 Hongjian Zhao†
Hongjian Zhao† Mingqing Zhou
Mingqing Zhou- Department of General Surgery, Zhabei Central Hospital of Jing'an District, Shanghai, China
Colorectal cancer (CRC) is one of the leading causes of cancer death worldwide, with most mortalities being caused by metastases. However, the underlying molecular mechanism of CRC metastases remains largely unknown. Emerging evidence has shown the role of the tripartite motif family, especially tripartite motif protein 6 (TRIM6), in carcinogenesis. In this study, we used CRC cell lines with TRIM6 knockdown and overexpression to investigate the function of TRIM6 in CRC metastasis. We found that TRIM6 promotes CRC cell migration and invasion both in vitro and in vivo. TRIM6 knockdown slows down the migration and invasion processes, whereas TRIM6 overexpression accelerates CRC cell migration and invasion. TRIM6 is potentially the upstream regulatory factor for signal transducer and activator of transcription 3 (STAT3) via the suppressor of cytokine signaling 2 (SOCS2). A total of 70 samples from patients with CRC further confirmed that TRIM6 expression level is positively correlated with STAT3 phosphorylation and negatively correlated with SOCS2 expression. Therefore, TRIM6 could be a potential therapeutic target for CRC metastasis.
Introduction
Colorectal cancer (CRC) accounts for 9% of all cancers worldwide, being the second most common cancer in women and the third most common cancer in men (1, 2). Majority (90%) of the CRC cases are adenocarcinomas originating from the colonic and rectal epithelium (3), while CRC metastasis in the liver is the leading cause of CRC mortality (4). Similar to other cancers, CRC metastasis is a highly integrated process, involving processes such as cancer cell migration, adhesion, and invasion (5). These processes require precise molecular and cellular regulation, including cell adhesion molecules (6), cell–matrix interactions (7), and epithelial–mesenchymal transition (EMT), among others (8). Multiple signaling pathways, including the transforming growth factor beta (TGF-β) (9), Wnt (10), Notch (11), signal transducer and activator of transcription 3 (STAT3) (12), and PI3K/AKT (13), have been linked to cancer metastasis. However, the underlying mechanisms of CRC metastasis remain largely unknown.
The tripartite motif family, also known as the TRIM family, consists of 70 members, most of which have the E3 ubiquitin ligase activity (14). Accumulating studies have indicated its function in regulating carcinogenesis and metastasis (14–16). Tripartite motif protein 6 (TRIM6) belongs to the TRIM family with the E3 ubiquitin ligase activity (17). Besides its antiviral effect (17–19), TRIM6 has also been selectively expressed in embryonic stem (ES) cells. It attenuates c-Myc activity via a direct interaction. Downregulating TRIM6 in mouse ES cells results in the increased transcriptional activity of c-Myc and ES cell differentiation. Regarding its mechanism, TRIM6 functions as a ubiquitin ligase for c-Myc, allowing it to maintain c-Myc at an appropriate level (20). Recently published data indicate that TRIM6 promotes CRC cell proliferation (16). This finding indicates the potential function of TRIM6 in carcinogenesis and cancer metastasis.
In this study, we investigated the role of TRIM6 in CRC cell migration and invasion and, thus, found that TRIM6 promotes CRC cell migration and invasion. We identified that the STAT3 singling pathway was positively correlated with TRIM6 expression in CRC samples via a gene set enrichment analysis (GSEA). STAT3 is a transcription factor and is activated by phosphorylation (p-STAT3). STAT3 promotes proliferation, angiogenesis, migration, and invasion in its constitutively active form (21–24). Reports have shown that STAT3-negative regulators, such as SHP-1, SHP-2, SOCS1, SOOCS2, SOCS3, and PIAS1, can potentially prevent cancer progression (25, 26). In CRC, STAT3 expression has been observed in the crypt epithelial cell cytoplasm, and p-STAT3 is found in both the cytoplasm and nucleus (22). Furthermore, the inhibition of STAT3 expression made therapy-resistant CRC cells sensitive to 5-fluorouracil (27). p-STAT3 inhibition induces CRC cell apoptosis and cell cycle arrest (28). Our data suggested that TRIM6 potentially promoted CRC migration and invasion by modulating the status of STAT3 activation. This TRIM6-STAT3 regulatory network might function through the STAT3 phosphatase suppressor of cytokine signaling 2 (SOCS2); decreased levels of ubiquitous SOCS2 were detected in TRIM6 knockdown cells. Data from 70 samples from patients with CRC further confirmed that TRIM6 expression is positively correlated with STAT3 expression and negatively correlated with SOCS2.
Materials And Methods
Cell Culture
SW620 and HCT116 cells were bought from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences. The cells were maintained in DMEM (HyClone, Logan, UT, USA) with 10% fetal bovine serum (FBS) (Life Technology, Grand Island, NY, USA) at 37°C with 5% CO2.
Manipulation of TRIM6 Expression by Lentivirus
Short hairpin RNA (shRNA) oligos targeting TRIM6 (shTRIM6-1: 5’-CCGGAGACAAGTGAGGTTT-3’; shTRIM6-2: 5’-GCTAAAGTATCTGGACCTT-3’; and shTRIM6-3: 5’-CCATGAATATAGGGCCTAT-3’) were cloned into the pLKO.1 (Addgene, Cambridge, MA, USA) vector between the AgeI and EcoRI sites. pLVX-puro (Clontech, Palo Alto, CA, USA) was used as the vector for a full-length TRIM6 construction. The corresponding lentivirus was produced in HEK-293T cells.
Quantitative RT-PCR
Total RNA was extracted using the TRIzol reagent (Life Technology). cDNA was synthesized using a reverse transcription kit according to the instructions of the manufacturer (Fermentas, Hanover, MD, USA). SYBR Green Mix (Thermo Fisher Scientific, Rockford, IL, USA) was used for qRT-PCR with three replications per sample. The primers used were SOCS2 (5’-TGCCTTGCCTTCTTAGGTTC-3’ and 5’-CTTGGTTCCTTCCCACTTCTTC-3’) and GAPDH (5’-AATCCCATCACCATCTTC-3’and 5’-AGGCTGTTGTCATACTTC-3’). GAPDH was used to normalize relative mRNA levels.
Western Blotting
RIPA buffer containing a protease inhibitor cocktail (Beyotime Biotech., Shanghai, China) was used to lyse cells. The cell was lysed for 30 min on ice. The extracted protein and Laemmli loading buffer were mixed at a 1:1 ratio before boiling for 5 min. Protein separation was then performed on 10% or 15% SDS-PAGE gel, and 5% skimmed milk was used for blocking for 1 h at room temperature. After the primary antibody (Appendix. Antibody list) incubation overnight, HRP-corresponding secondary antibodies (Beyotime Biotech.) were used. Signal exposure was conducted using the ECL kit (Pierce, Rockford, IL, USA).
