 Alyssa A. Leystra1
Alyssa A. Leystra1 Kristen N. Harvey1
Kristen N. Harvey1 Esther Kaunga1
Esther Kaunga1 Harvey Hensley2
Harvey Hensley2 Lisa A. Vanderveer1
Lisa A. Vanderveer1 Karthik Devarajan3Margie L. Clapper1*
Karthik Devarajan3Margie L. Clapper1*- 1Cancer Prevention and Control Program, Fox Chase Cancer Center, Philadelphia, PA, United States
- 2Biological Imaging Facility, Fox Chase Cancer Center, Philadelphia, PA, United States
- 3Biostatistics and Bioinformatics Facility, Fox Chase Cancer Center, Philadelphia, PA, United States
An urgent need exists to identify efficacious therapeutic preventive interventions for individuals who are at high risk of developing colorectal cancer. To maximize the benefits of preventive intervention, it is vital to identify the time interval during which the initiation of a preventive intervention will lead to an optimal outcome. The goal of the present study was to determine if oncogenic events can be detected in the nonneoplastic colonic mucosa of Apc+/Min-FCCC mice prior to formation of the first adenoma, thus defining an earlier point of intervention along the cancer continuum. Tissues taken at three potential points of intervention were characterized: prior to Apc mutation (wild type Apc+/+-FCCC mice); after initiation but prior to colon adenoma formation (tumor-free Apc+/Min-FCCC mice); and after formation of the first colon adenoma (tumor-bearing Apc+/Min-FCCC mice). Experimentation focused on molecular processes that are dysregulated in early colon lesions: 1) cellular proliferation (proliferative index and size of the proliferative zone); 2) cellular stemness (expression of Ascl2, Grem1, Lgr5 and Muc2); 3) EGFR signaling (expression of Ereg); and 4) inflammation (expression of Mmp9, Ptsg2, and Reg4, as well as secretion of 18 cytokines involved in immune activation and response). Interestingly, the nonneoplastic colonic mucosa of wild type, tumor-free Apc+/Min-FCCC, and tumor-bearing Apc+/Min-FCCC mice did not display significant differences in average epithelial cell proliferation (fold change 0.8–1.3, p≥0.11), mucosal gene expression (fold change 0.8–1.4, p≥0.22), or secretion of specific cytokines from colonic mucosa (fold change 0.2–1.5, p≥0.06). However, the level of cytokine secretion was highly variable, with many (22% of wild type, 31% of tumor-free Apc+/Min-FCCC, and 31% of tumor-bearing Apc+/Min-FCCC) mice categorized as outliers (> 1.5 x interquartile ranges below the first quartile or above the third quartile) due to elevated expression of at least one cytokine. In summary, no differences were observed in proliferation, stemness, and EGFR signaling in the colonic mucosa of wild type vs Apc+/Min-FCCC mice, with low baseline cytokine expression, prior to the formation of the first colon adenoma. The results of this study provide valuable baseline data to inform the design of future cancer prevention studies.
Introduction
Colorectal cancer (CRC) is the third-leading cause of cancer-related mortality in both men and women in the United States, with an estimated 147,950 new cases and 53,200 associated deaths expected this year (1). An alarming rise in CRC among younger adults (age ≤ 50) is emerging; incidence and mortality have increased 22% and 13%, respectively, over the past two decades (2). These statistics underscore the need to optimize preventive strategies to reduce CRC incidence.
The efficacy of several preventive agents varies depending on the time of intervention. Preclinical studies from this group and others indicate that some preventive agents work best when given prior to the formation of gross colon tumors (3–5). For example, thymoquinone exhibited more pronounced anti-tumor efficacy in rats when administered during initiation as opposed to afterwards (3). Administration of atorvastatin to Apc+/Min-FCCC mice that were tumor-free at the time of treatment initiation completely eliminated colon microadenomas and significantly decreased colon tumor incidence. In contrast, the regimen failed to alter tumor number or incidence in mice with pre-existing tumors (4). Likewise, a vaccine against Ascl2 decreased the number of colon microadenomas in mice when administered prior to tumor formation, but had a minimal effect when given to mice with existing tumors (5, 6). Thus, it is critical to identify the optimal time for treatment during tumorigenesis to maximize preventive efficacy.
The precise time when targetable molecular alterations arise during early colorectal carcinogenesis is not well defined. Most sporadic colon tumors are thought to arise via a stepwise process in which: (1) mutation of the APC colon tumor suppressor gene creates an ‘initiated’ epithelium that is predisposed to tumor formation; (2) loss of APC heterozygosity promotes the formation of an adenoma; and (3) additional mutations drive tumor progression (7, 8). In addition to mutational events, a number of other molecular changes occur at ill-defined time points before, during, or after adenoma formation. Compared to the normal epithelium, cells from early colon lesions demonstrate increased cellular proliferation (9), stemness (10–12), and EGFR (13, 14) and inflammatory signaling (15, 16). Importantly, each of these processes can be targeted with existing chemoprevention strategies. For example, treatment of Apc+/Min-FCCC mice with atorvastatin decreases expression of stem-related genes and subsequent tumor formation (4). The EGFR inhibitor erlotinib exhibits tumor-preventive efficacy in individuals with familial adenomatous polyposis, a syndrome caused by inheritance of a mutant allele of APC (17). Lastly, anti-inflammatory agents, including aspirin, sulindac, and celecoxib, reduce the risk of sporadic colon tumorigenesis (18). A greater understanding of the specific time when these processes are first disrupted during carcinogenesis will help define the optimal window for preventive intervention to achieve maximal efficacy.
The Apc+/Min-FCCC mouse model provides a unique opportunity to study early events in colon tumorigenesis. As in humans, adenomas arise sporadically within the colons of ApcMin mice following loss of Apc (19, 20), and exhibit increased cellular proliferation (21, 22), stemness (23), and EGFR (13, 24) and inflammatory signaling (25, 26). Thus, with respect to these events, the nonneoplastic colon tissue of the Apc+/Min-FCCC mouse mimics the initiated epithelium of an individual at high risk for CRC.
Others have hypothesized that Apc haploinsufficiency created by mutation of one allele of Apc might be sufficient to increase Wnt signaling in the colon crypt, expand the stem cell compartment, drive increased proliferation, and ultimately lead to adenoma formation (27–30). The goal of the present study was to test this hypothesis by determining if oncogenic events can be detected in the nonneoplastic colorectal epithelium that has been initiated through heterozygous mutation of Apc, thus defining an earlier point of intervention along the cancer continuum (Figure 1). Experimentation focused on molecular processes that can be targeted with existing preventive agents, including cellular proliferation (4), stemness (4), and EGFR (17) and inflammatory signaling (18). Three time points were modeled: prior to Apc mutation (wild type Apc+/+-FCCC mice); after initiation but prior to colon adenoma formation (tumor-free Apc+/Min-FCCC mice); and after formation of the first colon adenoma (tumor-bearing Apc+/Min-FCCC mice). A greater understanding of the timing of molecular changes during early colon tumorigenesis will aid in defining the optimal time interval (prior to initiation, after initiation but prior to formation of the first colon tumor, or after colon tumor formation) to initiate preventive interventions in high-risk individuals. In addition, the results of this study will provide valuable baseline data to inform the design of new therapeutic strategies for preventive intervention.
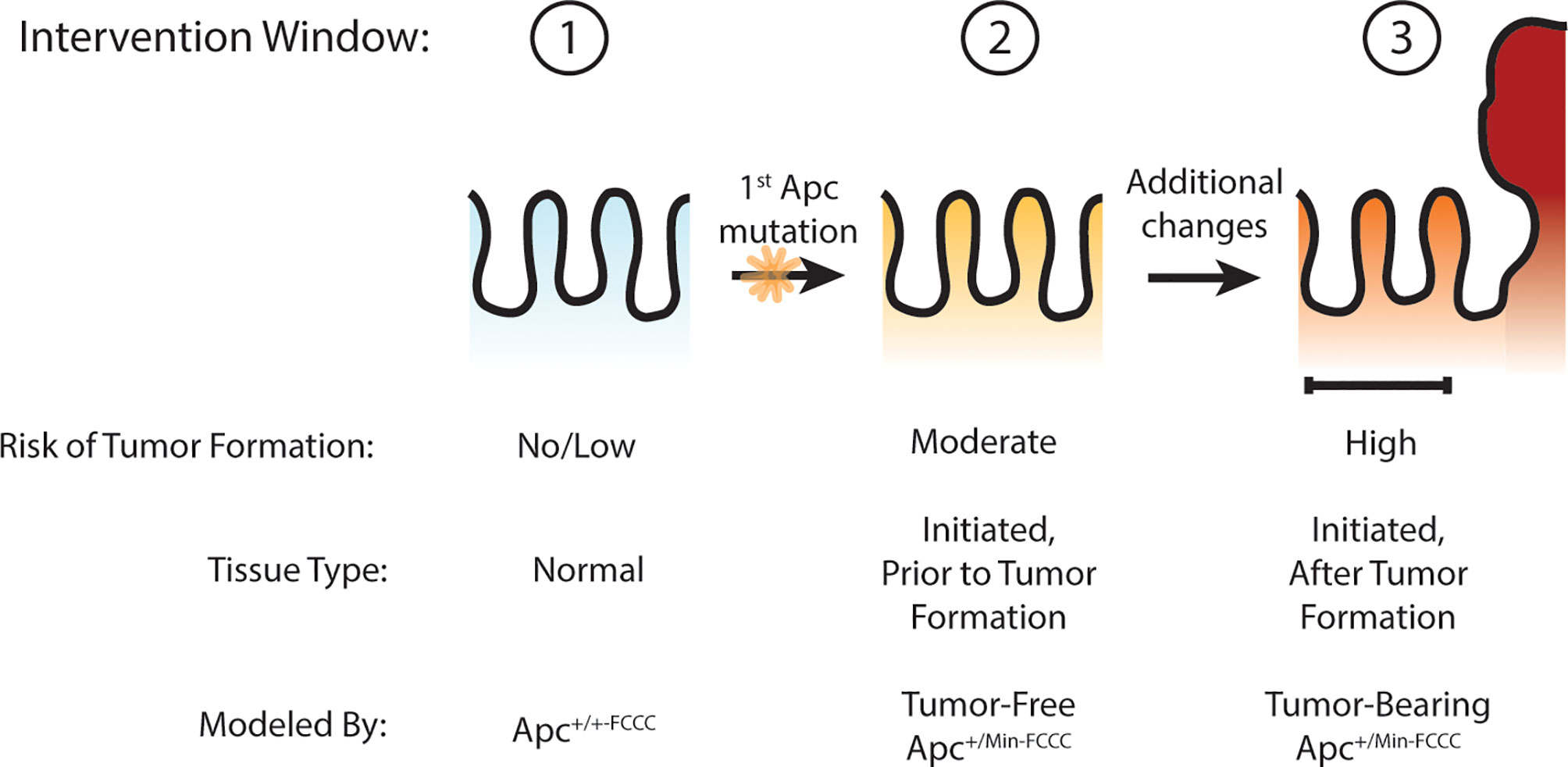
Figure 1 Windows of opportunity for preventive intervention for colon cancer. Three potential windows of opportunity were defined: (1) prior to tissue initiation (Apc mutation); (2) after initiation but prior to adenoma formation; (3) and after formation of the first adenoma. To examine these windows of opportunity, nonneoplastic tissue from mice representing each stage of early tumorigenesis was evaluated and pro-tumorigenic processes were characterized.
