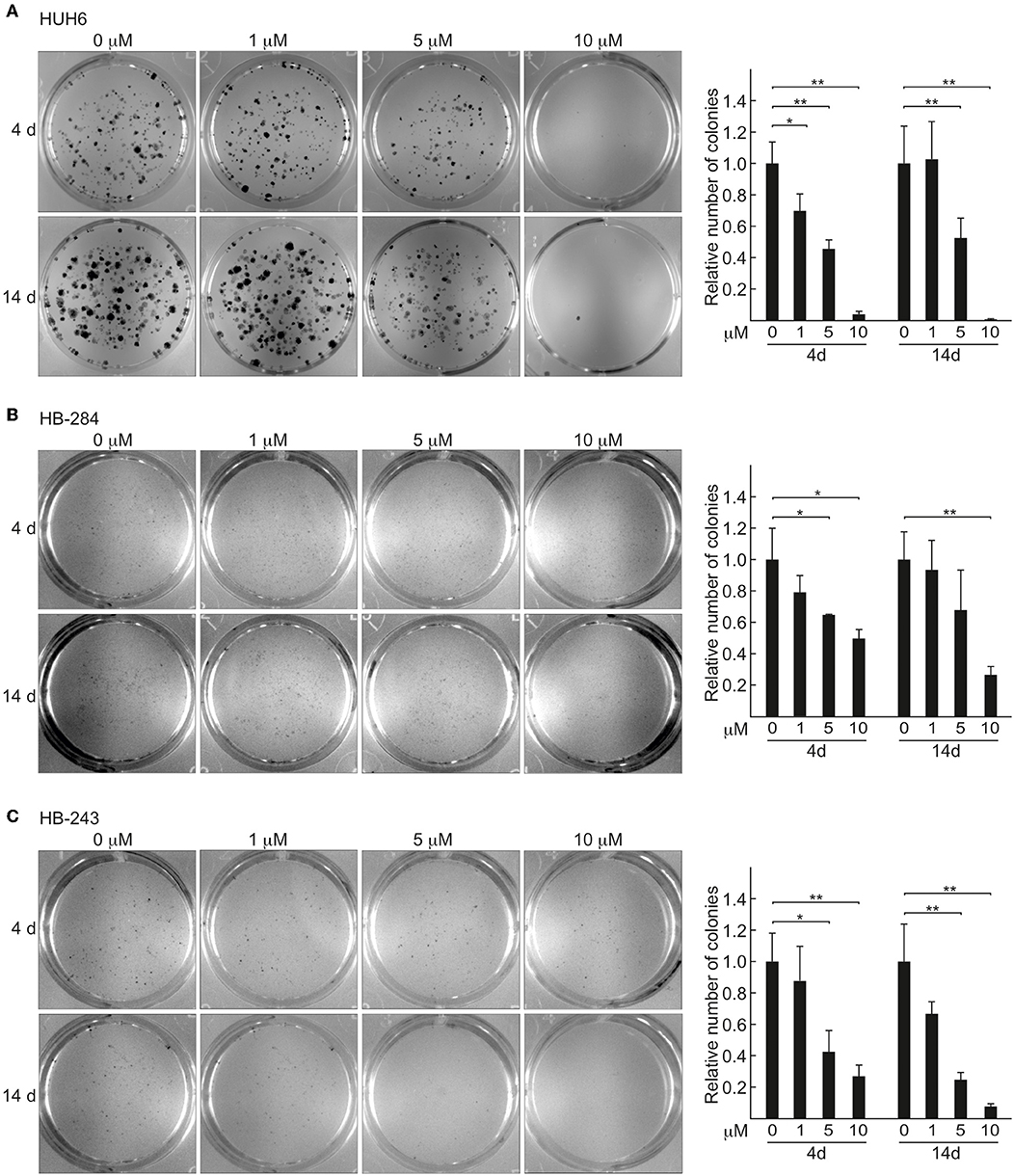
Figure 2. HB 2D cell cultures show decrease in viability and clonogenic potential after CQ exposure. Relative number of colonies after 4 and 14 days of CQ treatment: HUH6 (A), HB-284 (B), and HB-243 (C). Colonies stained with crystal violet after 4 or 14 days CQ administration. CQ; 1, 5, and 10 μM. *P < 0.05, **P < 0.01. One-way ANOVA was utilized to measure statistical significance. Bar plots are presented as relative values of mean ± RSD (N = 3).
CQ Treatment Depletes NAD+ and Aspartate in HUH6 Cells
Inhibition of autophagic flux is one of the known mechanisms of CQ action, and dysfunction in this process leads to a shortage of nutrients in cancer cells (53). Metabolomic profiling of HUH6 cell line treated with 5 μM CQ revealed a statistically significant decrease in 12 metabolites and an increase in 4 metabolites (Table 2). Metabolite enrichment set analysis denoted metabolites associated with malate-aspartate shuttle to be the most prominently altered (Figure 3A). Pathway analysis implicated alanine, aspartate, and glutamate metabolism as having the highest impact (Figure 3B). At the single metabolite level, nicotine adenine dinucleotide (NAD+) was the most significantly altered with a 60% decrease in concentration after CQ treatment (Figure 3C). In further investigation, NAD+/NADH ratio was noted to be 2.9-fold higher (p < 0.02) in untreated cells compared to CQ treated cells (Figure 3D). Aspartate, which was highlighted both in metabolite enrichment and pathway analyses, demonstrated 75% decrease in concentration when comparing CQ treated cells to control cells (Figure 3E). Closer evaluation demonstrated that aspartate supplementation was able to prevent cell death triggered by CQ treatment. Cell viability was 1.7-fold higher when cells were treated with 5 μM of CQ supplemented with aspartate compared to CQ treatment (Figure 3F). Aspartate supplementation alone had no effect on cell viability (Figure 3F). Normalized metabolite concentrations are shown in Supplementary Table 2.
