 Maria Dobre1
Maria Dobre1 Alessandro Salvi2
Alessandro Salvi2 Iulia Andreea Pelisenco3
Iulia Andreea Pelisenco3 Florina Vasilescu1
Florina Vasilescu1 Giuseppina De Petro2
Giuseppina De Petro2 Vlad Herlea4*
Vlad Herlea4* Elena Milanesi5
Elena Milanesi5- 1Laboratory of Histopathology and Immunohistochemistry, Victor Babes National Institute of Pathology, Bucharest, Romania
- 2Division of Biology and Genetics, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
- 3Faculty of Biology, University of Bucharest, Bucharest, Romania
- 4Department of Pathology, Fundeni Clinical Institute, Bucharest, Romania
- 5Laboratory of Radiobiology, Victor Babes National Institute of Pathology, Bucharest, Romania
Colorectal cancer (CRC) is often characterized by mutations and aberrant DNA methylation within the promoters of tumor suppressor genes and proto-oncogenes. The most frequent somatic mutations occur within KRAS and BRAF genes. Mutations of the KRAS gene have been detected in approximately 40% of patients, while mutations in BRAF have been detected less frequently at a rate of 10%. In this study, the DNA methylation levels of 22 candidate genes were evaluated in three types of tissue: mucosal tumoral tissue from 18 CRC patients, normal adjacent tissues from 10 CRC patients who underwent surgical resection, and tissue from a control group of six individuals with normal colonoscopies. A differential methylation profile of nine genes (RUNX3, SFRP1, WIF1, PCDH10, DKK2, DKK3, TMEFF2, OPCML, and SFRP2) presenting high methylation levels in tumoral compared to normal tissues was identified. KRAS mutations (codons 12 or 13) were detected in eight CRC cases, and BRAF mutations (codon 600) in four cases. One of the CRC patients presented concomitant mutations in KRAS codon 12 and BRAF, whereas seven patients did not present these mutations (WT). When comparing the methylation profile according to mutation status, we found that six genes (SFRP2, DKK2, PCDH10, TMEFF2, SFRP1, HS3ST2) showed a methylation level higher in BRAF positive cases than BRAF negative cases. The molecular sub-classification of CRC according to mutations and epigenetic modifications may help to identify epigenetic biomarkers useful in designing personalized strategies to improve patient outcomes.
Introduction
Colorectal cancer (CRC) is the third most common type of cancer and, even with advances in CRC screening and therapeutic strategies, it still remains the second deadliest malignancy for both sexes. CRC incidence has continued to increase in countries with medium to high human development indexes and in younger populations (1). This type of cancer is a highly heterogeneous disease that can be subtyped according to anatomical location or pathological and molecular signatures. The development of CRC is influenced by both environmental risk factors (such as obesity, a sedentary lifestyle, an unhealthy diet, alcohol consumption, and smoking) (2) and genetic risk factors, with less than 10% of patients presenting inherited mutations that increase the risk of CRC onset (3) and 25% of patients with “familial” CRC (4). About 70% of CRC cases are sporadic; these cases are most common in patients over the age of 50 and seem to depend mainly on dietary and environmental factors.
In general, the development of sporadic CRCs involves the “normal-adenoma-dysplasia-carcinoma” sequence as described by Vogelstein and Fearon (5). This sequence includes genetic and epigenetic changes in the colonic epithelium that transform normal glandular epithelium into invasive adenocarcinomas. The progression from adenoma to carcinoma is a multistep process that involves three molecular pathways: the Chromosomal Instability (CIN) pathway, the Microsatellite Instability (MSI) pathway, and the CpG Island Methylator Phenotype (CIMP) pathway. CIN is characterized by loss or gain of chromosomal segments, chromosomal translocations, or gene amplifications, which result in gene copy number variations. In addition, mutations in specific oncogenes, including KRAS and BRAF, and in tumor suppressor genes, such as APC and TP53, can be detected (6). MSI occurs in 15–20% of sporadic CRC cases and comprises recurrent alterations in the microsatellite zone without structural and numerical changes in the genome. CIMP, reported in 20–30% of sporadic CRCs (7), is characterized by hyper-methylation of CpG islands located in promoters that regulate the activity of several tumor suppressor genes and other CRC related genes. It is well known that specific mutations can modify DNA methylation (8) and that DNA methylation changes can cause an increase in mutation rate (9). In CRC, 10–40% lower levels of absolute methylation than normal colon tissue within the whole genome have been determined (10). However, CpG islands in the promoters of CRC related genes show a hyper-methylated profile, resulting in repression of transcription of tumor suppressor genes (11). Therefore, the first aim of this study was to identify methylation differences in the promoters of 22 candidate genes in CRC tissues compared with control tissues (from non-CRC individuals) and with paired normal adjacent tissues. The second aim was to examine the relationship between promoter DNA methylation and the presence of mutations in KRAS and BRAF genes.
Materials and Methods
Sample Collection
The DNA for the study was obtained from 18 CRC sporadic tumor tissues and 10 corresponding paired normal adjacent tissues (NAT) from CRC patients who underwent surgical resection. The NATs were collected approximatively 10 cm away from the tumors. For nine patients out of 10, the NAT was collected from the same anatomic segment. For one patient with tumor localization in the recto-sigmoid junction, the NAT was collected from the sigmoid. For five of the 18 CRC patients, blood samples were collected in EDTA tubes. The tissue specimens and the blood samples were collected at Fundeni Clinical Institute in Bucharest (Romania) and were stored at −80°C until DNA isolation. Six specimens from normal colonic mucosa as well as five blood samples from independent controls, from previous preclinical studies were available from the biobank of the Victor Babes Institute (Table 1). The present study was approved by the local ethics committee (registration number 291, 8 March 2016), carried out in accordance with the Declaration of Helsinki, and the individuals gave their written informed consent. Genomic DNA was isolated using the QIAmp DNA Mini Kit (Qiagen) according to the manufacturer’s instruction and quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific). All samples were examined by a pathologist, and clinical information of the patient’s cohort are reported in Table 2.