Materials and Methods
Animals
Male C57BL/6-Apc+/Min-FCCC mice (5 weeks old) were obtained from a closed colony that has been maintained at the Fox Chase Cancer Center for at least 50 generations (31). Apc+/Min-FCCC mice and Apc+/+-FCCC (wild type) littermates were provided autoclaved Rodent Breeder Diet (LabDiet #5013) and water ad libitum. At weaning, mice were assigned to cages by block randomization of litters, and switched to autoclaved 2018 Teklad 18% Protein/Extruded Global Rodent Diet (Envigo #2018SX) and autoclaved, double-distilled water for the duration of the study. Animals were housed in ventilated cages and maintained at 70 ± 2°F and 40-70% relative humidity with a 12-hour light/dark cycle. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the Fox Chase Cancer Center.
Colonoscopy
5-week-old mice weighing >15 g (33/36 Apc+/Min-FCCC mice and 9/10 wild type mice) were evaluated for colon tumors via colonoscopy, as described previously (4, 32). Colon tumors were detected in 3% (1/33) of the Apc+/Min-FCCC mice. As expected, no colon tumors were found in wild type mice.
Necropsy
At ~8 weeks of age, mice were injected with 100 mg/kg BrdU and euthanized one hour later by CO2 inhalation. Colons were excised, opened lengthwise, washed with PBS and examined grossly. The number, location, and size (length and width as measured with calipers) of each colon tumor was recorded. Colon tumor volume was calculated as a simplified approximation of an ellipsoid as follows:
Total tumor burden per mouse was calculated as the sum of all tumor volumes within the colon of that animal.
The distal portion of the colon (2.2 cm) was cut in half lengthwise and each half was divided into 2 mm sections (Supplemental Figure 1). Alternating pieces from one side were snap frozen in liquid nitrogen or placed in media for ex vivo cytokine analysis. Alternating pieces from the other side were embedded in OCT and frozen on dry ice or fixed in 4% paraformaldehyde for two days, equilibrated in a series of sucrose in 1x PBS (30% sucrose followed by 50% sucrose), embedded in OCT and frozen on dry ice. Any tissue section that contained a macroscopic colon tumor was excluded from molecular analysis.
BrdU Staining
OCT blocks of PFA-fixed tissue were cut (10 µM sections), thawed, and permeabilized in ice-cold methanol for 10 minutes. Antigen retrieval was performed with Citrate buffer (pH 6.0, 0.005% Tween) in a steamer for 20 minutes and cooled at room temperature for 40 minutes. Blocking, primary staining (rat anti-BrdU, 1:500; clone ICR1 Abcam ab6326), and secondary staining were completed with the VECTASTAIN ABC AP Kit, Rat IgG (Vector Laboratories), according to the manufacturer’s instructions. Sections were stained with DAB for 1-5 minutes, counterstained with hematoxylin, dehydrated in an ethanol series (70-100% ethanol), and cover slipped.
The total number of nuclei (stained and unstained) were counted from the right half of 6-20 full length crypts per mouse. The proliferative index was calculated as the proportion of stained nuclei normalized to the number of total nuclei in each crypt column (33). The size of the proliferative zone was calculated as the height of the highest BrdU+ cell normalized to the total height of the crypt column (33).
RT-qPCR
OCT blocks of frozen tissue were cut (10 µM sections) and thawed. The mucosal surface was microdissected from tissue sections using a razor blade. Total RNA was extracted from the microdissected tissue using the PicoPure™ RNA Isolation Kit (ThermoFisher, #KIT0214) according to the manufacturer’s instructions. Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Ref 4374966) per the manufacturer’s instructions. Quantitative PCR from the resulting cDNA was performed with iTaq Universal SYBR Green Supermix (Biorad). Amplification products were monitored using an ABI7900 Sequence Detection System and quantified using the comparative ΔΔCt method. Primer sequences are available in Supplemental Table 1.
Ex Vivo Cytokine Analysis
Tissues for ex vivo cytokine analyses were weighed and placed in 200 µL of RPMI containing 10% FBS, 100 U/mL Penicillin, and 100 µg/mL Streptomycin at 37°C. After 24 h, media was collected and spun at >16,000 x g at 4°C for 15 minutes. The supernatant was stored at -80°C until the time of analysis.
A Mouse Custom ProcartaPlex 18-plex chip (LifeTech, Assay ID MXGZFVD) was used to simultaneously assess the concentration of GM-CSF, GROα, IFNγ, IL-1β, IL-2, IL- 4, IL-5, IL-6, IL-9, IL-10, IL-12p70, IL-13, IL-17α, IL-18, IL-22, IL-23, IL-27, and TNFα in the supernatant from ex vivo cultures. Samples were loaded onto the chip according to the manufacturer’s instructions and analyzed using the Bioplex100/200 (Biorad).
Statistical Analyses
Mann-Whitney, Welch’s t-test, and linear regression tests were used as indicated throughout the text. All tests were two-sided and employed a type I error of 5% to determine significance. Outliers were defined as data points that were > 2 standard deviations from the mean in gene expression data or > 1.5 x interquartile ranges below the first quartile or above the third quartile in cytokine expression data. P-values from the comprehensive cytokine panel were adjusted to account for multiple testing using the Benjamini-Hochberg false discovery rate approach; both uncorrected and corrected values are reported. The R statistical software, including the dplyr package, was used to complete all analyses (34, 35).
Results
Timing of Macroscopic Tumor Formation in Apc+/Min-FCCC Mice
To identify the ideal age of mice to use in this study, a pilot study was conducted to characterize colon tumor incidence over a short time course. The colons of twenty male Apc+/Min-FCCC mice were examined by colonoscopy at 6, 8, and 10 weeks of age. Colon tumor incidence increased linearly over time; 25% of the mice had at least one colon tumor at 6 weeks of age, 45% by 8 weeks of age, and 65% at 10 weeks of age (Figure 2). Based on the results of the time course, 8-week-old mice were selected for further evaluation, as sexual maturity had been achieved and macroscopic colon tumors were identified in <50% of these animals.
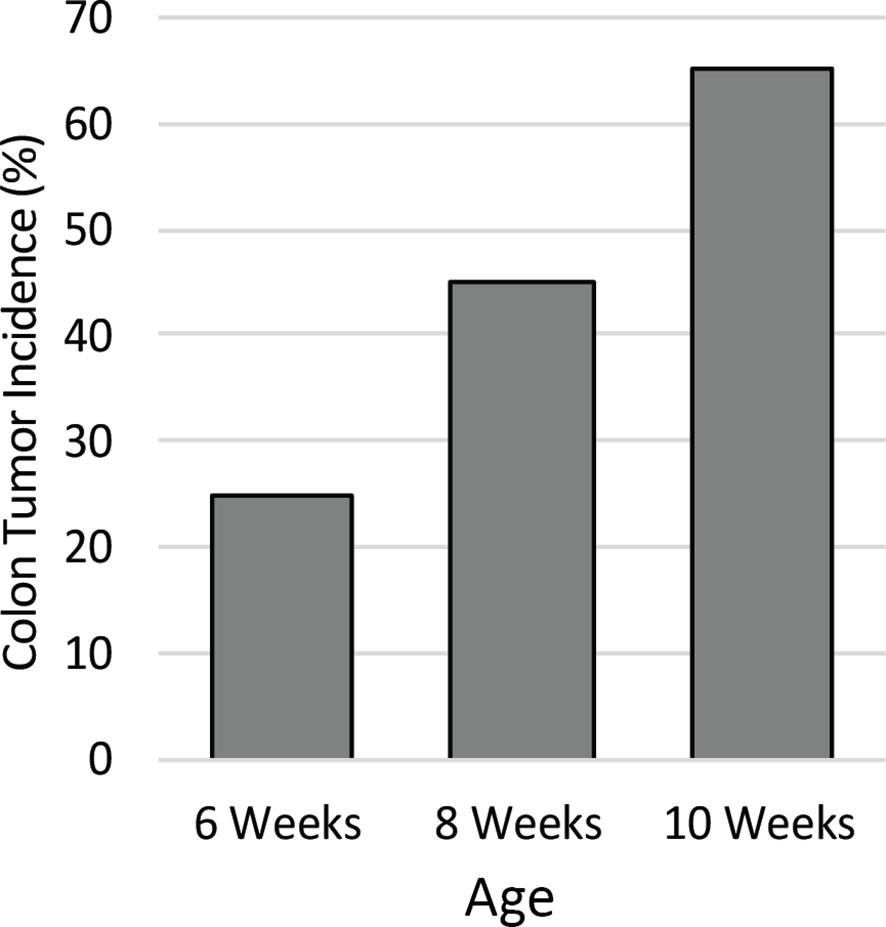
Figure 2 Colon tumor incidence in Apc+/Min-FCCC mice over time. Animals (n=20) assessed by colonoscopy exhibited a linear increase in colon tumor incidence from 6 to 10 weeks of age.
Animals
Apc+/Min-FCCC (n=36) and wild type (n=10) male mice were enrolled in the study. To standardize the impact of cage environment, animals were assigned to cages by block randomization of litters by 5 weeks of age. Apc+/Min-FCCC and wild type mice were cohoused.