Wound-Healing Assay
The treated cells were seeded in a 6-well dish (8 × 105 cells per well). Once cells achieved 85% confluence, a wound was scratched through the center of the dish. After washing with PBS, the cells were cultured in DMEM with 10% FBS. The images were taken at 0, 24, and 48 h post-wounding.
Cell Invasion Assay
For the cell invasion assay, 8-μm-pore filters (Corning, New York, NY, USA) were used, and 1 mg/ml Matrigel (BD Biosciences) was used to precoat the upper chamber. The cells were grown to 50% confluence. After a 24-h serum-free medium incubation, the cells were suspended, and 5 × 104 cells were seeded onto the upper chamber while the media with 10% FBS was filled in the lower chamber. At 24 h post-seeding, the non-migrating cells were washed away. The cells in the lower chamber were fixed in 4% PFA and stained with 0.5% crystal violet. The samples were imaged and counted with five random fields per chamber.
Immunoprecipitation Assays
The cell lysates were incubated with anti-TRIM6 (Bioss, Bs-9165R), anti- SOCS2 (Abcam, Ab3692), or control IgG (Santa Cruz Biotech., Santa Cruz, CA, USA) for 1 h at 4°C. The cell mixture was then incubated with protein A/G-agarose (150 μg protein A) for 3 h at 4°C. The precipitates were then detected via Western blot analysis.
In Vivo Metastasis Mouse Model
The experimental protocol was approved by the Animal Experimentation Ethics Committee of Zhabei Central Hospital of Jing'an District. We used 6-week-old male BALB/c nude mice housed under specific-pathogen-free conditions. To examine an experimental lung metastasis, HCT116 cells stably expressing TRIM6 shRNA (shTRIM6-1) or control shRNA (shNC) were introduced via lateral vein injection (1 × 106 cells per mouse, n = 5 per group). Images of the mice were taken weekly using IVIS to monitor the metastatic tumor growth. Mice were sacrificed after 4 weeks, and lung metastases were counted. The lung tissue was fixed using 10% formalin and embedded in paraffin. Histological analysis was performed via hematoxylin and eosin (H&E) staining.
Colorectal Cancer Tissue Samples and Immunohistochemical Staining
This study was approved by the Institutional Review Board of Zhabei Central Hospital of Jing'an District. Paraffin-embedded CRC specimens from patients treated at Zhabei Central Hospital of Jing'an District were enrolled in this study with their written consent. None of these patients had received radiotherapy or chemotherapy prior to surgery.
CRC specimen sections were deparaffinized and dehydrated using sequential diluted xylene and ethanol wash. The antigen was retrieved by treating slides in 0.01 M sodium citrate buffer (pH 6) for 15–20 min. The slides were then permeabilized in 3% hydrogen peroxide and blocked using 5% bovine serum albumin. Primary antibody anti-TRIM6 (ProteinTech, 11953-1-AP), anti-SOCS2 (Abcam, ab109245), or anti-p-STAT3 (Abcam, ab76315) was incubated overnight. The corresponding HRP secondary antibodies (Long Island Bio, Shanghai, China) were then used. The signal was developed using the DAB staining kit (Long Island Bio) along with hematoxylin staining. The intensity grading was calculated as follows: IRS = staining intensity (SI) × percentage of positive cells. The SI was ranked from 1 to 3 (negative to strong). Overexpression was defined as having a staining index >3. Staining was independently assessed by two investigators.
Bioinformatics Analysis
