 Kinjal Shah1,2
Kinjal Shah1,2 Julhash U. Kazi1,2*
Julhash U. Kazi1,2*- 1Division of Translational Cancer Research, Department of Laboratory Medicine, Lund University, Lund, Sweden
- 2Lund Stem Cell Center, Department of Laboratory Medicine, Lund University, Lund, Sweden
WNT/β-catenin signaling is a highly complex pathway that plays diverse roles in various cellular processes. While WNT ligands usually signal through their dedicated Frizzled receptors, the decision to signal in a β-catenin-dependent or -independent manner rests upon the type of co-receptors used. Canonical WNT signaling is β-catenin-dependent, whereas non-canonical WNT signaling is β-catenin-independent according to the classical definition. This still holds true, albeit with some added complexity, as both the pathways seem to cross-talk with intertwined networks that involve the use of different ligands, receptors, and co-receptors. β-catenin can be directly phosphorylated by various kinases governing its participation in either canonical or non-canonical pathways. Moreover, the co-activators that associate with β-catenin determine the output of the pathway in terms of induction of genes promoting proliferation or differentiation. In this review, we provide an overview of how protein phosphorylation controls WNT/β-catenin signaling, particularly in human cancer.
Introduction
WNT/β-catenin signaling is a tightly controlled and highly conserved pathway that regulates cell fate during embryogenesis, hepatobiliary development, liver homeostasis, repair in adulthood, cell proliferation, differentiation, and cell polarity (1, 2). The intracellular responses that WNT ligands trigger can be classified into canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) signaling (2, 3). WNT is a family of nineteen hydrophobic cysteine-rich secreted glycoproteins which serve as ligands for ten members of the Frizzled (Fz) family of 7-transmembrane receptors, the co-receptors low-density lipoprotein receptor-related proteins 5/6 (LRP 5/6), and non-classical WNT receptors like RYK and ROR (4–8). Abnormal WNT/β-catenin signaling is involved in many diseases including Alzheimer’s disease, heart disease, osteoarthritis, and cancer (9, 10). β-catenin is one of the core molecules in the canonical WNT signaling pathway. It is also involved in E-cadherin and cytoskeleton-associated cell-cell adhesion when localized to the plasma membrane (11, 12). However, cytosolic β-catenin acts as the molecular effector of the WNT ligands (1, 13). This review discusses the phosphorylation-dependent regulations of β-catenin and WNT signaling in cancer.
Structure, Location, and Function of β-Catenin
β-catenin is a multifunctional protein encoded by the CTNNB1 gene in humans and is the vertebrate homolog of the Drosophila Armadillo (14). It is a 781-amino-acid-long protein consisting of the N-terminal domain (NTD), twelve armadillo (ARM) domains in the middle of the protein, and the C-terminal domain (CTD) (Figure 1). Each ARM domain contains three α-helices and, together, all twelve ARM domains create a compact superhelix with a positively charged groove spanning all the ARM domains (15, 16). This core ARM domain structure serves as a scaffold and interacts with various β-catenin binding partners that are critical for both WNT signaling and the formation of adherens junctions (16, 17). β-catenin can exist in three distinct pools inside the cell: membranous, cytoplasmic, and nuclear (18). β-catenin normally interacts with E-cadherin at the cell membrane and plays an important structural role in the adherens junctions. β-catenin that is free in the cytoplasm is captured by the destruction complex for degradation. However, when some of the components of the destruction complex are compromised, β-catenin evades degradation; instead, it translocates to the nucleus and contributes to the transcriptional regulation of genes (18). β-catenin thus acts as both an adaptor protein and a transcriptional coregulator (19). This spatial separation of β-catenin at the plasma membrane, cytoplasm, and the nucleus is regulated by specific phosphorylation mechanisms.