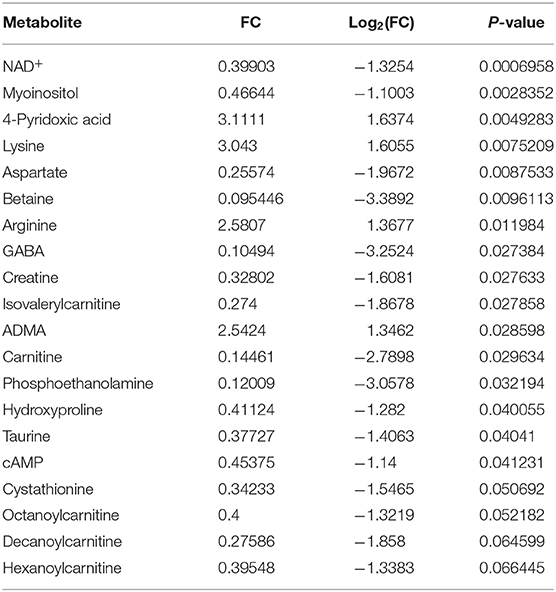
Table 2. The most altered metabolites after 96 h CQ (5 μM) exposure.
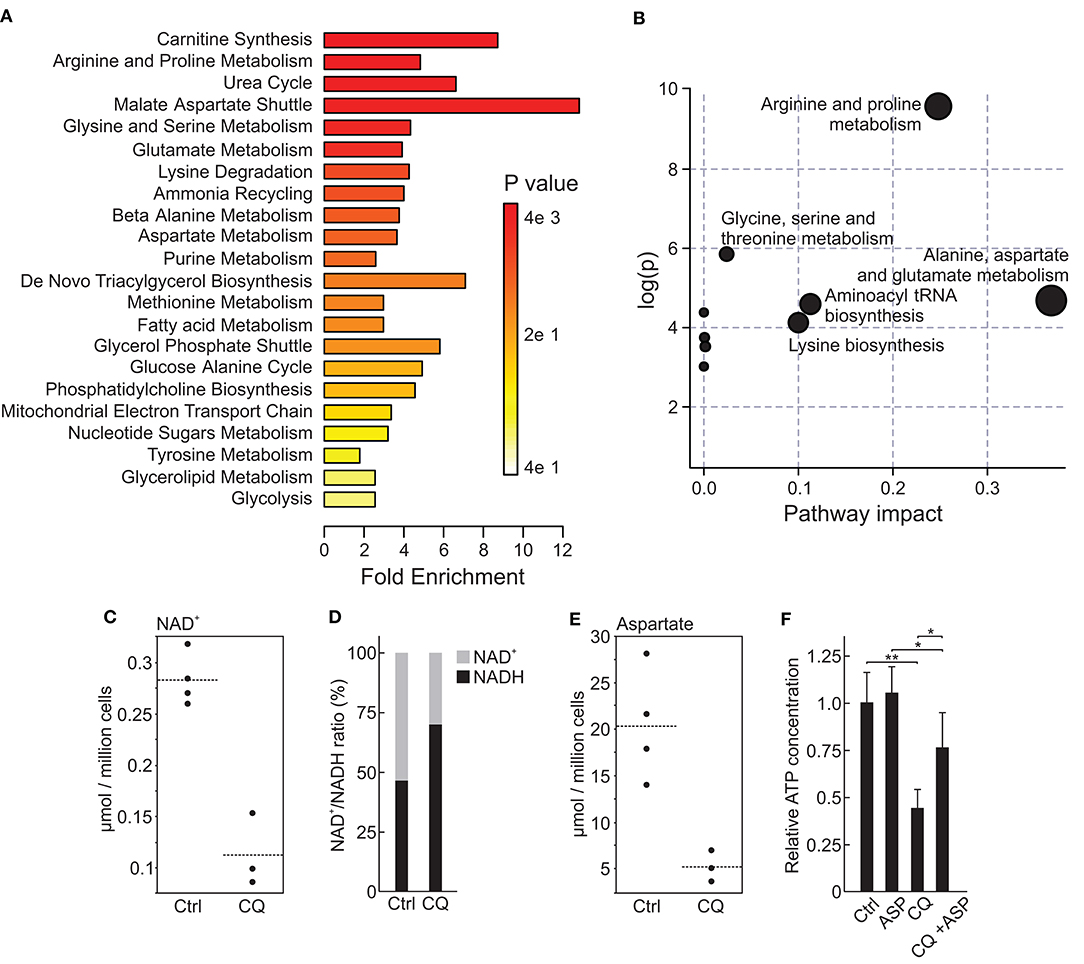
Figure 3. Metabolomic alterations in HUH6 cells treated with 5 μM CQ. Enrichment analysis showing the most abundantly altered metabolite sets (A). Pathway impact analysis demonstrating the importance of altered metabolites for pathway functionality (B). NAD+ concentrations (C) and NAD+/NADH (D) ratio in CQ treated HUH6 cells compared to untreated control cells. Aspartate concentrations (E) in CQ treated cells compared to untreated control cells. Impact of aspartate supplementation on CQ treated or untreated HUH6 cell viability (F). Single metabolites values are given as normalized concentration (μmol/million cells). NAD+/NADH ratio is presented as percentages of normalized total NAD concentrations. Bar plots are presented as relative values of mean ± RSD, (N = 3). ASP; 10 mM aspartate. Metabolite analyses were performed with MetaboAnalyst 4.0. *P < 0.05, **P < 0.01.