Pathway and gene analyses were performed based on a CRC dataset (GSE14333) (29) using the GSEA version 2.0 from the Broad Institute at MIT. In our analysis, gene sets of fewer than 10 genes were excluded. The permutation test had a cutoff of 1,000 times. Significance was set at p < 0.01.
Statistical Analysis
Statistical analysis was conducted using the GraphPad Prism software (version 6.0, San Diego, CA, USA). For the statistical evaluation of two groups or more, two-tailed Student’s t-test and one-way analysis of variance were used, respectively (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
TRIM6 Knockdown Inhibits Colorectal Cancer Cell Migration and Invasion Both In Vitro and In Vivo
To investigate the function of TRIM6 in CRC cell migration, we knocked down the TRIM6 expression in HCT116 cells using lentivirus-mediated shRNAs. Western blot validation showed that both shRNAs (shRNA-1 and shRNA-2) successfully downregulated the TRIM6 protein expression (Figures 1A; S1A). Wound-healing was significantly delayed in both TRIM6 knockdown groups (Figure 1B). Quantitatively, the healing gaps in the TRIM6 knockdown groups were around 20% larger than that of the shNC group (Figure 1B). Transwell invasion assay was then performed to identify the role of TRIM6 in cell invasion. Around half as much invaded cells were found in the TRIM6 knockdown groups compared with the control cells (Figure 1C). Moreover, we also detected the downregulation of the EMT markers, such as Snail and Twist1, and the invasion and migration marker MMP2 (Figure 1D). These results indicate that TRIM6 knockdown potentially inhibits CRC cell migration and invasion.
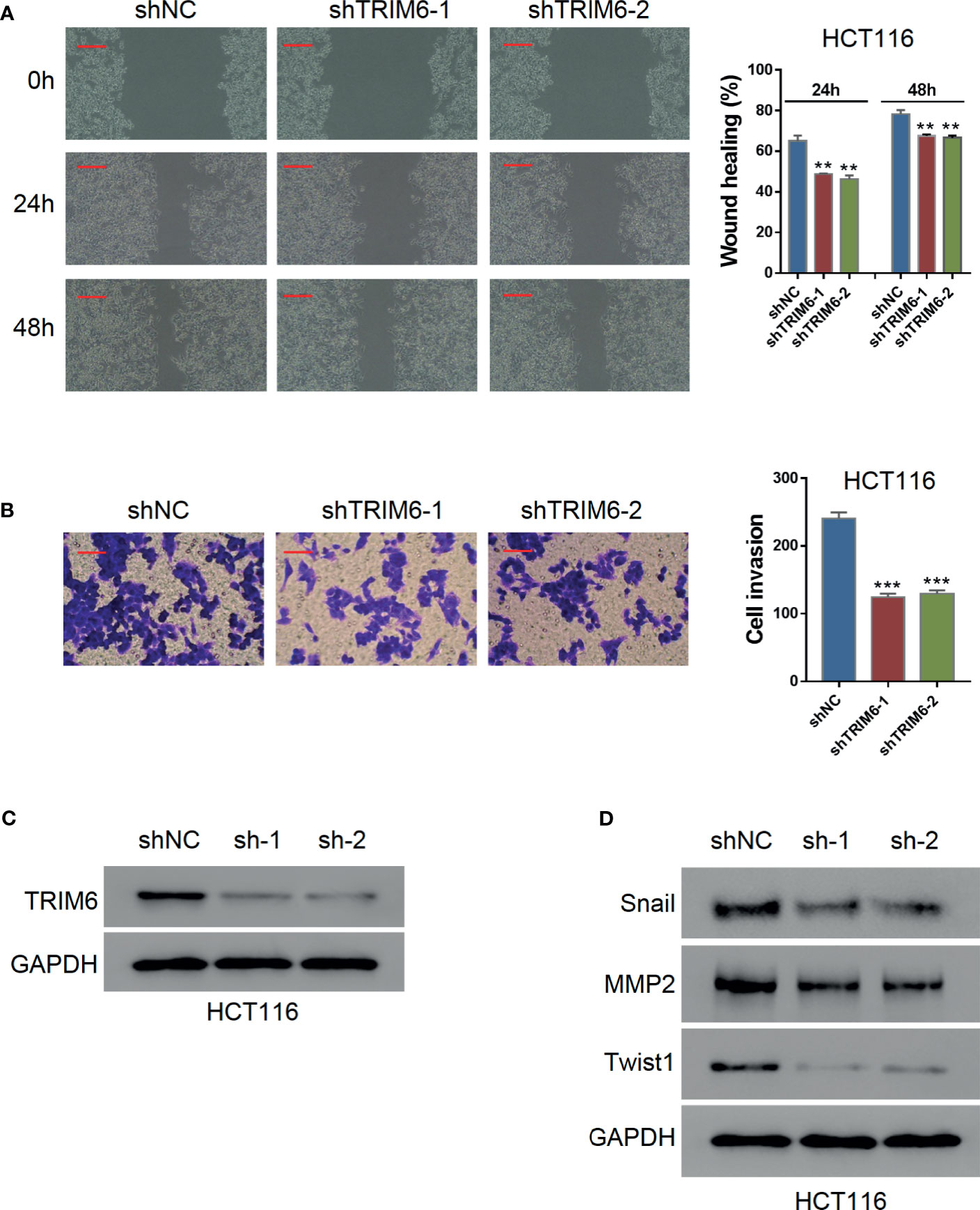
Figure 1 TRIM6 knockdown inhibits CRC cell migration and invasion in HCT116 cells. (A) Western blots images for validating TRIM6 knockdown by shRNAs. (B) Representative images and quantitative analysis of wound-healing assay at 0, 24, and 48 h post-scratching. shNC, negative control; shTRIM6-1, shRNA-1; shTRIM6-2, shRNA-2. Scale bar: 100 µm. (C) Representative images and quantitative analysis of the cell invasion assay. Scale bar: 100 µm. (D) Detecting the expression of Snail, MMP2, and Twist. The results are from at least three separate experiments. **P < 0.01, ***P < 0.001 vs. shNC.
We then explored the effect of TRIM6 in cell migration in vivo by monitoring the lung metastasis in mice injected with HCT116 stably expressing shRNA-1 and shNC. On IVIS imaging, shRNA-1-injected mice had lower lung metastatic signals (Figure 2A). Histologically, lung metastatic sites revealed cancerous features that were distinct from those of the surrounding lung tissues, such as nuclear and cytoplasmic enlargement, nuclear pleomorphism, and hyperchromatic and frequent multinucleation (Figure 2B). These metastatic sites were less frequently seen in the shRNA-1-injected group than in the shNC-injected group. The number of metastatic sites in the shRNA-1-injected group was around three times lower than those in the shNC-injected group (Figure 2C). These results indicate that TRIM6 dysfunction could potentially inhibit CRC metastasis.
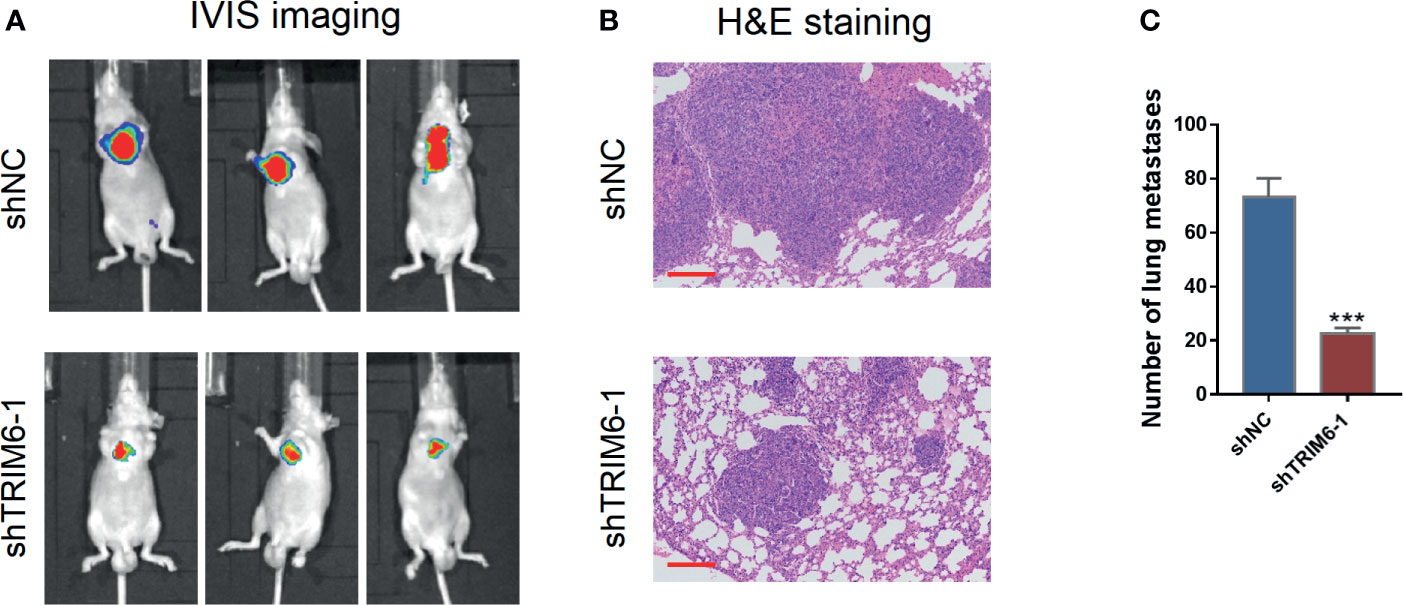
Figure 2 TRIM6 knockdown inhibits CRC lung metastasis in mice injected with HCT116 cells. (A) Representative bioluminescent images of mice (n = 5 per group) injected with the indicated cells taken at day 28. (B) Hematoxylin and eosin (H&E)-stained sections revealed that tumor cells metastasized to the alveoli of the lung in mice injected with the indicated cells. Scale bar: 200 µm. (C) Number of lung metastases. ***P < 0.001 vs. shNC.
