 Laura Ondrisova1,2
Laura Ondrisova1,2 Marek Mraz1,2*
Marek Mraz1,2*- 1Molecular Medicine, CEITEC Masaryk University, Brno, Czechia
- 2Department of Internal Medicine, Hematology and Oncology, University Hospital Brno and Faculty of Medicine, Masaryk University, Brno, Czechia
The approval of BTK and PI3K inhibitors (ibrutinib, idelalisib) represents a revolution in the therapy of B cell malignancies such as chronic lymphocytic leukemia (CLL), mantle-cell lymphoma (MCL), diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL), or Waldenström’s macroglobulinemia (WM). However, these “BCR inhibitors” function by interfering with B cell pathophysiology in a more complex way than anticipated, and resistance develops through multiple mechanisms. In ibrutinib treated patients, the most commonly described resistance-mechanism is a mutation in BTK itself, which prevents the covalent binding of ibrutinib, or a mutation in PLCG2, which acts to bypass the dependency on BTK at the BCR signalosome. However, additional genetic aberrations leading to resistance are being described (such as mutations in the CARD11, CCND1, BIRC3, TRAF2, TRAF3, TNFAIP3, loss of chromosomal region 6q or 8p, a gain of Toll-like receptor (TLR)/MYD88 signaling or gain of 2p chromosomal region). Furthermore, relative resistance to BTK inhibitors can be caused by non-genetic adaptive mechanisms leading to compensatory pro-survival pathway activation. For instance, PI3K/mTOR/Akt, NFkB and MAPK activation, BCL2, MYC, and XPO1 upregulation or PTEN downregulation lead to B cell survival despite BTK inhibition. Resistance could also arise from activating microenvironmental pathways such as chemokine or integrin signaling via CXCR4 or VLA4 upregulation, respectively. Defining these compensatory pro-survival mechanisms can help to develop novel therapeutic combinations of BTK inhibitors with other inhibitors (such as BH3-mimetic venetoclax, XPO1 inhibitor selinexor, mTOR, or MEK inhibitors). The mechanisms of resistance to PI3K inhibitors remain relatively unclear, but some studies point to MAPK signaling upregulation via both genetic and non-genetic changes, which could be co-targeted therapeutically. Alternatively, drugs mimicking the BTK/PI3K inhibition effect can be used to prevent adhesion and/or malignant B cell migration (chemokine and integrin inhibitors) or to block the pro-proliferative T cell signals in the microenvironment (such as IL4/STAT signaling inhibitors). Here we review the genetic and non-genetic mechanisms of resistance and adaptation to the first generation of BTK and PI3K inhibitors (ibrutinib and idelalisib, respectively), and discuss possible combinatorial therapeutic strategies to overcome resistance or to increase clinical efficacy.
Introduction
The BCR signaling pathway plays a central role in the onset and progression of mature B cell malignancies, such as chronic lymphocytic leukemia (CLL), mantle-cell lymphoma (MCL), diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL), or Waldenström’s macroglobulinemia (WM). Activating mutations in the BCR signaling pathway are commonly found in DLBCL, FL, or WM (1). Though these mutations are usually missing in CLL and MCL, BCR signaling is constitutively activated and is a key player in their pathogenesis (2–5). Introducing “B cell receptor (BCR) inhibitors” in recent years has marked a revolution in treating B cell malignancies since many patients are responsive to the inhibitors of BCR-associated kinases BTK or PI3K, such as ibrutinib and idelalisib, respectively. They are now widely used as a first-line treatment or to treat relapsed/refractory diseases. However, the patient response to them varies across B cell malignancies in clinical trials as well as in real-world setting, and a large percentage of patients develop resistance or have to stop the therapy due to toxicities associated with these inhibitors’ long-term use (6–14).
In this review, we summarize the genetic and non-genetic mechanisms of resistance and adaptation to the first generation of BTK and PI3K inhibitors (ibrutinib and idelalisib, respectively), and discuss possible therapeutic strategies to overcome resistance or increase clinical efficacy by using combinatorial therapeutic strategies. We also discuss the complexity of the mechanisms of action of “BCR inhibitors” and how this affects the choice of potential combinatorial therapy.
BCR Signaling and Its Cross-Talk With Other Pathways
Cell-surface immunoglobulin does not have any kinase activity itself. It is non-covalently connected to disulphide-linked heterodimers Igα and Igβ (CD79A, CD79B). After recognition and antigen binding, BCRs start to aggregate and change their conformation, which concludes in phosphorylation of tyrosine-based activation motifs (ITAMs) on Igα and Igβ’s cytoplasmic domains. This phosphorylation, mediated by Src-family kinase LYN, creates a docking site for spleen tyrosine kinase (SYK) (15). The activated SYK then phosphorylates the B cell linker protein (BLNK), an adaptor protein helpful in recruiting other molecules such as Bruton tyrosine kinase (BTK). BCR stimulation also leads to phosphorylation of co-receptor CD19 and PI3K adaptor BCAP by LYN and SYK, which afterwards activates phosphoinositide 3-kinase (PI3K) leading to PIP3 generation (16, 17) (Figure 1).
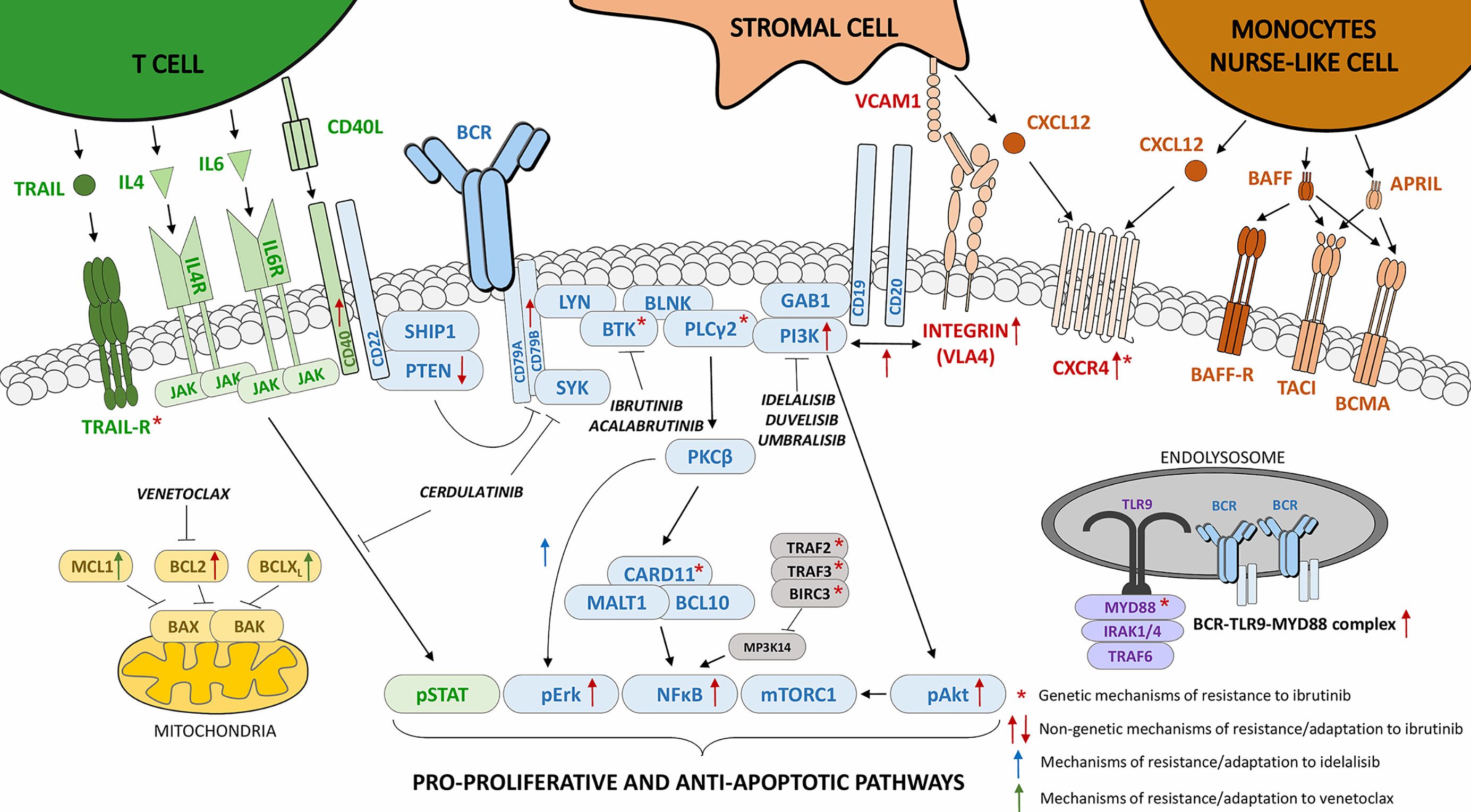
Figure 1 The genetic and non-genetic mechanisms of resistance to BTK or PI3K inhibition in B cell malignancies. The “*” indicates genetic mechanisms of resistance to ibrutinib (BTK inhibitor), the red arrows indicate non-genetic mechanisms of resistance/adaptation to ibrutinib, the blue arrows indicate mechanisms of resistance/adaptation to idelalisib (PI3K inhibitor), and the green arrows indicate mechanisms of resistance/adaptation to venetoclax (BH3-mimetic [BCL2 inhibition]).
PIP3 helps to recruit GRB2-associated-binding protein 1 (GAB1), 3-phosphoinositide-dependent protein kinase 1 (PDK1, also known as PDPK1), protein kinase B (PKB, Akt), and BTK to the plasma membrane via their pleckstrin homology (PH) domain. Here, Akt is phosphorylated on S473 by mTORC2 which also facilitates Akt phosphorylation on T308 by PDK1 leading to full Akt activation (18). PI3K signaling is further positively regulated by the adaptor protein GAB1, which recruits additional PI3K molecules generating more PIP3 (19, 20). On the other hand, the amount of PIP3 is negatively balanced by the activity of phosphatases such as SHIP1, SHP1, and PTEN.
PIP3 is also needed for optimal BTK activation, since it helps to translocate BTK to the cell membrane and via the interaction with its PH domain, it allows the activation of BTK’s kinase activity (21). For full BTK activation after the recruitment to the cell membrane, phosphorylation at two sites is needed. Firstly, BTK gets phosphorylated by SYK or LYN at tyrosine Y551, which then leads to autophosphorylation at Y223 (22, 23). Fully activated BTK phosphorylates phospholipase Cγ2 (PLCγ2). PLCγ2 hydrolyses PIP2 into secondary messengers inositol triphosphate (IP3), which controls intracellular Ca2+ levels, and diacylglycerol (DAG) which, via protein kinase Cβ (PKCβ) activation, induces cRaf-MEK-Erk pathway activation. PKCβ also activates CARD11, which then forms a complex with MALT1 and BCL10 to activate TAK1 (24). Afterwards, TAK1 phosphorylates IKKβ which initiates the NFκB pathway (25). Apart from this, PKCβ plays a role in negative feedback regulation of BCR signaling by removing BTK from the plasma membrane by phosphorylating BTK on S180 (26). Non-redundant negative regulation is also mediated by LYN kinase, since mouse B cells with LYN knockout have a surprisingly stronger BCR signaling suggesting that LYN has a specific role in negatively regulating the pathway (27). BCR signaling propensity is also affected by levels of cell-surface molecules that act as docking sites for positive or negative BCR pathway regulators, which include molecules such as CD19, CD22, and CD32. Recently, we have shown that a notorious therapeutic target in B cell malignancies, CD20, is also a positive BCR signaling regulator (28). When CD20 is silenced, response to BCR stimulation is weaker, as underscored by the lower phosphorylation of BCR-associated kinases and impaired calcium flux (29, 30). Moreover, an additional layer of regulation involves small non-coding RNAs (microRNAs) that influence both the positive and negative regulation of BCR signaling propensity (20, 31–38).