Mouse weight represents one surrogate of overall health of the animal. To examine whether a single mutation in the Apc gene caused a difference in body weight, animals were weighed at 8 weeks of age. On average, Apc+/Min-FCCC mice weighed slightly less than wild type mice; Apc+/Min-FCCC weighed 21.5 ± 0.3 g (Mean ± S.E.M.) whereas wild type mice weighed 22.9 ± 0.2 g (Mean ± S.E.M.; Mann-Whitney p=0.04; Supplemental Figure 2).
At the time of euthanasia, colons were examined grossly to identify potential tumors. As seen in 8-week-old animals in the time course study, approximately half (42%; 15/36) of Apc+/Min-FCCC mice and 0% (0/10) of wild type mice had macroscopic colon adenomas. Mice with tumors had an average of 1.6 ± 1.0 (Mean ± S.D.; Range 1-4) colon tumors per animal. As expected, tumors arose primarily in the distal and medial colon (Supplemental Figure 3).
Notably, factors associated with colon tumorigenesis vary naturally along the length of the colon, including cellular proliferation (36), as well as the expression of stem cell-associated genes (28) and inflammatory cytokines (37). To control for this natural variation, all comparisons of these factors among different mouse groups were made using tissue from the distal colon, as this region is: 1) where >20% of colon tumors eventually form in Apc+/Min-FCCC mice (Supplemental Figure 3) can be evaluated by colonoscopy during preclinical chemo- and immunoprevention studies (4, 38, 39).
Analysis of Cellular Proliferation in the Nonneoplastic Colonic Epithelium of Apc+/Min-FCCC and Wild Type Mice
The rate of cellular proliferation in the initiated epithelium of Apc+/Min-FCCC mice was compared to that of the uninitiated epithelium of wild type mice. Proliferating cells were labeled via BrdU incorporation and a 2 cm segment of the distal colon was examined (Supplemental Figure 1). Both the proliferative index (the proportion of epithelial cells within each crypt that are actively proliferating) and the size of the proliferative zone (the region at the base of the crypt that contains proliferating cells) within nonneoplastic colonic crypts were quantified. No significant difference in the proliferative index of nonneoplastic colonic crypts of Apc+/Min-FCCC and wild type mice was detected (Mean ± S.E.M. - 19 ± 6% and 24 ± 6%, respectively; Welch’s t-test p = 0.55) (Figure 3). Likewise, the size of the proliferative zones within the nonneoplastic crypts of Apc+/Min-FCCC and wild type mice was comparable (Welch’s t-test p = 0.56, Figure 3); the average size of the proliferative zone of nonneoplastic crypts from Apc+/Min-FCCC mice was 40 ± 2% (Mean ± S.E.M.), compared to 44 ± 4% for wild type mice. Thus, cellular proliferation is similar in the nonneoplastic colonic epithelium of Apc+/Min-FCCC and wild type animals.
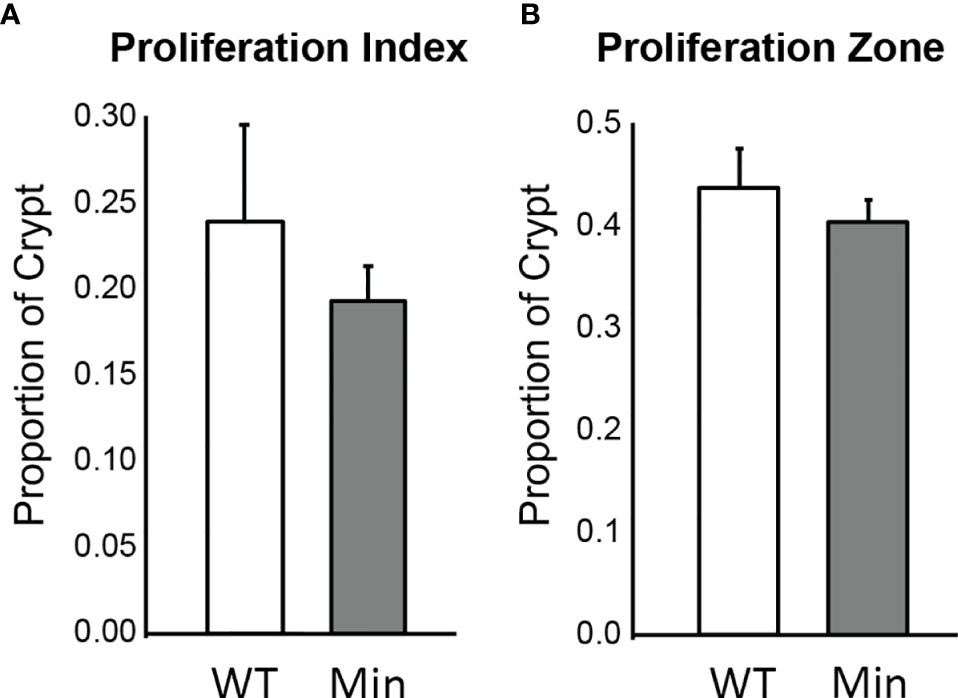
Figure 3 Epithelial cell proliferation is similar in the distal colon of Apc+/Min-FCCC (‘Min’; n=12) vs. Apc+/+-FCCC wild type (‘WT’; n=4) mice. Bars represent mean ± S.E.M. (A) Fold change in mean proliferative index = 0.8; Welch’s t-test p = 0.55. (B) Fold change in mean size of the proliferative zone = 0.9; Welch’s t-test p = 0.56.
Expression of Adenoma-Associated Genes in the Nonneoplastic Colonic Mucosa of Apc+/Min-FCCC and Wild Type Mice
To determine whether gene transcription was impacted by a single Apc mutation in the nonneoplastic mucosa of young Apc+/Min-FCCC mice, a panel of genes known to be modulated during early tumorigenesis was selected for analysis. The gene set included stem-related genes [Ascl2 (12, 40, 41), Grem1 (42, 43), and Lgr5 (40, 44)], a differentiation-associated gene [Muc2 (44, 45)], inflammation-associated molecules [Mmp9 (46), Ptsg2 (47), and Reg4 (48, 49)]; and an EGFR ligand [Ereg (13, 50)]. Relative RNA expression levels in tissues from Apc+/Min-FCCC and wild type mice were compared by qPCR. Overall, the mean relative expression of all genes analyzed was comparable in the nonneoplastic mucosa of Apc+/Min-FCCC and wild type mice (fold change 0.9 – 1.4, Welch’s t-test p≥0.22; Table 1; Supplemental Table 2).

Table 1 Fold change in mean gene transcription in the initiated colonic mucosa of Apc+/Min-FCCC (Min) vs wild type (WT) mice after removing outliers.
Cytokine Expression in the Nonneoplastic Colon Tissue of Mice
Inflammation is associated with an increased risk of colon tumorigenesis. As observed in humans, inflammatory cytokine production can increase colon tumor risk in Apc+/Min mice (15, 51, 52). Thus, the basal levels of cytokines involved in Th1, Th2, Th9, and Th17 immune activation and response were evaluated in nonnoneoplastic colon tissue from Apc+/Min-FCCC mice and compared to those of the uninitiated mucosa of wild type mice at 8 weeks of age. Most (14/18) cytokines were secreted at lower median levels in colon tissue from Apc+/Min-FCCC vs. wild type mice, although no comparisons reached statistical significance (fold change 0.2 – 1.5, Mann-Whitney p≥0.32; Table 2 and Figure 4).
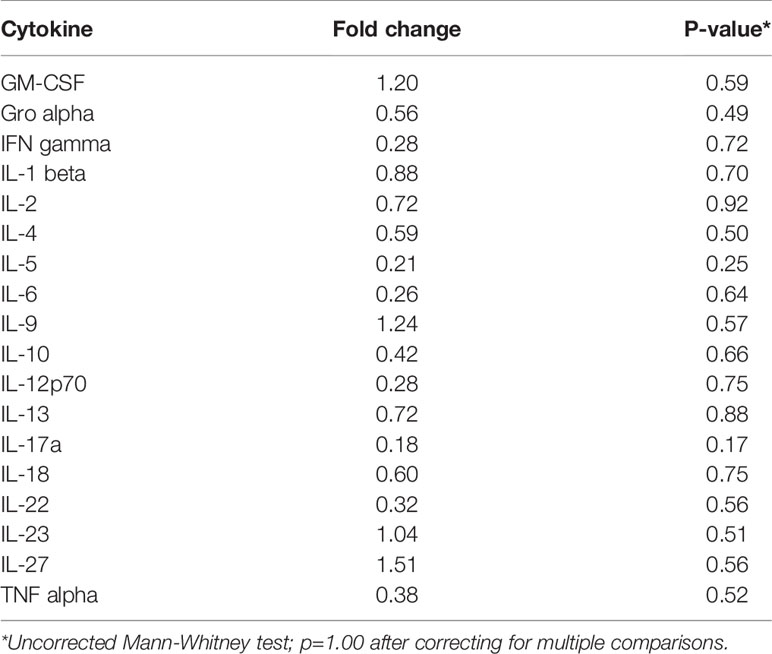
Table 2 Median fold change in secretion of cytokines involved in Th1, Th2, Th9, and Th17 signaling from nonneoplastic colon tissue from Apc+/Min-FCCC vs. normal colon tissue from wild type mice.