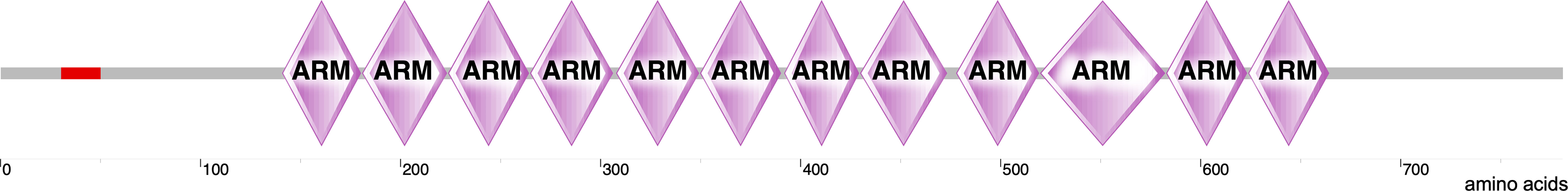
Figure 1 Structure of β-catenin. The structure of β-catenin was generated by SMART (http://smart.embl-heidelberg.de/smart/show_motifs.pl?ID=P35222) and modified using Canvas X draw. It includes an N-terminal domain where several regulatory phosphorylation sites are located (red). This domain follows twelve ARM domains and a long C-terminal domain.
Stabilization of β-Catenin at the Plasma Membrane as an Intracellular Adhesion Regulator
β-catenin acts as an adaptor protein and binds to the intracellular part of E-cadherin present at the plasma membrane via its C-terminal region. Apart from β-catenin, the cytoplasmic tail of E-cadherin can interact with various molecules such as γ-catenin and other regulatory proteins, while its extracellular part interacts with other cadherins present on adjacent cells (20, 21). The N-terminus of β-catenin interacts with α-catenin, which links β-catenin to the actin cytoskeleton. This entire structure of actin filaments-α-catenin-β-catenin-E-cadherin interactions promote clustering of the adhesion junction proteins, thereby stabilizing cell adhesion (22). In the absence of WNT, the majority of β-catenin localizes to the plasma membrane, building an epithelial barrier and restricting cell invasion and metastasis (23). However, the β-catenin-E-cadherin complex gets weakened by the phosphorylation of β-catenin at the plasma membrane (24). Tyrosine phosphorylation in the different armadillo repeats probably specifies interactions with α-catenin and E-cadherin. Phosphorylation of Tyr142/Tyr654 results in the dissociation of the adherens junctions, which leads to the cytoplasmic accumulation of β-catenin followed by its nuclear translocation to promote gene transcription (25).
Transcriptional Regulations Through Stabilization of β-Catenin in the Cytoplasm and Nucleus
In the absence of WNT signaling, β-catenin is sequestered in the cytoplasm by the ‘destruction complex’ composed of the scaffolding protein AXIN, tumor suppressor adenomatous polyposis coli (APC), and two serine/threonine kinases: casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β) (Figure 2A). AXIN directly binds with APC, GSK3β, CK1, and β-catenin and holds the destruction complex (26–29). Besides interaction with kinases, AXIN has an interaction site for protein phosphatase 2A (PP2A) that induces dephosphorylation of AXIN (30). Thus, AXIN is the core protein of the destruction complex that mediates the whole assembly of the destruction complex. Although AXIN can hold all proteins, the interaction between APC and β-catenin is required for active complex formation (31). GSK3β phosphorylates both AXIN (32, 33) and APC, which further increases its β-catenin binding affinity (34). CK1α binds to the first ARM domain of β-catenin (35) and mediates the first regulatory phosphorylation at Ser45 (36); this process requires AXIN-CK1α complex formation (29). Additionally, α-catenin must be dissociated from β-catenin to provide CK1 access. This dissociation is achieved by Tyr142 phosphorylation, which is mediated by tyrosine kinases FEline Sarcoma (FES)-related (FER) and FYN (35, 37). FYN is a member of SRC family of protein tyrosine kinases (SFKs). CK1α-induced Ser45 phosphorylation creates a priming site for GSK3β, which is necessary and sufficient for GSK3β-mediated phosphorylation at Thr41, Ser37, and Ser33 (29, 36). Ser33 and Ser37 in β-catenin create docking sites for the E3 ubiquitin ligase, β-transducing repeat-containing protein (β-TRCP) (Figure 2B) that ubiquitinates β-catenin and targets it for proteasomal degradation after forming a complex with Skp1 and Cullin (24, 38). Mutation of Ser37 thus results in stabilization of β-catenin (39).