BCR signaling is activated in the lymphatic tissue microenvironment and is closely intertwined with the pathways responsible for the cell homing and adhesion (5). BCR activation affects adhesion via integrin VLA4 formed by CD49d and CD29 (integrin β1); together BCR and VLA4 provide B lymphocytes with adhesion and enhanced signaling (39). CD49d activation causes SYK phosphorylation and, on the other hand, BCR stimulation leads to VLA4 activation (40–42). BCR stimulation also increases chemotaxis towards chemokines such as CXCL12 produced in the microenvironment. Binding of CXCL12 to its receptor CXCR4 activates PI3K, MAPK, and STAT3, and leads to actin polymerization and cell migration (43–45). In CLL, cell-surface IgM levels and BCR signaling is increased by the IL4 produced by T cells which also activates the JAK1-3/STAT6 pathway and upregulates the levels of anti-apoptotic proteins from BCL2 family, resulting in partial malignant B cell protection from the effects of “BCR inhibitors” (46, 47). The importance of the microenvironment can be well illustrated in CLL, where malignant B cells are dependent on constant re-circulation between the peripheral blood and lymph nodes, where they are supported by pro-survival signals from mesenchymal stromal cells, monocyte-derived nurse-like cells, and T lymphocytes (29, 43, 48–50). The supportive stromal cells produce not only chemoattractants CXCL12 and CXCL13 but also BAFF, APRIL, CD31, and plexin B1 which protect CLL cells from spontaneous and induced apoptosis by activating BCR and NFκB signaling (43, 49, 51, 52). Kinases of the BCR pathway BTK and PI3Kδ together with JAK are also involved in T cell dependent proliferation induced by CD40L and IL21, which can be inhibited by ibrutinib, idelalisib or JAK inhibitor (53).
Overall, there is crosstalk between the BCR, chemokine signaling and cell adhesion pathways. Therefore, the success of “BCR inhibitors” lies not only in inhibiting the BCR pathway itself but also in inhibiting other processes. In CLL and some lymphomas, BTK/PI3K inhibition results in malignant B cells egressing from the lymph nodes, causing transient lymphocytosis in patients (8, 54, 55).
Mechanistic Effects of Ibrutinib Action
Ibrutinib is an orally administered small-molecule inhibitor targeting BTK. It binds to BTK covalently, selectively, and irreversibly, inhibiting its phosphorylation and enzymatic activity. BTK is an important kinase in BCR signaling needed for B cells to properly develop (56, 57). Inhibiting it with ibrutinib leads to a loss of pro-survival signals from BCR activation by ligands, and also impairs the “tonic” BCR signals that sustain B cell survival. BTK inhibition decreases cell proliferation as well as interferes with the activation of downstream molecules in BCR pathway such as PLCγ2, Akt and Erk irrespective of BCR stimulation (58–61). As BTK is not only involved in BCR signaling (see above), ibrutinib also disrupts CXCR4 internalization, impairs migration toward CXCL12 and also indirectly decreases total BTK levels (62). Ibrutinib further disrupts signaling from CXCR5 and integrins, molecules that allow B lymphocyte migration and adhesion (63, 64). Altogether, ibrutinib inhibits BCR stimulation, B cell proliferation, and migration toward homing chemokines such as CXCL12 and CXCL13. It also blocks BCR-dependent CCL3 and CCL4 chemokine release in CLL and decreases CCL4, CCL22, and CXCL13 levels in the serum of ibrutinib-treated MCL patients (54, 61). As mentioned above, inhibiting the adhesion and homing capacity causes transient lymphocytosis in CLL and MCL patients (54, 55). In most of the patients, this resolves within 8 months after starting therapy (55). CLL cells together with non-malignant immune cells after ibrutinib treatment are of a quiescent phenotype as shown by the expression of the genes involved in senescence and/or cell quiescence (30, 55, 65). Apart from the mentioned mechanisms of ibrutinib action, it also affects the microRNAs’ expression, resulting in higher levels of several tumor suppressors and inhibition of cell proliferation (66, 67).
Ibrutinib has been approved for therapy of CLL, MCL, WM, and marginal zone lymphoma (MZL). Though it is a potent drug, not all patients are responsive to ibrutinib and a significant number of them acquire resistance to the treatment or discontinue the therapy due to toxicities that are most likely caused by ibrutinib off-target inhibition of molecules such as BLK, JAK3, EGFR, and several TFK members (for a list of off-targets of ibrutinib and other BTK inhibitors see Supplementary Table 1). In the following sections, we will summarize the genetic mechanisms of ibrutinib resistance, the non-genetic mechanisms of adaptation/resistance by activating compensatory pro-survival pathways and describe possible solutions to different types of ibrutinib resistance (Figure 1).
Genetic Mechanisms of Ibrutinib Resistance
Genetic mechanisms of primary or acquired resistance to ibrutinib have been widely studied and recurrent mutations associated with resistance have been described in B cell malignancies (Table 1, Figure 1). Whole-exome sequencing revealed mutations in BCR-involved proteins BTK and PLCγ2 in ~80% of CLL patients with acquired resistance to ibrutinib (7, 96), however, some studies have reported a much lower frequency of these mutations (97, 98). The most common mutation in BTK is a C481S point mutation which interferes with the binding of ibrutinib to BTK (7, 68). Other mutations in the BTK gene were also found in ibrutinib-resistant patients and have been suggested to affect either ibrutinib binding to BTK or BLNK binding to BTK, altogether leading to PLCγ2 activation even in the presence of ibrutinib (93, 99–101) (Supplementary Table 2). PLCG2, a gene coding for PLCγ2, seems to mostly harbor gain-of-function mutations when PLCγ2 can be activated by RAC2 or SYK and LYN without BTK (7, 78, 79, 100) (Supplementary Table 2). As mutations in BTK and PLCG2 occur early before relapse on ibrutinib, they may serve as a biomarker to indicate a need to change the therapy before disease progression (102). They can also co-occur, which may bring complications to solving the resistance with next-generation BTK inhibitors (103). Apart from the mentioned mutations, resistance to ibrutinib in CLL has also been associated with chromosomal aberrations such as 8p deletion and gain in the 2p region (92, 93). The 8p region contains a gene for TRAIL receptor and its deletion results in insensitivity to TRAIL-induced cell death (93). On the other hand, 2p gain causes exportin-1 overexpression (XPO1), regulating the transport of proteins between the nucleus and cytoplasm (92). Additionally, CLL patients that progress or develop Richter’s transformation on ibrutinib recurrently harbor mutations of tumor-suppressor TP53, splicing factor SF3B1, or NFκB pathway regulator CARD11, however, whether these genetic aberrations may directly impact response to ibrutinib during Richter’s transformation remains unclear (80). Though, despite good initial response in most of CLL patients, it has been shown that mutations in TP53 are responsible for a worse prognosis in a long-term ibrutinib treatment and they also partially protect CLL cells in vitro from ibrutinib-induced apoptosis and inhibition of proliferation (104–107). This might be related to the recently described role of p53 in the negative regulation of BCR signalling (31, 32).
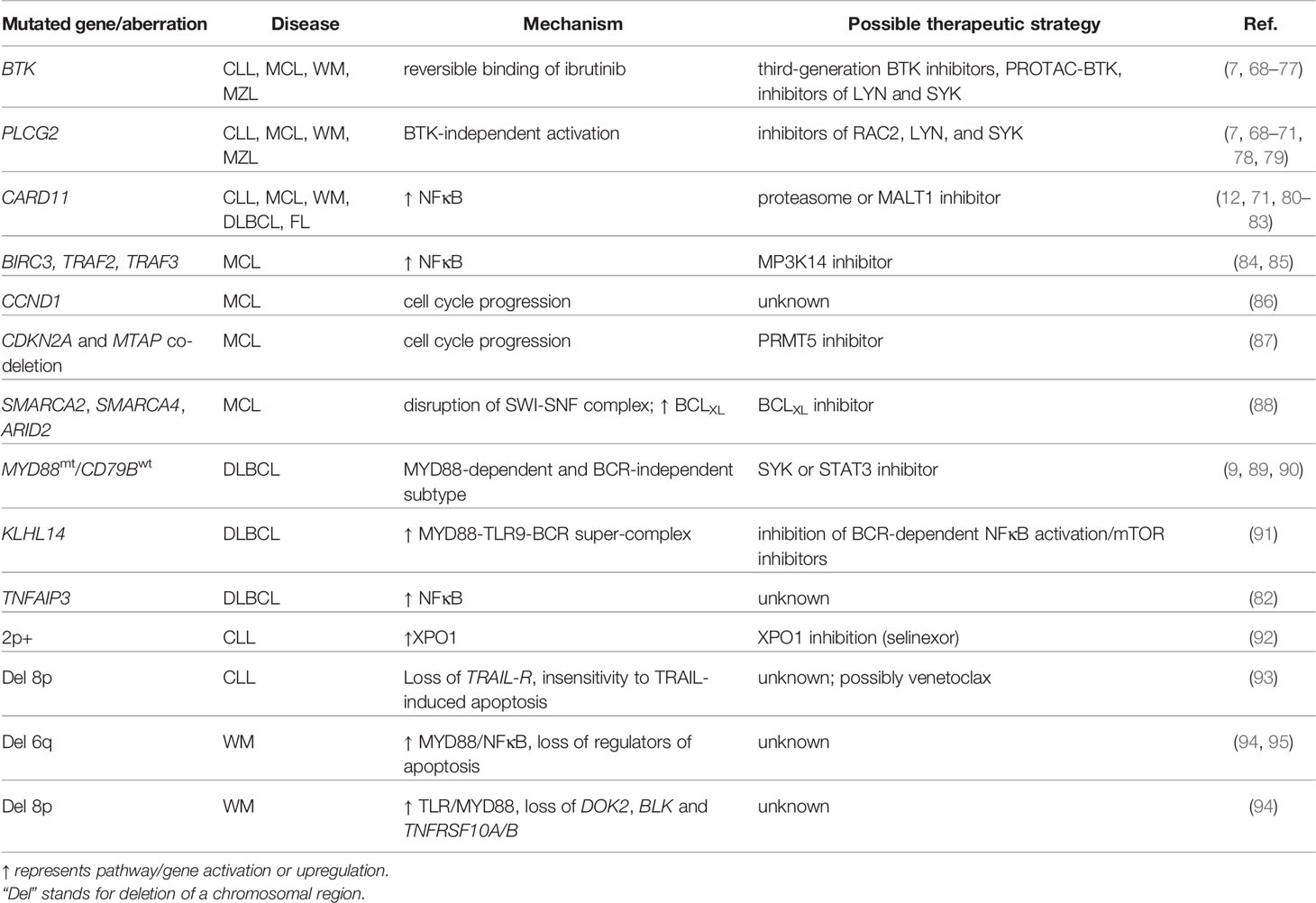
Table 1 Recurrent mutations in ibrutinib-resistant patients and possible therapeutic strategies to overcome them.
Mutations in BTK and PLCG2 have also been found in MCL and WM patients with acquired ibrutinib resistance as well as in one MZL patient (69–71). List of the most common BTK and PLCG2 mutations is provided in the Supplementary Table 2. Though CLL cells on ibrutinib have a decreased NFκB binding to DNA elements, activating an alternative NFκB pathway by genetic changes is another mechanism responsible for ibrutinib resistance, mostly in MCL (65). This is caused by mutations in BIRC3, TRAF2, or TRAF3, whose absence leads to MP3K14 enzyme stabilization and constitutive activation of alternative NFκB pathway (Figure 1) (84, 85, 108, 109). Recurrent mutations in MCL patients who have relapsed on ibrutinib have also been found in CARD11, a protein responsible for BCR-induced NFκB activation, or in CCND1, a cyclin that promotes G1-S cell cycle progression (24, 81, 86). CDKN2A and MTAP co-deletion was also observed in ibrutinib-resistant MCL tumors (87). Additionally, loss and/or mutations in the SWI-SNF chromatin remodeling complex lead to the upregulation of anti-apoptotic BCLXL and cause a primary or acquired resistance to the combination of ibrutinib and venetoclax in MCL (88).
In Waldenström’s macroglobulinemia (WM), mutations in CARD11 also lead to ibrutinib resistance, and in WM patients the ibrutinib resistance may be accompanied by 6q and 8p chromosome region deletions that expand from pre-existing clones or emerge during treatment (71, 94). These chromosomal regions contain important signaling pathway regulators. The 6q region loss involves negative regulators of MYD88/NFκB (TNFAIP3, HIVEP2, TRAF3IP2, IRAK1BP1), an inhibitor of BTK (IBTK), and regulators of apoptosis (FOXO3, BCLAF1, PERP). The genes deleted on 8p include DOK2, a TLR/MYD88 signaling inhibitor, BLK, another target of ibrutinib that is important for B cell proliferation and differentiation, and TNFRSF10A/B, a gene encoding for TRAIL receptor (94, 95). Common mutations in WM are WHIM-like mutations in CXCR4 and L265P mutations in MYD88, a mediator of Toll-like receptor signaling (110). In WM, MYD88L265P activates NFκB via BTK and IRAK1/4, making these cells sensitive to ibrutinib (110, 111). WHIM-like mutations in CXCR4 are responsible for impaired CXCR4 internalization upon stimulation and result in constant Akt and Erk activation. However, mutations in MYD88, which occur in 90% of WM patients, seem to have a more profound effect on the WM cell survival than CXCR4WHIM, as MYD88 inhibition could not be rescued by the CXCR4WHIM mutation (112). These facts might explain why, even though WHIM-like mutations promote ibrutinib resistance in vitro, MYD88wt/CXCR4wt patients have a lower response rate to ibrutinib therapy than MYD88L265P/CXCR4wt or MYD88L265P/CXCR4WHIM patients (13, 113).