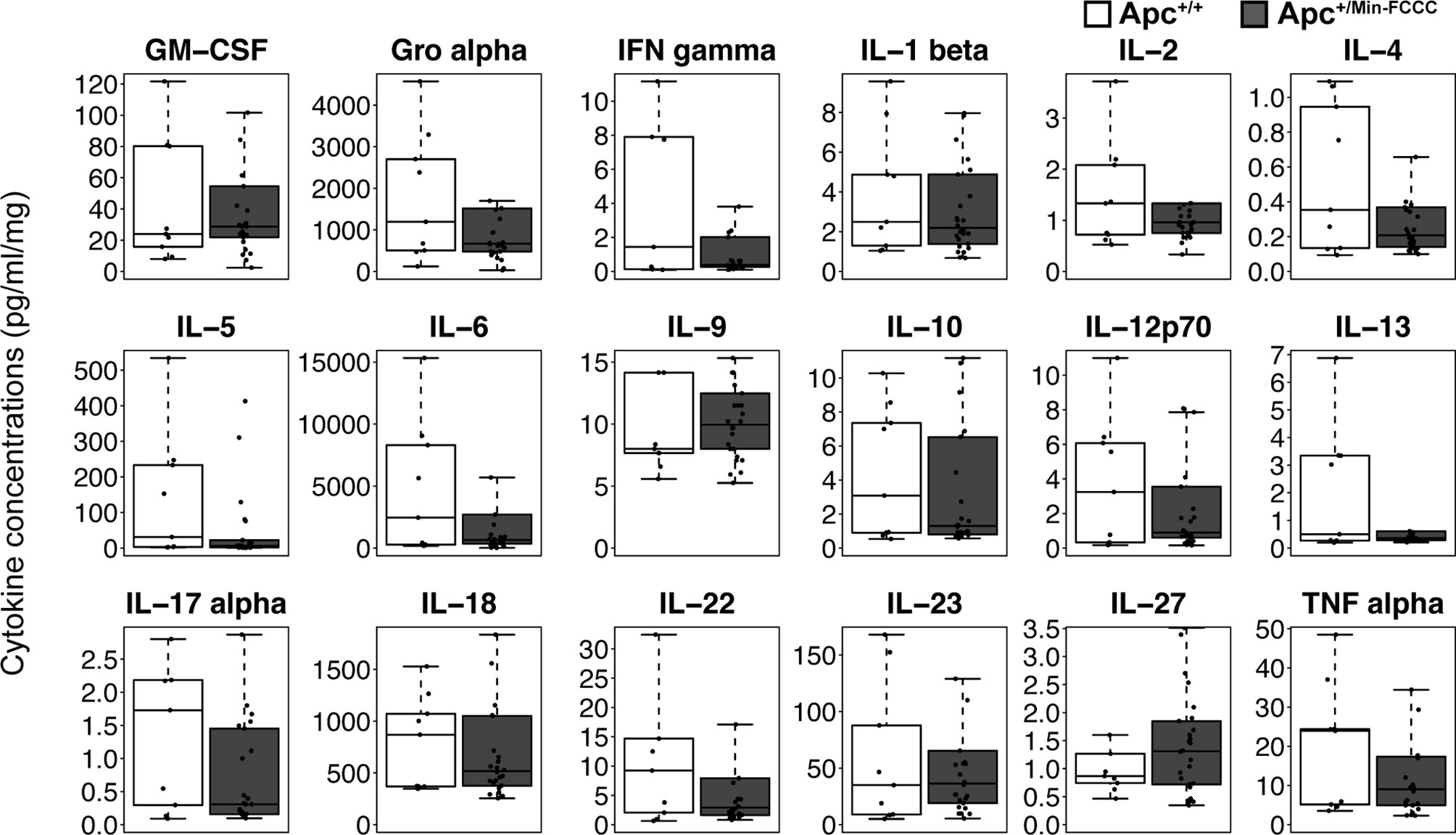
Figure 4 Cytokines involved in Th1, Th2, Th9, and Th17 signaling in colon tissues from Apc+/Min-FCCC and Apc+/+-FCCC wild type mice. Boxplots depict cytokine concentration in media (pg/ml) normalized to mass of colon tissue (mg) from Apc+/+-FCCC wild type mice (white; n=9) and Apc+/Min-FCCC mice (gray; n=26). Outlying data points (>3rd quartile + 1.5 x interquartile range) are omitted; all data points are shown in Supplemental Figure 4. Median fold change = 0.2 – 1.5; Mann Whitney p≥0.17; p=1.00 after adjusting for multiple comparisons.
A high degree of variability in cytokine expression was observed among the mice. In general, the interquartile range and median absolute deviation of cytokine expression was greater in wild type mice, while the number of outliers (> 1.5 x interquartile ranges below the first quartile or above the third quartile) was greater in Apc+/Min-FCCC mice (Table 3 and Supplemental Figure 4). Variability was high even among animals from the same litter, batch, or cage (Supplemental Figure 5). Although production of inflammatory cytokines is increased in obese animals (53–55), cytokine expression did not correlate with the weight of the animals in this study (linear model R2 ≤ 0.08; p≥0.13; Supplemental Figure 6). Thus, no single genetic or environmental factor was identified that accounted for the inter-mouse variability in cytokine expression.
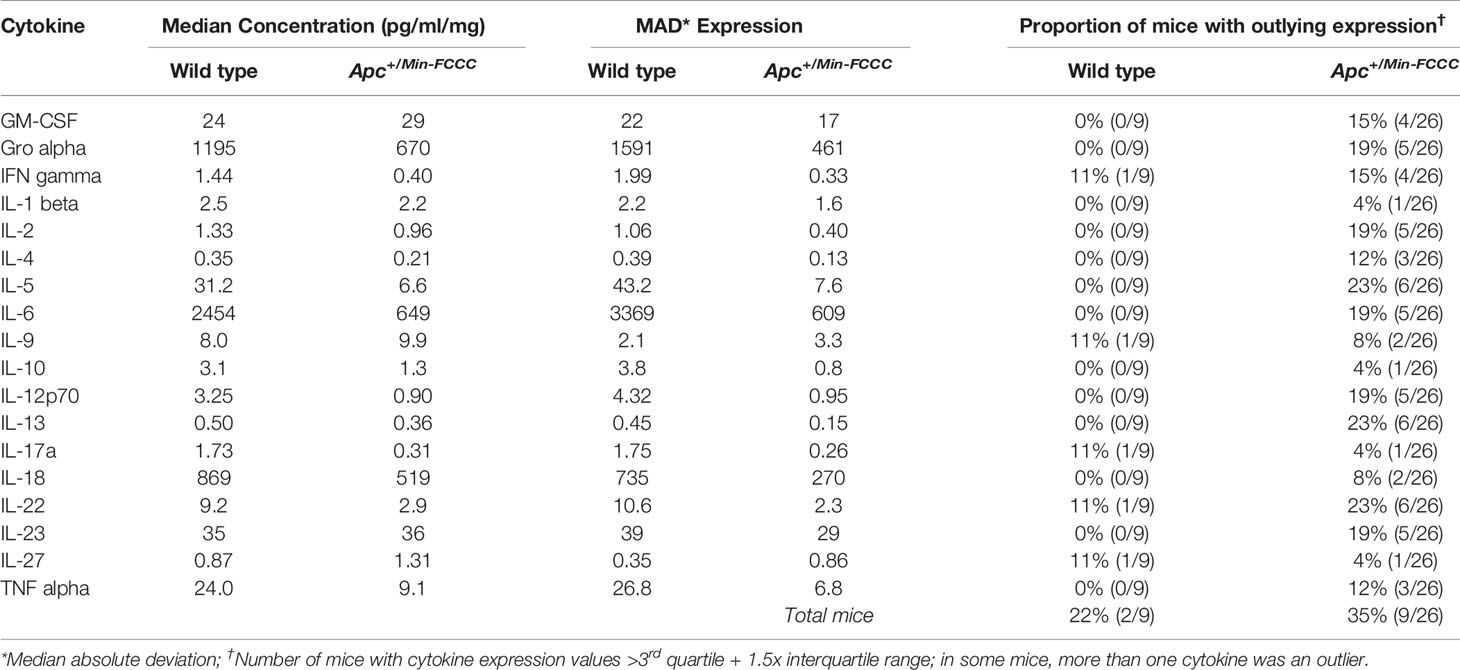
Table 3 Intra-group variability in secretion of cytokines involved in Th1, Th2, Th9, and Th17 signaling from initiated colon tissues from Apc+/Min-FCCC mice vs normal colon tissues from wild type mice.
Comparison of Initiated Colon Tissue From 8-Week-Old Tumor-Free vs. Tumor-Bearing Apc+/Min-FCCC Mice
Given that chemo- and immunopreventive interventions appear to be more effective at reducing tumor incidence and burden in Apc+/Min-FCCC mice when administered prior to polyp formation vs. after development of the first polyp (4, 5), we reasoned that the initiated colonic mucosa of tumor-free Apc+/Min-FCCC mice might differ from that of Apc+/Min-FCCC mice bearing at least one macroscopic tumor. Therefore, Apc+/Min-FCCC mice were classified as ‘tumor-free’ if no lesions were grossly visible within the colon at necropsy, or ‘tumor-bearing’ if at least one lesion was detected. As above, nonneoplastic colon tissue from tumor-free and tumor-bearing Apc+/Min-FCCC mice were evaluated for perturbations in processes associated with early tumorigenesis, including cellular proliferation (9), stemness (10–12), and EGFR (13, 14) and inflammatory signaling (15, 16).
The proliferation rates of the initiated epithelium of tumor-free and tumor-bearing Apc+/Min-FCCC mice were compared. The average proliferative index for initiated, nonneoplastic tissue in tumor-free Apc+/Min-FCCC mice was 20 ± 3% (Mean ± S.E.M.), whereas the average proliferative index of nonneoplastic tissue from tumor-bearing Apc+/Min-FCCC mice was 18 ± 3%. The average size of the proliferative zone of nonneoplastic crypts from tumor-free Apc+/Min-FCCC mice was 44 ± 4% (Mean ± S.E.M.), whereas the average size of the proliferative zone of nonneoplastic crypts from tumor-bearing Apc+/Min-FCCC mice was 37 ± 2% (Mean ± S.E.M.). Thus, cellular proliferation did not differ in tumor-free vs tumor-bearing Apc+/Min-FCCC mice (Welch’s t-test p = 0.65 for the proliferative index and p = 0.15 for proliferative zone; Figure 5).