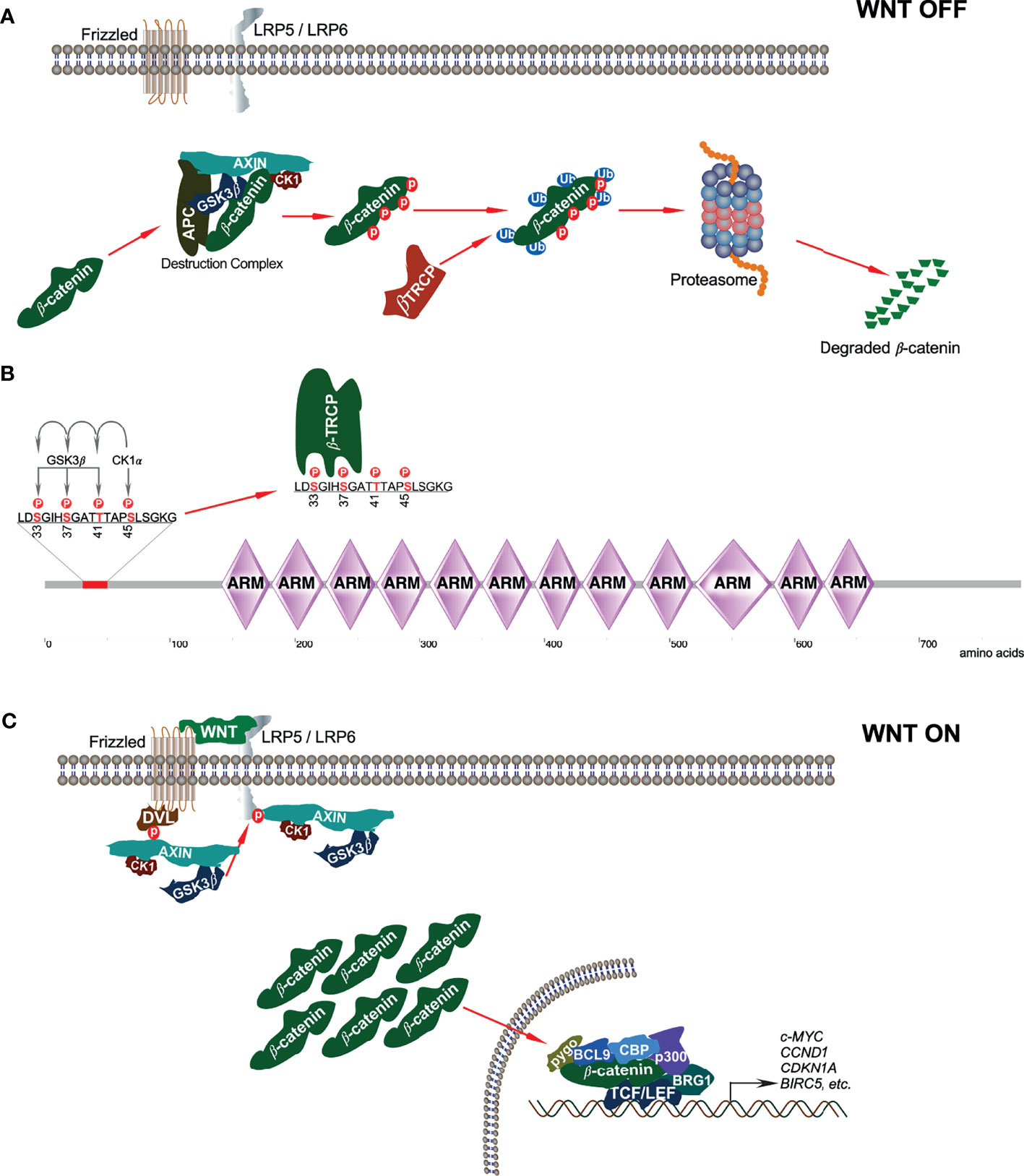
Figure 2 Canonical WNT signaling. (A) In absence of canonical WNT ligands, β-catenin is associated with the destruction complex. This interaction leads to phosphorylation-dependent ubiquitination of β-catenin and thereby its degradation in the proteasome. (B) In the destruction complex, CK1α phosphorylates β-catenin on Ser45 residue that initiates sequential phosphorylation of Thr41, Ser37, and Ser33 phosphorylation by GSK3β. Ser33 and Ser37 phosphorylation sites facilitate β-TRCP interaction with β-catenin. (C) In the presence of classical WNT ligands, a destruction complex cannot be formed and thus, β-catenin is stabilized from degradation.