Death-Associated Gene Expression Changes in HUH6 Cells Exposed to CQ
RT2 Profiler Cell Death Pathway Finder array recognized 16 out 84 genes (Figure 4) to be statistically significantly differentially expressed in HUH6 cells treated with 5 μM CQ for 96 h. Six of these genes were classified by the manufacturer as pro-apoptotic genes (Figures 4A–F: CD40LG, CD40, TNF, TP53, IFNG, and ABL1), five as autophagy related (Figures 4G–K: ESR1, IGF1, SQSTM1, BECN1, and CTSS), and five as necroptotic (Figures 4L–P: PARP1, PARP2, FOXI1, TXNL4B, and DPYSL4). Normalized gene expression of the whole array panel is shown in Supplementary Table 2.
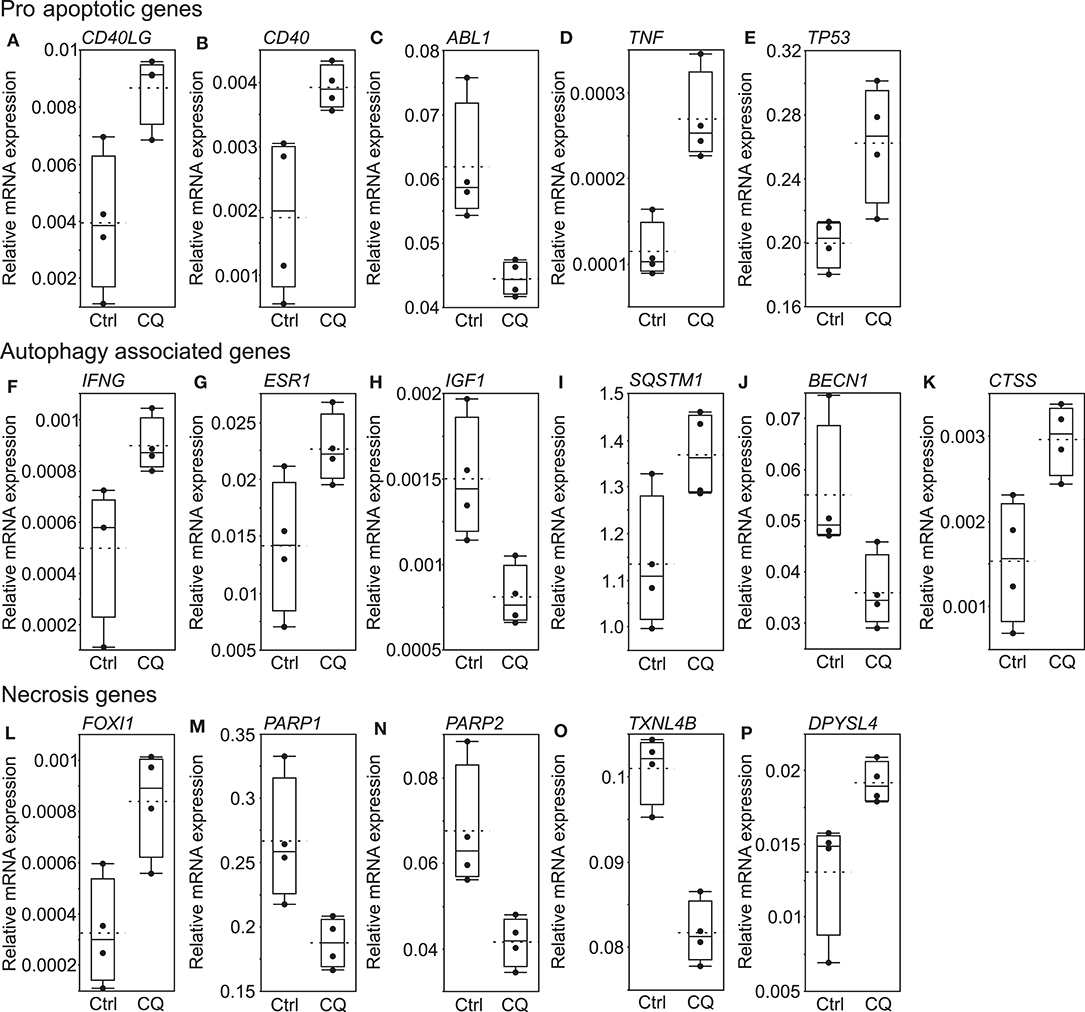
Figure 4. Significantly altered genes detected by Cell Death Pathway Finder array. Pro-apoptotic genes (A–E), autophagy associated genes (F–K), necrosis genes (L–P). Black dots represent normalized expression of independent samples, the box represents the interquartile range, and the whiskers represent the first and fourth quartile. The solid line is median and dashed line shows mean expression of each gene after 96 h of 5 μM CQ or control treatment (N = 4).
CQ Inhibits PARPs in HB Cells
Given that the most significant alteration found by metabolomic profiling was a drastic decrease in NAD+ concentration and that poly(ADP)-ribose polymerase (PARP) function is highly NAD+-dependent (54), we chose PARP1 and PARP2 (Figures 4M,N) for further analyses. HUH6, HB-284, and HB-243 cells were treated with 5 μM of CQ for 96 h and subsequently quantitative western blotting analysis was performed. Statistically significant decrease in PARP1 protein expression was detected in HUH6 and HB-243 cell lines (Figure 5A), over 60 and 30% reduction, respectively. Furthermore, immunofluorescence staining revealed decreased nuclear expression of PARP1 after CQ treatment compared to control cells (Supplementary Figures 2D–F). Similarly, PARP2 expression was observed to be markedly decreased in cells treated with CQ (Figure 5B).