In DLBCL, ibrutinib seems to be more effective in patients with an activated B cell-like DLBCL (ABC-DLBCL) subtype rather than in patients with germinal center B cell-like DLBCL (GC-DLBCL) due to constitutively active BCR signaling in ABC-DLBCL. However, even amongst ABC-DLBCL, complete or partial response was only detected in 37% of patients (9, 82), and a phase III clinical trial confirmed the benefit of adding ibrutinib to R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) therapy only in younger patients with non-GC-DLBCL (114). As for resistance, it has been shown that DLBCL patients carrying mutations in MYD88 and simultaneously having wild-type CD79B are primarily resistant to ibrutinib. As other combinations (MYD88mt/CD79Bmt, MYD88wt/CD79Bmt, MYD88wt/MYD88wt) are sensitive to ibrutinib, there is a possibility of an MYD88-dependent but BCR-independent ABC-DLBCL subtype (9). These findings might be explained by the formation of a multiprotein super-complex consisting of MYD88, TLR9 and BCR. It activates the NFκB pathway and is found in ABC-DLBCL tumors that respond to ibrutinib (Figure 1) (115). On the other hand, cells with inactivated KLHL14, a negative BCR component regulator often mutated in DLBCL, induce the NFκB pathway by activating the MYD88-TLR9-BCR super-complex, which partially protects them from ibrutinib-induced cell death (91). The role of knockout of the KLHL14 tumor suppressor was demonstrated in rescuing the ABC-DLBCL cell line from apoptosis when IgM and CD79A were knocked down, which is a manipulation normally lethal to the DLBCL cells (91). Lastly, the above mentioned mutations in CARD11 and inactivating mutations in TNFAIP3, a negative NFκB regulator, were also found in DLBCL patients not responding to ibrutinib treatment (82).
The genetic mechanisms of resistance, especially the BTK mutations that develop during ibrutinib therapy, are a clear indication of the on-target effects of the utilized small molecular inhibitors. The other mutations in the BCR signaling pathway members such as PLCG2 or CARD11 also demonstrate that malignant B cells try to gain or lose gene activity in order to overcome the inhibition of the key BCR-associated kinase (Table 1). However, it has been shown that cancer cells can also utilize non-genetic mechanisms to bypass the inhibition of a key pathway (116–120). In the following section we will review these potential mechanisms for ibrutinib/BTK-inhibition based therapy, and suggest the implications for combinatorial therapeutic strategies (Table 2).
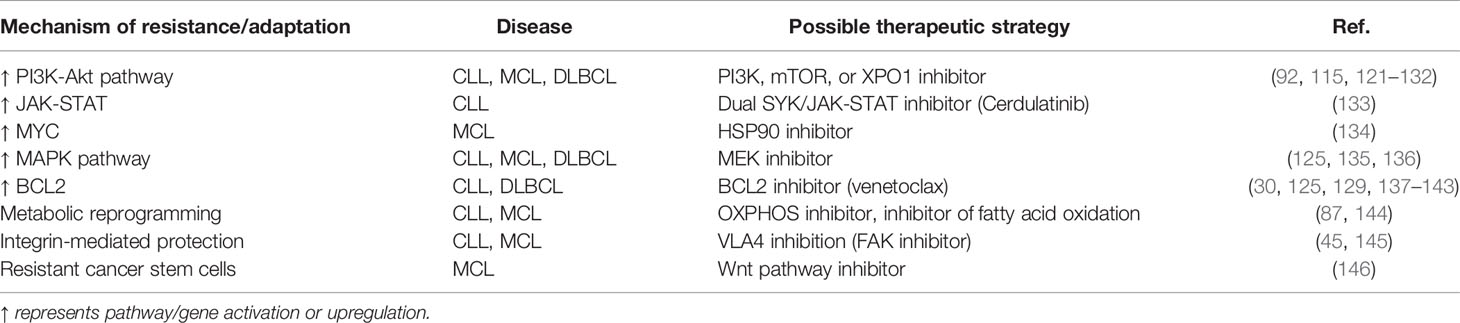
Table 2 Non-genetic mechanisms of resistance/adaptation to ibrutinib and possible therapeutic strategies to overcome them.
Non-Genetic Mechanisms of Adaptation to Ibrutinib
Patients resistant to ibrutinib that harbor the above-mentioned mutations often have these mutations subclonally in a relatively small fraction of surviving malignant B cells. Malignant non-mutated cells that co-exist with BTKC418S mutated cells might be protected by the mutated cells though this has only been conceptually demonstrated in MYD88mt WM and ABC-DLBCL. Mutated cells showed Erk1/2 activation, which led to pro-survival chemokine release and protection of BTKwt cells (147). Even if this could explain the survival of the non-mutated sub-populations of cells, there are still ibrutinib-resistant patients (>20% of ibrutinib-resistant patients in CLL) that do not show any genetic mutations responsible for the resistance (96). This, together with slow cell apoptosis during ibrutinib therapy, suggest that there are non-genetic mechanisms of resistance and cells are able to partially adapt to ibrutinib and BTK inhibition (Table 2, Figure 1). Collectively, it seems that malignant B cells activate BTK-independent compensatory survival pathways under ibrutinib treatment, mainly PI3K/mTOR/Akt pathway and adhesion. Activating the NFκB pathway also induces some degree of ibrutinib resistance. This was described mainly by genetic mechanisms as mentioned before, but the non-canonical NFκB pathway together with MAPK signaling can also be activated and thus protect the cells from ibrutinib in MCL cells by activating CD40 (148, 149).
Activating the PI3K pathway is a well-known mechanism that rescues BCR deficient mature B cells from apoptosis (150, 151). It is therefore not surprising that more and more studies see a similar mechanism in malignant B cells by which they overcome BTK inhibition. Activated Akt has been observed in ibrutinib-resistant CLL and DLBCL cell lines, together with downregulated FoxO3a and PTEN levels (Figure 1) (121). Activating Akt/MAPK via CD79B overexpression has been sufficient to induce ibrutinib resistance in primary ABC-DLBCL (152). Ibrutinib also synergizes with CRISPR-Cas9 knockout of PI3Kδ (91). PI3K pathway activation was observed in B cell lymphoma patient-derived xenograft models with acquired resistance to ibrutinib and the growth of these tumors was blocked by combination of ibrutinib and idelalisib (122).
However, activation of PI3K/mTOR/Akt signaling after ibrutinib has been best described in MCL. Activated Akt and Erk levels, but not BTK, correlate with the response to ibrutinib in MCL cell lines and furthermore, ibrutinib-responsive patients have dephosphorylated Akt as opposed to non-responsive patients (69, 153). Ibrutinib in MCL strengthens cell adhesion via integrin dimer VLA4 formed by β1-integrin and CD49d and activates the PI3K/Akt pathway in this way (Figure 1). Zhao et al. described this phenomenon by showing that ibrutinib-resistant cells have a higher β1-integrin expression that helps to form an ILK-Rictor complex that activates a pro-survival mTORC2/Akt pathway. This was disrupted by ibrutinib in combination with dual mTOR1/2 inhibitor AZD8055 or dual PI3K-mTOR1/2 inhibitor BEZ235 (123). Here, it is also worth mentioning that ibrutinib-resistant MCL samples upregulate the mTOR signaling pathway (as well as genes involved in cell cycle regulation and MYC targets) compared to ibrutinib-sensitive cells (87). A similar compensatory survival mechanism was seen by Guan and colleagues who demonstrated that stromal cells protect MCL cells from ibrutinib-induced death via their interaction with VLA4. A combination of ibrutinib with VLA4 blockage or with an inhibitor of PI3K catalytic p110α subunit disrupted the interaction and overcame the resistance (124).
In CLL, the cells lose the ability to adhere to fibronectin almost completely and partly to stromal cells when treated with ibrutinib in vitro for a short period of time (63, 154). Interestingly though, BCR stimulation activates VLA4 in CLL cells exposed to ibrutinib for an extended time via a BTK-independent manner involving PI3K. Also, higher CD49d levels in patients prevent ibrutinib-induced lymphocytosis and cause a lower nodal response. This translates into a shorter progression-free survival in ibrutinib treated patients (42). Analogically, it has been described that ibrutinib induces CXCR4 expression on cell-surface, which might translate to a paradoxically increased responsiveness of these cells to chemokine ligands, however, it is likely that the response to chemokines in this context is not completely physiological since BTK is involved in the chemokine pathway (30, 155). Time to progression can also be predicted by cell-surface IgM levels prior to ibrutinib treatment, suggesting another non-genetic mechanism of ibrutinib resistance in CLL, potentially similar to the CD79B overexpression in DLBCL (Figure 1) (152, 156). In CLL, the mechanism might be explained by BCR signaling bypassing BTK upon BCR crosslinking as ibrutinib is not able to properly inhibit Ca2+ mobilization and Erk1/2 phosphorylation when surface IgM levels are high (156). Moreover, cell-surface IgM levels rise during ibrutinib treatment in CLL patients, although this might depend on the time since the start of therapy (30, 157).
Activating the MAPK pathway might also be one of the compensatory mechanisms for BTK inhibition, as noted in a recent study. Upregulation of the genes involved in MAPK was observed by Forestieri et al. in residual CLL cells after ibrutinib treatment and in addition, acquired mutations in BRAF, NRAS, and KRAS were found in a fraction of patients (135). This might explain the observed synergy between MEK inhibitors with “BCR inhibitors” in B cell lymphomas (125, 136).
Besides the described pathways, other ibrutinib resistance mechanisms are also possible and fit the biology of B cells. It has been shown that MYC acts as a key downstream BCR effector, and its over-expression can rescue the absence of BCR activity in some B cells (158, 159). Indeed, upregulation of MYC has been observed in ibrutinib-resistant MCL cell lines and this resistance can be reversed by inhibiting HSP90 (134). Protection from ibrutinib can also be provided by cells in the microenvironment (133, 160, 161). On the other hand, CLL cells resistant to BTK inhibition recover the ability to produce and respond to IL4 and require less T cell help for growth (162). Lymphoma relapse can potentially also arise from cancer stem cells described in MCL and FL. Their quiescent phenotype, together with high ABC-transporter activity gives them general drug-resistant properties (163–166). Indeed, the MCL-initiating cells were found to be resistant to ibrutinib and could be eliminated by inhibiting Wnt signaling pathway whose genes were overexpressed in these cancer stem cells (146).
Targeting Ibrutinib Resistance
There are several potential ways to overcome ibrutinib resistance such as i) in cases with specific BTK mutations using third generation BTK inhibitors which do not target C481, or PROTAC mediated BTK degradation, ii) using different molecular targets once a patient is resistant to ibrutinib, or iii) preventing the resistance by a more rapid B cell elimination that would lower the chance of developing resistance or activating compensatory survival pathways (Figure 1, Tables 1 and 2). Additionally, time-limited or more selective treatment would likely lower toxicities in patients as up to 40% of CLL patients discontinue ibrutinib therapy, which is caused mostly by the toxicities (11, 167).
Unfortunately, second-generation BTK inhibitors such as acalabrutinib, zanubrutinib, or tirabrutinib are not able to overcome the resistance caused by BTKC481S since they bind to the same protein region as ibrutinib. Their advantage is their higher selectivity with less off-targets (Supplementary Table 1) and lesser toxicities than ibrutinib, making them more feasible for patients intolerant to ibrutinib (168–172). Acalabrutinib has recently been approved for CLL and MCL and zanubrutinib for MCL (171, 173). The solution to BTKC481S mutations could be the use of non-covalent third-generation BTK inhibitors that are able to inhibit the kinase’s activity independently of C481S (Supplementary Table 1). BTK inhibitors such as fenebrutinib (GDC-0853), LOXO-305, or vecabrutinib are currently in the early phases of clinical testing even for patients with BTKC481S (72–74). ARQ 531 has shown better efficacy than ibrutinib in murine models resembling Richter transformation, targets CLL not only with BTK but also PLCG2 mutations and has off-target activity against kinases in Erk signaling and kinases in the Src family (75). The off-targets of various BTK inhibitors are summarized in the Supplementary Table 1. A different but also promising therapeutic strategy is provided by PROTAC which degrades its target with E3 ligase. PROTAC-induced BTK degradation is highly selective and effective in treating BTKC481S ibrutinib-resistant mouse models (76, 77). CLL cells with R665W and L845F mutations in PLCG2 are sensitive to RAC2 or SYK and LYN inhibition (78, 79). Inhibiting RAC2 or its binding partner VAV1 is synergistic with ibrutinib also in DLBCL just like inhibiting STAT3 or SYK together with ibrutinib in MYD88 mutated DLBCL (89–91).