TRIM6 Overexpression Induces Colorectal Cancer Cell Migration via the Signal Transducer and Activator of Transcription 3 Pathway
TRIM6 knockdown showed an inhibitory effect on CRC cell migration. To further test how TRIM6 promotes cell migration, we conducted a GSEA analysis on TRIM6. Interestingly, TRIM6 expression was positively correlated with several cancer-related pathways (Table S1), including NF-κB, CTNNB1, and STAT3 signaling (Figure 3A). Our preliminary data showed that TRIM6 knockdown had little effects on the phosphorylation of NF-κB p65 and the nuclear translocation of β-catenin (data not shown). Furthermore, although STAT3 expression was not affected in TRIM6 knockdown HCT116 cells, the phosphorylated STAT3 level was significantly lower than that in the control cells (Figure 3B). Thus, we supposed that TRIM6 may promote cell migration via the STAT3 signaling pathway. The genes included in DAUER_STAT3_TARGETS_UP affected by TRIM6 expression levels are listed in Table S2. To test this hypothesis, SW620 cells were overexpressed with TRIM6 (Figure S1B) and treated with STAT3 phosphorylation inhibitor HO-3867. TRIM6 overexpression in SW620 was able to induce cell migration in both the wound-healing and cell invasion assays (Figures 3C, D). After inhibiting STAT3 phosphorylation, the SW620 cells had a delayed cell migration (Figures 3C, D). Interestingly, HO-3867 rescued the TRIM6 overexpression-induced CRC cell migration phenotype.
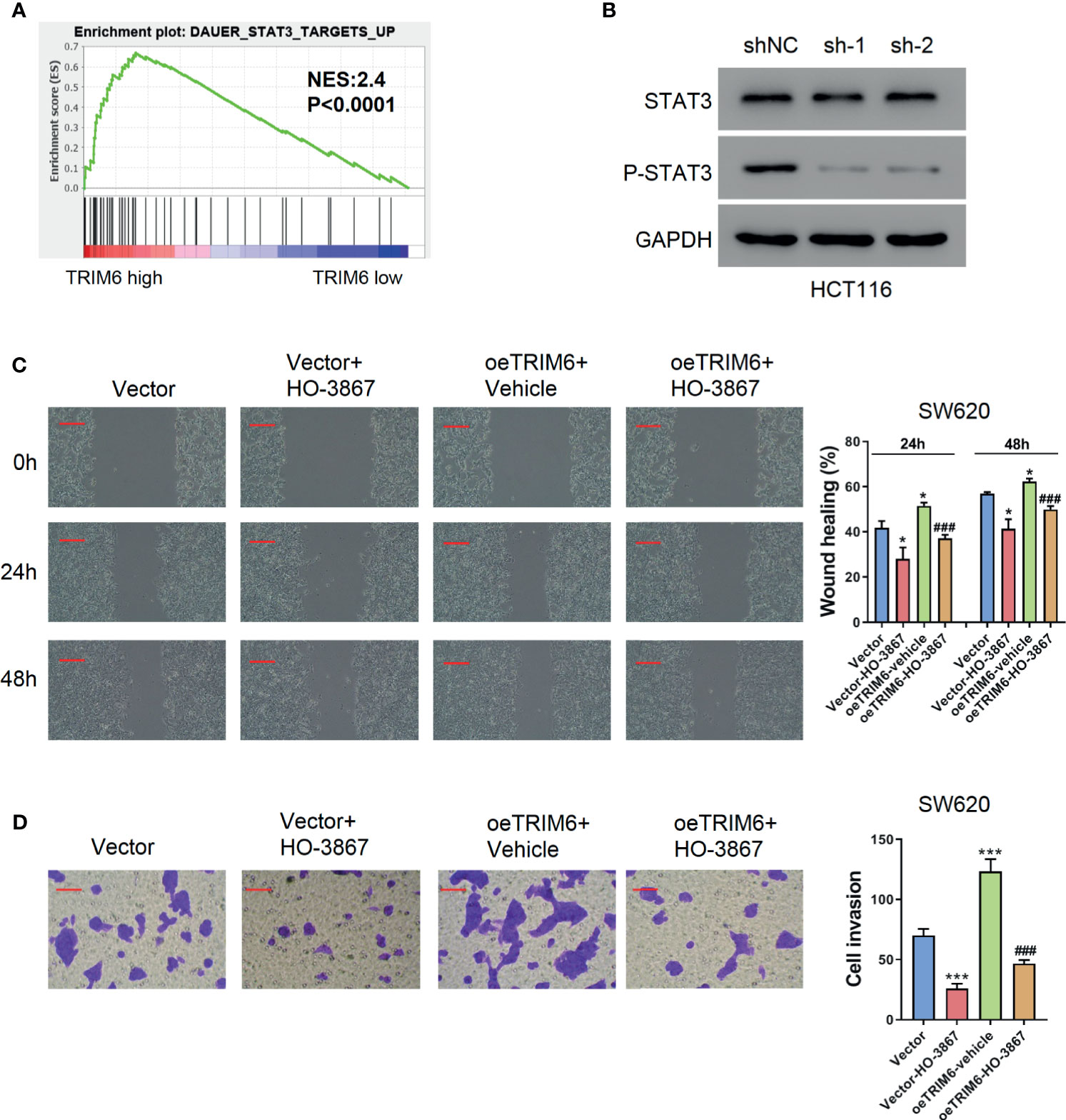
Figure 3 TRIM6 overexpression induces CRC cell migration via the STAT3 pathway in the SW620 cell line. (A) Enrichment plot of GSEA of the TRIM6 target. (B) STAT3 and p-STAT3 expression level detected via Western blot in TRIM6 knockdown HCT116 cells. (C) Representative images and quantitative analysis of the wound-healing assay in TRIM6-overexpressed SW620 cells with/without HO-3867 treatment at 0, 24, and 48 h post-scratching. Scale bar: 100 µm. (D) Representative images and quantitative analysis of the cell invasion assay in TRIM6-overexpressed SW620 cells with/without HO-3867 treatment. Scale bar: 100 µm. *P < 0.05, ***P < 0.001 vs. Vector; ###P < 0.001 vs. oeTRIM6 + Vehicle. The results are from at least three separate experiments.
TRIM6 May Promote Signal Transducer and Signal Transducer and Activator of Transcription 3 Phosphorylation via Suppressor of Cytokine Signaling 2
Our previous results indicated that TRIM6 promotes CRC cell migration via STAT3 signaling. To further investigate the regulatory mechanism of TRIM6 on STAT3, we examined the expression level of STAT3-negative regulators (SHP-1, SHP-2, SOCS1, SOCS2, SOCS3, and PIAS1). Among these, only the SOCS2 protein expression was decreased in TRIM6 knockdown HCT116 cells (Figure 4A). Thus, we focused on SOCS2. The mRNA expression level of SOCS2 remained unchanged (Figure 4B), whereas the protein expression of SOCS2 was upregulated in TRIM6-overexpressed SW620 cells without altering its mRNA expression level (Figures 4C, D). Meanwhile, the expression level of other STAT3-negative regulators remained unchanged.