Table 1 Socio-demographic data of the individuals involved in the study.
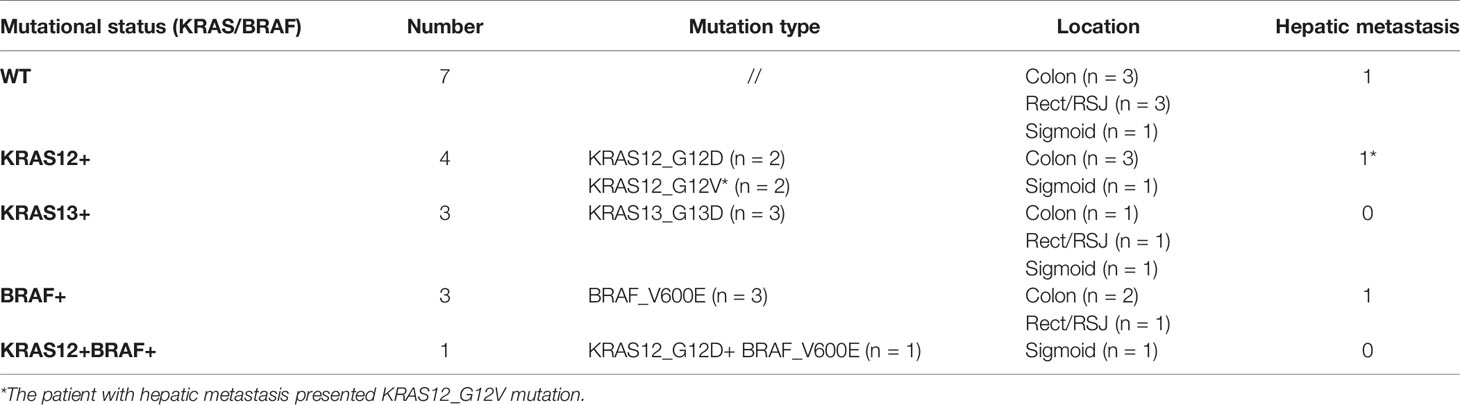
Table 2 Available clinical data and mutation status of CRC patients involved in the study.
Detection of KRAS and BRAF Mutations Detection and DNA Methylation Analysis
KRAS gene mutations (in codons 12, 13, 61) were detected by pyrosequencing as previously described (12). Mutations in codons 600 and 601 of the BRAF gene were assessed using the BRAF 600/601 StripAssay (ViennaLab Diagnostic GmbH, Vienna, Austria) by PCR followed by reverse hybridization according to the manufacturer’s protocol. The DNA methylation levels of the promoters of the 22 genes involved in colorectal cancer (APC, CDH1, CDKN2A, DKK2, DKK3, HIC1, HNF1B, HS3ST2, MGMT, MLH1, OPCML, PCDH10, RASSF1, RUNX3, SFRP1, SFRP2, SFRP5, SPARC, TMEFF2, UCHL1, WIF1, and WT1) were analyzed using the EpiTect Methyl II PCR Array (Qiagen, Hilden, Germany) according to manufacturer’s protocol. This array system is based on the detection of the input DNA that remains after digestion with a methylation-sensitive and/or methylation-dependent restriction enzyme using the EpiTect Methyl II DNA Restriction kit (Qiagen, Hilden, Germany). The relative amount of un-methylated (UM) and methylated (M) DNA was quantified by qPCR using the comparative cycle threshold method.
Statistical Analysis
The statistical analysis was conducted using the Statistical Package for Social Science (SPSS version 17.0). Categorical variables were tested using the chi-square test and continuous variables were tested using a t-test or Mann-Whitney U test. The Mann-Whitney U test was used to identify differences in promoter methylation levels between CRC patients (T) and controls (C), normal adjacent tissue (NAT) and controls (C), as well as to compare samples from patients with different mutation status for KRAS and BRAF. Receiver operating characteristic (ROC) curves were created and the area under the curve (AUC) was calculated to determine the role of methylation in differentiating T from C. Logistic regression was used to build a diagnostic model that could explore whether varying combinations of differentially methylated genes could better differentiate T from C. A gene promoter was considered to be methylated if the methylation level was over 20% according to the instructions of the manufacturer. A further analysis of the 10 tumoral samples with their matched NAT samples was performed using the Wilcoxon Signed Rank test. All reported significant p values were two-sided, with p < 0.05.
Results
In this study, colonic mucosa sample from 18 patients with CRC and six controls were investigated to evaluate the levels of DNA methylation of the promoters of 22 target genes. The tumors were located in the ascending and transverse colon (n = 9), in the sigmoid (n = 4), and in the rectum or the rectosigmoid junction (RSJ) (n = 5). At the moment of sample collection, three cases presented hepatic metastasis. Seven out of the 18 analyzed tumoral tissues presented mutations in the KRAS gene: four occurred in codon 12 and three in codon 13. Three patients had mutations in the BRAF gene, and one patient showed a double mutation, i.e., mutations in both BRAF and KRAS genes. KRAS and BRAF mutations were not found in the 10 normal adjacent tissues or in the seven tumoral tissues (WT) (Table 2).
Statistical analysis of the DNA methylation levels of the promoters was performed for 16 genes out of 22, because in six genes, the detected DNA methylation level in the tumoral tissue was below 20% (Figure 1 and Table 3).