Another approach would be combining ibrutinib with compensatory survival pathway inhibitors such as PI3K/mTOR/Akt or NFκB (Table 2). As mentioned before, ibrutinib synergizes with PI3K/mTOR/Akt pathway inhibitors in MCL, CLL and DLBCL (121–126). A combination of ibrutinib and umbralisib, a next-generation PI3Kδ inhibitor, was studied in a clinical trial in CLL and MCL patients with promising results (127). It has been shown that ibrutinib increases CLL-cell sensitivity to mTOR inhibitors as well as proteasome and PLK1 inhibitors (128). Targeting mTOR combined with ibrutinib was also suggested in ABC-DLBCL by Phelan et al. as mTORC1/2 inhibitor AZD2014 further attenuates formation of MYD88/TLR9/BCR super-complex when compared to ibrutinib alone (115). CC-115, a dual mTOR/DNA-dependent protein kinase inhibitor, is now in a clinical trial and is able to revert CD40-mediated resistance to venetoclax and also inhibits BCR signaling in CLL patients with acquired idelalisib resistance (174). Inhibiting the PI3K/mTOR/Akt pathway has been shown to be successful also in in vitro drug screening in DLBCL, where PI3K inhibitors synergized with ibrutinib (129). The same study, confirmed by others, proved a synergy between ibrutinib and inhibition of IRAK4, a mediator for TLR and NFκB activation whose targeting is studied not only in DLBCL but in CLL as well (129, 175, 176). Furthermore, the synergy with ibrutinib was also seen in combination with selinexor, an XPO1 inhibitor, although this seems to be regulated again via the PI3K/mTOR/Akt pathway as selinexor restores a nuclear abundance of FoxO3a and PTEN after ibrutinib treatment, resulting in the inhibition of PI3K/mTOR/Akt signaling activation (Figure 1) (121, 129). Absence of tumor suppressors FoxO3a and PTEN in the nucleus could be an explanation for ibrutinib resistance in 2p+ CLL patients who overexpress XPO1 and why selinexor and next-generation XPO1 inhibitors seem to be efficient in preclinical CLL and MCL models where it reduces NFκB binding to DNA (92, 130–132). Another molecule that could be targeted together with BTK is the MP3K14 enzyme, a member of an alternative NFκB pathway constitutively activated in ibrutinib-resistant MCL patients due to mutations in BIRC3, TRAF2, or TRAF3 (Figure 1) (84, 85).
An attractive strategy to increase the treatment efficacy is combining drugs that are already approved in therapy. Recently, ibrutinib has been shown to be efficient with venetoclax, a BH3 mimetic that inhibits anti-apoptotic molecule BCL2 and is approved for CLL treatment (125, 137, 138, 177). This combination is rational as ibrutinib induces BCL2 expression and on the other hand, decreases anti-apoptotic MCL1 levels, which can be a cause of venetoclax resistance (Figure 1) (30, 139–141). Higher BCL2 levels have also been found in DLBCL patients with poorer response to ibrutinib therapy (142). Furthermore, ibrutinib inhibits malignant cell proliferation while venetoclax targets preferentially resting sub-populations, potentially explaining the synergy of these two drugs in B cell malignancies (129, 143). Combining ibrutinib with fludarabine, a purine analog commonly used together with cyclophosphamide and anti-CD20 monoclonal antibody rituximab to treat CLL patients, has been shown to be synergic ex vivo (125). Ibrutinib with immune modulator lenalidomide and rituximab is under investigation in DLBCL and MCL but has not been successful in CLL (178–181). A profoundly studied possibility for therapy is combining “BCR inhibitors” with widely-used anti-CD20 monoclonal antibodies (28). However, adding rituximab to ibrutinib did not bring any clinical benefit and this is likely due to ibrutinib downregulating CD20 levels and/or interfering with effector cell functions (28, 30, 182, 183). Furthermore, ibrutinib has been shown to negatively regulate anti-CD20 induced apoptosis in MCL cell lines (184). A combination of ibrutinib with a more efficient anti-CD20 antibody, obinutuzumab, is now approved for CLL therapy. However, the real benefit of obinutuzumab still remains unclear as the control arm of the clinical trial was chlorambucil with obinutuzumab (185). In the ELEVATE clinical trial, acalabrutinib or acalabrutinib plus obinutuzumab were both superior to chlorambucil plus obinutuzumab (170). Even though it is not yet clear whether this combination reduces the occurrence of acquired BTK inhibition resistance, it is true that re-distributing malignant cells to the peripheral blood makes malignant cells more susceptible to monoclonal antibodies (145, 186). An interesting option is ibrutinib combined with anti-ROR1 monoclonal antibody in CLL, which is expected to have a great specificity for malignant B cells, and ROR1 levels are not reduced during ibrutinib therapy (187). Promising results have also been obtained from a fixed-duration treatment with venetoclax and obinutuzumab in CLL (188). Therapy nowadays aims for a time-limited treatment setting as it would lower the selection pressure, leave shorter time for cells to compensate for inhibition of targeted pathway, and reduce toxicities in patients.
It has been reported that patients who relapse or are intolerant to one kinase-inhibitor benefit from a change to different small-molecule inhibitors rather than chemotherapy (189). Novel therapeutic targets and drugs are therefore being investigated. Promising results have been obtained by targeting different players in BCR signaling such as PKCβ or MALT1 whose inhibition is effective in ABC-DLBCL, MCL, and naïve as well as ibrutinib-resistant CLL (190–193). Cerdulatinib, a dual SYK/JAK-STAT inhibitor, targets BCR signaling and is also able to overcome microenvironmental protection and blocks proliferation in ibrutinib-resistant primary CLL samples and BTKC481S lymphoma cell lines (Figure 1) (133). Directly targeting microenvironmental interactions, migration and adhesion could also have potential use in therapy of ibrutinib-resistant patients via the use of natalizumab or CXCR4 inhibitor plerixafor (45, 145). Lastly, malignant cells of ibrutinib-resistant CLL and MCL patients show metabolic reprogramming, which has also been suggested as a possible therapeutic target (87, 144).
Effects of Idelalisib and Mechanisms of Resistance
Another molecule in BCR signaling widely therapeutically targeted in B cell malignancies is PI3K. Activated PI3K/Akt axis in B cell malignancies and its signaling pathway’s role in cell survival makes it an attractive therapeutic target (3, 151, 194, 195). PI3K exists in four catalytic isoforms: p110α, p110β, p110γ, and p110δ. p110α (PI3Kα) and p110δ (PI3Kδ) are both needed for “tonic” (antigen-independent) BCR signaling, while only p110δ is needed for antigen-induced BCR signaling (196). The PI3Kδ isoform is targeted by widely-used idelalisib, which has been approved for the treatment of CLL, FL, and non-Hodgkin lymphomas. Idelalisib not only thwarts the PI3K/Akt/mTOR pathway but also inhibits cell migration towards chemokines and adhesion to stromal cells, which, just like with ibrutinib, leads to an initial increase in the number of lymphocytes in the peripheral blood caused by lymphocyte migration out of the tissues in CLL. This is accompanied by a reduction in Akt phosphorylation and other downstream effectors as well as by apoptosis induction (8, 197–199). Unfortunately, due to serious adverse effects and infections, it has been suggested to primarily give idelalisib to CLL patients with progression on ibrutinib or indolent NHL patients with progression on two prior therapies (14, 200, 201). As already mentioned, promising results have recently been seen in a clinical trial of a combination of ibrutinib and a next-generation dual PI3Kδ/CK1ϵ inhibitor, umbralisib (127). Furthermore, the importance of the p110γ subunit is now emerging in CLL. Its activation does not respond to BCR stimulation but increases in response to CD40L/IL4 and cells with overexpressed PI3Kγ show an enhanced cell migration towards CXCL12. A dual PI3Kδ/γ inhibitor, duvelisib, seems to have a bigger impact on cell migration than idelalisib alone and is now approved to treat CLL/SLL and FL (197). Another PI3K inhibitor, copanlisib, is approved for relapsed/refractory FL. It is a pan-class I PI3K inhibitor that inhibits all four PI3K isoforms with higher selectivity against PI3Kα and PI3Kδ (202). Its advantage lies not only in its molecular mechanism as inhibition of other PI3K isoforms can lower viability and migration of B cells, but it also seems to cause fewer adverse events in patients when compared to idelalisib (197, 203–205).
Despite idelalisib’s and other PI3K inhibitors’ ability to initially control the disease in some patients, a fraction of patients develop resistance. Unfortunately, unlike in ibrutinib treated patients, the mechanisms of resistance remain mostly unclear (8, 206). No recurrent mutations were found in patients progressing on idelalisib nor have they been found in a mouse model resistant to PI3Kδ inhibition (207, 208). Two studies have shown the role of MAPK signaling in resisting PI3Kδ inhibition. Firstly, Murali et al. confirmed activating mutations in MAPK pathway leading to Erk phosphorylation in patients resistant to PI3K inhibition and suggested that blocking Erk might sensitize patients to PI3K inhibitors (Figure 1) (209). Secondly, a non-genetic mechanism of resistance was found in PI3Kδ resistant mice, where the upregulation of insulin-like growth factor 1 receptor (IGF1R) led to MAPK signaling activation. IGF1R upregulation was caused by FoxO1 and GSK3β and the resistance was resolved by inhibiting the receptor with linsitinib (208). Initial experiments with copanlisib point to IL6 signaling as a main player in copanlisib resistance; levels of IL6 and phosphorylation of STAT5, Akt, p70S6K, and MAPK were increased in copanlisib-resistant B cell lymphoma cell lines and the resistance was reversible by JAK inhibitor (210).
Extensive research is needed in order to reveal the resistance mechanisms for PI3K inhibitors and it seems that they might be more complex than in BTK inhibition. Despite their adverse effects in patients and the emergence of BCL2 inhibitors and next-generation BTK inhibitors, the PI3K/mTOR/Akt signaling pathway plays an important role in B cell malignancy pathogenesis and its inhibition might find its therapeutic place.
Discussion
Targeting BCR signaling is now a commonly used therapy strategy for B cell malignancies. Unfortunately, some patients are primarily resistant to “BCR inhibitors” or develop resistance during the course of treatment. Furthermore, the now required continuous treatment often leads to toxicities and forces a change of therapy. BTK and PLCG2 mutations are the most common and best-described mechanisms of resistance to BTK inhibitor ibrutinib, although recurrent mutations in other genes or aberrations in larger chromosomal regions have been described as being responsible for the resistance across the B cell malignancies. Interestingly, malignant B cells are able to overcome BTK inhibition by non-genetic mechanisms as well. These include activation of compensatory survival pathway, such as PI3K/mTOR/Akt, NFkB, or MAPK signaling pathways. Compensatory survival is also provided by the upregulation of anti-apoptotic BCL2, MYC or adhesion involved integrins. Even though a lot is known about BTK inhibition, PI3K inhibitor resistance remains largely unclear. Several studies point to MAPK pathway activation as a compensatory mechanism to PI3K inhibition, but further research is needed in this area.