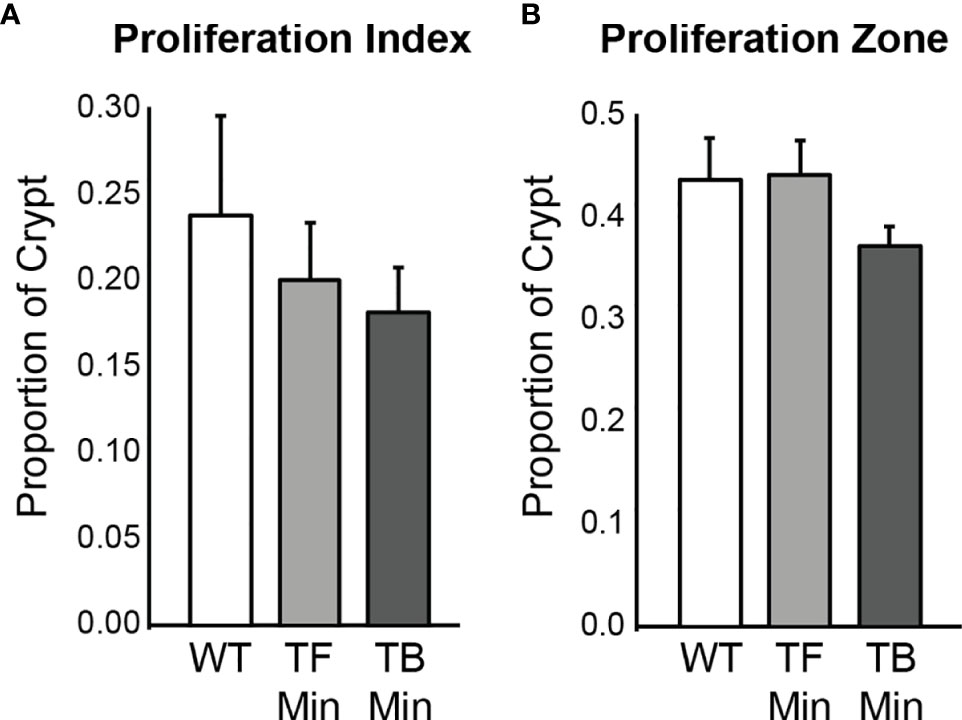
Figure 5 Epithelial cell proliferation in the distal colon of Apc+/+-FCCC wild type mice (‘WT’; n=4), tumor-free Apc+/Min-FCCC mice (‘TF Min’; n=6) and tumor-bearing Apc+/Min-FCCC mice (‘TB Min’; n=6). Bars represent mean ± S.E.M. (A) Fold change in proliferative index in tumor-bearing vs tumor-free mice = 0.9; Welch’s t-test p = 0.65. (B) Fold change in size of the proliferative zone in tumor-bearing vs tumor-free mice = 0.8; Welch’s t-test p = 0.15.
To examine if pro-tumorigenic gene expression changes in the nonneoplastic colon tissue accompany the formation of a macroscopic tumor, the expression of 8 adenoma-associated genes was compared within the initiated mucosa of tumor-free vs. tumor-bearing Apc+/Min-FCCC mice. To circumvent tumor-elicited effects, only samples located at least 1 cm from the nearest macroscopic colon tumor were analyzed. Overall, the relative mean expression of all genes analyzed was comparable between the nonneoplastic mucosa of tumor-free and tumor-bearing Apc+/Min-FCCC mice (fold change 0.8 – 1.3, Welch’s t-test p≥0.25; Table 4 and Supplemental Table 3).

Table 4 Mean fold change in gene transcription in the initiated colonic mucosa of tumor-bearing vs tumor-free Apc+/Min-FCCC mice after removing outliers.
To assess if increased pro-inflammatory signaling within nonneoplastic colon tissue accompanies the formation of a macroscopic tumor, the secretion of inflammatory cytokines from ex vivo segments of nonneoplastic, initiated colon tissue was examined in tumor-free and tumor-bearing Apc+/Min-FCCC mice. Interestingly, cytokine expression did not differ significantly between the initiated tissues of tumor-bearing vs tumor-free Apc+/Min-FCCC mice (fold change 0.4 – 0.9, Mann-Whitney p≥0.06; Figure 6 and Table 5). However, interindividual variability in the level of cytokine secretion was high, irrespective of the presence or absence of a macroscopic tumor (Supplemental Figure 7 and Table 6); 31% (4/13) of tumor-free Apc+/Min-FCCC mice and 31% (4/13) of tumor-bearing Apc+/Min-FCCC mice were defined as outliers due to high expression of one or more cytokines (Table 6). Within tumor-bearing animals, the concentration of most cytokines did not correlate with either overall tumor burden (linear model R2 ≤ 0.02; Supplemental Figure 8) or distance from the nearest detectable tumor (linear model R2 ≤ 0.38; Supplemental Figure 9). There was a trend toward increasing concentration of cytokine with greater distance from the tumor. This relationship was statistically significant in the case of IL-27 (p=0.03), but significance was lost after correcting for multiple comparisons (adjusted p=0.58).
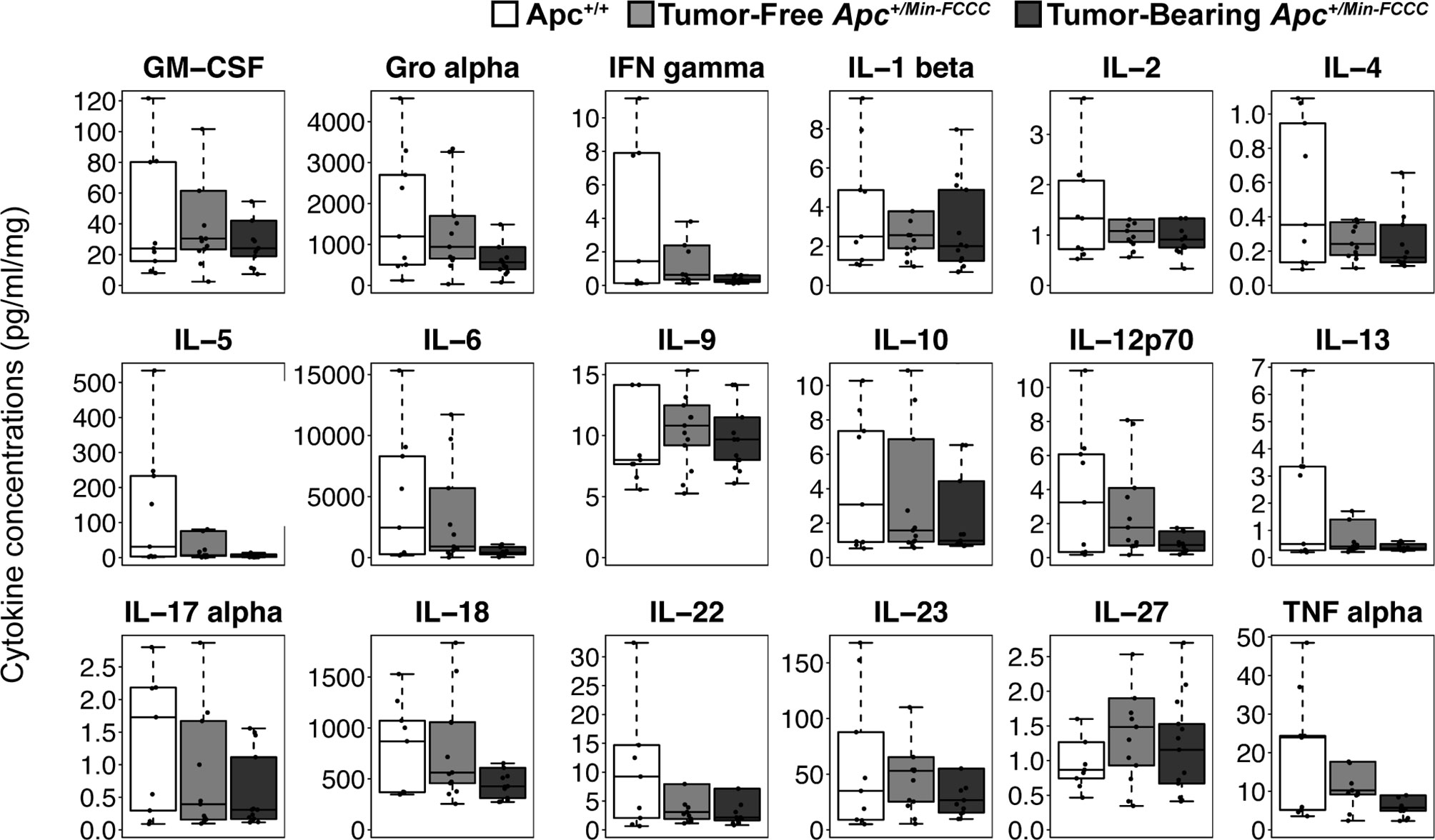
Figure 6 Expression of cytokines involved in Th1, Th2, Th9, and Th17 signaling in nonneoplastic colon tissue from Apc+/+-FCCC wild type (white; n=9), tumor-free Apc+/Min-FCCC (light gray; n=13), and tumor-bearing Apc+/Min-FCCC (dark gray; n=13) mice. Boxplots depict the cytokine concentration in media (pg/ml) normalized to mass of colon tissue (mg) cultured from each mouse. Outlying data points (>3rd quartile + 1.5 x interquartile range) are omitted; all data points are shown in Supplemental Figure 7. Fold change in median cytokine expression in tumor-bearing tumor-free = 0.4 – 0.9; Mann Whitney p≥0.06; p=1.00 after adjusting for multiple comparisons.
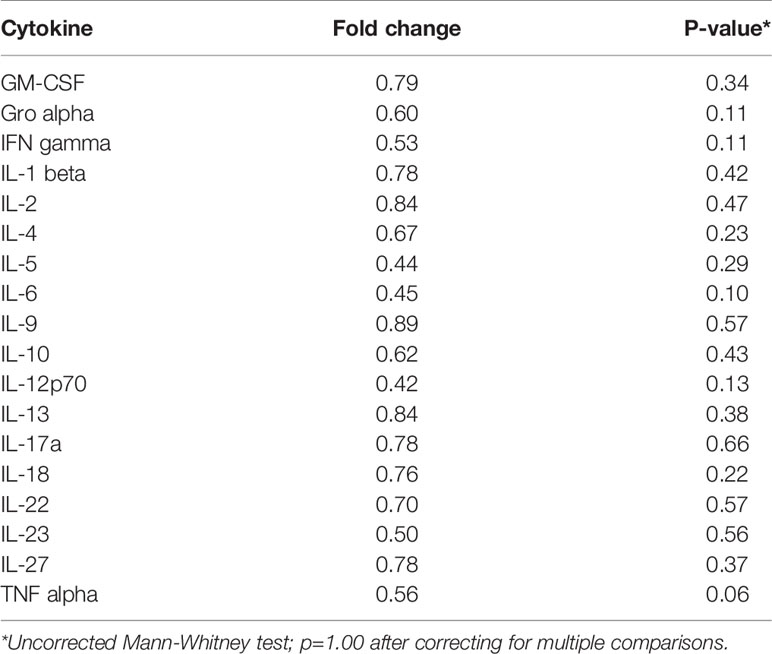
Table 5 Median fold change in secretion of cytokines involved in Th1, Th2, Th9, and Th17 signaling from initiated colon tissues from tumor-bearing vs tumor-free Apc+/Min-FCCC mice.