The destruction complex is ultimately responsible for the phosphorylation of β-catenin, thereby priming it for ubiquitin-mediated proteasomal degradation (31). However, when canonical WNT ligands bind to their respective Fz receptor and LRP5/6 co-receptors, the result is the activation of the canonical WNT signaling pathway (40). Due to this, the phosphoprotein dishevelled (DVL, DVL1/2/3, segment polarity protein) is activated and recruited to the plasma membrane where it interacts with the cytoplasmic domain of Fz (41). DVL then recruits the destruction complex to the plasma membrane, thereby promoting interaction between LRP5/6 and AXIN (42, 43). GSK3β and CDK14 facilitate LRP5/6 phosphorylation that further enables AXIN-CK1-GSK3β complex recruitment (9). Recruitment of this complex to WNT receptors disrupts it and thereby inhibits CK1-GSK3β-mediated β-catenin phosphorylation, resulting in the stabilization and accumulation of β-catenin in the cytoplasm (Figure 2C).
Inhibition of CK1, WNT signaling activation, and DVL overexpression suppresses Ser45 phosphorylation (29). Moreover, GSK3β can be inactivated through Ser9 phosphorylation by AKT. These events result in the stabilization and accumulation of β-catenin in the cytoplasm (44). β-catenin activity in the canonical WNT pathway can also be regulated in a GSK3β and β-TrCP-independent manner.
It was observed in Drosophila that upon canonical WNT signaling, Arrow (LRP5/6) recruits AXIN to the membrane, which leads to degradation of AXIN. Thus, the scaffolding member of the destruction complex, AXIN, is no longer present to form the destruction complex (45). A more recent study demonstrated that an E3 ubiquitin ligase, tripartite motif-containing protein 11 (TRIM11), serves as an oncogene in lymphomas by promoting cell proliferation through activation of the β-catenin signaling. This regulation is brought about by TRIM11-mediated ubiquitination and degradation of AXIN1, part of the destruction complex of β-catenin (46). Thus, inactivation or degradation of any of the components forming the β-catenin destruction complex results in the stabilization and accumulation of β-catenin in the cytoplasm.
Stabilized β-catenin then translocates to the nucleus, where it interacts with different transcription factors, notably T-cell factor (TCF), and lymphoid enhancing factor (LEF). The repressor of the TCF/LEF complex, Groucho, is displaced upon this interaction, whose function is to compact chromatin (44). Thereafter, transcriptional co-activators and histone modifiers are recruited, which is sometimes referred to as WNT enhanceosome. These include the cyclic adenosine mono-phosphate response element (CREB)-binding protein (CBP), its closely related homolog p300, B-cell lymphoma 9 (BCL9), pygopus, and ATP-dependent helicase Brahma-related gene 1 (BRG1, also known as SMARCA4) (44, 47). Chromatin is remodeled by the WNT enhanceosome and results in the transcription of WNT/β-catenin target genes that are involved in cell survival and growth such as c-MYC, CCND1, CDKN1A, and BIRC5 (40). C-MYC is a proto-oncogene that further activates cyclin D1 and also inhibits the tumor suppressors p21 and p27, thereby leading to uncontrolled cell proliferation (48, 49).