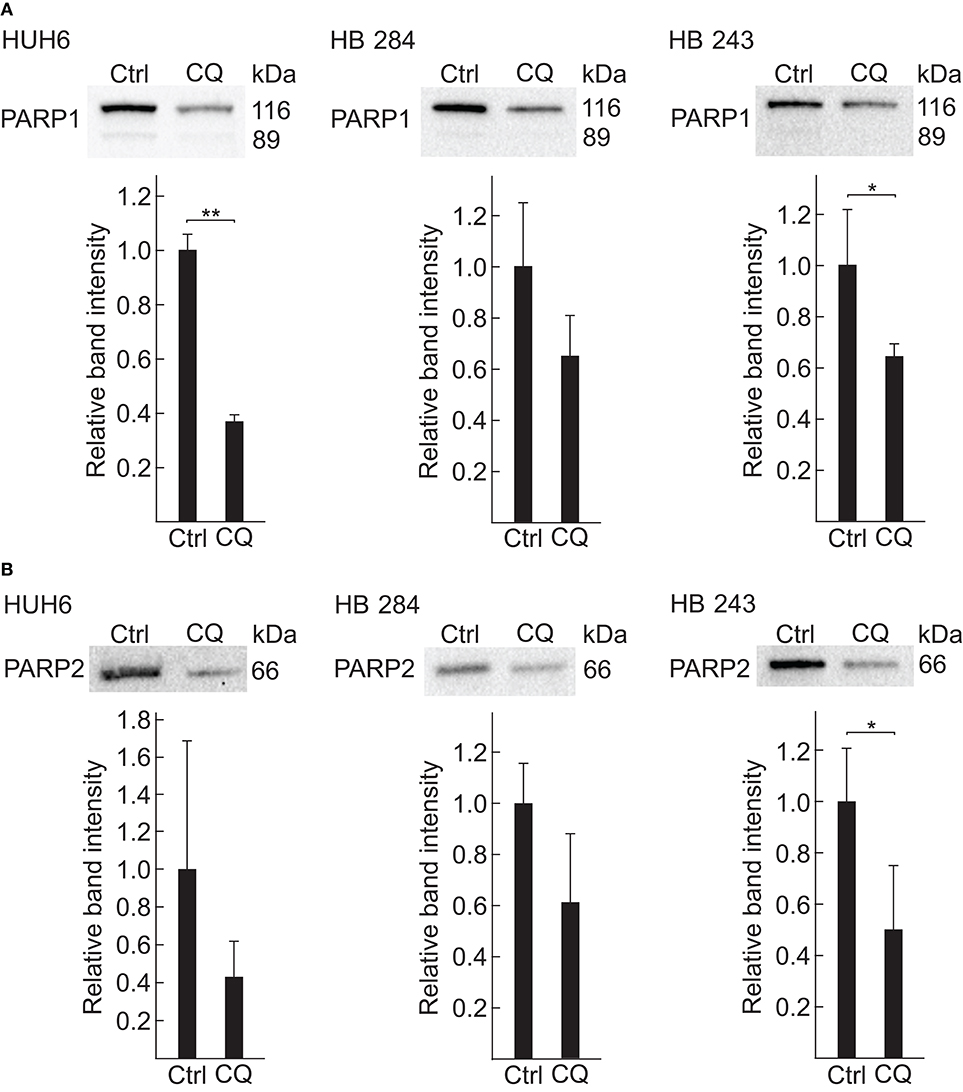
Figure 5. PARP1 and PARP2 are downregulated after 96 h CQ exposure in HB cells. Relative PARP1 (A) and PARP2 (B) protein expression in HUH6 (left panel), HB-284 (middle panel), and HB-243 (right panel) cells treated with 5 μM CQ detected with western blotting. Normalized band intensity of three independent sample in each group were used to calculate relative protein expression. Student's t-test was exploited for statistical analysis. *P < 0.05, **P < 0.01. Bar plots are presented as relative values of mean ± RSD.
Discussion
Chemotherapy resistance is the major obstacle limiting HB patient survival (10). Therefore, novel treatment strategies are needed for patients with advanced and chemoresistant HB. We found robust antineoplastic activity of CQ monotherapy in HB tumor spheroid models. Further, our results shed light on the molecular mechanisms of the CQ. To this end, our findings describe the CQ administration associated gene expression alterations in cell death related pathways, especially the inhibition of PARP1 and PARP2, as well as metabolomic perturbations leading to failure in NAD+ balance and aspartate availability.
Spheroids mimic 3D architecture of solid tumors and offer tissue-like environment for cancer drug research (55). The patient-derived HB models used here recapitulate the characteristics of advanced and aggressive original HB tumors (50). Taken together, our results demonstrate decreased cell viability and activation of apoptotic cell death as response to CQ treatment in these HB cell models. Although spheroids have multiple advantages compared to the traditional 2D cell cultures, they still lack responses produced by multicellular organ system. Organoids may better represent the complexity of tumor and neighboring tissues (56), and in the future, these findings should be confirmed in HB organoids.
Aspartate depletion induced by CQ treatment was recently demonstrated in pancreatic cancer cells restricting nucleotide biosynthesis and subsequently predisposing cells to the replication stress (57). Additionally, in oxygen deprived environment, typical for solid tumors, aspartate has been suggested to be a limiting metabolite for proliferation (58, 59). In line with these previous observations, the present findings demonstrate reduced aspartate availability and simultaneous decrease in cell viability of HB cells treated with CQ. Moreover, aspartate supplementation rescued cells from CQ induced cell death suggesting that aspartate availability may be one the crucial targets of this treatment.