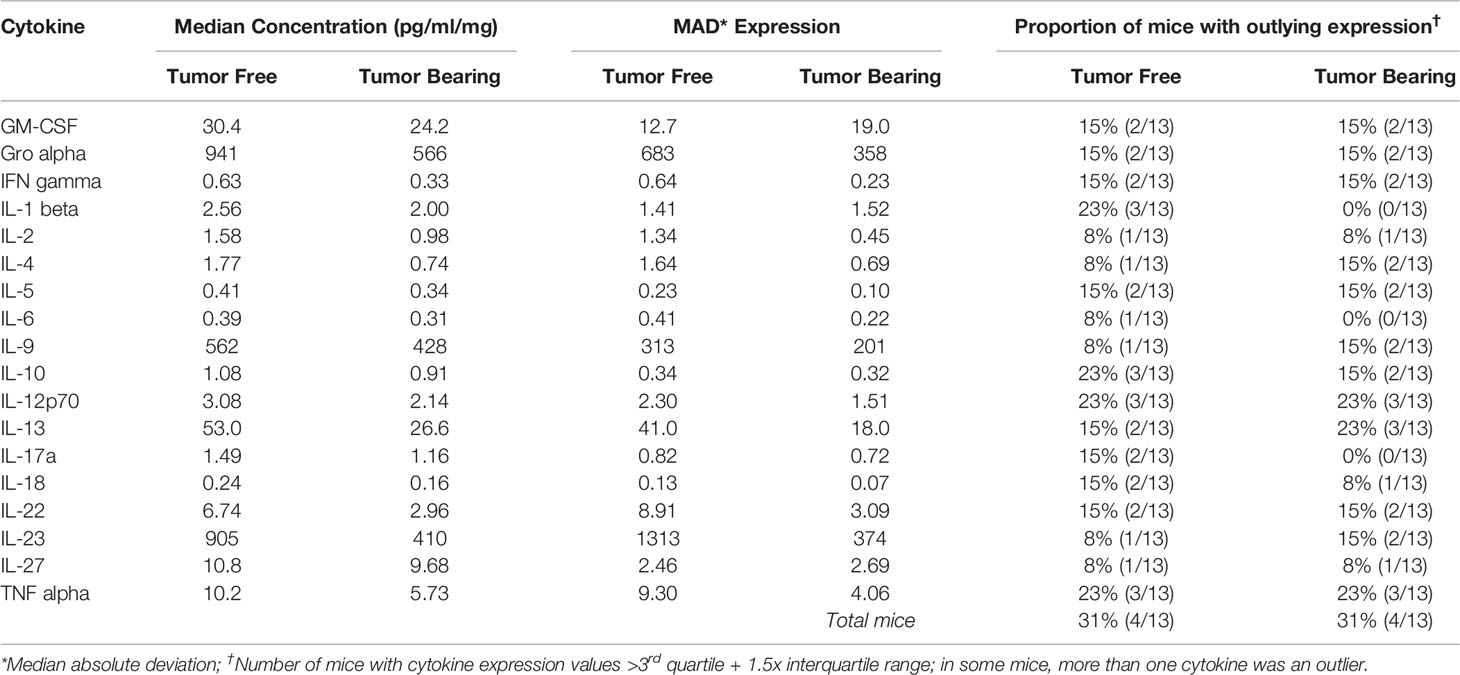
Table 6 Intra-group variability in secretion of cytokines involved in Th1, Th2, Th9, and Th17 signaling from initiated colon tissue from tumor-free and tumor-bearing Apc+/Min-FCCC mice.
Discussion
This study provides the first highly controlled characterization of early carcinogenic processes within the nonneoplastic colon of ApcMin mice: (1) prior to tissue initiation, (2) after initiation but prior to adenoma formation, and (3) after initiation and adenoma formation (Figure 1). To maximize the benefits of preventive intervention while minimizing the risk of side effects, it is critical to identify the ideal time to intervene. The Apc+/Min-FCCC mouse model is a powerful system with which to study the timing of molecular events during early colon tumorigenesis. A methodical approach was employed to characterize transformational processes in the colonic mucosa. Confounding variables from genetics and cage environment were controlled through block randomization of age-matched litters of Apc+/Min-FCCC mice and wild type animals. Small (2 mm) regions of only the distal colon were examined to control for variation in cellular proliferation, gene expression, and cytokine production along the length of the colon. The resulting data establish the baseline levels of cellular proliferation, relative gene expression, and cytokine production in the nonneoplastic mucosa of young adult mice (regardless of Apc mutational status) and may inform the design of new preclinical prevention studies.
To determine the impact of a heterozygous APC mutation on molecular characteristics associated with early tumorigenesis, “initiated”, at-risk nonneoplastic tissue from the distal colon of Apc+/Min-FCCC mice was compared to uninitiated tissue from the distal colon of wild type littermates. Interestingly, no significant differences were identified between nonneoplastic tissues from tumor-bearing Apc+/Min-FCCC, tumor-free Apc+/Min-FCCC, and wild type animals, with minimal changes observed in cellular proliferation, transcription of select genes related to cellular stemness, EGFR signaling, and inflammation, or secretion of select cytokines indicative of an inflamed microenvironment. Thus, we did not find evidence that the 8 genes or 18 cytokines examined in the present study would be sufficient targets for preventive intervention initiated prior to the formation of the first colon tumor in individuals who are at elevated risk of developing CRC.
Other groups have observed that a single truncating mutation in Apc (as seen in Apc+/Min-FCCC mice) may increase Wnt signaling at the base of the nonneoplastic colon crypt; enhanced signaling may then drive an expansion of colon epithelial stem cells, increase cellular proliferation along the length of the crypt (27), and ultimately promote adenoma formation (30, 56). In the present study, however, no changes in gene expression or proliferation rates were identified in the nonneoplastic epithelium among wild type, tumor-free Apc+/Min-FCCC, and tumor-bearing Apc+/Min-FCCC mice. It is possible that the initiating mutation in Apc+/Min-FCCC mice was insufficient to drive even modest changes in gene transcription within the colons of 8-week-old Apc+/Min-FCCC mice. Notably, however, only a small number of genes were examined in this study. Thus, a more comprehensive RNA-Seq analysis may reveal transcriptional changes in other genes. Alternatively, it is possible that gene expression and proliferative changes arise within a specific cellular compartment of the crypt. Since the entire mucosa was examined in the present study, molecular alterations specific to a subpopulation of cells in each crypt would likely be masked. Interestingly, the transcript levels of genes associated with Wnt signaling and cellular stemness, including Ascl2 and Lgr5, were slightly elevated in the nonneoplastic mucosa of Apc+/Min-FCCC vs. wild type mice (Table 1). Although the differences were modest and did not reach statistical significance, these trends may indicate subtle changes within the stem cell compartment of young adult Apc+/Min-FCCC mice compared to wild type animals. This shift is consistent with the observation that chemo- and immunoprevention strategies that target the stem cell compartment, including atorvastatin and a vaccine against Ascl2, are most effective when administered prior to tumor formation (4, 5).
Inflammation drives tumorigenesis within the colon, and may be an early event in the initiation of many sporadic and hereditary colon cancers (57–60). Mutations in Apc are associated with shifts in the gut microbiome, which may in turn induce inflammation (61). In the present study, no changes in cytokine secretion or the expression of inflammation-associated genes were identified in the nonneoplastic epithelium among 8-week-old wild type, tumor-free Apc+/Min-FCCC, and tumor-bearing Apc+/Min-FCCC mice. However, pro-inflammatory signaling may become more pronounced at a timepoint later than what was examined in this study. Consistent with this hypothesis, the production of inflammatory cytokines increases significantly within the initiated intestinal epithelium of C57BL6 ApcMin mice as animals approach the end of their life span, as compared to normal intestinal tissue from age-matched wild type C57BL6 mice (albeit animals from a different colony), and correlates with the number of large adenomas in each animal (25). Notably, expression of MCP-1, IL-1β, IL-6 and TNF-alpha within the small intestine of ApcMin mice did not differ from that of age-matched wild type mice at 8 weeks of age, but did increase significantly by 12-16 weeks of age (25).
In the present study, some animals (irrespective of Apc mutational status) had higher baseline production of inflammatory cytokines within the colonic mucosa than others at 8 weeks of age. The biological variability among mice within each group was typically larger than the average differences between groups. This was unexpected as extensive measures were taken to standardize experimental conditions across the groups. The source of this variability remains unclear; no single genetic or environmental factor, including litter, age, or cage environment, appeared to account for the large inter-individual variability. Regardless of what causes this interindividual heterogeneity, it must occur early and before the animals reach sexual maturity at 8 weeks of age. This heterogeneity may contribute to the phenotypic differences observed at older ages. Notably, in a separate study from this group, Apc+/Min-FCCC mice that did not have colon tumors at 6-8 weeks of age developed significantly fewer tumors by 20-22 weeks of age (1.8 ± 1.3 Mean ± S.D.) than animals that had a tumor at 6-8 weeks of age (6.4 ± 3.9; Mean ± S.D.; p=0.002) (4). These data suggest that the number of colon tumors that Apc+/Min-FCCC mice develop, while highly variable, is influenced by events that occur early in the lifespan of the animal (by or before 8 weeks of age).
Given that inflammation promotes tumor formation, growth, and progression following loss of Apc (51, 57, 62, 63), longitudinal studies are needed in the future to determine if Apc+/Min-FCCC mice with high baseline levels of cytokine activity represent a subpopulation that is at highest risk of developing colorectal tumors. Colon tissue biopsies could be collected at 8 weeks of age to assess cytokine production within nonneoplastic colon tissue prior to tumor formation. However, tissue damage from a biopsy might trigger an inflammatory response, thus confounding the analyses. Alternatively, cytokine levels could be measured in proxy samples, such as serum or feces (64, 65). Ultimately, high baseline inflammatory signaling in the normal colonic mucosa, serum, or feces of healthy young adults may prove to be a novel biomarker of colon cancer risk.