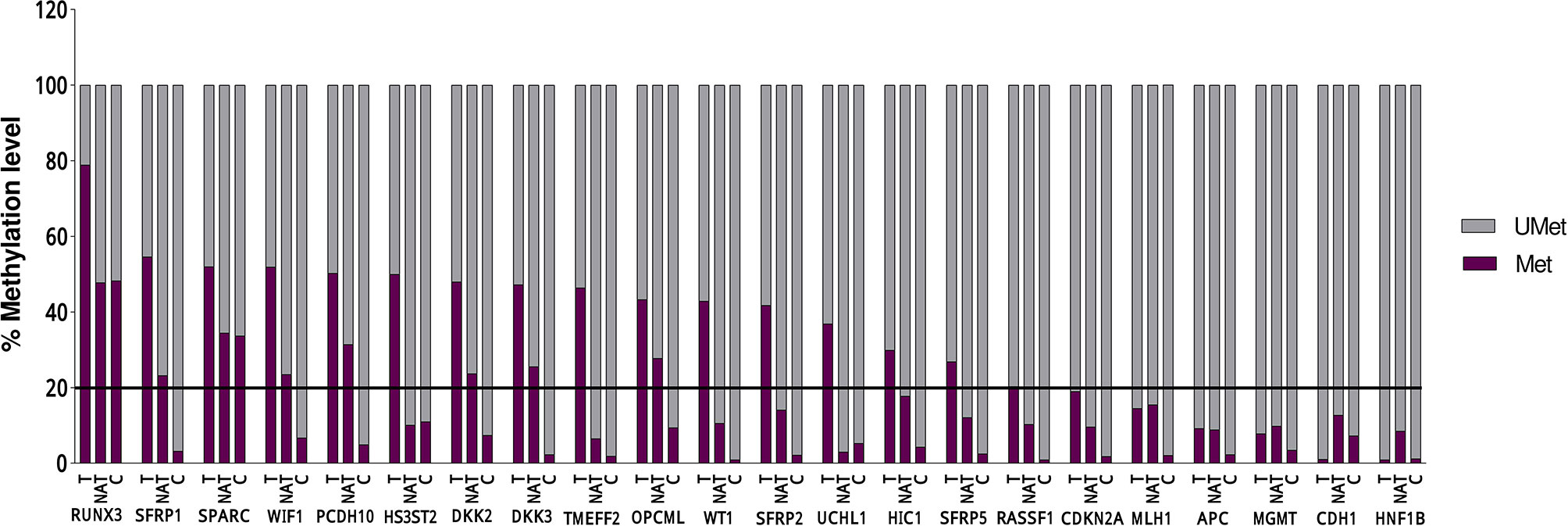
Figure 1 DNA methylation levels in 18 Tumoral (T), 10 Normal Adjacent Tissue l (NAT), and 6 Control (C) of each of the 22 evaluated genes. The line represent the threshold of 20% of CpG dinucleotides modified by a methyl group, above which the promoter is defined as methylated.
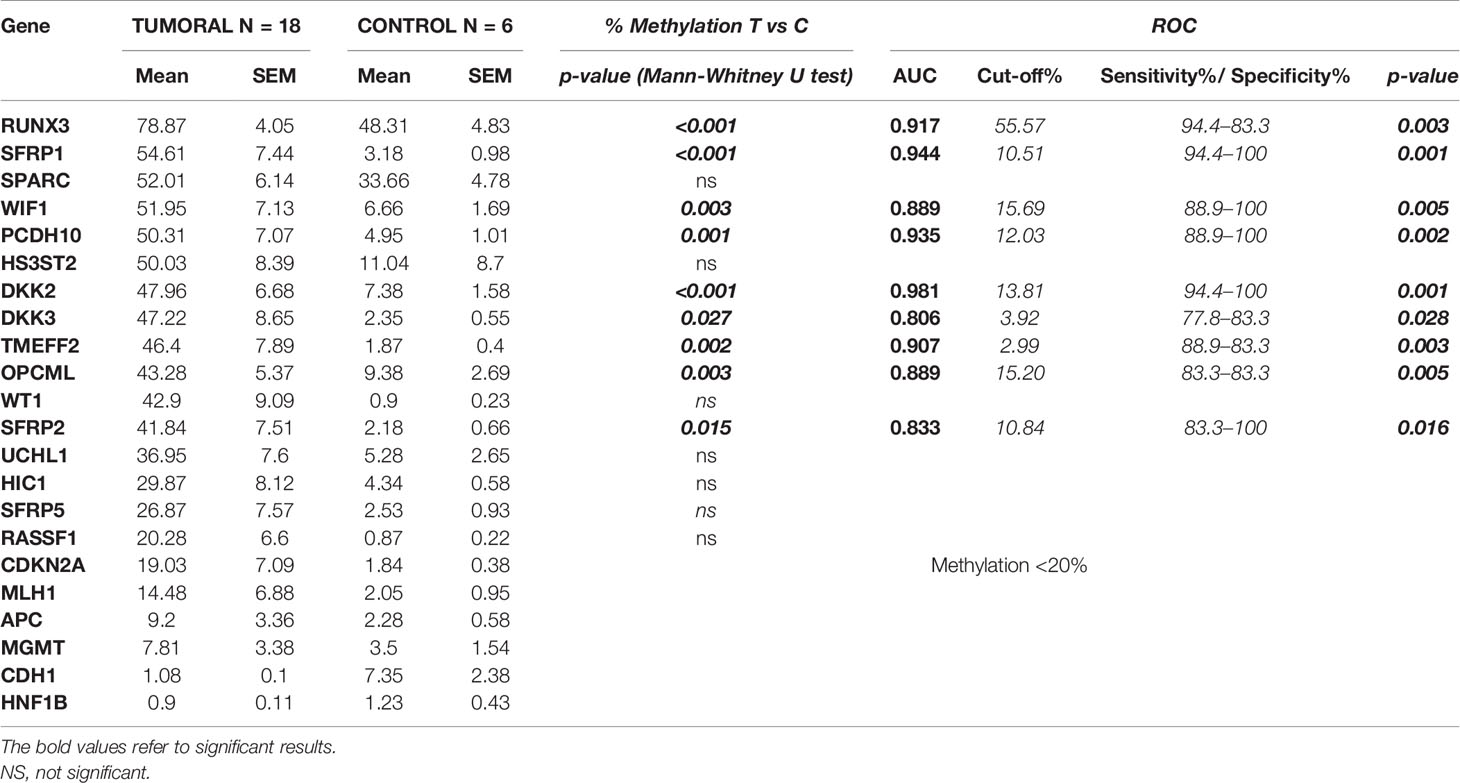
Table 3 Average of the methylation percentage (± SEM) in tumoral and control tissues along with the statistical results between the two groups and the ROC curve analysis.