Mutations of Components Involved in the Canonical WNT/β-Catenin Pathway
After having understood the regulation of β-catenin in the canonical WNT pathway, we now know that mutations in any of the components of this pathway can lead to its deregulation, which can contribute to a variety of diseases. Herein, we will focus on such mutations contributing to cancer. APC plays an important role in β-catenin degradation, and mutation in APC impairs destruction complex formation. Over 70% of colorectal adenocarcinoma patients carry mutations in the APC gene (Figure 3A) that lead to the stabilization of β-catenin. APC has long been known to be an important initiator gene for the majority of colorectal cancers. However, a recent study suggests that colorectal cancer tumors with a single APC mutation can have a survival benefit, whereas tumors lacking any APC mutations convey a worse prognosis (50). Other cancers display a lower number of APC mutations, whereas endometrial carcinoma, esophagogastric adenocarcinoma, and melanoma exhibit over 10% APC mutations. An aberrant APC promoter mutation is found in early endometrial carcinoma, which decreases with cancer progression (51), suggesting that loss of APC function is an early event in endometrial carcinoma. β-catenin mutations in N-terminal serine/threonine residues or adjacent residues that interrupt CK1- and GSK3β-induced regulatory serine/threonine phosphorylations have been found in several cancers (Figure 3B). In endometrial carcinoma, 20-40% of patients carry mutations in β-catenin (52). It is also frequently mutated in hepatocellular carcinoma (15-33%) (53).
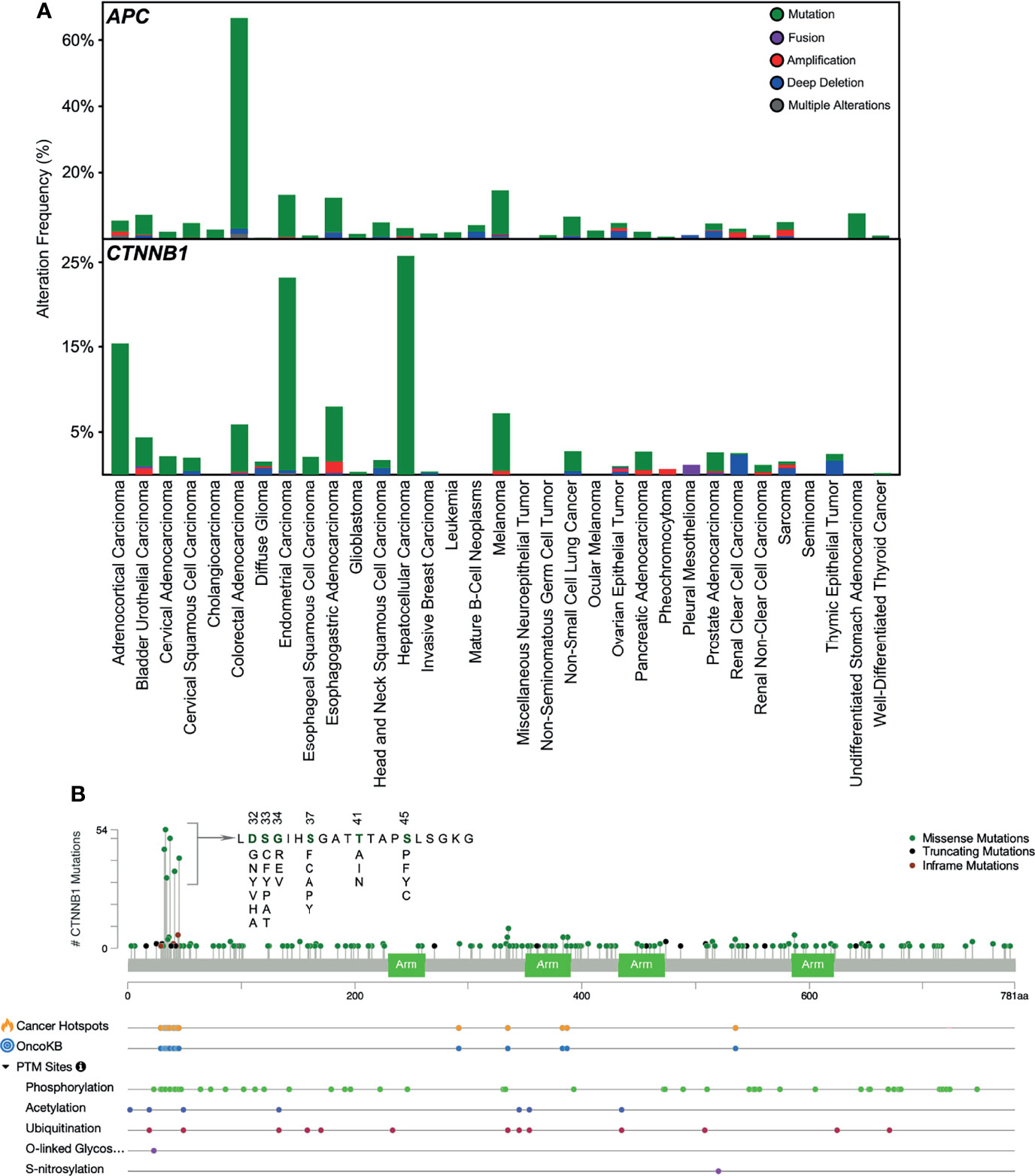
Figure 3 Mutations in APC and β-catenin. (A) Mutations in APC and CTNNB1 (β-catenin) in different cancers have been collected from cbioportal. (B) Mutation frequency in CTNNB1 gene and other post-translational modifications collected from cbioportal. TCGA PanCancer Atlas Studies (http://www.cbioportal.org/study/summary?id=5c8a7d55e4b046111fee2296) was used.