Aspartate biosynthesis requires electron acceptors, e.g., NAD+, and in the presence of oxygen their pools are maintained by electron transport chain (ETC) reactions (58, 59). ETC is carried out by four complexes (I–IV) and ATP synthase. Complex I regenerates NAD+ from NADH and pharmacological inhibition of that reaction is linked with disturbed NAD+/NADH balance and subsequent reduction in aspartate synthesis (59). We noted a contemporaneous drop in aspartate and NAD+ concentrations in HB cells treated with CQ, implying that limited aspartate availability may be a consequence of ETC malfunction. In addition, NAD+/NADH balance shifted toward NADH following CQ treatment, suggesting that CQ impacts NAD oxidation. Further investigations are needed to determine whether inhibition of mitochondrial complex I is a bona fide mechanism of CQ action in HB cells.
PARPs are multifunctional enzymes involved in epigenetic modifications, signal transduction, stress sensing, and DNA repair (60–62). Interestingly, in a context of aggressive HB, PARP1 was recently shown to be aberrantly activated, promoting expression of nonfunctional tumor suppressor proteins (63). Herein, we demonstrated decreases in PARP1 and PARP2 expression in HB cells both at mRNA and protein level after CQ treatment. Since the catalytic activities of PARP1 and PARP2 are NAD+ dependent (54), we suggest that reduced NAD+ pools trigger degradation of PARPs. Moreover, CQ may downregulate aberrantly expressed tumor suppressor proteins by PARP inhibition compromising HB cell survival. The PARP inhibitor (PARPi) talazoparib has been shown to synergize with CQ in a pediatric chronic myeloid leukemia mouse model (64). Another PARPi, niraparib, combined with CQ exhibited increased cytotoxicity in hepatocellular carcinoma in vitro and in vivo (65). It would be of interest to study whether a similar synergism exists also in HB models.
All in all, our results suggest that CQ has potential as an additional treatment modality for aggressive HB through the mechanisms summarized in Figure 6. Thus, this pre-clinical study sets the basis for further investigations in HB and offers novel potential applications for CQ re-purposing strategies.
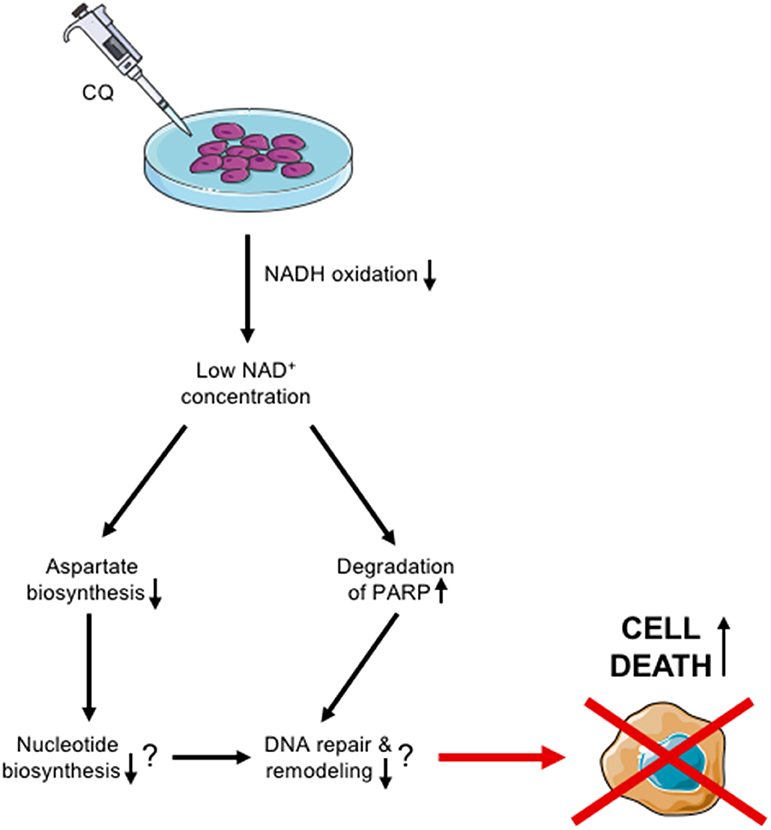
Figure 6. Schematic view of proposed CQ action in HUH6 cells.
Conclusions
CQ treatment in relevant concentrations decreases viability of HB spheroids. Further, the spheroids established from chemoresistant tumors exhibited increased apoptotic activity after CQ treatment, suggesting that CQ holds potential in treatment of aggressive HB. Moreover, our study demonstrates disturbance in NAD+ and aspartate metabolism exposing cells to impaired DNA repair and histone remodeling by PARPs.
Data Availability Statement
All datasets presented in this study are included in the article/Supplementary Material.
Author Contributions
KE, MH, and MP: conceptualization. KE, SC, DW, and MP: methodology. KE, EL, TS, AK, and MP: acquisition, analysis, or interpretation of data. MH: funding acquisition. SC and J-GJ: resources. DW, MH, and MP: supervision. KE: writing-first draft. SC, EL, TS, AK, J-GJ, DW, MH, and MP: writing-review and editing. KE, SC, EL, TS, AK, J-GJ, DW, MH, and MP: final approval of the manuscript version to be published. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Cancer Foundation Finland, Finska Läkaresällskapet, Helsinki University Central Hospital Research Grants, Päivikki, and Sakari Sohlberg Foundation, Sigrid Jusélius Foundation, and Doctoral Program in Clinical Research at University of Helsinki Funds.