Resistance caused by BTK mutations can be solved by third-generation BTK inhibitors now in clinical trials or by BTK degradation by PROTAC-based compounds; mutations in PLCG2 by combining BTK inhibition with RAC2 or SYK and LYN inhibition. Compensatory survival by upregulating other signaling pathways could be solved by combining several inhibitors as well. The synergy between blockage of BTK and the PI3K/mTOR/Akt pathway has been shown repeatedly in CLL, DLBCL and MCL. It remains largely unknown how the non-coding part of the genome influencing BCR signaling is affected by “BCR inhibitors” and if (de)regulation of these molecules could contribute to therapy resistance or be directly used as a therapeutic target (20, 31, 34, 35). Novel therapeutic targets and strategies are still being investigated and their inhibition is tested alone or in combination with “BCR inhibitors”. These combinations should be supported by analyzing the responses of malignant cells to individual drugs and using multi-omics to identify possible compensatory signaling pathways to be co-targeted by small-molecule inhibitors. Furthermore, co-targeting two kinases in seemingly the same pathway can also have a synergistic effect, as observed by BTK and PI3K inhibition’s synergy. Analyzing individual patient-to-patient response to “BCR inhibitors” could help to identify specific compensatory pathways and this could help to define a personalized combinatorial therapy. However, it is likely that there might be some universal responses to “BCR inhibitors” in each B cell malignancy and this could follow pre-existing mechanisms that allow B cells to survive for an extended time without antigen encounter. Single cell analysis of the response to BTK/PI3K inhibitors in multiple patients could help to understand if the response or adaptation to therapy follows a generally uniform course or if there are major intra- and inter- patient specific mechanisms. Besides transcriptomics and proteomics, research should focus on describing the cells’ immunophenotypic profiles after “BCR inhibition” to identify surface molecules that could be targeted by monoclonal antibodies, since this can be of high specificity for cancer cells and low toxicity in general. The time dynamics of changes during therapy should also be studied in order to describe the mechanisms of early and long-term adaptation and potentially identify an optimal time point for adding a second drug in combination. Improving clinical efficacy of drug combinations containing “BCR inhibitors” should allow a time-limited treatment with a deep molecular response, decreased chance of resistance and limited toxicities associated with long-term therapy.
Author Contributions
LO and MM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Czech Science Foundation (project No. 20-02566S). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 802644). Supported by MH CZ - DRO (FNBr, 65269705). This research was carried out under the project CEITEC 2020 (LQ1601) with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustainability Programme II. LO is a Brno Ph.D. Talent Scholarship Holder - Funded by the Brno City Municipality.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Lenka Kostalova (MU) for her help with figure preparation, and to Vaclav Seda (MU) for interesting discussions on the topic.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.591577/full#supplementary-material
References
1. Devan J, Janikova A, Mraz M. New concepts in follicular lymphoma biology: From BCL2 to epigenetic regulators and non-coding RNAs. Semin Oncol (2018) 45:291–302. doi: 10.1053/j.seminoncol.2018.07.005
2. Dühren-von Minden M, Übelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature (2012) 489:309–12. doi: 10.1038/nature11309
3. Ringshausen I, Schneller F, Bogner C, Hipp S, Duyster J, Peschel C, et al. Constitutively activated phosphatidylinositol-3 kinase (PI-3K) is involved in the defect of apoptosis in B-CLL: association with protein kinase Cdelta. Blood (2002) 100:3741–8. doi: 10.1182/blood-2002-02-0539
4. Saba NS, Liu D, Herman SEM, Underbayev C, Tian X, Behrend D, et al. Pathogenic role of B-cell receptor signaling and canonical NF-κB activation in mantle cell lymphoma. Blood (2016) 128:82–92. doi: 10.1182/blood-2015-11-681460
5. Seda V. Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol (2015) 94:193–205. doi: 10.1111/ejh.12427
6. Lew TE, Anderson MA, Seymour JF. Promises and pitfalls of targeted agents in chronic lymphocytic leukemia. Cancer Drug Resist (2020) 3:415–44. doi: 10.20517/cdr.2019.108
7. Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med (2014) 370:2286–94. doi: 10.1056/NEJMoa1400029
8. Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood (2014) 123:3390–7. doi: 10.1182/blood-2013-11-535047
9. Wilson WH, Gerecitano JF, Goy A, de Vos S, Kenkre VP, Barr PM, et al. The Bruton’s Tyrosine Kinase (BTK) Inhibitor, Ibrutinib (PCI-32765), Has Preferential Activity in the ABC Subtype of Relapsed/Refractory De Novo Diffuse Large B-Cell Lymphoma (DLBCL): Interim Results of a Multicenter, Open-Label, Phase 2 Study. Blood (2012) 120:686–6. doi: 10.1182/blood.V120.21.686.686
10. Jain P, Kanagal-Shamanna R, Zhang S, Ahmed M, Ghorab A, Zhang L, et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br J Haematol (2018) 183:578–87. doi: 10.1111/bjh.15567
11. Mato AR, Nabhan C, Thompson MC, Lamanna N, Brander DM, Hill B, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica (2018) 103:874–9. doi: 10.3324/haematol.2017.182907
12. Bartlett NL, Costello BA, LaPlant BR, Ansell SM, Kuruvilla JG, Reeder CB, et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: a phase 2 consortium trial. Blood (2018) 131:182–90. doi: 10.1182/blood-2017-09-804641
13. Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. New Engl J Med (2015) 372:1430–40. doi: 10.1056/NEJMoa1501548
14. Salles G, Schuster SJ, de Vos S, Wagner-Johnston ND, Viardot A, Blum KA, et al. Efficacy and safety of idelalisib in patients with relapsed, rituximab- and alkylating agent-refractory follicular lymphoma: a subgroup analysis of a phase 2 study. Haematologica (2017) 102:e156–9. doi: 10.3324/haematol.2016.151738
15. Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WWA, Zürn C, et al. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol Cell (2002) 10:1057–69. doi: 10.1016/S1097-2765(02)00739-6
16. Aiba Y, Kameyama M, Yamazaki T, Tedder TF, Kurosaki T. Regulation of B-cell development by BCAP and CD19 through their binding to phosphoinositide 3-kinase. Blood (2008) 111:1497–503. doi: 10.1182/blood-2007-08-109769
17. Okada T, Maeda A, Iwamatsu A, Gotoh K, Kurosaki T. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity (2000) 13:817–27. doi: 10.1016/S1074-7613(00)00079-0
18. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307:1098–101. doi: 10.1126/science.1106148
19. Ingham RJ, Santos L, Dang-Lawson M, Holgado-Madruga M, Dudek P, Maroun CR, et al. The Gab1 docking protein links the b cell antigen receptor to the phosphatidylinositol 3-kinase/Akt signaling pathway and to the SHP2 tyrosine phosphatase. J Biol Chem (2001) 276:12257–65. doi: 10.1074/jbc.M010590200
20. Mraz M, Chen L, Rassenti LZ, Ghia EM, Li H, Jepsen K, et al. miR-150 influences B-cell receptor signaling in chronic lymphocytic leukemia by regulating expression of GAB1 and FOXP1. Blood (2014) 124:84–95. doi: 10.1182/blood-2013-09-527234
21. Saito K, Scharenberg AM, Kinet JP. Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates Btk. J Biol Chem (2001) 276:16201–6. doi: 10.1074/jbc.M100873200
22. Park H, Wahl MI, Afar DE, Turck CW, Rawlings DJ, Tam C, et al. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity (1996) 4:515–25. doi: 10.1016/S1074-7613(00)80417-3
23. Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science (1996) 271:822–5. doi: 10.1126/science.271.5250.822
24. Shinohara H, Yasuda T, Aiba Y, Sanjo H, Hamadate M, Watarai H, et al. PKC beta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J Exp Med (2005) 202:1423–31. doi: 10.1084/jem.20051591
25. Shinohara H, Kurosaki T. Comprehending the complex connection between PKCbeta, TAK1, and IKK in BCR signaling. Immunol Rev (2009) 232:300–18. doi: 10.1111/j.1600-065X.2009.00836.x
26. Kang SW, Wahl MI, Chu J, Kitaura J, Kawakami Y, Kato RM, et al. PKCbeta modulates antigen receptor signaling via regulation of Btk membrane localization. EMBO J (2001) 20:5692–702. doi: 10.1093/emboj/20.20.5692
27. Chan VW, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity (1997) 7:69–81. doi: 10.1016/S1074-7613(00)80511-7
28. Pavlasova G, Mraz M. The regulation and function of CD20: an “enigma” of B-cell biology and targeted therapy. Haematologica (2020) 105:1494–506. doi: 10.3324/haematol.2019.243543
29. Pavlasova G, Borsky M, Svobodova V, Oppelt J, Cerna K, Novotna J, et al. Rituximab primarily targets an intra-clonal BCR signaling proficient CLL subpopulation characterized by high CD20 levels. Leukemia (2018) 32:2028–31. doi: 10.1038/s41375-018-0211-0
30. Pavlasova G, Borsky M, Seda V, Cerna K, Osickova J, Doubek M, et al. Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis. Blood (2016) 128:1609–13. doi: 10.1182/blood-2016-04-709519
31. Cerna K, Oppelt J, Chochola V, Musilova K, Seda V, Pavlasova G, et al. MicroRNA miR-34a downregulates FOXP1 during DNA damage response to limit BCR signalling in chronic lymphocytic leukaemia B cells. Leukemia (2019) 33:403–14. doi: 10.1038/s41375-018-0230-x
32. Cerna K, Mraz M. p53 limits B cell receptor (BCR) signalling: a new role for miR-34a and FOXP1. Oncotarget (2018) 9:36409–10. doi: 10.18632/oncotarget.26376
33. Cui B, Chen L, Zhang S, Mraz M, Fecteau J-F, Yu J, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood (2014) 124:546–54. doi: 10.1182/blood-2014-03-559690
34. Musilova K, Devan J, Cerna K, Seda V, Pavlasova G, Sharma S, et al. miR-150 downregulation contributes to the high-grade transformation of follicular lymphoma by upregulating FOXP1 levels. Blood (2018) 132:2389–400. doi: 10.1182/blood-2018-06-855502
35. Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia (2015) 29:1004–17. doi: 10.1038/leu.2014.351
36. Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev (2009) 23:2839–49. doi: 10.1101/gad.1861409
37. Palacios F, Abreu C, Prieto D, Morande P, Ruiz S, Fernández-Calero T, et al. Activation of the PI3K/AKT pathway by microRNA-22 results in CLL B-cell proliferation. Leukemia (2015) 29:115–25. doi: 10.1038/leu.2014.158
38. Psathas JN, Doonan PJ, Raman P, Freedman BD, Minn AJ, Thomas-Tikhonenko A. The Myc-miR-17-92 axis amplifies B-cell receptor signaling via inhibition of ITIM proteins: a novel lymphomagenic feed-forward loop. Blood (2013) 122:4220–9. doi: 10.1182/blood-2012-12-473090
39. Carrasco YR. Batista FD. B-cell activation by membrane-bound antigens is facilitated by the interaction of VLA-4 with VCAM-1. EMBO J (2006) 25:889–99. doi: 10.1038/sj.emboj.7600944
40. Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, et al. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood (2010) 115:4497–506. doi: 10.1182/blood-2009-07-233692
41. Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S, et al. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med (2003) 198:1539–50. doi: 10.1084/jem.20011866
42. Tissino E, Benedetti D, Herman SEM, Ten Hacken E, Ahn IE, Chaffee KG, et al. Functional and clinical relevance of VLA-4 (CD49d/CD29) in ibrutinib-treated chronic lymphocytic leukemia. J Exp Med (2018) 215:681–97. doi: 10.1084/jem.20171288
43. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood (2000) 96:2655–63. doi: 10.1182/blood.V96.8.2655.h8002655_2655_2663
44. Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood (1999) 94:3658–67. doi: 10.1182/blood.V94.11.3658.423k11_3658_3667
45. Burger M, Hartmann T, Krome M, Rawluk J, Tamamura H, Fujii N, et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood (2005) 106:1824–30. doi: 10.1182/blood-2004-12-4918
46. Aguilar-Hernandez MM, Blunt MD, Dobson R, Yeomans A, Thirdborough S, Larrayoz M, et al. IL-4 enhances expression and function of surface IgM in CLL cells. Blood (2016) 127:3015–25. doi: 10.1182/blood-2015-11-682906
47. Steele AJ, Prentice AG, Cwynarski K, Hoffbrand AV, Hart SM, Lowdell MW, et al. The JAK3-selective inhibitor PF-956980 reverses the resistance to cytotoxic agents induced by interleukin-4 treatment of chronic lymphocytic leukemia cells: potential for reversal of cytoprotection by the microenvironment. Blood (2010) 116:4569–77. doi: 10.1182/blood-2009-09-245811
48. Ghia P, Strola G, Granziero L, Geuna M, Guida G, Sallusto F, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol (2002) 32:1403–13. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403<3.0.CO;2-Y
49. Herishanu Y, Pérez-Galán P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The Lymph Node Microenvironment Promotes B-cell Receptor Signaling, NF-kappaB Activation, and Tumor Proliferation in Chronic Lymphocytic Leukemia. Blood (2011) 117:563–74. doi: 10.1182/blood-2010-05-284984
50. Ruan J, Hyjek E, Kermani P, Christos PJ, Hooper AT, Coleman M, et al. Magnitude of stromal hemangiogenesis correlates with histologic subtype of non-Hodgkin’s lymphoma. Clin Cancer Res (2006) 12:5622–31. doi: 10.1158/1078-0432.CCR-06-1204
51. Burger JA, Quiroga MP, Hartmann E, Bürkle A, Wierda WG, Keating MJ, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood (2009) 113:3050–8. doi: 10.1182/blood-2008-07-170415
52. Bürkle A, Niedermeier M, Schmitt-Gräff A, Wierda WG, Keating MJ, Burger JA. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood (2007) 110:3316–25. doi: 10.1182/blood-2007-05-089409
53. Slinger E, Thijssen R, Kater AP, Eldering E. Targeting antigen-independent proliferation in chronic lymphocytic leukemia through differential kinase inhibition. Leukemia (2017) 31:2601–7. doi: 10.1038/leu.2017.129
54. Chang BY, Francesco M, De Rooij MFM, Magadala P, Steggerda SM, Huang MM, et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood (2013) 122:2412–24. doi: 10.1182/blood-2013-02-482125
55. Woyach JA, Smucker K, Smith LL, Lozanski A, Zhong Y, Ruppert AS, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood (2014) 123:1810–7. doi: 10.1182/blood-2013-09-527853
56. Satterthwaite AB, Cheroutre H, Khan WN, Sideras P, Witte ON. Btk dosage determines sensitivity to B cell antigen receptor cross-linking. Proc Natl Acad Sci USA (1997) 94:13152–7. doi: 10.1073/pnas.94.24.13152
57. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell (1993) 72:279–90. doi: 10.1016/0092-8674(93)90667-F
58. Burger JA, Li KW, Keating MJ, Sivina M, Amer AM, Garg N, et al. Leukemia cell proliferation and death in chronic lymphocytic leukemia patients on therapy with the BTK inhibitor ibrutinib. JCI Insight (2017) 2:e89904. doi: 10.1172/jci.insight.89904
59. Cheng S, Ma J, Guo A, Lu P, Leonard JP, Coleman M, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia (2014) 28:649–57. doi: 10.1038/leu.2013.358
60. Krysov S, Steele AJ, Coelho V, Linley A, Sanchez Hidalgo M, Carter M, et al. Stimulation of surface IgM of chronic lymphocytic leukemia cells induces an unfolded protein response dependent on BTK and SYK. Blood (2014) 124:3101–9. doi: 10.1182/blood-2014-04-567198
61. Ponader S, Chen S-S, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood (2012) 119:1182–9. doi: 10.1182/blood-2011-10-386417
62. Chen S-S, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia (2016) 30:833–43. doi: 10.1038/leu.2015.316
63. de Rooij MFM, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood (2012) 119:2590–4. doi: 10.1182/blood-2011-11-390989
64. Ortolano S, Hwang I-Y, Han S-B, Kehrl JH. Roles for phosphoinositide 3-kinases, Bruton’s tyrosine kinase, and Jun kinases in B lymphocyte chemotaxis and homing. Eur J Immunol (2006) 36:1285–95. doi: 10.1002/eji.200535799
65. Rendeiro AF, Krausgruber T, Fortelny N, Zhao F, Penz T, Farlik M, et al. Chromatin mapping and single-cell immune profiling define the temporal dynamics of ibrutinib response in CLL. Nat Commun (2020) 11:577. doi: 10.1038/s41467-019-14081-6
66. Guinn D, Ruppert AS, Maddocks K, Jaglowski S, Gordon A, Lin TS, et al. miR-155 expression is associated with chemoimmunotherapy outcome and is modulated by Bruton’s tyrosine kinase inhibition with Ibrutinib. Leukemia (2015) 29:1210–3. doi: 10.1038/leu.2014.344
67. Saleh LM, Wang W, Herman SEM, Saba NS, Anastas V, Barber E, et al. Ibrutinib downregulates a subset of miRNA leading to upregulation of tumor suppressors and inhibition of cell proliferation in chronic lymphocytic leukemia. Leukemia (2017) 31:340–9. doi: 10.1038/leu.2016.181
68. Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. New Engl J Med (2014) 370:2352–4. doi: 10.1056/NEJMc1402716
69. Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discovery (2014) 4:1022–35. doi: 10.1158/2159-8290.CD-14-0098
70. Epperla N, Shana’ah AY, Jones D, Christian BA, Ayyappan S, Maddocks K, et al. Resistance mechanism for ibrutinib in marginal zone lymphoma. Blood Adv (2019) 3:500–2. doi: 10.1182/bloodadvances.2018029058
71. Xu L, Tsakmaklis N, Yang G, Chen JG, Liu X, Demos M, et al. Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood (2017) 129:2519–25. doi: 10.1182/blood-2017-01-761726
72. Brandhuber B, Gomez E, Smith S, Eary T, Spencer S, Rothenberg SM, et al. LOXO-305, A Next Generation Reversible BTK Inhibitor, for Overcoming Acquired Resistance to Irreversible BTK Inhibitors. Clin Lymphoma Myeloma Leukemia (2018) 18:S216. doi: 10.1016/j.clml.2018.07.081
73. Byrd JC, Smith S, Wagner-Johnston N, Sharman J, Chen AI, Advani R, et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget (2018) 9:13023–35. doi: 10.18632/oncotarget.24310
74. Neuman LL, Ward R, Arnold D, Combs DL, Gruver D, Hill W, et al. First-in-Human Phase 1a Study of the Safety, Pharmacokinetics, and Pharmacodynamics of the Noncovalent Bruton Tyrosine Kinase (BTK) Inhibitor SNS-062 in Healthy Subjects. Blood (2016) 128:2032–2. doi: 10.1182/blood.V128.22.2032.2032
75. Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discovery (2018) 8:1300–15. doi: 10.1158/2159-8290.CD-17-1409
76. Sun Y, Ding N, Song Y, Yang Z, Liu W, Zhu J, et al. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia (2019) 33:2105–10. doi: 10.1038/s41375-019-0440-x
77. Sun Y, Zhao X, Ding N, Gao H, Wu Y, Yang Y, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res (2018) 28:779–81. doi: 10.1038/s41422-018-0055-1
78. Liu T-M, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood (2015) 126:61–8. doi: 10.1182/blood-2015-02-626846
79. Walliser C, Hermkes E, Schade A, Wiese S, Deinzer J, Zapatka M, et al. The Phospholipase Cγ2 Mutants R665W and L845F Identified in Ibrutinib-resistant Chronic Lymphocytic Leukemia Patients Are Hypersensitive to the Rho GTPase Rac2 Protein. J Biol Chem (2016) 291:22136–48. doi: 10.1074/jbc.M116.746842
80. Kanagal-Shamanna R, Jain P, Patel KP, Routbort M, Bueso-Ramos C, Alhalouli T, et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor-resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer (2019) 125:559–74. doi: 10.1002/cncr.31831
81. Wu C, de Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget (2016) 7:38180–90. doi: 10.18632/oncotarget.9500
82. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med (2015) 21:922–6. doi: 10.1038/nm.3884
83. Xue L, Apatira M, Sirisawad M, Chang B. Abstract 1742: Ibrutinib plus proteasome or MALT1 inhibitors overcome resistance to BCR antagonists in CARD11 mutant-expressing B-lymphoma cells. In: Experimental and Molecular Therapeutics. Philadelphia, PA: American Association for Cancer Research (2015). p. 1742–2. doi: 10.1158/1538-7445.AM2015-1742
84. Lenz G, Balasubramanian S, Goldberg J, Rizo A, Schaffer M, Phelps C, et al. Sequence variants in patients with primary and acquired resistance to ibrutinib in the phase 3 MCL3001 (RAY) trial. J Clin Oncol (2016) 34:7570–0. doi: 10.1200/JCO.2016.34.15_suppl.7570
85. Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chan FC, et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat Med (2014) 20:87–92. doi: 10.1038/nm.3435
86. Mohanty A, Sandoval N, Das M, Pillai R, Chen L, Chen RW, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget (2016) 7:73558–72. doi: 10.18632/oncotarget.12434
87. Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med (2019) 11(491):eaau1167. doi: 10.1126/scitranslmed.aau1167
88. Agarwal R, Chan Y-C, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med (2019) 25:119–29. doi: 10.1038/s41591-018-0243-z
89. Mondello P, Brea EJ, De Stanchina E, Toska E, Chang AY, Fennell M, et al. Panobinostat acts synergistically with ibrutinib in diffuse large B cell lymphoma cells with MyD88 L265 mutations. JCI Insight (2017) 2:1–14. doi: 10.1172/jci.insight.90196
90. Munshi M, Liu X, Chen JG, Xu L, Tsakmaklis N, Demos MG, et al. SYK is activated by mutated MYD88 and drives pro-survival signaling in MYD88 driven B-cell lymphomas. Blood Cancer J (2020) 10:12. doi: 10.1038/s41408-020-0277-6
91. Choi J, Phelan JD, Wright GW, Häupl B, Huang DW, Shaffer AL, et al. Regulation of B cell receptor-dependent NF-κB signaling by the tumor suppressor KLHL14. Proc Natl Acad Sci USA (2020) 117:6092–102. doi: 10.1073/pnas.1921187117
92. Cosson A, Chapiro E, Bougacha N, Lambert J, Herbi L, Cung H-A, et al. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: analysis of the central role of XPO1. Leukemia (2017) 31:1625–9. doi: 10.1038/leu.2017.100
93. Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun (2016) 7:11589. doi: 10.1038/ncomms11589
94. Jiménez C, Chan GG, Xu L, Tsakmaklis N, Kofides A, Demos MG, et al. Genomic evolution of ibrutinib-resistant clones in Waldenström macroglobulinaemia. Br J Haematol (2020) 189(6):1165–70. doi: 10.1111/bjh.16463
95. Guerrera ML, Tsakmaklis N, Xu L, Yang G, Demos M, Kofides A, et al. MYD88 mutated and wild-type Waldenström’s Macroglobulinemia: characterization of chromosome 6q gene losses and their mutual exclusivity with mutations in CXCR4. Haematologica (2018) 103:e408–11. doi: 10.3324/haematol.2018.190181
96. Ahn IE, Underbayev C, Albitar A, Herman SEM, Tian X, Maric I, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood (2017) 129:1469–79. doi: 10.1182/blood-2016-06-719294
97. Quinquenel A, Fornecker L-M, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood (2019) 134:641–4. doi: 10.1182/blood.2019000854
98. Scarfò L, Bonfiglio S, Sutton L-A, Ljungström V, Pandzic T, Cortese D, et al. BTK and PLCG2 Mutations In Patients With Chronic Lymphocytic Leukemia Relapsing On Ibrutinib: A European Research Initiative On CLL (ERIC) Study Based On Real-World Evidence. In: 25th Congress of the European Hematology Association (2020). Abstract S161. Available at: https://library.ehaweb.org/eha/2020/eha25th/294981/lydia.scarf.btk.and.plcg2.mutations.in.patients.with.chronic.lymphocytic.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Ace_id%3D1766%2Amarker%3D756.