However, β-catenin is not mutated in pediatric T-ALL, and even if it is found to be mutated in any T-ALL cell line or patient sample, this holds no clinical significance (54). The most common activating missense mutation found in the endometroid carcinoma subtype of epithelial ovarian cancer (EOC) is in the β-catenin gene, CTNNB1, accounting for 54% of cases (55). This mutation occurred within the amino-terminal domain of β-catenin (55), which is positively correlated with its nuclear localization and expression of the β-catenin target genes. GSK3β phosphorylates the amino-terminal domain of β-catenin, leading to its degradation. Thus, mutations within this domain help β-catenin in evading degradation instead of accumulating in the nucleus (56). Moreover, loss-of-function mutations in genes encoding several components of the destruction complex, such as AXIN, APC, and GSK3β, were also reported in EOC, although not frequently (57). Thus, β-catenin target genes, such as cMyc, CCND1, and VEGF, were constitutively activated due to the disrupted WNT pathways contributed by the various mutations in its different components, which aided in cancer progression (58).
Non-Canonical WNT Signaling Pathway
Non-canonical WNT ligands such as WNT4, WNT5A, WNT5B, WNT7A, WNT7B, and WNT11 bind to Frizzled receptors (Fzd2, Fzd3, Fzd4, Fzd5, and Fzd6), and ROR1/ROR2 (receptor tyrosine kinase-like orphan receptor) or RYK acts as co-receptors to initiate non-canonical signaling (Figure 4). This pathway has always been defined as the one where β-catenin does not accumulate in the nucleus (59). It generally governs intercalation, cellular movement, and directed migration culminating in convergence and extension along the anterior/posterior axis of the organism (60, 61). Based on the phenotypic response, non-canonical signaling can be classified into two branches: WNT/PCP (Planar Cell Polarity) and the WNT-cGMP (cyclic guanosine monophosphate)/Ca2+ pathways (61, 62).