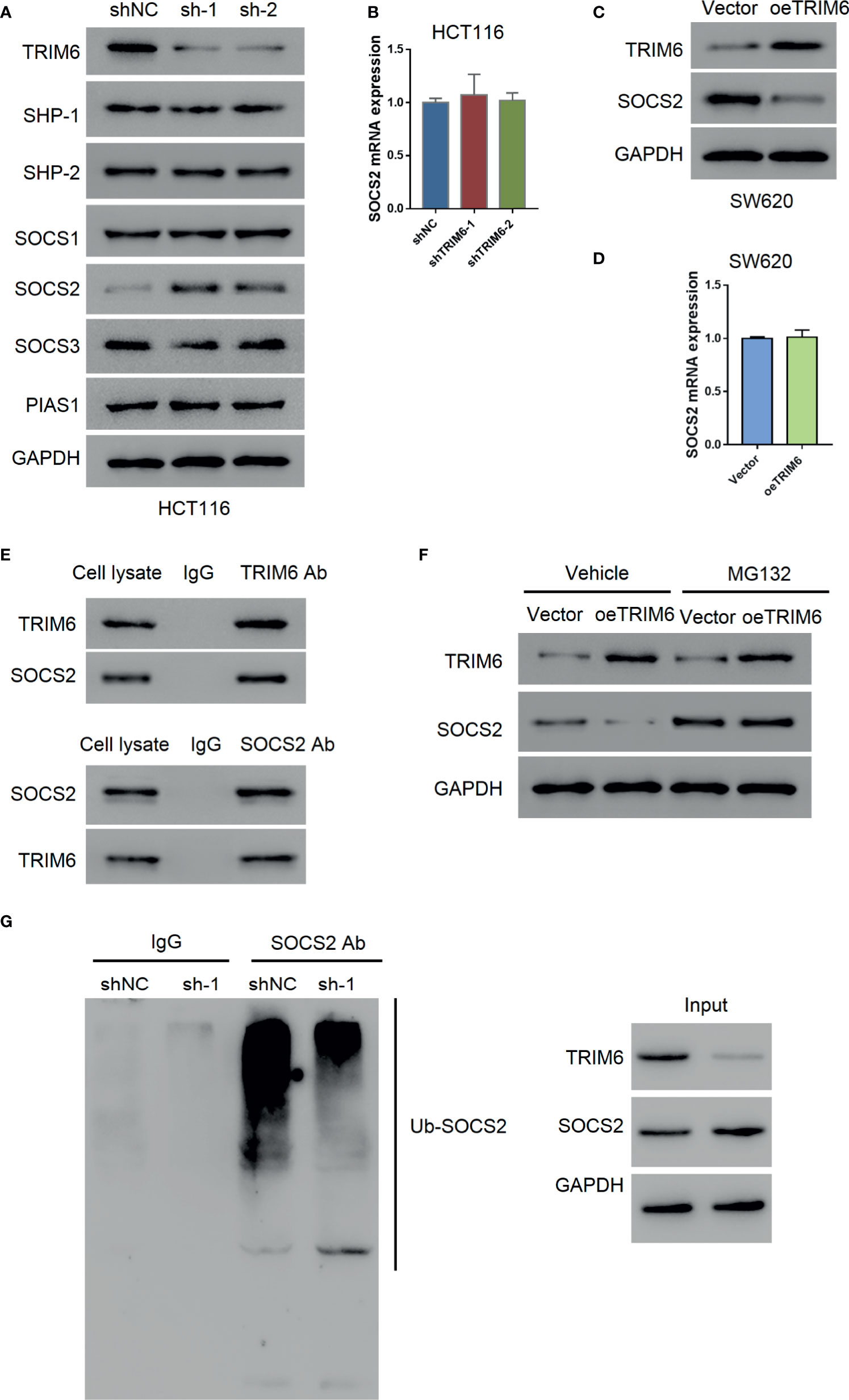
Figure 4 TRIM6 may promote STAT3 phosphorylation via SOCS2. (A) Protein expression of STAT3 phosphatases (SHP-1, SHP-2), SOCS1, SOCS2, SOCS3, and PIAS1 in TRIM6 knockdown HCT116 cells. (B) SOCS2 mRNA expression level measured via qRT-PCR in TRIM6 knockdown HCT116 cells. SOCS2 protein (C) and mRNA expression levels (D) in TRIM6-overexpressed SW620 cells detected via Western blot and qRT-PCR, respectively. (E) CO-IP analysis between TRIM6 and SOCS2. (F) TRIM6 and SOCS2 expression levels in TRIM6-overexpressed SW620 cells with/without MG132 treatment. (G) IP analysis of SOCS2 ubiquitination in TRIM6 knockdown HCT116 cells. The results are from at least three separate experiments.
SOCS2 plays a key role in inhibiting JAK-STAT signaling by degrading the signaling molecules via ubiquitination (30). CO-IP analysis with HCT116 cell lysate revealed a potential interaction between TRIM6 and SOCS2 (Figure 4E). To further understand the relationship between TRIM6 and SOCS2, we used the proteasome inhibitor MG132 to treat the TRIM6-overexpressed SW620 cells. We observed that MG132 treatment blocked the TRIM6 overexpression-induced reduction of SOCS2 protein expression in SW620 cells (Figure 4F). In TRIM6 knockdown HCT116 cells, because of the increase in the SOCS2 protein expression, SOCS2 ubiquitination was largely inhibited (Figure 4G). These results indicate that TRIM6 can negatively promote SOCS2 expression through ubiquitination.
TRIM6 Expression Positively Correlates With p-Signal Transducer and Activator of Transcription 3 and Negatively Correlates With SOCS2 in Samples From Patients With Colorectal Cancer
To further investigate the function of TRIM6 in patients with CRC, we collected 70 paraffin-embedded CRC specimens from patients treated at Zhabei Central Hospital of Jing'an District between 2012 and 2014. Patient information is shown in Table S3. Immunohistochemical staining and statistical analysis further confirmed our previous results. In the majority of the CRC specimens collected, TRIM6 expression was positively correlated with a phosphorylated STAT3 expression, whereas TRIM6 expression was negatively correlated with SOCS2 expression (Figure 5).