99. Gángó A, Alpár D, Galik B, Marosvári D, Kiss R, Fésüs V, et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int J Cancer (2020) 146:85–93. doi: 10.1002/ijc.32502
100. Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol (2015) 1:80. doi: 10.1001/jamaoncol.2014.218
101. Sharma S, Galanina N, Guo A, Lee J, Kadri S, Van Slambrouck C, et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget (2016) 7:68833–41. doi: 10.18632/oncotarget.11932
102. Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTKC481S-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J Clin Oncol (2017) 35:1437–43. doi: 10.1200/JCO.2016.70.2282
103. Jones D, Woyach JA, Zhao W, Caruthers S, Tu H, Coleman J, et al. PLCG2 C2 domain mutations co-occur with BTK and PLCG2 resistance mutations in chronic lymphocytic leukemia undergoing ibrutinib treatment. Leukemia (2017) 31:1645–7. doi: 10.1038/leu.2017.110
104. Guarini A, Peragine N, Messina M, Marinelli M, Ilari C, Cafforio L, et al. Unravelling the suboptimal response of TP53 -mutated chronic lymphocytic leukaemia to ibrutinib. Br J Haematol (2019) 184:392–6. doi: 10.1111/bjh.15613
105. Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood (2015) 125:2497–506. doi: 10.1182/blood-2014-10-606038
106. Brown JR, Hillmen P, O’Brien S, Barrientos JC, Reddy NM, Coutre SE, et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia (2018) 32:83–91. doi: 10.1038/leu.2017.175
107. O’Brien S, Furman RR, Coutre S, Flinn IW, Burger JA, Blum K, et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood (2018) 131:1910–9. doi: 10.1182/blood-2017-10-810044
108. Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng P-H, Keats JJ, Wang H, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol (2008) 9:1364–70. doi: 10.1038/ni.1678
109. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol (2008) 9:1371–8. doi: 10.1038/ni.1676
110. Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood (2014) 123:1637–46. doi: 10.1182/blood-2013-09-525808
111. Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood (2013) 122:1222–32. doi: 10.1182/blood-2012-12-475111
112. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia (2015) 29:169–76. doi: 10.1038/leu.2014.187
113. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88(L265P) -directed survival signalling in Waldenström macroglobulinaemia cells. Br J Haematol (2015) 168:701–7. doi: 10.1111/bjh.13200
114. Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J Clin Oncol (2019) 37:1285–95. doi: 10.1200/JCO.18.02403
115. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature (2018) 560:387–91. doi: 10.1038/s41586-018-0290-0
116. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell (2013) 155:1309–22. doi: 10.1016/j.cell.2013.11.012
117. Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell (2015) 27:240–56. doi: 10.1016/j.ccell.2014.11.018
118. Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell (2012) 21:227–39. doi: 10.1016/j.ccr.2011.12.024
119. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature (2010) 468:973–7. doi: 10.1038/nature09626
120. Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell (2015) 27:397–408. doi: 10.1016/j.ccell.2015.02.005
121. Kapoor I, Li Y, Sharma A, Zhu H, Bodo J, Xu W, et al. Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis (2019) 10:924. doi: 10.1038/s41419-019-2158-0
122. Zhang L, Nomie K, Zhang H, Bell T, Pham L, Kadri S, et al. B-Cell Lymphoma Patient-Derived Xenograft Models Enable Drug Discovery and Are a Platform for Personalized Therapy. Clin Cancer Res (2017) 23:4212–23. doi: 10.1158/1078-0432.CCR-16-2703
123. Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun (2017) 8:14920. doi: 10.1038/ncomms14920
124. Guan J, Huang D, Yakimchuk K, Okret S. p110α Inhibition Overcomes Stromal Cell–Mediated Ibrutinib Resistance in Mantle Cell Lymphoma. Mol Cancer Ther (2018) 17:1090–100. doi: 10.1158/1535-7163.MCT-17-0784
125. Lukas M, Velten B, Sellner L, Tomska K, Hüellein J, Walther T, et al. Survey of ex vivo drug combination effects in chronic lymphocytic leukemia reveals synergistic drug effects and genetic dependencies. Leukemia (2020). doi: 10.1038/s41375-020-0846-5
126. de Rooij MFM, Kuil A, Kater AP, Kersten MJ, Pals ST, Spaargaren M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: a rationale for combination therapy. Blood (2015) 125:2306–9. doi: 10.1182/blood-2014-12-619163
127. Davids MS, Kim HT, Nicotra A, Savell A, Francoeur K, Hellman JM, et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: a multicentre phase 1-1b study. Lancet Haematol (2019) 6:e38–47. doi: 10.1016/S2352-3026(18)30196-0
128. Schmidl C, Vladimer GI, Rendeiro AF, Schnabl S, Krausgruber T, Taubert C, et al. Combined chemosensitivity and chromatin profiling prioritizes drug combinations in CLL. Nat Chem Biol (2019) 15:232–40. doi: 10.1038/s41589-018-0205-2
129. Schaffer M, Chaturvedi S, Davis C, Aquino R, Stepanchick E, Versele M, et al. Identification of potential ibrutinib combinations in hematological malignancies using a combination high-throughput screen. Leuk Lymphoma (2018) 59:931–40. doi: 10.1080/10428194.2017.1349899
130. Hing ZA, Fung HYJ, Ranganathan P, Mitchell S, El-Gamal D, Woyach JA, et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia (2016) 30:2364–72. doi: 10.1038/leu.2016.136
131. Hing ZA, Mantel R, Beckwith KA, Guinn D, Williams E, Smith LL, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood (2015) 125:3128–32. doi: 10.1182/blood-2015-01-621391
132. Ming M, Wu W, Xie B, Sukhanova M, Wang W, Kadri S, et al. XPO1 Inhibitor Selinexor Overcomes Intrinsic Ibrutinib Resistance in Mantle Cell Lymphoma via Nuclear Retention of IκB. Mol Cancer Ther (2018) 17:2564–74. doi: 10.1158/1535-7163.MCT-17-0789-ATR
133. Guo A, Lu P, Coffey G, Conley P, Pandey A, Wang YL. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: Cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget (2017) 8:12953–67. doi: 10.18632/oncotarget.14588
134. Lee J, Zhang LL, Wu W, Guo H, Li Y, Sukhanova M, et al. Activation of MYC, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv (2018) 2:2039–51. doi: 10.1182/bloodadvances.2018016048
135. Forestieri G, Terzi di Bergamo L, Loh JW, Spina V, Zucchetto A, Condoluci A, et al. Mechanisms Of Adaptation To Ibrutinib In High Risk Chronic Lymphocytic Leukemia. In: 25th Congress of the European Hematology Association (2020). Abstract S154. Available at: https://library.ehaweb.org/eha/2020/eha25th/294974/gabriela.forestieri.mechanisms.of.adaptation.to.ibrutinib.in.high.risk.chronic.html?f=listing%3D4%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Aspeaker%3D663813.
136. Gaudio E, Tarantelli C, Kwee I, Barassi C, Bernasconi E, Rinaldi A, et al. Combination of the MEK inhibitor pimasertib with BTK or PI3K-delta inhibitors is active in preclinical models of aggressive lymphomas. Ann Oncol (2016) 27:1123–8. doi: 10.1093/annonc/mdw131
137. Hillmen P, Rawstron AC, Brock K, Muñoz-Vicente S, Yates FJ, Bishop R, et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J Clin Oncol (2019) 37:2722–9. doi: 10.1200/JCO.19.00894
138. Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, Borthakur G, et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med (2019) 380:2095–103. doi: 10.1056/NEJMoa1900574
139. Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, et al. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clin Cancer Res (2015) 21:3705–15. doi: 10.1158/1078-0432.CCR-14-2809
140. Kater AP, Seymour JF, Hillmen P, Eichhorst B, Langerak AW, Owen C, et al. Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study. J Clin Oncol (2019) 37:269–77. doi: 10.1200/JCO.18.01580
141. Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, et al. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer (2017) 17:399. doi: 10.1186/s12885-017-3383-5
142. Kuo H-P, Ezell SA, Schweighofer KJ, Cheung LWK, Hsieh S, Apatira M, et al. Combination of Ibrutinib and ABT-199 in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma. Mol Cancer Ther (2017) 16:1246–56. doi: 10.1158/1535-7163.MCT-16-0555
143. Wang YLL, Franzen C, Wang S, Venkataraman G, Li L, Niu N, et al. Ibrutinib and Venetoclax Target Distinct Subpopulation of CLL Cells: Rationale for Drug Combination and Implication of Minimal Residual Disease Eradication. Blood (2019) 134:475–5. doi: 10.1182/blood-2019-125396
144. Galicia-Vázquez G, Aloyz R. Ibrutinib Resistance Is Reduced by an Inhibitor of Fatty Acid Oxidation in Primary CLL Lymphocytes. Front Oncol (2018) 8:411. doi: 10.3389/fonc.2018.00411
145. Mraz M, Zent CS, Church AK, Jelinek DF, Wu X, Pospisilova S, et al. Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin α-4-β-1 (VLA-4) with natalizumab can overcome this resistance. Br J Haematol (2011) 155:53–64. doi: 10.1111/j.1365-2141.2011.08794.x
146. Mathur R, Sehgal L, Braun FK, Berkova Z, Romaguerra J, Wang M, et al. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J Hematol Oncol (2015) 8:63. doi: 10.1186/s13045-015-0161-1
147. Chen JG, Liu X, Munshi M, Xu L, Tsakmaklis N, Demos MG, et al. BTKCys481Ser drives ibrutinib resistance via ERK1/2 and protects BTKwild-type MYD88-mutated cells by a paracrine mechanism. Blood (2018) 131:2047–59. doi: 10.1182/blood-2017-10-811752
148. Rauert-Wunderlich H, Rudelius M, Berberich I, Rosenwald A. CD40L mediated alternative NFκB-signaling induces resistance to BCR-inhibitors in patients with mantle cell lymphoma. Cell Death Dis (2018) 9:86. doi: 10.1038/s41419-017-0157-6
149. Sun Z, Luo L. Abstract 1298: CD40L-CD40 signaling on B-cell lymphoma response to BTK inhibitors. In: Experimental and Molecular Therapeutics. New Orleans, LA: American Association for Cancer Research (2016). p. 1298–8. doi: 10.1158/1538-7445.AM2016-1298
150. Lam K-P, Kühn R, Rajewsky K. In Vivo Ablation of Surface Immunoglobulin on Mature B Cells by Inducible Gene Targeting Results in Rapid Cell Death. Cell (1997) 90:1073–83. doi: 10.1016/S0092-8674(00)80373-6
151. Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell (2009) 139:573–86. doi: 10.1016/j.cell.2009.08.041
152. Kim JH, Kim WS, Ryu K, Kim SJ, Park C. CD79B limits response of diffuse large B cell lymphoma to ibrutinib. Leukemia Lymphoma (2016) 57:1413–22. doi: 10.3109/10428194.2015.1113276
153. Ma J, Lu P, Guo A, Cheng S, Zong H, Martin P, et al. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol (2014) 166:849–61. doi: 10.1111/bjh.12974
154. Herman SEM, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with Ibrutinib Inhibits BTK- and VLA-4-Dependent Adhesion of Chronic Lymphocytic Leukemia Cells In Vivo. Clin Cancer Res (2015) 21:4642–51. doi: 10.1158/1078-0432.CCR-15-0781
155. Chen L, Ouyang J, Wienand K, Bojarczuk K, Hao Y, Chapuy B, et al. CXCR4 upregulation is an indicator of sensitivity to B-cell receptor/PI3K blockade and a potential resistance mechanism in B-cell receptor-dependent diffuse large B-cell lymphomas. Haematologica (2020) 105:1361–8. doi: 10.3324/haematol.2019.216218
156. Chiodin G, Dutton D, Martino EA, Drennan S, Tracy I, Ondrisova L, et al. High Surface IgM Levels Associate with Shorter Response Duration and Bypass of the BTK Blockade during Ibrutinib Therapy in CLL Patients. Blood (2019) 134:1752–2. doi: 10.1182/blood-2019-128899
157. Drennan S, Chiodin G, D’Avola A, Tracy I, Johnson PW, Trentin L, et al. Ibrutinib Therapy Releases Leukemic Surface IgM from Antigen Drive in Chronic Lymphocytic Leukemia Patients. Clin Cancer Res (2019) 25:2503–12. doi: 10.1158/1078-0432.CCR-18-1286
158. Filip D, Mraz M. The role of MYC in the transformation and aggressiveness of “indolent” B-cell malignancies. Leuk Lymphoma (2020) 61:510–24. doi: 10.1080/10428194.2019.1675877