In conclusion, the data from the present study provide valuable insight into the basal levels of cellular proliferation and cytokine production in the nonneoplastic mucosa of young adult mice. These data can be used to inform the design of future preclinical prevention studies. Notably, a high degree of variability was observed in the baseline levels of cytokine production in age-matched, genetically identical animals. These subpopulations of animals with elevated cytokine signaling in the nonneoplastic colonic mucosa may represent a high-risk group that would benefit from earlier intervention. Further study is needed to better understand the contribution of high baseline cytokine production within the nonneoplastic colonic mucosa to long-term colon cancer risk.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available upon request by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by The Institutional Animal Care and Use Committee at the Fox Chase Cancer Center.
Author Contributions
AL and MC contributed to the conception and design of the study. KH performed animal husbandry. AL, KH, EK, and HH conducted the colonoscopies. AL, MC, KH, and LV performed necropsies. AL performed the remaining experiments. AL and KD performed the statistical analyses. AL wrote the first draft of the manuscript, and MC wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by grants from the National Cancer Institute, National Institutes of Health (P30 CA006927 and T32 CA009035), and by the Timothy and Aurora Hughes Colon Cancer Research Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The following core facilities at Fox Chase Cancer Center contributed to this study: Biostatistics and Bioinformatics, Cell Culture, Genotyping and Real-Time PCR, High Throughput Screening, Laboratory Animal, and the Small Animal Imaging Component of the Biological Imaging Facility. The authors would like to thank Glenn Rall of Fox Chase Cancer Center for the use of his cryostat, Margret Einarson, Ekaterina Koltsova, and Sergei Grivennikov for their assistance in designing the 18-plex cytokine analysis experiments, and Darlene Curran for her assistance in preparing the manuscript for publication.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.705562/full#supplementary-material
References
2. Society AC. Colorectal Cancer Facts & Figures 2020-2022 Vol. 2020. Atlanta: American Cancer Society (2020).
3. Jrah-Harzallah H, Ben-Hadj-Khalifa S, Almawi WY, Maaloul A, Houas Z, Mahjoub T. Effect of Thymoquinone on 1,2-Dimethyl-Hydrazine-Induced Oxidative Stress During Initiation and Promotion of Colon Carcinogenesis. Eur J Cancer (2013) 49:1127–35. doi: 10.1016/j.ejca.2012.10.007
4. Chang WL, Jackson C, Riel S, Cooper HS, Devarajan K, Hensley HH, et al. Differential Preventive Activity of Sulindac and Atorvastatin in Apc(+/Min-FCCC)Mice With or Without Colorectal Adenomas. Gut (2018) 67:1290–8. doi: 10.1136/gutjnl-2017-313942
5. Rioux CR, Clapper ML, Cooper HS, Michaud J, St-Amant N, Koohsari H, et al. Self-Antigen MASH2 Combined With the AS15 Immunostimulant Induces Tumor Protection in Colorectal Cancer Mouse Models. PloS One (2019) 14:e0210261. doi: 10.1371/journal.pone.0210261
6. Belnoue E LA, Carboni S, Cooper HS, Macedo RT, Harvey KN, Colby KB, et al. Novel Protein-Based Vaccine Against Self-Antigen Reduces the Formation of Sporadic Colon Adenomas in Mice. Cancers (2021) 13:845. doi: 10.3390/cancers13040845
7. Kinzler KW, Vogelstein B. Lessons From Hereditary Colorectal Cancer. Cell (1996) 87:159–70. doi: 10.1016/S0092-8674(00)81333-1
8. Chung DC. The Genetic Basis of Colorectal Cancer: Insights Into Critical Pathways of Tumorigenesis. Gastroenterology (2000) 119:854–65. doi: 10.1053/gast.2000.16507
9. Clapper ML, Chang WL, Cooper HS. Dysplastic Aberrant Crypt Foci: Biomarkers of Early Colorectal Neoplasia and Response to Preventive Intervention. Cancer Prev Res (Phila) (2020) 13:229–40. doi: 10.1158/1940-6207.CAPR-19-0316
10. Prasetyanti PR, Zimberlin CD, Bots M, Vermeulen L, Melo Fde S, Medema JP. Regulation of Stem Cell Self-Renewal and Differentiation by Wnt and Notch are Conserved Throughout the Adenoma-Carcinoma Sequence in the Colon. Mol Cancer (2013) 12:126. doi: 10.1186/1476-4598-12-126
11. Kim JH PS, Jun Y, Nam JS. Roles of Wnt Target Genes in the Journey of Cancer Stem Cells. International Journal of Molecular Sciences. Int J Mol Sci (2017) 18:1604. doi: 10.3390/ijms18081604
12. Jang BG, Kim HS, Kim KJ, Rhee YY, Kim WH, Kang GH. Distribution of Intestinal Stem Cell Markers in Colorectal Precancerous Lesions. Histopathology (2016) 68:567–77. doi: 10.1111/his.12787
13. Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ, et al. Importance of Epidermal Growth Factor Receptor Signaling in Establishment of Adenomas and Maintenance of Carcinomas During Intestinal Tumorigenesis. Proc Natl Acad Sci USA (2002) 99:1521–6. doi: 10.1073/pnas.032678499
14. Ivancic MM, Irving AA, Jonakin KG, Dove WF, Sussman MR. The Concentrations of EGFR, LRG1, ITIH4, and F5 in Serum Correlate With the Number of Colonic Adenomas in Apcpirc/+ Rats. Cancer Prev Res (Phila) (2014) 7:1160–9. doi: 10.1158/1940-6207.CAPR-14-0056
15. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
16. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-Linked Barrier Defects and Microbial Products Drive IL-23/IL-17-Mediated Tumour Growth. Nature (2012) 491:254–8. doi: 10.1038/nature11465
17. Samadder NJ, Kuwada SK, Boucher KM, Byrne K, Kanth P, Samowitz W, et al. Association of Sulindac and Erlotinib vs Placebo With Colorectal Neoplasia in Familial Adenomatous Polyposis: Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol (2018) 4:671–7. doi: 10.1001/jamaoncol.2017.5431
18. Umezawa S, Higurashi T, Komiya Y, Arimoto J, Horita N, Kaneko T, et al. Chemoprevention of Colorectal Cancer: Past, Present, and Future. Cancer Sci (2019) 110:3018–26. doi: 10.1111/cas.14149
19. Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in Intestinal Adenomas From Min Mice. Cancer Res (1994) 54:5947–52.
20. Yamada Y, Hata K, Hirose Y, Hara A, Sugie S, Kuno T, et al. Microadenomatous Lesions Involving Loss of Apc Heterozygosity in the Colon of Adult Apc(Min/+) Mice. Cancer Res (2002) 62:6367–70.
21. Moser AR, Dove WF, Roth KA, Gordon JI. The Min (Multiple Intestinal Neoplasia) Mutation: Its Effect on Gut Epithelial Cell Differentiation and Interaction With a Modifier System. J Cell Biol (1992) 116:1517–26. doi: 10.1083/jcb.116.6.1517
22. Gutierrez LS, Suckow M, Lawler J, Ploplis VA, Castellino FJ. Thrombospondin 1–a Regulator of Adenoma Growth and Carcinoma Progression in the APC(Min/+) Mouse Model. Carcinogenesis (2003) 24:199–207. doi: 10.1093/carcin/24.2.199
23. Van der Flier LG, Sabates-Bellver J, Oving I, Haegebarth A, De Palo M, Anti M, et al. The Intestinal Wnt/Tcf Signature. Gastroenterology (2007) 132:628–32. doi: 10.1053/j.gastro.2006.08.039
24. Moran AE, Hunt DH, Javid SH, Redston M, Carothers AM, Bertagnolli MM. Apc Deficiency Is Associated With Increased Egfr Activity in the Intestinal Enterocytes and Adenomas of C57BL/6J-Min/+ Mice. J Biol Chem (2004) 279:43261–72. doi: 10.1074/jbc.M404276200
25. McClellan JL, Davis JM, Steiner JL, Day SD, Steck SE, Carmichael MD, et al. Intestinal Inflammatory Cytokine Response in Relation to Tumorigenesis in the Apc(Min/+) Mouse. Cytokine (2012) 57:113–9. doi: 10.1016/j.cyto.2011.09.027
26. Shapiro M, Nandi B, Pai C, Samur MK, Pelluru D, Fulciniti M, et al. Deficiency of IL-17A, But Not the Prototypical Th17 Transcription Factor Rorgammat, Decreases Murine Spontaneous Intestinal Tumorigenesis. Cancer Immunol Immunother (2016) 65:13–24. doi: 10.1007/s00262-015-1769-2
27. Boman BM, Fields JZ. An APC : WNT Counter-Current-Like Mechanism Regulates Cell Division Along the Human Colonic Crypt Axis: A Mechanism That Explains How APC Mutations Induce Proliferative Abnormalities That Drive Colon Cancer Development. Front Oncol (2013) 3:244. doi: 10.3389/fonc.2013.00244
28. Leedham SJ, Rodenas-Cuadrado P, Howarth K, Lewis A, Mallappa S, Segditsas S, et al. A Basal Gradient of Wnt and Stem-Cell Number Influences Regional Tumour Distribution in Human and Mouse Intestinal Tracts. Gut (2013) 62:83–93. doi: 10.1136/gutjnl-2011-301601
29. Aceto GM, Fantini F, De Iure S, Di Nicola M, Palka G, Valanzano R, et al. Correlation Between Mutations and Mrna Expression of APC and MUTYH Genes: New Insight Into Hereditary Colorectal Polyposis Predisposition. J Exp Clin Cancer Res (2015) 34:131. doi: 10.1186/s13046-015-0244-4
30. Amos-Landgraf JM, Irving AA, Hartman C, Hunter A, Laube B, Chen X, et al. Monoallelic Silencing and Haploinsufficiency in Early Murine Intestinal Neoplasms. Proc Natl Acad Sci USA (2012) 109:2060–5. doi: 10.1073/pnas1120753109
31. Cooper HS, Chang W-CL, Coudry R, Gary MA, Everley L, Spittle CS, et al. Generation of a Unique Strain of Multiple Intestinal Neoplasia (Apc+/Min-FCCC) Mice With Significantly Increased Numbers of Colorectal Adenomas. Mol Carcinog (2005) 44:31–41. doi: 10.1002/mc.20114
32. Hensley HH, Merkel CE, Chang WC, Devarajan K, Cooper HS, Clapper ML. Endoscopic Imaging and Size Estimation of Colorectal Adenomas in the Multiple Intestinal Neoplasia Mouse. Gastrointest Endosc (2009) 69(3 Suppl):742–9. doi: 10.1016/j.gie.2008.09.054
33. Hong MY, Nulton E, Shelechi M, Hernandez LM, Nemoseck T. Effects of Dark Chocolate on Azoxymethane-Induced Colonic Aberrant Crypt Foci. Nutr Cancer (2013) 65:677–85. doi: 10.1080/01635581.2013.789542