Conflict of Interest
SC is employed by the company XenTech and J-GJ is the president of the company XenTech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Metabolomics analyses were carried out at the Metabolomics Unit, Technology Center, Institute for Molecular Medicine Finland FIMM, University of Helsinki.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01138/full#supplementary-material
Abbreviations
ABL1, ABL Proto-Oncogene 1, Non-Receptor Tyrosine Kinase; ASP, Aspartate; B2M, Beta-2-Microglobulin; BECN1, Beclin-1; CD40, CD40 Molecule; CD40LG, CD40 Ligand; CQ, Chloroquine; CTSS, Cathepsin S; DPYSL4, Dihydropyrimidinase Like 4; ESR1, Estrogen Receptor 1; ETC, Electron transport chain; FAP, Familial adenomatous polyposis; FOXI1, Forkhead Box I1; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; HB, Hepatoblastoma; HPRT1, Hypoxanthine Phosphoribosyltransferase 1; IFNG, Interferon gamma; IGF1, Insulin Like Growth Factor 1; NAD+, Nicotinamide adenine dinucleotide; NADH, Nicotinamide adenine dinucleotide hydrogen; PARP1, Poly(ADP)-ribose polymerase 1; PARP2, Poly(ADP)-ribose polymerase 2; PARPi, Poly(ADP)-ribose polymerase inhibitor; SQSTM1, Sequestosome 1; TNF, Tumor necrosis factor; TP53, Tumor Protein P53; TXNL4B, Thioredoxin Like 4B.
References
1. Finegold MJ, Egler RA, Goss JA, Guillerman RP, Karpen SJ, Krishnamurthy R, et al. Liver tumors: pediatric population. Liver Transpl. (2008) 14:1545–56. doi: 10.1002/lt.21654
2. Czauderna P, Haeberle B, Hiyama E, Rangaswami A, Krailo M, Maibach R, et al. The Children's Hepatic tumors International Collaboration (CHIC): novel global rare tumor database yields new prognostic factors in hepatoblastoma and becomes a research model. Eur J Cancer. (2016) 52:92–101. doi: 10.1016/j.ejca.2015.09.023
3. Tanimura M, Matsui I, Abe J, Ikeda H, Kobayashi N, Ohira M, et al. Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res. (1998) 58:3032–5.
4. Sotelo-Avila C, Gonzalez-Crussi F, Fowler JW. Complete and incomplete forms of Beckwith-Wiedemann syndrome: their oncogenic potential. J Pediatr. (1980) 96:47–50. doi: 10.1016/S0022-3476(80)80322-2
5. Hughes LJ, Michels VV. Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet. (1992) 43:1023–5. doi: 10.1002/ajmg.1320430621
6. Raney B. Hepatoblastoma in children: a review. J Pediatr Hematol Oncol. (1997) 19:418–22. doi: 10.1097/00043426-199709000-00002
7. Pritchard J, Brown J, Shafford E, Perilongo G, Brock P, Dicks-Mireaux C, et al. Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: a successful approach–results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol. (2000) 18:3819–28. doi: 10.1200/JCO.2000.18.22.3819
8. Fuchs J, Rydzynski J, Hecker H, Mildenberger H, Burger D, Harms D, et al. The influence of preoperative chemotherapy and surgical technique in the treatment of hepatoblastoma–a report from the German Cooperative Liver Tumour Studies HB 89 and HB 94. Eur J Pediatr Surg. (2002) 12:255–61. doi: 10.1055/s-2002-34484
9. Zsiros J, Maibach R, Shafford E, Brugieres L, Brock P, Czauderna P, et al. Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol. (2010) 28:2584–90. doi: 10.1200/JCO.2009.22.4857
10. De Ioris M, Brugieres L, Zimmermann A, Keeling J, Brock P, Maibach R, et al. Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: the SIOPEL group experience. Eur J Cancer. (2008) 44:545–50. doi: 10.1016/j.ejca.2007.11.022
11. López-Terrada DH. Hepatoblastoma. Diagn Histopathol. (2014) 20:67–75. doi: 10.1016/j.mpdhp.2014.01.002
12. Volkova M, Russell R 3rd. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. (2011) 7:214–20. doi: 10.2174/157340311799960645
13. Rynes RI. Antimalarial drugs in the treatment of rheumatological diseases. Br J Rheumatol. (1997) 36:799–805. doi: 10.1093/rheumatology/36.7.799
14. Coatney GR. Pitfalls in a discovery: the chronicle of chloroquine. Am J Trop Med Hyg. (1963) 12:121–8. doi: 10.4269/ajtmh.1963.12.121
15. Lee SJ, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat Rev Nephrol. (2011) 7:718–29. doi: 10.1038/nrneph.2011.150
16. Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology. (2015) 23:231–69. doi: 10.1007/s10787-015-0239-y
17. Hu T, Li P, Luo Z, Chen X, Zhang J, Wang C, et al. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol Rep. (2016) 35:43–9. doi: 10.3892/or.2015.4380
18. Zou Y, Ling YH, Sironi J, Schwartz EL, Perez-Soler R, Piperdi B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J Thorac Oncol. (2013) 8:693–702. doi: 10.1097/JTO.0b013e31828c7210
19. Zheng Y, Zhao YL, Deng X, Yang S, Mao Y, Li Z, et al. Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Invest. (2009) 27:286–92. doi: 10.1080/07357900802427927
20. Lin YC, Lin JF, Wen SI, Yang SC, Tsai TF, Chen HE, et al. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J Med Sci. (2017) 33:215–23. doi: 10.1016/j.kjms.2017.01.004