159. Varano G, Raffel S, Sormani M, Zanardi F, Lonardi S, Zasada C, et al. The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3β inhibition. Nature (2017) 546:302–6. doi: 10.1038/nature22353
160. Boissard F, Fournié J-J, Quillet-Mary A, Ysebaert L, Poupot M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J (2015) 5:e355–5. doi: 10.1038/bcj.2015.74
161. Jayappa KD, Portell CA, Gordon VL, Capaldo BJ, Bekiranov S, Axelrod MJ, et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv (2017) 1:933–46. doi: 10.1182/bloodadvances.2016004176
162. Chen S-S, Tam CS, Ramsay AG, Ravichandran P, Couto-Francisco NC, Ibrahim M, et al. CLL B Cells Develop Resistance to Ibrutinib By Reinvigorating the IL-4R - IL-4 Axis Blocked By Bruton’s Tyrosine Kinase Inhibitors Including Acalabrutinib and Zanubrutinib. Blood (2019) 134:477–7. doi: 10.1182/blood-2019-127255
163. Chen Z, Ayala P, Wang M, Fayad L, Katz RL, Romaguera J, et al. Prospective isolation of clonogenic mantle cell lymphoma-initiating cells. Stem Cell Res (2010) 5:212–25. doi: 10.1016/j.scr.2010.07.003
164. Jung HJ, Chen Z, McCarty N. Stem-like tumor cells confer drug resistant properties to mantle cell lymphoma. Leuk Lymphoma (2011) 52:1066–79. doi: 10.3109/10428194.2011.562570
165. Lee C-G, Das B, Lin TL, Grimes C, Zhang X, Lavezzi T, et al. A rare fraction of drug-resistant follicular lymphoma cancer stem cells interacts with follicular dendritic cells to maintain tumourigenic potential. Br J Haematol (2012) 158:79–90. doi: 10.1111/j.1365-2141.2012.09123.x
166. Medina DJ, Abass-Shereef J, Walton K, Goodell L, Aviv H, Strair RK, et al. Cobblestone-area forming cells derived from patients with mantle cell lymphoma are enriched for CD133+ tumor-initiating cells. PloS One (2014) 9:e91042. doi: 10.1371/journal.pone.0091042
167. O’Brien SM, Furman RR, Coutre SE, Flinn IW, Burger J, Blum K, et al. Five-Year Experience with Single-Agent Ibrutinib in Patients with Previously Untreated and Relapsed/Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Leukemia. Blood (2016) 128:233–3. doi: 10.1182/blood.V128.22.233.233
168. Awan FT, Schuh A, Brown JR, Furman RR, Pagel JM, Hillmen P, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv (2019) 3:1553–62. doi: 10.1182/bloodadvances.2018030007
169. Barf T, Covey T, Izumi R, van de Kar B, Gulrajani M, van Lith B, et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J Pharmacol Exp Ther (2017) 363:240–52. doi: 10.1124/jpet.117.242909
170. Sharman JP, Egyed M, Jurczak W, Skarbnik A, Pagel JM, Flinn IW, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet (2020) 395:1278–91. doi: 10.1016/S0140-6736(20)30262-2
171. Tam CS, Trotman J, Opat S, Burger JA, Cull G, Gottlieb D, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood (2019) 134:851–9. doi: 10.1182/blood.2019001160
172. Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, et al. Durable response with single-agent acalabrutinib in patients with relapsed or refractory mantle cell lymphoma. Leukemia (2019) 33:2762–6. doi: 10.1038/s41375-019-0575-9
173. George B, Mullick Chowdhury S, Hart A, Sircar A, Singh SK, Nath UK, et al. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers (2020) 12:1328. doi: 10.3390/cancers12051328
174. Thijssen R, Ter Burg J, Garrick B, van Bochove GGW, Brown JR, Fernandes SM, et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood (2016) 128:574–83. doi: 10.1182/blood-2016-02-700328
175. Delvecchio VS, Sana I, Mantione ME, Vilia MG, Ranghetti P, Rovida A, et al. Interleukin-1 receptor-associated kinase 4 inhibitor interrupts toll-like receptor signalling and sensitizes chronic lymphocytic leukaemia cells to apoptosis. Br J Haematol (2020) 189:475–88. doi: 10.1111/bjh.16386
176. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470:115–9. doi: 10.1038/nature09671
177. Roberts AW, Ma S, Kipps TJ, Coutre SE, Davids MS, Eichhorst B, et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood (2019) 134:111–22. doi: 10.1182/blood.2018882555
178. Goy A, Ramchandren R, Ghosh N, Munoz J, Morgan DS, Dang NH, et al. Ibrutinib plus lenalidomide and rituximab has promising activity in relapsed/refractory non–germinal center B-cell–like DLBCL. Blood (2019) 134:1024–36. doi: 10.1182/blood.2018891598
179. Jerkeman M, Eskelund CW, Hutchings M, Räty R, Wader KF, Laurell A, et al. Ibrutinib, lenalidomide, and rituximab in relapsed or refractory mantle cell lymphoma (PHILEMON): a multicentre, open-label, single-arm, phase 2 trial. Lancet Haematol (2018) 5:e109–16. doi: 10.1016/S2352-3026(18)30018-8
180. Ujjani C, Wang H, Skarbnik A, Trivedi N, Ramzi P, Khan N, et al. A phase 1 study of lenalidomide and ibrutinib in combination with rituximab in relapsed and refractory CLL. Blood Adv (2018) 2:762–8. doi: 10.1182/bloodadvances.2017015263
181. Yang Y, Shaffer AL, Emre NCT, Ceribelli M, Zhang M, Wright G, et al. Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell (2012) 21:723–37. doi: 10.1016/j.ccr.2012.05.024
182. Burger JA, Sivina M, Jain N, Kim E, Kadia T, Estrov Z, et al. Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood (2019) 133:1011–9. doi: 10.1182/blood-2018-10-879429
183. Skarzynski M, Niemann CU, Lee YS, Martyr S, Maric I, Salem D, et al. Interactions between Ibrutinib and Anti-CD20 Antibodies: Competing Effects on the Outcome of Combination Therapy. Clin Cancer Res (2016) 22:86–95. doi: 10.1158/1078-0432.CCR-15-1304
184. Albertsson-Lindblad A, Freiburghaus C, Jerkeman M, Ek S. Ibrutinib inhibits antibody dependent cellular cytotoxicity induced by rituximab or obinutuzumab in MCL cell lines, not overcome by addition of lenalidomide. Exp Hematol Oncol (2019) 8:16. doi: 10.1186/s40164-019-0141-1
185. Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol (2019) 20:43–56. doi: 10.1016/S1470-2045(18)30788-5
186. Buchner M, Brantner P, Stickel N, Prinz G, Burger M, Bär C, et al. The microenvironment differentially impairs passive and active immunotherapy in chronic lymphocytic leukaemia - CXCR4 antagonists as potential adjuvants for monoclonal antibodies. Br J Haematol (2010) 151:167–78. doi: 10.1111/j.1365-2141.2010.08316.x
187. Choi MY, Widhopf GF, Ghia EM, Kidwell RL, Hasan MK, Yu J, et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell (2018) 22:951–9.e3. doi: 10.1016/j.stem.2018.05.018
188. Fischer K, Al-Sawaf O, Bahlo J, Fink A-M, Tandon M, Dixon M, et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. New Engl J Med (2019) 380:2225–36. doi: 10.1056/NEJMoa1815281
189. Mato AR, Hill BT, Lamanna N, Barr PM, Ujjani CS, Brander DM, et al. Optimal sequencing of ibrutinib, idelalisib, and venetoclax in chronic lymphocytic leukemia: results from a multicenter study of 683 patients. Ann Oncol (2017) 28:1050–6. doi: 10.1093/annonc/mdx031
190. Dai B, Grau M, Juilland M, Klener P, Höring E, Molinsky J, et al. B-cell receptor-driven MALT1 activity regulates MYC signaling in mantle cell lymphoma. Blood (2017) 129:333–46. doi: 10.1182/blood-2016-05-718775
191. El-Gamal D, Williams K, LaFollette TD, Cannon M, Blachly JS, Zhong Y, et al. PKC-β as a therapeutic target in CLL: PKC inhibitor AEB071 demonstrates preclinical activity in CLL. Blood (2014) 124:1481–91. doi: 10.1182/blood-2014-05-574830
192. Fontán L, Qiao Q, Hatcher JM, Casalena G, Us I, Teater M, et al. Specific covalent inhibition of MALT1 paracaspase suppresses B cell lymphoma growth. J Clin Invest (2018) 128:4397–412. doi: 10.1172/JCI99436
193. Saba NS, Wong DH, Tanios G, Iyer JR, Lobelle-Rich P, Dadashian EL, et al. MALT1 Inhibition Is Efficacious in Both Naïve and Ibrutinib-Resistant Chronic Lymphocytic Leukemia. Cancer Res (2017) 77:7038–48. doi: 10.1158/0008-5472.CAN-17-2485
194. Gobessi S, Laurenti L, Longo PG, Carsetti L, Berno V, Sica S, et al. Inhibition of constitutive and BCR-induced Syk activation downregulates Mcl-1 and induces apoptosis in chronic lymphocytic leukemia B cells. Leukemia (2009) 23:686–97. doi: 10.1038/leu.2008.346
195. Zhuang J, Hawkins SF, Glenn MA, Lin K, Johnson GG, Carter A, et al. Akt is activated in chronic lymphocytic leukemia cells and delivers a pro-survival signal: the therapeutic potential of Akt inhibition. Haematologica (2010) 95:110–8. doi: 10.3324/haematol.2009.010272
196. Ramadani F, Bolland DJ, Garcon F, Emery JL, Vanhaesebroeck B, Corcoran AE, et al. The PI3K Isoforms p110 and p110 Are Essential for Pre-B Cell Receptor Signaling and B Cell Development. Sci Signaling (2010) 3:ra60–0. doi: 10.1126/scisignal.2001104
197. Ali AY, Wu X, Eissa N, Hou S, Ghia J-E, Murooka TT, et al. Distinct roles for phosphoinositide 3-kinases γ and δ in malignant B cell migration. Leukemia (2018) 32:1958–69. doi: 10.1038/s41375-018-0012-5
198. Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood (2011) 118:3603–12. doi: 10.1182/blood-2011-05-352492
199. Lannutti BJ, Meadows SA, Herman SEM, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood (2011) 117:591–4. doi: 10.1182/blood-2010-03-275305
200. Lampson BL, Brown JR. PI3Kδ-selective and PI3Kα/δ-combinatorial inhibitors in clinical development for B-cell non-Hodgkin lymphoma. Expert Opin Invest Drugs (2017) 26:1267–79. doi: 10.1080/13543784.2017.1384815
201. Zelenetz AD, Barrientos JC, Brown JR, Coiffier B, Delgado J, Egyed M, et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol (2017) 18:297–311. doi: 10.1016/S1470-2045(16)30671-4
202. Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther (2013) 12:2319–30. doi: 10.1158/1535-7163.MCT-12-0993-T
203. de Frias M, Iglesias-Serret D, Cosialls AM, González-Gironès DM, Pérez-Perarnau A, Rubio-Patiño C, et al. Isoform-selective phosphoinositide 3-kinase inhibitors induce apoptosis in chronic lymphocytic leukaemia cells. Br J Haematol (2010) 150:108–11. doi: 10.1111/j.1365-2141.2010.08151.x
204. Krause G, Hassenrück F, Hallek M. Copanlisib for treatment of B-cell malignancies: the development of a PI3K inhibitor with considerable differences to idelalisib. DDDT (2018) 12:2577–90. doi: 10.2147/DDDT.S142406
205. Mensah F, Blaize J-P, Bryan L. Spotlight on copanlisib and its potential in the treatment of relapsed/refractory follicular lymphoma: evidence to date. OTT (2018) 11:4817–27. doi: 10.2147/OTT.S142264
206. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med (2014) 370:997–1007. doi: 10.1056/NEJMoa1315226
207. Ghia P, Ljungström V, Tausch E, Agathangelidis A, Scheffold A, Scarfo L, et al. Whole-Exome Sequencing Revealed No Recurrent Mutations within the PI3K Pathway in Relapsed Chronic Lymphocytic Leukemia Patients Progressing Under Idelalisib Treatment. Blood (2016) 128:2770–0. doi: 10.1182/blood.V128.22.2770.2770
208. Scheffold A, Jebaraj BMC, Tausch E, Bloehdorn J, Ghia P, Yahiaoui A, et al. IGF1R as druggable target mediating PI3K-δ inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood (2019) 134:534–47. doi: 10.1182/blood.2018881029
209. Murali I, Kasar S, McWilliams EM, Itchaki G, Tyekucheva S, Livitz D, et al. Activating MAPK Pathway Mutations Mediate Primary Resistance to PI3K Inhibitors in Chronic Lymphocytic Leukemia (CLL). Blood (2018) 132:587–7. doi: 10.1182/blood-2018-99-115304
Keywords: B cell malignancies, ibrutinib, resistance, adaptation, targeted therapy, B cell receptor, BCR inhibitor
Citation: Ondrisova L and Mraz M (2020) Genetic and Non-Genetic Mechanisms of Resistance to BCR Signaling Inhibitors in B Cell Malignancies. Front. Oncol. 10:591577. doi: 10.3389/fonc.2020.591577
Received: 04 August 2020; Accepted: 24 September 2020;
Published: 26 October 2020.
Edited by:
Jérôme Paggetti, Luxembourg Institute of Health, LuxembourgReviewed by:
Michele Merli, University of Insubria, ItalyTeresa Sadras, Beckman Research Institute, City of Hope, United States
Copyright © 2020 Ondrisova and Mraz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marek Mraz, bWFyZWsubXJhekBlbWFpbC5jeg==