34. Team RC. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing (2019).
35. Wickham H, François R, Henry L, Müller K. Dplyr: A Grammar of Data Manipulation. (2019). R package version 0.8.2.
36. Chaudhary M, Mandir N, FitzGerald AJ, Howard JK, Lord GM, Ghatei MA, et al. Starvation, Leptin and Epithelial Cell Proliferation in the Gastrointestinal Tract of the Mouse. Digestion (2000) 61:223–9. doi: 10.1159/000007762
37. Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides Fragilis Toxin Coordinates a Pro-Carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe (2018) 23:203–14.e5. doi: 10.1016/j.chom.2018.01.007
38. Huang EH, Carter JJ, Whelan RL, Liu YH, Rosenberg JO, Rotterdam H, et al. Colonoscopy in Mice. Surg Endosc (2002) 16:22–4. doi: 10.1007/s004640080168
39. Durkee BY, Shinki K, Newton MA, Iverson CE, Weichert JP, Dove WF, et al. Longitudinal Assessment of Colonic Tumor Fate in Mice by Computed Tomography and Optical Colonoscopy. Acad Radiol (2009) 16:1475–82. doi: 10.1016/j.acra.2009.07.023
40. Ziskin JL, Dunlap D, Yaylaoglu M, Fodor IK, Forrest WF, Patel R, et al. In Situ Validation of an Intestinal Stem Cell Signature in Colorectal Cancer. Gut (2013) 62:1012–23. doi: 10.1136/gutjnl-2011-301195
41. Schuijers J, Junker JP, Mokry M, Hatzis P, Koo BK, Sasselli V, et al. Ascl2 Acts as an R-Spondin/Wnt-Responsive Switch to Control Stemness in Intestinal Crypts. Cell Stem Cell (2015) 16:158–70. doi: 10.1016/j.stem.2014.12.006
42. Segditsas S, Sieber O, Deheragoda M, East P, Rowan A, Jeffery R, et al. Putative Direct and Indirect Wnt Targets Identified Through Consistent Gene Expression Changes in APC-Mutant Intestinal Adenomas From Humans and Mice. Hum Mol Genet (2008) 17:3864–75. doi: 10.1093/hmg/ddn286
43. Davis H, Irshad S, Bansal M, Rafferty H, Boitsova T, Bardella C, et al. Aberrant Epithelial GREM1 Expression Initiates Colonic Tumorigenesis From Cells Outside the Stem Cell Niche. Nat Med (2015) 21:62–70. doi: 10.1038/nm.3750
44. Dame MK, Attili D, McClintock SD, Dedhia PH, Ouillette P, Hardt O, et al. Identification, Isolation and Characterization of Human LGR5-Positive Colon Adenoma Cells. Development (2018) 145:dev153049. doi: 10.1242/dev.153049
45. Krishn SR, Kaur S, Smith LM, Johansson SL, Jain M, Patel A, et al. Mucins and Associated Glycan Signatures in Colon Adenoma-Carcinoma Sequence: Prospective Pathological Implication(s) for Early Diagnosis of Colon Cancer. Cancer Lett (2016) 374:304–14. doi: 10.1016/j.canlet.2016.02.016
46. Clapper ML, Hensley HH, Chang WC, Devarajan K, Nguyen MT, Cooper HS. Detection of Colorectal Adenomas Using a Bioactivatable Probe Specific for Matrix Metalloproteinase Activity. Neoplasia (2011) 13:685–91. doi: 10.1593/neo.11400
47. Wang J, Cho NL, Zauber AG, Hsu M, Dawson D, Srivastava A, et al. Chemopreventive Efficacy of the Cyclooxygenase-2 (Cox-2) Inhibitor, Celecoxib, is Predicted by Adenoma Expression of Cox-2 and 15-PGDH. Cancer Epidemiol Biomarkers Prev (2018) 27:728–36. doi: 10.1158/1055-9965.EPI-17-0573
48. Glebov OK, Rodriguez LM, Soballe P, DeNobile J, Cliatt J, Nakahara K, et al. Gene Expression Patterns Distinguish Colonoscopically Isolated Human Aberrant Crypt Foci From Normal Colonic Mucosa. Cancer Epidemiol Biomarkers Prev (2006) 15:2253–62. doi: 10.1158/1055-9965.EPI-05-0694
49. Sasaki N, Sachs N, Wiebrands K, Ellenbroek SIJ, Fumagalli A, Lyubimova A, et al. Reg4+ Deep Crypt Secretory Cells Function as Epithelial Niche for Lgr5+ Stem Cells in Colon. Proc Natl Acad Sci USA (2016) 113:E5399–407. doi: 10.1073/pnas.1607327113
50. Qu X, Sandmann T, Frierson H, Fu L, Feuntes E, Walter K, et al. Integrated Genomic Analysis of Colorectal Cancer Progression Reveals Activation of EGFR Through Demethylation of the EREG Promoter. Oncogene (2016) 35:6403–15. doi: 10.1038/onc.2016.170
51. Cooper HS, Everley L, Chang W-C, Pfeiffer G, Lee B, Murthy S, et al. The Role of Mutant Apc in the Development of Dysplasia and Cancer in the Mouse Model of Dextran Sulfate Sodium-Induced Colitis. Gastroenterology (2001) 121:1407–16. doi: 10.1053/gast.2001.29609
52. Tomkovich S, Yang Y, Winglee K, Gauthier J, Muhlbauer M, Sun X, et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res (2017) 77:2620–32. doi: 10.1158/0008-5472.CAN-16-3472
53. Caër C RC, Le Roy T, Poitou C, Aron-Wisnewsky J, Torcivia A, Bichet J-C, et al. Immune Cell-Derived Cytokines Contribute to Obesity-Related Inflammation, Fibrogenesis and Metabolic Deregulation in Human Adipose Tissue. Sci Rep (2017) 7:3000. doi: 10.1038/s41598-017-02660-w
54. Winer S PG, Chan Y, Tsui H, Engleman E, Winer D, Dosch HM. Obesity Predisposes to Th17 Bias. Eur J Immunol (2009) 39:2629–35. doi: 10.1002/eji.200838893
55. Riondino S, Roselli M, Palmirotta R, Della-Morte D, Ferroni P, Guadagni F. Obesity and Colorectal Cancer: Role of Adipokines in Tumor Initiation and Progression. World J Gastroenterol (2014) 20:5177–90. doi: 10.3748/wjg.v20.i18.5177
56. Boman BM, Walters R, Fields JZ, Kovatich AJ, Zhang T, Isenberg GA, et al. Colonic Crypt Changes During Adenoma Development in Familial Adenomatous Polyposis: Immunohistochemical Evidence for Expansion of the Crypt Base Cell Population. Am J Pathol (2004) 165:1489–98. doi: 10.1016/S0002-9440(10)63407-4
57. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and Colon Cancer. Gastroenterology (2010) 138:2101–14.e5. doi: 10.1053/j.gastro.2010.01.058
58. Chen LC, Hao CY, Chiu YS, Wong P, Melnick JS, Brotman M, et al. Alteration of Gene Expression in Normal-Appearing Colon Mucosa of APC(Min) Mice and Human Cancer Patients. Cancer Res (2004) 64:3694–700. doi: 10.1158/0008-5472.CAN-03-3264
59. Li SKH, Martin A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol Med (2016) 22:274–89. doi: 10.1016/j.molmed.2016.02.003
60. Derikx LA, Smits LJ, van Vliet S, Dekker E, Aalfs CM, van Kouwen MCA, et al. Colorectal Cancer Risk in Patients With Lynch Syndrome and Inflammatory Bowel Disease. Clin Gastroenterol Hepatol (2017) 15:454–8.e1. doi: 10.1016/j.cgh.2016.08.005
61. Son JS, Khair S, Pettet DW, Ouyang N, Tian X, Zhang Y, et al. Altered Interactions Between the Gut Microbiome and Colonic Mucosa Precede Polyposis in Apcmin/+ Mice. PloS One (2015) 10:e0127985. doi: 10.1371/journal.pone.0127985
62. Paul Olson TJ, Hadac JN, Sievers CK, Leystra AA, Deming DA, Zahm CD, et al. Dynamic Tumor Growth Patterns in a Novel Murine Model of Colorectal Cancer. Cancer Prev Res (Phila) (2014) 7:105–13. doi: 10.1158/1940-6207.CAPR-13-0163
63. Nugent KP, Farmer KC, Spigelman AD, Williams CB, Phillips RK. Randomized Controlled Trial of the Effect of Sulindac on Duodenal and Rectal Polyposis and Cell Proliferation in Patients With Familial Adenomatous Polyposis. Br J Surg (1993) 80:1618–9. doi: 10.1002/bjs.1800801244
64. Poutahidis T, Cappelle K, Levkovich T, Lee CW, Doulberis M, Ge Z, et al. Pathogenic Intestinal Bacteria Enhance Prostate Cancer Development via Systemic Activation of Immune Cells in Mice. PloS One (2013) 8:e73933. doi: 10.1371/journal.pone.0073933
Keywords: colorectal cancer prevention, cancer prevention, cytokine signaling, tumor initiation, chemoprevention, immunoprevention
Citation: Leystra AA, Harvey KN, Kaunga E, Hensley H, Vanderveer LA, Devarajan K and Clapper ML (2021) High Variability in Cellular Proliferation, Gene Expression, and Cytokine Production in the Nonneoplastic Colonic Epithelium of Young Apc+/Min-FCCC Mice. Front. Oncol. 11:705562. doi: 10.3389/fonc.2021.705562
Received: 05 May 2021; Accepted: 09 August 2021;
Published: 27 August 2021.
Edited by:
Qingfeng Zhu, Johns Hopkins Medicine, United StatesReviewed by:
Ding Ding, Johns Hopkins Medicine, United StatesTao Xia, Zhejiang Provincial People’s Hospital, China
Copyright © 2021 Leystra, Harvey, Kaunga, Hensley, Vanderveer, Devarajan and Clapper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margie L. Clapper, TWFyZ2llLkNsYXBwZXJAZmNjYy5lZHU=