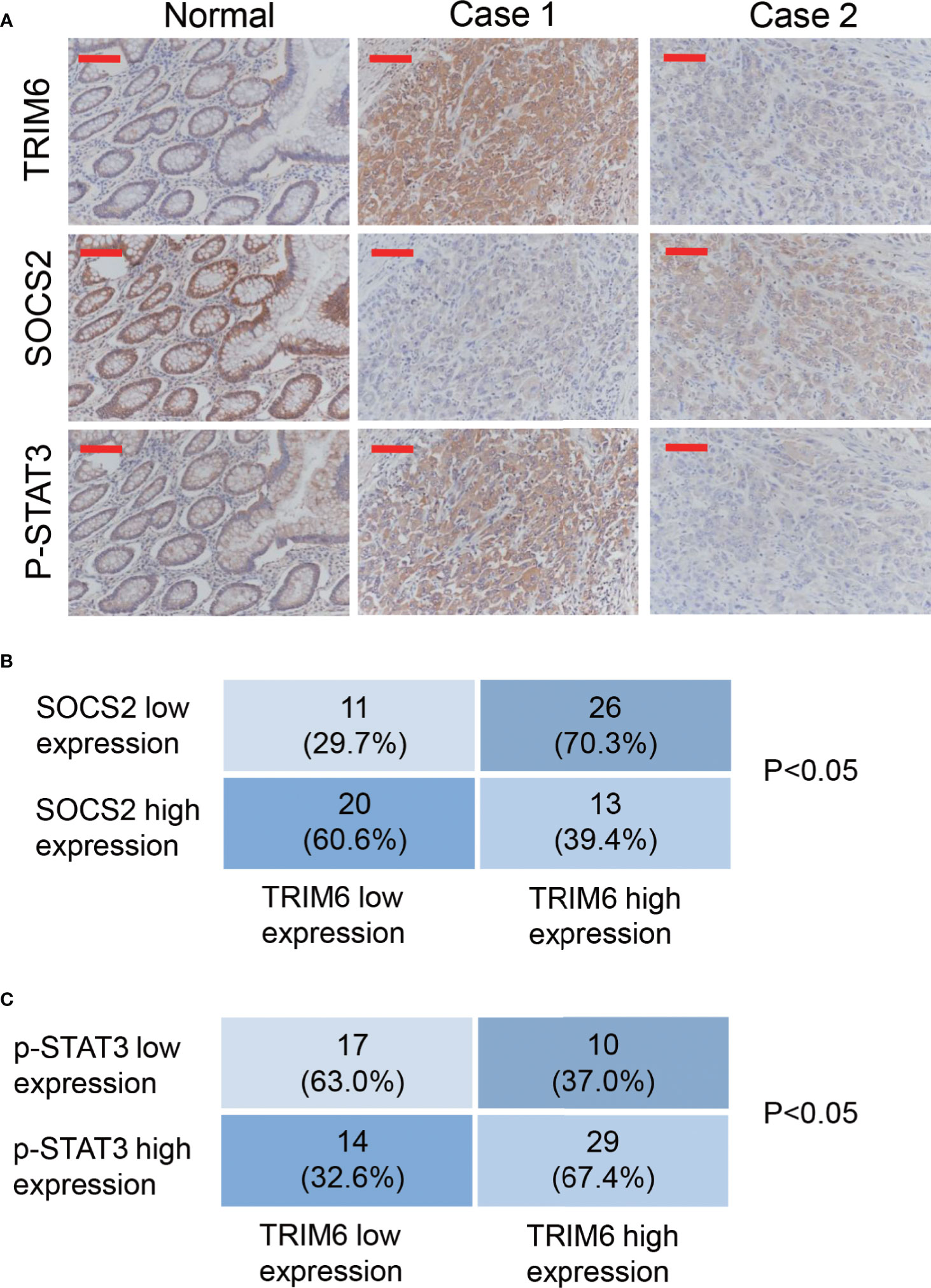
Figure 5 TRIM6 expression positively correlated with p-STAT3 and negatively correlated with SOCS2 in the CRC samples. (A) Representative immunohistochemical staining of TRIM6, SOCS2, and p-STAT3 in paraffin-embedded normal and CRC specimens (Case 1 and Case 2). n = 70. Scale bar: 100 µm. (B, C) Statistical analysis of CRC tissues under different staining conditions.
Discussion
TRIM6 protein has been reported to promote cell cycle progression and CRC cell proliferation (16). In the in vitro and in vivo functional assays, we observed that TRIM6 promotes CRC migration. Our findings, combined with that of a previous study (16), support the important roles of TRIM6 in CRC growth and metastasis.
In the current study, we used two independent CRC cell lines, HCT116 and SW620, which have previously established their intravasation capabilities and suitability for building a CRC metastasis model (31). Studies have also shown that the HCT116 cell lines have a mutation in TGFβRII, which inactivates kinase activity (32). However, the TRIM family has also been shown to affect cancer progression via TGF-β signaling (33). Moreover, a previous study comparing 10 CRC cell lines found that SW620 has the lowest TRIM6 expression while being at a physiologically relevant level (34). Therefore, to investigate the function of TRIM6 in CRC metastasis under restored TGF-β signaling, we chose to use SW620 cells for TRIM6 overexpression studies. In vitro, TRIM6 knockdown in HCT116 cells inhibited cell migration and invasion (Figure 1). Furthermore, the difference caused by TRIM6 knockdown was more noticeable at 24 h post wound creation, suggesting the involvement of other migration related factors at later times post the scratch formation. Meanwhile, whether secondary pathways or TRIM6 directly results in the downstream effects needs to be further verified. Epithelial-mesenchymal transition (EMT) is strongly implicated in tumor cell migration, invasion, and metastasis (35). Snail1 and Twist1, two markers of EMT, and MMP2, a marker of invasion and migration, which could degrade the extracellular matrix, were downregulated in TRIM6 knockdown CRC cells, which is consistent with a study on TRIM6 in renal fibrosis (36). In this perspective, TRIM6 stimulates the EMT and production of MMP2, which could facilitate tumor cell migration, invasion, and metastasis, thus, causes tumor recurrence. In vivo, we observed fewer CRC lung metastatic sites in the TRIM6 knockdown HCT116 cell-injected mouse CRC model (Figure 2). In contrast, TRIM6-overexpressed SW620 cells induced cell migration and invasion (Figure 3). Both in vitro and in vivo experiments revealed that TRIM6 promotes CRC migration. Evidence suggested that cell hyperproliferation is another key event contributing to the anticancer response (16). The possible effect of TRIM6 on cell proliferation could not be ruled out and further work will be necessary.
Regarding downstream signaling, TRIM6 expression was positively correlated with STAT3 signaling on GSEA (Table S1). Evidence has supported the critical role of STAT3 singling in the migration and invasion of cancer cells (24). In line with this, we found that the STAT3 phosphorylation inhibitor can rescue the TRIM6 overexpression phenotype, showing a delayed migration and decreased cell invasion (Figure 3). We further showed that TRIM6 expression level was correlated with the protein expression level of SOCS2 (a negative regulator of STAT3) whose expression was reduced in CRC tissues (37). TRIM family proteins are reported to have a E3 ubiquitin ligase activity (38). Recent studies have reported that TRIM6 promoted the ubiquitination of TIS21 (16) and TSC1-TSC2 (36). TRIM6 interacts with SOCS2, and the level of SOCS2 ubiquitination was lower in TRIM6 knockdown HCT116 cells (Figure 4), whereas the ubiquitination site on SOCS2 is to be identified. Furthermore, previous studies in dendritic cells have shown that SOCS2 silencing caused a hyperphosphorylation of STAT3 (39). STAT3 hyperactivation was also reported to correlate with SOCS2 silencing in ovarian and breast cancers (25). Based on these findings, we propose the possibility of a TRIM6-STAT3-SOCS2 regulatory signaling network involved in CRC metastasis. TRIM6 dysfunction causes an increased SOCS2 expression, resulting in a decreased level of STAT3 activation, CRC cell migration, and invasion. On the other hand, TRIM6 overexpression causes SOCS2 expression to be downregulated. This results in an increased cell migration and invasion, as well as CRC metastasis. Inhibition of the STAT3 signaling could decrease the expression of Snail1, Twist1, and MMP2 (40, 41). Here, TRIM6 knockdown decreased the expression of Snail1, Twist1, and MMP2 (Figure 1), which may be due to the decreased phosphorylation of STAT3 through inhibiting the ubiquitination of SOCS2.