A significant differential methylation profile of nine genes (RUNX3, SFRP1, WIF1, PCDH10, DKK2, DKK3, TMEFF2, OPCML, and SFRP2) was detected in tumoral tissues when compared to control tissues (Table 3). No statistical difference was observed when comparing the methylation levels of NAT to C (p > 0.05).To assess the potential diagnostic value in discriminating tumoral from normal tissues from controls, ROC analysis was performed. All the significant genes presented an AUC above 0.80 and a specificity and sensitivity between 77.8 and 100% (Table 3). When combining three selected potential biomarkers (SFRP1, PCDH10, and DKK2) the value of the AUC was 0.972 and p = 0.001 (Figure 2).
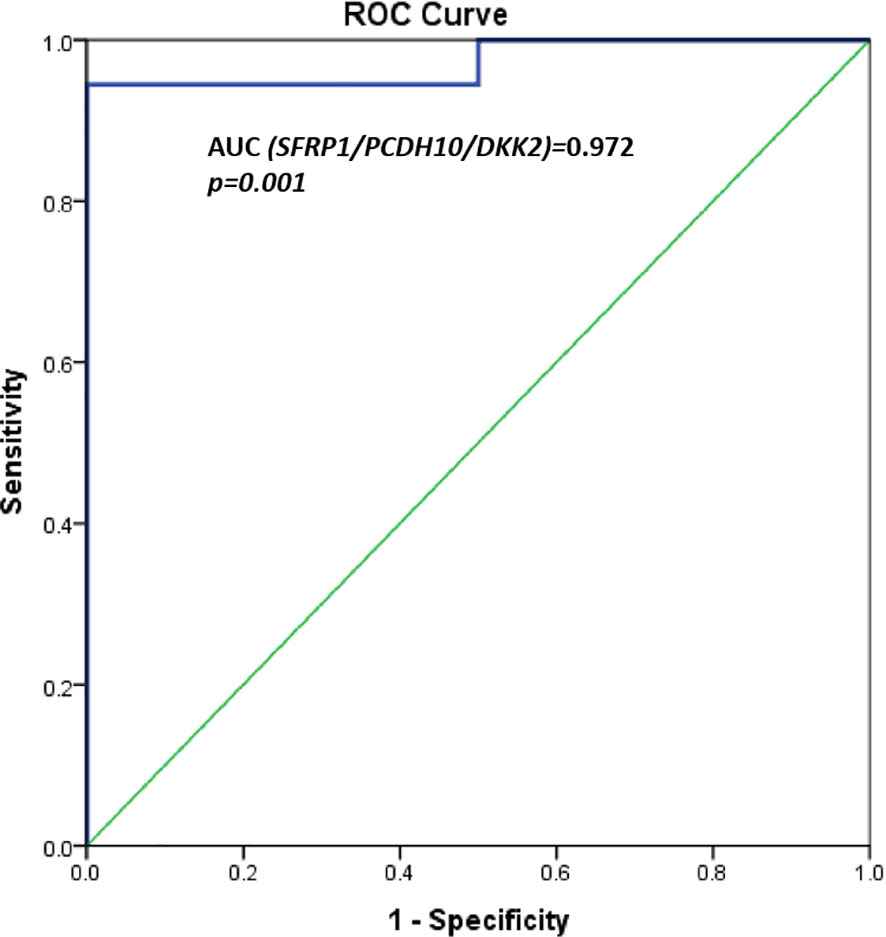
Figure 2 ROC curves for three potential biomarkers combined (SFRP1, PCDH10, and DKK2). AUC = 0.972, p = 0.001.
We also performed a paired analysis for the 10 tumoral samples with their matched NATs. The results showed that RUNX3 and TMEFF2 promoters were significantly hyper-methylated in tumoral tissue (p = 0.017 and p = 0.037, respectively) (Figure 3). SFRP1, WIF1, PCDH10, DKK2, DKK3, OPCML, and SFRP2 maintained the same trend of hyper-methylation in T vs. C cases, without reaching statistical significance in the comparison with NAT.
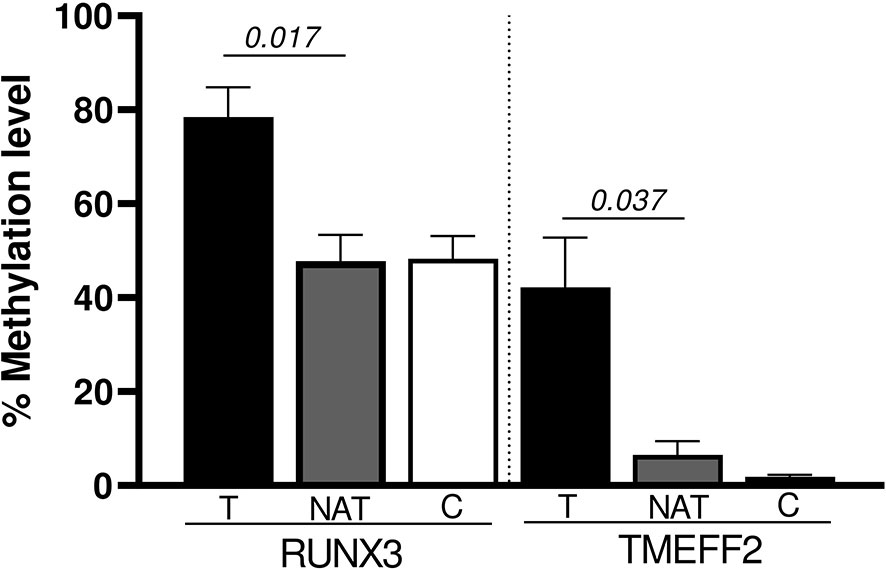
Figure 3 Significant promoter genes hyper-methylated in the paired analysis (Wilcoxon Signed Rank test): T vs NAT.