21. Molejon MI, Swayden M, Fanale D, Bintz J, Gayet O, Soubeyran P, et al. Chloroquine plays a cell-dependent role in the response to treatment of pancreatic adenocarcinoma. Oncotarget. (2018) 9:30837–46. doi: 10.18632/oncotarget.25745
22. Geng Y, Kohli L, Klocke BJ, Roth KA. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro Oncol. (2010) 12:473–81. doi: 10.1093/neuonc/nop048
23. Ye H, Chen M, Cao F, Huang H, Zhan R, Zheng X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. (2016) 16:178. doi: 10.1186/s12883-016-0700-6
24. Liu X, Sun K, Wang H, Dai Y. Inhibition of autophagy by chloroquine enhances the antitumor efficacy of sorafenib in glioblastoma. Cell Mol Neurobiol. (2016) 36:1197–208. doi: 10.1007/s10571-015-0318-z
25. Qu X, Sheng J, Shen L, Su J, Xu Y, Xie Q, et al. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS One. (2017) 12:e0173712. doi: 10.1371/journal.pone.0173712
26. Helmy SA, El-Mesery M, El-Karef A, Eissa LA, El Gayar AM. Chloroquine upregulates TRAIL/TRAILR2 expression and potentiates doxorubicin anti-tumor activity in thioacetamide-induced hepatocellular carcinoma model. Chem Biol Interact. (2018) 279:84–94. doi: 10.1016/j.cbi.2017.11.009
27. Samaras P, Tusup M, Nguyen-Kim TDL, Seifert B, Bachmann H, von Moos R, et al. Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother Pharmacol. (2017) 80:1005–12. doi: 10.1007/s00280-017-3446-y
28. Goncalves RM, Agnes JP, Delgobo M, de Souza PO, Thome MP, Heimfarth L, et al. Late autophagy inhibitor chloroquine improves efficacy of the histone deacetylase inhibitor SAHA and temozolomide in gliomas. Biochem Pharmacol. (2019) 163:440–50. doi: 10.1016/j.bcp.2019.03.015
29. Ojha R, Bhattacharyya S, Singh SK. Autophagy in cancer stem cells: a potential link between chemoresistance, recurrence, and metastasis. Biores Open Access. (2015) 4:97–108. doi: 10.1089/biores.2014.0035
30. Egger ME, Huang JS, Yin W, McMasters KM, McNally LR. Inhibition of autophagy with chloroquine is effective in melanoma. J Surg Res. (2013) 184:274–81. doi: 10.1016/j.jss.2013.04.055
31. Liang X, Tang J, Liang Y, Jin R, Cai X. Suppression of autophagy by chloroquine sensitizes 5-fluorouracil-mediated cell death in gallbladder carcinoma cells. Cell Biosci. (2014) 4:10. doi: 10.1186/2045-3701-4-10
32. Schonewolf CA, Mehta M, Schiff D, Wu H, Haffty BG, Karantza V, et al. Autophagy inhibition by chloroquine sensitizes HT-29 colorectal cancer cells to concurrent chemoradiation. World J Gastrointest Oncol. (2014) 6:74–82. doi: 10.4251/wjgo.v6.i3.74
33. Roy LO, Poirier MB, Fortin D. Chloroquine inhibits the malignant phenotype of glioblastoma partially by suppressing TGF-β. Invest New Drugs. (2015) 33:1020–31. doi: 10.1007/s10637-015-0275-x
34. Balic A, Sorensen MD, Trabulo SM, Sainz B Jr, Cioffi M, Vieira CR, et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. (2014) 13:1758–71. doi: 10.1158/1535-7163.MCT-13-0948
35. Zhang Y, Cao Y, Sun X, Feng Y, Du Y, Liu F, et al. Chloroquine (CQ) exerts anti-breast cancer through modulating microenvironment and inducing apoptosis. Int Immunopharmacol. (2017) 42:100–7. doi: 10.1016/j.intimp.2016.11.027
36. Chen P, Luo X, Nie P, Wu B, Xu W, Shi X, et al. CQ synergistically sensitizes human colorectal cancer cells to SN-38/CPT-11 through lysosomal and mitochondrial apoptotic pathway via p53-ROS cross-talk. Free Radic Biol Med. (2017) 104:280–97. doi: 10.1016/j.freeradbiomed.2017.01.033
37. Lakhter AJ, Sahu RP, Sun Y, Kaufmann WK, Androphy EJ, Travers JB, et al. Chloroquine promotes apoptosis in melanoma cells by inhibiting BH3 domain-mediated PUMA degradation. J Invest Dermatol. (2013) 133:2247–54. doi: 10.1038/jid.2013.56
38. Jiang PD, Zhao YL, Shi W, Deng XQ, Xie G, Mao YQ, et al. Cell growth inhibition, G2/M cell cycle arrest, and apoptosis induced by chloroquine in human breast cancer cell line Bcap-37. Cell Physiol Biochem. (2008) 22:431–40. doi: 10.1159/000185488
39. Monma H, Iida Y, Moritani T, Okimoto T, Tanino R, Tajima Y, et al. Chloroquine augments TRAIL-induced apoptosis and induces G2/M phase arrest in human pancreatic cancer cells. PLoS One. (2018) 13:e0193990. doi: 10.1371/journal.pone.0193990
40. Kim EL, Wustenberg R, Rubsam A, Schmitz-Salue C, Warnecke G, Bucker EM, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol. (2010) 12:389–400. doi: 10.1093/neuonc/nop046
41. Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. (2018) 9:873. doi: 10.1038/s41467-018-04169-w
42. Maes H, Kuchnio A, Carmeliet P, Agostinis P. How to teach an old dog new tricks: autophagy-independent action of chloroquine on the tumor vasculature. Autophagy. (2014) 10:2082–4. doi: 10.4161/auto.36259