Our findings were also supported by the samples from patients with CRC. From the 70 collected samples, Fisher’s exact test showed that the TRIM6 levels were correlated with the tumor size, clinical stage, and metastasis (Table S4). In addition, we found that TRIM6 expression was positively correlated with STAT3 phosphorylation and negatively correlated with SOCS2 expression (Figure 5). According to the analysis of GSE14333 (Figure S2A) and a recent report (16), TRIM6 expression was negatively correlated with the overall survival of CRC. By using GEPIA (42), we found that TRIM6 expression was not correlated with the overall survival of CRC (Figure S2B), but it was negatively correlated with the disease-free survival of CRC (Figure S2C), suggesting that TRIM6 could constitute a useful prognostic marker to identify patients who are more likely to have disease recurrence and are, thus, good candidates to receive an aggressive adjuvant treatment. However, future prospective studies are needed to determine its accuracy and efficiency on predicting the recurrence of patients with CRC in order to tailor adjuvant treatment. The restricted patient sample size limited our findings. Further retrospective analysis with a larger sample size can be carried out to investigate the role of TRIM6 in the overall survival of patients. Furthermore, the cell line with a lower TRIM6 expression (SW620) was metastatic, indicating the complicated regulation of cancer cell metastasis. Our study suggested that TRIM6 may serve as a CRC therapeutic target for those patients with a high expression of TRIM6 in the future. For those with a low expression of TRIM6, other therapeutic targets, such as Wnt/β-catenin and NF-κB (43, 44), should be considered.
In conclusion, our study was the first to reveal that TRIM6 promotes CRC cell migration and invasion via the SOCS2-STAT3 signaling. Our results uncover the potential of TRIM6 as a potential CRC therapeutic target.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of Zhabei Central Hospital of Jing'an District. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Animal Experimentation Ethics Committee in Zhabei Central Hospital of Jing'an District.
Author Contributions
MZ, XC, and HZ designed the experiments. HZ, JH, and MC performed the experiments. HZ and BL performed the statistical analysis. HZ and JH wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by 2019 Research Projects of Shanghai Municipal Health Commission (201940198).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.695525/full#supplementary-material
References
1. Ghoncheh M, Mohammadian M, Mohammadian-Hafshejani A, Salehiniya H. The Incidence and Mortality of Colorectal Cancer and Its Relationship With the Human Development Index in Asia. Ann Glob Health (2016) 82(5):726–37. doi: 10.1016/j.aogh.2016.10.004
2. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal Cancer Statistics, 2020. CA: Cancer J Clin (2020) 70(3):145–64. doi: 10.3322/caac.21601
3. Fleming M, Ravula S, Tatishchev SF, Wang HL. Colorectal Carcinoma: Pathologic Aspects. J Gastrointest Oncol (2012) 3(3):153–73. doi: 10.3978/j.issn.2078-6891.2012.030
4. Tsitskari M, Filippiadis D, Kostantos C, Palialexis K, Zavridis P, Kelekis N, et al. The Role of Interventional Oncology in the Treatment of Colorectal Cancer Liver Metastases. Ann Gastroenterol (2019) 32(2):147. doi: 10.20524/aog.2019.0338
5. Tahtamouni L, Ahram M, Koblinski J, Rolfo C. Molecular Regulation of Cancer Cell Migration, Invasion, and Metastasis. Anal Cell Pathol (Amst) (2019) 2019:1356508. doi: 10.1155/2019/1356508
6. Harjunpää H, Llort Asens M, Guenther C, Fagerholm SC. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front Immunol (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
7. Gkretsi V, Stylianopoulos T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front Oncol (2018) 8:145. doi: 10.3389/fonc.2018.00145
8. Cao H, Xu E, Liu H, Wan L, Lai M. Epithelial–mesenchymal Transition in Colorectal Cancer Metastasis: A System Review. Pathol-Research Pract (2015) 211(8):557–69. doi: 10.1016/j.prp.2015.05.010
9. Hao Y, Baker D, Ten Dijke P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci (2019) 20(11):2767. doi: 10.3390/ijms20112767
10. Zhan T, Rindtorff N, Boutros M. Wnt Signaling in Cancer. Oncogene (2017) 36(11):1461–73. doi: 10.1038/onc.2016.304
11. Li L, Tang P, Li S, Qin X, Yang H, Wu C, et al. Notch Signaling Pathway Networks in Cancer Metastasis: A New Target for Cancer Therapy. Med Oncol (2017) 34(10):1–10. doi: 10.1007/s12032-017-1039-6
12. Jin W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells (2020) 9(1):217. doi: 10.3390/cells9010217
13. Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT Signaling Pathway and Cancer: An Updated Review. Ann Med (2014) 46(6):372–83. doi: 10.3109/07853890.2014.912836
14. Hatakeyama S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem Sci (2017) 42(4):297–311. doi: 10.1016/j.tibs.2017.01.002
15. Hatakeyama S. TRIM Proteins and Cancer. Nat Rev Cancer (2011) 11(11):792–804. doi: 10.1038/nrc3139
16. Zheng S, Zhou C, Wang Y, Li H, Sun Y, Shen Z. TRIM6 Promotes Colorectal Cancer Cells Proliferation and Response to Thiostrepton by TIS21/Foxm1. J Exp Clin Cancer Res (2020) 39(1):23. doi: 10.1186/s13046-019-1504-5
17. Bharaj P, Atkins C, Luthra P, Giraldo MI, Dawes BE, Miorin L, et al. The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. J Virol (2017) 91(18):e00833–17. doi: 10.1128/JVI.00833-17
18. Bharaj P, Wang YE, Dawes BE, Yun TE, Park A, Yen B, et al. The Matrix Protein of Nipah Virus Targets the E3-Ubiquitin Ligase TRIM6 to Inhibit the IKKepsilon Kinase-Mediated Type-I IFN Antiviral Response. PloS Pathog (2016) 12(9):e1005880. doi: 10.1371/journal.ppat.1005880