When comparing methylation profiles according to BRAF mutation status, we found that SFRP2, DKK2, PCDH10, TMEF2, SFRP1, and HS3ST2 showed methylation levels that were significantly higher in the BRAF positive cases (n = 4, including the case with BRAF+ and KRAS12+) than in BRAF negative cases (WT and KRAS+; n = 14) (Figure 4A). No changes were identified comparing BRAF+ vs WT (n = 7).
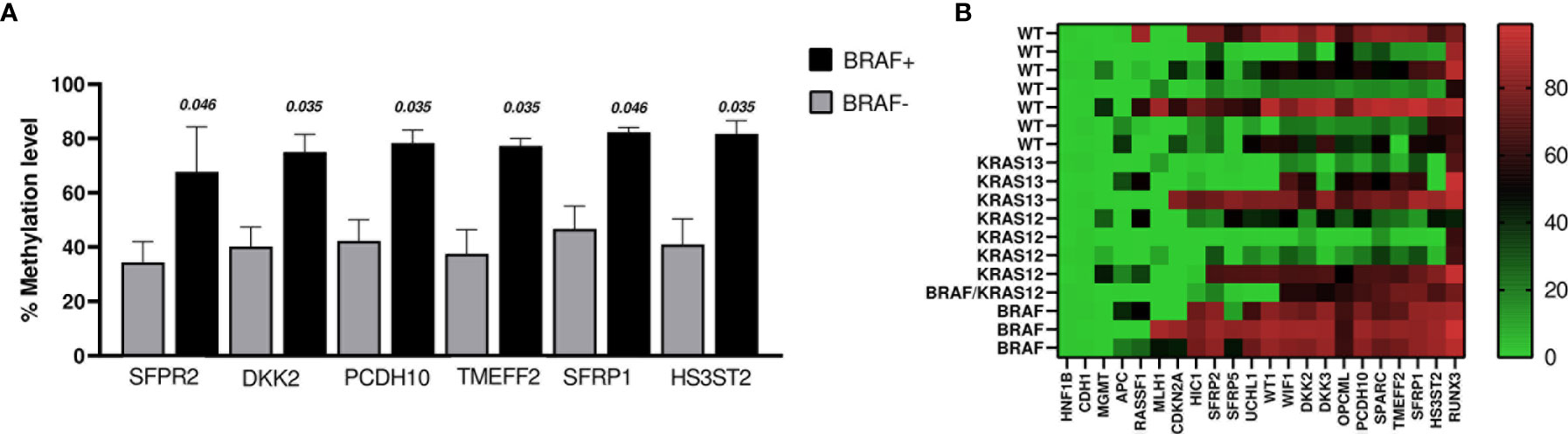
Figure 4 (A) The graph shows the genes whose promoters methylation percentage were significantly higher in BRAF+ cases (n = 4) vs BRAF− cases (n = 14). The bars indicate the mean ± SEM. (B) Heat map of DNA promoter methylation according to mutation status. Methylation levels vary according to the color scale, ranging from light green (low) to red (high).
Regarding KRAS mutation status, we found that the methylation level of SFRP2 was lower in KRAS12+/13+ (n = 8) versus KRAS12−/13− (n = 10) (p = 0.027). Even when the case with double mutation (BRAF+ and KRAS12+) was excluded, the comparison remained significant (p = 0.043). Moreover, OPCML was less methylated in the comparison of KRAS12+ (n = 4) to KRAS12− (n = 14), (p = 0.035). No significant difference in the percentage levels of methylation was found in the following comparisons: KRAS12+ (n = 4) with WT (n = 7), KRAS13+ (n = 3) with WT (n = 7), KRAS12+/13+ (n = 8) with WT (n = 7). The methylation results of the tumoral samples grouped by mutational status are presented in the heat map of Figure 4B.
Furthermore, we quantified the methylation levels of the promoters in peripheral blood mononuclear cells (PBMC) from five controls and five CRC patients whose methylation profiles were performed in the tumoral tissue. None of the five patients presented BRAF/KRAS mutations in their blood. Except for RUNX3, which was highly methylated both in patients and controls, the other 21 gene promoters did not show a significant general level of methylation (data not shown).
Discussion
In this study, we profiled DNA methylation of the promoters of 22 candidate genes in tumoral tissue, normal adjacent tissue from CRC patients, and normal mucosa from controls. In all tumoral samples, we also assessed the mutation status of KRAS and BRAF to investigate a possible association between hyper-methylation of these candidate genes and KRAS and BRAF mutations. Our study revealed the hyper-methylation of nine genes out of 22 in tumoral tissue from CRC patients compared to normal mucosa and identified a specific methylation profile for patients with the BRAF V600E mutation.
In the case-control comparison, we found hyper-methylation of genes belonging to the Wnt signaling pathway (SFRP1, SFRP2, DKK1, DKK2, and WIF1), the TGF-Beta;/SMAD pathway (RUNX3), two tumor suppressor genes (PCDH10 and OPCML), and a gene that acts both as an oncogene and a tumor suppressor, depending on the cellular context (TMEFF2). Many CRCs feature methylation of the extracellular inhibitors of Wnt signaling, such as SFRP1, SFRP2 (13–15), and genes belonging to the dickkopf (Dkk) family. Although genes from the Dkk family largely have an inhibitory effect on this pathway, there is evidence that DKK2 can also activate Wnt signaling (16). In particular, DKK2 and DKK3 were hyper-methylated in tumoral tissue from CRC versus control tissues or paired adjacent healthy tissues (13, 17, 18). Furthermore, DKK3 expression levels were negatively correlated with the rate of promoter methylation (19). Moreover, it has been suggested that DKK2 and DKK3 co hyper-methylation might be considered as an independent prognostic predictor (20). Aberrant methylation of WIF1 was found in CRC (13, 14, 20, 21) with a statistically significant association with increasing tumor stage and tumor differentiation (22).