43. Quan M, Liu S, Wang Q, Li G, Zhang Y, Feng S, et al. NS5ATP9 promotes beclin 1-dependent starvation-induced autophagy of hepatoblastoma cells. J Cell Biochem. (2015) 116:1574–82. doi: 10.1002/jcb.25111
44. Chang Y, Chen L, Liu Y, Hu L, Li L, Tu Q, et al. Inhibition of autophagy may suppress the development of hepatoblastoma. FEBS J. (2011) 278:4811–23. doi: 10.1111/j.1742-4658.2011.08381.x
45. Dzieran J, Beck JF, Sonnemann J. Differential responsiveness of human hepatoma cells versus normal hepatocytes to TRAIL in combination with either histone deacetylase inhibitors or conventional cytostatics. Cancer Sci. (2008) 99:1685–92. doi: 10.1111/j.1349-7006.2008.00868.x
46. Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. (2014) 9:e92444. doi: 10.1371/journal.pone.0092444
47. Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. (2018) 46:W486–W94. doi: 10.1093/nar/gky310
48. Roman-Garcia P, Quiros-Gonzalez I, Mottram L, Lieben L, Sharan K, Wangwiwatsin A, et al. Vitamin B(1)(2)-dependent taurine synthesis regulates growth and bone mass. J Clin Invest. (2014) 124:2988–3002. doi: 10.1172/JCI72606
49. Ren JG, Seth P, Everett P, Clish CB, Sukhatme VP. Induction of erythroid differentiation in human erythroleukemia cells by depletion of malic enzyme 2. PLoS One. (2010) 5:e12520. doi: 10.1371/journal.pone.0012520
50. Kats D, Ricker CA, Berlow NE, Noblet B, Nicolle D, Mevel K, et al. Volasertib preclinical activity in high-risk hepatoblastoma. Oncotarget. (2019) 10:6403–17. doi: 10.18632/oncotarget.27237
51. Pascolo S. Time to use a dose of Chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol. (2016) 771:139–44. doi: 10.1016/j.ejphar.2015.12.017
52. Masud Alam M, Kariya R, Kawaguchi A, Matsuda K, Kudo E, Okada S. Inhibition of autophagy by chloroquine induces apoptosis in primary effusion lymphoma in vitro and in vivo through induction of endoplasmic reticulum stress. Apoptosis. (2016) 21:1191–201. doi: 10.1007/s10495-016-1277-7
53. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. (2018) 14:1435–55. doi: 10.1080/15548627.2018.1474314
54. Yelamos J, Farres J, Llacuna L, Ampurdanes C, Martin-Caballero J. PARP-1 and PARP-2: New players in tumour development. Am J Cancer Res. (2011) 1:328–46. Available online at: http://www.ajcr.us/ajcr0000027A.html
55. LaBarbera DV, Reid BG, Yoo BH. The multicellular tumor spheroid model for high-throughput cancer drug discovery. Expert Opin Drug Discov. (2012) 7:819–30. doi: 10.1517/17460441.2012.708334
56. Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. (2018) 18:407–18. doi: 10.1038/s41568-018-0007-6
57. Elliott IA, Dann AM, Xu S, Kim SS, Abt ER, Kim W, et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc Natl Acad Sci U S A. (2019) 116:6842–7. doi: 10.1073/pnas.1812410116
58. Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. (2018) 20:775–81. doi: 10.1038/s41556-018-0118-z
59. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. (2015) 162:552–63. doi: 10.1016/j.cell.2015.07.017
60. Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. (2012) 13:411–24. doi: 10.1038/nrm3376
61. Quenet D, El Ramy R, Schreiber V, Dantzer F. The role of poly(ADP-ribosyl)ation in epigenetic events. Int J Biochem Cell Biol. (2009) 41:60–5. doi: 10.1016/j.biocel.2008.07.023
62. Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. (2012) 26:417–32. doi: 10.1101/gad.183509.111
63. Valanejad L, Cast A, Wright M, Bissig KD, Karns R, Weirauch MT, et al. PARP1 activation increases expression of modified tumor suppressors and pathways underlying development of aggressive hepatoblastoma. Commun Biol. (2018) 1:67. doi: 10.1038/s42003-018-0077-8
64. Liu Y, Song H, Song H, Feng X, Zhou C, Huo Z. Targeting autophagy potentiates the anti-tumor effect of PARP inhibitor in pediatric chronic myeloid leukemia. AMB Express. (2019) 9:108. doi: 10.1186/s13568-019-0836-z
Keywords: liver cancer, hepatoblastoma, pediatric oncology, poly(ADP)-ribose polymerase, metabolomics, aspartate, NAD+
Citation: Eloranta K, Cairo S, Liljeström E, Soini T, Kyrönlahti A, Judde J-G, Wilson DB, Heikinheimo M and Pihlajoki M (2020) Chloroquine Triggers Cell Death and Inhibits PARPs in Cell Models of Aggressive Hepatoblastoma. Front. Oncol. 10:1138. doi: 10.3389/fonc.2020.01138
Received: 08 April 2020; Accepted: 05 June 2020;
Published: 17 July 2020.
Edited by:
Carlos Pérez-Plasencia, National Autonomous University of Mexico, MexicoReviewed by:
Luis Enrique Gomez-Quiroz, Autonomous Metropolitan University, MexicoNadia Judith Jacobo-Herrera, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), Mexico
Copyright © 2020 Eloranta, Cairo, Liljeström, Soini, Kyrönlahti, Judde, Wilson, Heikinheimo and Pihlajoki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marjut Pihlajoki, bWFyanV0LnBpaGxham9raUBoZWxzaW5raS5maQ==
†These authors have contributed equally to this work