19. Rajsbaum R, Versteeg GA, Schmid S, Maestre AM, Belicha-Villanueva A, Martinez-Romero C, et al. Unanchored K48-Linked Polyubiquitin Synthesized by the E3-Ubiquitin Ligase TRIM6 Stimulates the Interferon-IKKepsilon Kinase-Mediated Antiviral Response. Immunity (2014) 40(6):880–95. doi: 10.1016/j.immuni.2014.04.018
20. Sato T, Okumura F, Ariga T, Hatakeyama S. TRIM6 Interacts With Myc and Maintains the Pluripotency of Mouse Embryonic Stem Cells. J Cell Sci (2012) 125(Pt 6):1544–55. doi: 10.1242/jcs.095273
21. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 Activation in Colon Cancer Is Associated With Enhanced Cell Proliferation and Tumor Growth. Neoplasia (2005) 7(6):545–55. doi: 10.1593/neo.04571
22. Lassmann S, Schuster I, Walch A, Gobel H, Jutting U, Makowiec F, et al. STAT3 mRNA and Protein Expression in Colorectal Cancer: Effects on STAT3-Inducible Targets Linked to Cell Survival and Proliferation. J Clin Pathol (2007) 60(2):173–9. doi: 10.1136/jcp.2005.035113
23. Yu H, Pardoll D, Jove R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat Rev Cancer (2009) 9(11):798–809. doi: 10.1038/nrc2734
24. Avalle L, Camporeale A, Camperi A, Poli V. STAT3 in Cancer: A Double Edged Sword. Cytokine (2017) 98:42–50. doi: 10.1016/j.cyto.2017.03.018
25. Sutherland KD, Lindeman GJ, Choong DY, Wittlin S, Brentzell L, Phillips W, et al. Differential Hypermethylation of SOCS Genes in Ovarian and Breast Carcinomas. Oncogene (2004) 23(46):7726–33. doi: 10.1038/sj.onc.1207787
26. Wu M, Song D, Li H, Yang Y, Ma X, Deng S, et al. Negative Regulators of STAT3 Signaling Pathway in Cancers. Cancer Manag Res (2019) 11:4957–69. doi: 10.2147/CMAR.S206175
27. Spitzner M, Roesler B, Bielfeld C, Emons G, Gaedcke J, Wolff HA, et al. STAT3 Inhibition Sensitizes Colorectal Cancer to Chemoradiotherapy In Vitro and In Vivo. Int J Cancer (2014) 134(4):997–1007. doi: 10.1002/ijc.28429
28. Lin Q, Lai R, Chirieac LR, Li C, Thomazy VA, Grammatikakis I, et al. Constitutive Activation of JAK3/STAT3 in Colon Carcinoma Tumors and Cell Lines: Inhibition of JAK3/STAT3 Signaling Induces Apoptosis and Cell Cycle Arrest of Colon Carcinoma Cells. Am J Pathol (2005) 167(4):969–80. doi: 10.1016/S0002-9440(10)61187-X
29. Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients With Dukes Stage B and C Colorectal Cancer. Clin Cancer Res (2009) 15(24):7642–51. doi: 10.1158/1078-0432.CCR-09-1431
30. Croker BA, Kiu H, Nicholson SE. SOCS Regulation of the JAK/STAT Signalling Pathway. Semin Cell Dev Biol (2008) 19(4):414–22. doi: 10.1016/j.semcdb.2008.07.010
31. Xu Y, Zhang L, Wang Q, Zheng M. Comparison of Different Colorectal Cancer With Liver Metastases Models Using Six Colorectal Cancer Cell Lines. Pathol Oncol Res (2020) 26(4):2177–83. doi: 10.1007/s12253-020-00805-3
32. Carethers JM, Pham TT. Mutations of Transforming Growth Factor Beta 1 Type II Receptor, BAX, and Insulin-Like Growth Factor II Receptor Genes in Microsatellite Unstable Cell Lines. In Vivo (2000) 14(1):13–20. doi: 10.1089/10430340050016283
33. Lee HJ. The Role of Tripartite Motif Family Proteins in TGF-Beta Signaling Pathway and Cancer. J Cancer Prev (2018) 23(4):162–9. doi: 10.15430/JCP.2018.23.4.162
34. Papageorgis P, Cheng K, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM, et al. Smad4 Inactivation Promotes Malignancy and Drug Resistance of Colon Cancer. Cancer Res (2011) 71(3):998–1008. doi: 10.1158/0008-5472.CAN-09-3269
35. Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell (2018) 45(6):681–95.e4. doi: 10.1016/j.devcel.2018.05.027
36. Liu W, Yi Y, Zhang C, Zhou B, Liao L, Liu W, et al. The Expression of TRIM6 Activates the Mtorc1 Pathway by Regulating the Ubiquitination of TSC1-TSC2 to Promote Renal Fibrosis. Front Cell Dev Biol (2020) 8:616747. doi: 10.3389/fcell.2020.616747
37. Letellier E, Schmitz M, Baig K, Beaume N, Schwartz C, Frasquilho S, et al. Identification of SOCS2 and SOCS6 as Biomarkers in Human Colorectal Cancer. Br J Cancer (2014) 111(4):726–35. doi: 10.1038/bjc.2014.377
38. Short KM, Cox TC. Subclassification of the RBCC/TRIM Superfamily Reveals a Novel Motif Necessary for Microtubule Binding. J Biol Chem (2006) 281(13):8970–80. doi: 10.1074/jbc.M512755200
39. Posselt G, Schwarz H, Duschl A, Horejs-Hoeck J. Suppressor of Cytokine Signaling 2 Is a Feedback Inhibitor of TLR-Induced Activation in Human Monocyte-Derived Dendritic Cells. J Immunol (2011) 187(6):2875–84. doi: 10.4049/jimmunol.1003348
40. Azare J, Doane A, Leslie K, Chang Q, Berishaj M, Nnoli J, et al. Stat3 Mediates Expression of Autotaxin in Breast Cancer. PloS One (2011) 6(11):e27851. doi: 10.1371/journal.pone.0027851
41. Liang Z, Liu H, Zhang Y, Xiong L, Zeng Z, He X, et al. Cyr61 From Adipose-Derived Stem Cells Promotes Colorectal Cancer Metastasis and Vasculogenic Mimicry Formation via Integrin α(V) β(5). Mol Oncol (2021). doi: 10.1002/1878-0261.12998
42. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45(W1):W98–102. doi: 10.1093/nar/gkx247
43. Cheng X, Xu X, Chen D, Zhao F, Wang W. Therapeutic Potential of Targeting the Wnt/β-Catenin Signaling Pathway in Colorectal Cancer. Biomed Pharmacother (2019) 110:473–81. doi: 10.1016/j.biopha.2018.11.082
Keywords: colorectal cancer, metastasis, TRIM6, STAT3, SOCS2
Citation: Zhao H, Huang J, Chen M, Li B, Chen X and Zhou M (2021) Tripartite Motif Protein 6 Promotes Colorectal Cancer Cell Migration and Metastasis via SOCS2-STAT3 Signaling. Front. Oncol. 11:695525. doi: 10.3389/fonc.2021.695525
Received: 15 April 2021; Accepted: 24 August 2021;
Published: 13 September 2021.
Edited by:
Kenneth K.W. To, The Chinese University of Hong Kong, ChinaReviewed by:
Smrithi Rajendiran, University of California, Santa Cruz, United StatesChristos K. Kontos, National and Kapodistrian University of Athens, Greece
Copyright © 2021 Zhao, Huang, Chen, Li, Chen and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingqing Zhou, emhvdW1pbmdxaW5nMTk2NkAxNjMuY29t; Xinran Chen, cG93ZXIyMDAwMDExQHNpbmEuY29t
†These authors have contributed equally to this work