A high percentage of colorectal cancers contain mutations that disrupt signaling in the pathways of the TGF-β family that regulate the proliferation, differentiation, adhesion, and migration of cells (23). One of the transcriptional effectors involved in TGF-β/SMAD signaling is RUNX3. Its promoter has been found to be methylated in approximately 30% of CRCs (15) with high levels both in CRC tissues (17, 24) and in serum, where methylation levels increase with the advancement of pathological stage (25). PCDH10 belongs to the protocadherin gene family and acts as tumor suppressor gene inhibiting cell proliferation and cell invasion in colorectal cancer development. The rate of PCDH10 methylation in CRC tissue was significantly higher compared with normal mucosa in different studies (26, 27) as well as in other types of cancer such as lymphomas (28, 29), breast cancer (30) and medulloblastoma (31). Another tumor suppressor gene found hyper-methylated in our study was OPCML (Opioid Binding Protein/Cell Adhesion Molecule Like). It is involved in cell growth, invasion, and metastasis. This gene exhibits high promoter methylation and reduced expression levels in different cancers, including ovarian, bladder, nasopharyngeal, and cervical cancers, as well as hepatocellular carcinomas, and colorectal cancer (32, 33). Finally, our comparison between CRC tissue and controls revealed hyper-methylation of TMEFF2. In this case, our results are also in line with the literature as it shows aberrant methylation of this gene in CRC (34), suggesting its role as prognostic marker (35).
Mutations in BRAF and KRAS genes occur in about 10 and 40% of CRC cases respectively (36, 37) and affect different biological pathways. Functionally, these genes are linked to dysregulated DNA methylation (38) and miRNA expression (39). We found a specific methylation profile of tumoral tissues with the BRAF V600E mutation that showed increased methylation levels of SFRP2, DKK2, PCDH10, TMEFF2, SFRP1, and HS3ST2 compared with tissues without this mutation.
The association of the BRAF V600E mutation and SFRP2 methylation levels has been already shown by Bagci and colleagues (40). In line with the results of this research group, the association between KRAS mutations in codon 12 and codon 13 and hyper-methylation of SFRP2 did not show significant results. Regarding the other identified genes, a specific association between BRAF V600E mutation and methylation levels has not, to our knowledge, been reported in the literature.
Epigenetic aberrations are reversible and therefore represent promising targets for novel approaches for cancer therapies (41, 42). The observation that BRAF-mutant CRCs displayed six hyper-methylated genes with tumor suppressive functions respect to BRAF-negative CRCs may shed light on their possible re-expression following treatment with demethylating agents. In CRC, epigenetic alterations may promote resistance to systemic drugs such as 5-fluorouracil (5-FU), oxaliplatin, and irinotecan (43). For this reason, it has been suggested that combined therapies with epigenetic agents may reverse drug resistance. It has been demonstrated that the use of a demethylating compound, such as 5-azacitidine (5-AC) improves sensitivity and reduces resistance in BRAF-mutant CRCs that are usually characterized by very aggressive behavior, poor prognosis, and resistance to therapies with 5-FU or irinotecans (44). Mao et al. demonstrated that treatment with demethylating agents might prime CRC for BRAF inhibitor treatment. In a xenograft model of CRC, these authors showed that pretreatment with 5-AC significantly increased the efficacy of subsequent treatments with a BRAF inhibitor (45).
Our results indicate a panel of genes that could be considered for the identification of an epigenetic molecular signature of BRAF-mutant CRCs and thus potential molecular targets for selective epigenetic treatments. Several epigenetic drugs are currently under study in clinical trials for the treatment of CRC (46); epigenetic target-based therapy might be a promising approach in the future to improve the curative treatments of CRC.
Clearly, there were some limitations to this study. Firstly, the sample size was relatively small; secondly, mRNA data, which is useful to perform a correlation between methylation and gene expression, was not available.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Victor Babes National Institute of Pathology. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MD designed and coordinated the study, performed the laboratory experiments, contributed to data processing and writing. VH and FV were responsible of the collection and diagnosis of the samples included in the study. EM was responsible of data processing, statistical analysis, and wrote the first draft of the manuscript. AS and GP gave their contribution in critical revision, data interpretation, and writing. IAP was involved in scientific writing. All authors discussed the results and commented on the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Ministry of Research, Innovation and Digitalization in Romania under grant no. PN 1N/2019_19.29.01.05.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patients involved in the study for the generous collaboration.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.697409/full#supplementary-material
References
1. Wong MCS, Huang J, Lok V, Wang J, Fung F, Ding H, et al. Differences in Incidence and Mortality Trends of Colorectal Cancer Worldwide Based on Sex, Age, and Anatomic Location. Clin Gastroenterol Hepatol (2020) 19(5):955–66.e61. doi: 10.1016/j.cgh.2020.02.026
2. Keum N, Giovannucci E. Global Burden of Colorectal Cancer: Emerging Trends, Risk Factors and Prevention Strategies. Nat Rev Gastroenterol Hepatol (2019) 16:713–32. doi: 10.1038/s41575-019-0189-8
3. Yamagishi H, Kuroda H, Imai Y, Hiraishi H. Molecular Pathogenesis of Sporadic Colorectal Cancers. Chin J Cancer (2016) 35:4. doi: 10.1186/s40880-015-0066-y
4. Montazeri Z, Li X, Nyiraneza C, Ma X, Timofeeva M, Svinti V, et al. Systematic Meta-Analyses, Field Synopsis and Global Assessment of the Evidence of Genetic Association Studies in Colorectal Cancer. Gut (2019) 69(8):1460–71. doi: 10.1136/gutjnl-2019-319313
5. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic Alterations During Colorectal-Tumor Development. N Engl J Med (1988) 319:525–32. doi: 10.1056/NEJM198809013190901
6. Nguyen HT, Duong H-Q. The Molecular Characteristics of Colorectal Cancer: Implications for Diagnosis and Therapy. Oncol Lett (2018) 16:9–18. doi: 10.3892/ol.2018.8679
7. Cionca FL, Dobre M, Dobrea CM, Iosif CI, Comănescu MV, Ardeleanu CM. Mutational Status of KRAS and MMR Genes in a Series of Colorectal Carcinoma Cases. Rom J Morphol Embryol (2018) 59:121–9.
8. Lee C-J, Ahn H, Jeong D, Pak M, Moon JH, Kim S. Impact of Mutations in DNA Methylation Modification Genes on Genome-Wide Methylation Landscapes and Downstream Gene Activations in Pan-Cancer. BMC Med Genomics (2020) 13:27. doi: 10.1186/s12920-020-0659-4
9. Robertson KD, Jones PA. DNA Methylation: Past, Present and Future Directions. Carcinogenesis (2000) 21:461–7. doi: 10.1093/carcin/21.3.461
10. Feinberg AP, Vogelstein B. Hypomethylation Distinguishes Genes of Some Human Cancers From Their Normal Counterparts. Nature (1983) 301:89–92. doi: 10.1038/301089a0
11. Kerachian MA, Kerachian M. Long Interspersed Nucleotide Element-1 (LINE-1) Methylation in Colorectal Cancer. Clin Chim Acta (2019) 488:209–14. doi: 10.1016/j.cca.2018.11.018
12. Dobre M, Comănescu M, Arsene D, Iosif C. Bussolati G. K-Ras Gene Mutation Status in Colorectal Cancer: Comparative Analysis of Pyrosequencing and PCR-RFLP. Rom J Morphol Embryol (2013) 54:567–74.
13. Voorham QJM, Janssen J, Tijssen M, Snellenberg S, Mongera S, van Grieken NCT, et al. Promoter Methylation of Wnt-Antagonists in Polypoid and Nonpolypoid Colorectal Adenomas. BMC Cancer (2013) 13:603. doi: 10.1186/1471-2407-13-603
14. Patai ÁV, Valcz G, Hollósi P, Kalmár A, Péterfia B, Patai Á, et al. Comprehensive DNA Methylation Analysis Reveals a Common Ten-Gene Methylation Signature in Colorectal Adenomas and Carcinomas. PloS One (2015) 10:e0133836. doi: 10.1371/journal.pone.0133836
15. Tse JWT, Jenkins LJ, Chionh F, Mariadason JM. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target? Trends Cancer (2017) 3:698–712. doi: 10.1016/j.trecan.2017.08.003
16. Niehrs C. Function and Biological Roles of the Dickkopf Family of Wnt Modulators. Oncogene (2006) 25:7469–81. doi: 10.1038/sj.onc.1210054
17. Silva TD, Vidigal VM, Felipe AV, DE Lima JM, Neto RA, Saad SS, et al. DNA Methylation as an Epigenetic Biomarker in Colorectal Cancer. Oncol Lett (2013) 6:1687–92. doi: 10.3892/ol.2013.1606
18. Winther K. The Effect of Beta-Blockade on Platelet Function and Fibrinolytic Activity. J Cardiovasc Pharmacol (1987) 10 Suppl 2:S94–8. doi: 10.1097/00005344-198710011-00018
19. Zhao S, Hao C-L, Zhao E-H, Jiang H-M, Zheng H-C. The Suppressing Effects of Dkk3 Expression on Aggressiveness and Tumorigenesis of Colorectal Cancer. Front Oncol (2020) 10:600322. doi: 10.3389/fonc.2020.600322
20. Liu X, Fu J, Bi H, Ge A, Xia T, Liu Y, et al. DNA Methylation of SFRP1, SFRP2, and WIF1 and Prognosis of Postoperative Colorectal Cancer Patients. BMC Cancer (2019) 19:1212. doi: 10.1186/s12885-019-6436-0
21. Jalilvand A, Soltanpour MS. Promoter Hypermethylation of Wnt/Beta;-Catenin Signaling Pathway Inhibitor WIF-1 Gene and Its Association With MTHFR C677T Polymorphism in Patients With Colorectal Cancer. Oman Med J (2020) 35:e131. doi: 10.5001/omj.2020.49
22. Samaei NM, Yazdani Y, Alizadeh-Navaei R, Azadeh H, Farazmandfar T. Promoter Methylation Analysis of WNT/β-Catenin Pathway Regulators and its Association With Expression of DNMT1 Enzyme in Colorectal Cancer. J BioMed Sci (2014) 21:73. doi: 10.1186/s12929-014-0073-3
23. Jung B, Staudacher JJ, Beauchamp D. Transforming Growth Factor Beta; Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology (2017) 152:36–52. doi: 10.1053/j.gastro.2016.10.015
24. Shin EJ, Kim HJ, Son MW, Ahn TS, Lee HY, Lim DR, et al. Epigenetic Inactivation of RUNX3 in Colorectal Cancer. Ann Surg Treat Res (2018) 94:19–25. doi: 10.4174/astr.2018.94.1.19
25. Nishio M, Sakakura C, Nagata T, Komiyama S, Miyashita A, Hamada T, et al. RUNX3 Promoter Methylation in Colorectal Cancer: Its Relationship With Microsatellite Instability and its Suitability as a Novel Serum Tumor Marker. Anticancer Res (2010) 30:2673–82.
26. Zhong X, Zhu Y, Mao J, Zhang J, Zheng S. Frequent Epigenetic Silencing of PCDH10 by Methylation in Human Colorectal Cancer. J Cancer Res Clin Oncol (2013) 139:485–90. doi: 10.1007/s00432-012-1353-5
27. Li M, Yan DG, Liu JL. Methylation Status of PCDH10 and RASSF1A Gene Promoters in Colorectal Cancer. Zhonghua Yi Xue Za Zhi (2016) 96:456–9. doi: 10.3760/cma.j.issn.0376-2491.2016.06.010
28. Narayan G, Xie D, Freddy AJ, Ishdorj G, Do C, Satwani P, et al. PCDH10 Promoter Hypermethylation Is Frequent in Most Histologic Subtypes of Mature Lymphoid Malignancies and Occurs Early in Lymphomagenesis. Genes Chromosomes Cancer (2013) 52:1030–41. doi: 10.1002/gcc.22098
29. Huang W, Xue X, Shan L, Qiu T, Guo L, Ying J, et al. Clinical Significance of PCDH10 Promoter Methylation in Diffuse Large B-Cell Lymphoma. BMC Cancer (2017) 17:815. doi: 10.1186/s12885-017-3810-7
30. de Ruijter TC, van der Heide F, Smits KM, Aarts MJ, van Engeland M, Heijnen VCG. Prognostic DNA Methylation Markers for Hormone Receptor Breast Cancer: A Systematic Review. Breast Cancer Res (2020) 22:13. doi: 10.1186/s13058-020-1250-9
31. Bertrand KC, Mack SC, Northcott PA, Garzia L, Dubuc A, Pfister SM, et al. PCDH10 is a Candidate Tumour Suppressor Gene in Medulloblastoma. Childs Nerv Syst (2011) 27:1243–9. doi: 10.1007/s00381-011-1486-x
32. Li C, Tang L, Zhao L, Li L, Xiao Q, Luo X, et al. OPCML is Frequently Methylated in Human Colorectal Cancer and its Restored Expression Reverses EMT via Downregulation of Smad Signaling. Am J Cancer Res (2015) 5:1635–48.
33. Abdel-Rahman WM, Lotsari-Salomaa JE, Kaur S, Niskakoski A, Knuutila S, Järvinen H, et al. The Role of Chromosomal Instability and Epigenetics in Colorectal Cancers Lacking β-Catenin/TCF Regulated Transcription. Gastroenterol Res Pract (2016) 2016:6089658. doi: 10.1155/2016/6089658
34. Gonzalo V, Lozano JJ, Muñoz J, Balaguer F, Pellisé M, Rodríguez de Miguel C, et al. Aberrant Gene Promoter Methylation Associated With Sporadic Multiple Colorectal Cancer. PloS One (2010) 5:e8777. doi: 10.1371/journal.pone.0008777
35. Lam K, Pan K, Linnekamp JF, Medema JP, Kandimalla R. DNA Methylation Based Biomarkers in Colorectal Cancer: A Systematic Review. Biochim Biophys Acta (2016) 1866:106–20. doi: 10.1016/j.bbcan.2016.07.001
36. Caputo F, Santini C, Bardasi C, Cerma K, Casadei-Gardini A, Spallanzani A, et al. BRAF-Mutated Colorectal Cancer: Clinical and Molecular Insights. Int J Mol Sci (2019) 20:5369. doi: 10.3390/ijms20215369
37. Dobre M, Dinu DE, Panaitescu E, Bîrlă RD, Iosif CI, Boeriu M, et al. KRAS Gene Mutations - Prognostic Factor in Colorectal Cancer? Rom J Morphol Embryol (2015) 56:671–8. doi: 10.26226/morressier.57a2f938d462b8028d88feff
38. Tanaka N, Huttenhower C, Nosho K, Baba Y, Shima K, Quackenbush J, et al. Novel Application of Structural Equation Modeling to Correlation Structure Analysis of Cpg Island Methylation in Colorectal Cancer. Am J Pathol (2010) 177:2731–40. doi: 10.2353/ajpath.2010.100361
39. Milanesi E, Dobre M, Bucuroiu AI, Herlea V, Manuc TE, Salvi A, et al. MiRNAs-Based Molecular Signature for KRAS Mutated and Wild Type Colorectal Cancer: An Explorative Study. J Immunol Res (2020) 2020:4927120. doi: 10.1155/2020/4927120
40. Bagci B, Sari M, Karadayi K, Turan M, Ozdemir O, Bagci G. KRAS, BRAF Oncogene Mutations and Tissue Specific Promoter Hypermethylation of Tumor Suppressor SFRP2, DAPK1, MGMT, HIC1 and P16 Genes in Colorectal Cancer Patients. Cancer Biomark (2016) 17:133–43. doi: 10.3233/CBM-160624
41. Suvà ML, Riggi N, Bernstein BE. Epigenetic Reprogramming in Cancer. Science (2013) 339:1567–70. doi: 10.1126/science.1230184
42. Baretti M, Azad NS. The Role of Epigenetic Therapies in Colorectal Cancer. Curr Probl Cancer (2018) 42:530–47. doi: 10.1016/j.currproblcancer.2018.03.001
43. Crea F, Nobili S, Paolicchi E, Perrone G, Napoli C, Landini I, et al. Epigenetics and Chemoresistance in Colorectal Cancer: An Opportunity for Treatment Tailoring and Novel Therapeutic Strategies. Drug Resist Update (2011) 14:280–96. doi: 10.1016/j.drup.2011.08.001
44. Sharma A, Vatapalli R, Abdelfatah E, Wyatt McMahon K, Kerner Z, A Guzzetta A, et al. Hypomethylating Agents Synergize With Irinotecan to Improve Response to Chemotherapy in Colorectal Cancer Cells. PloS One (2017) 12:e0176139. doi: 10.1371/journal.pone.0176139
45. Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Dayyani F, et al. Resistance to BRAF Inhibition in BRAF-Mutant Colon Cancer can be Overcome With PI3K Inhibition or Demethylating Agents. Clin Cancer Res (2013) 19:657–67. doi: 10.1158/1078-0432.CCR-11-1446
Keywords: colorectal cancer, DNA methylation, KRAS, BRAF, mutations
Citation: Dobre M, Salvi A, Pelisenco IA, Vasilescu F, De Petro G, Herlea V and Milanesi E (2021) Crosstalk Between DNA Methylation and Gene Mutations in Colorectal Cancer. Front. Oncol. 11:697409. doi: 10.3389/fonc.2021.697409
Received: 19 April 2021; Accepted: 14 June 2021;
Published: 01 July 2021.
Edited by:
Octav Ginghina, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Paul Willemsen, Hospital Network Antwerp (ZNA), BelgiumJames N. Luo, Brigham and Women’s Hospital and Harvard Medical School, United States
Copyright © 2021 Dobre, Salvi, Pelisenco, Vasilescu, De Petro, Herlea and Milanesi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vlad Herlea, aGVybGVhMjAwMkB5YWhvby5jb20=