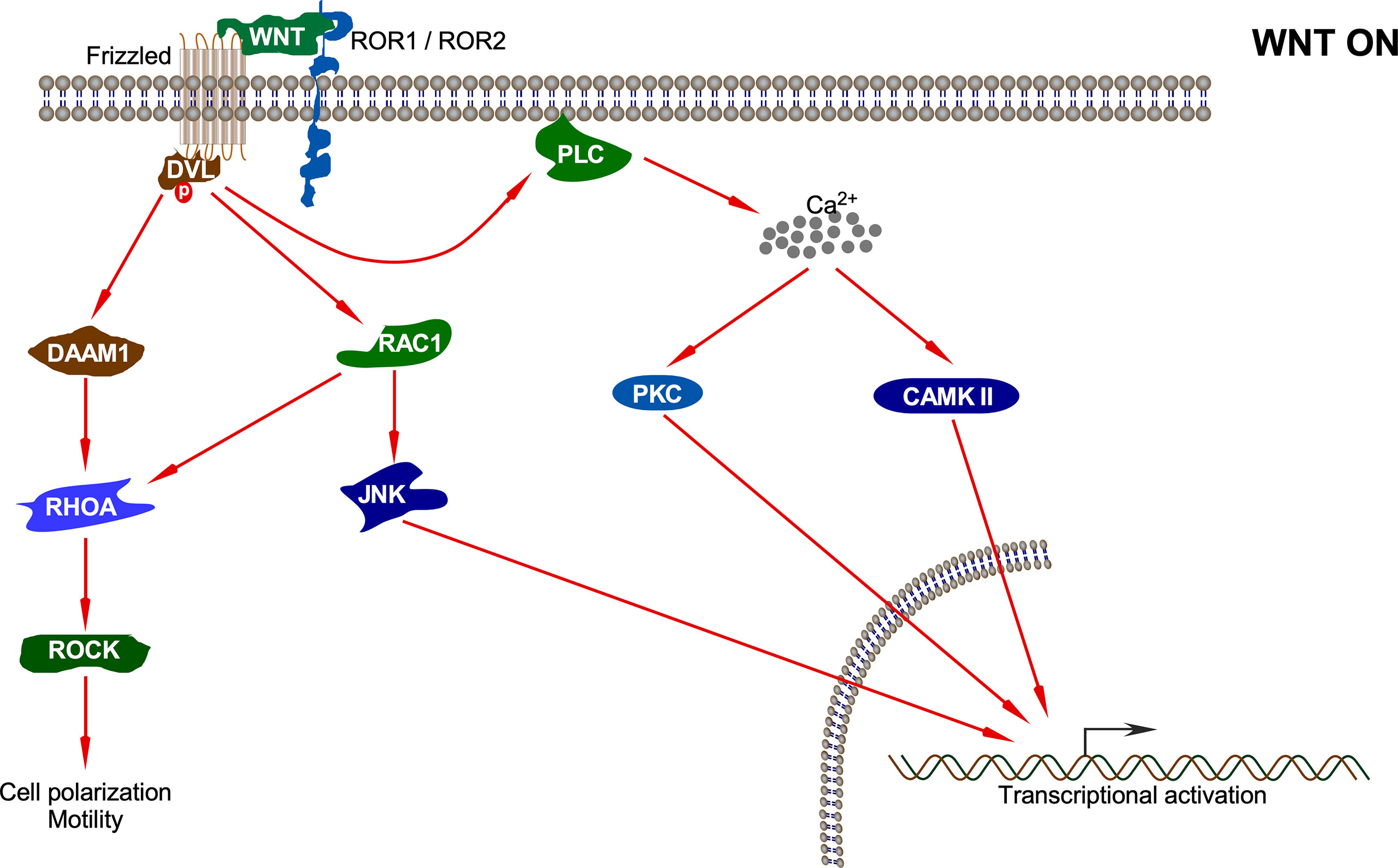
Figure 4 Non-canonical WNT pathway. Upon interaction of the non-canonical WNT ligand with Fz and ROR1/ROR2, several pathways including DAAM1-RHOA-ROCK, RAC1-JNK, PKC, and CAMKII pathways are activated which results in different cellular processes and transcriptional activation of different sets of genes.
The first branch of the non-canonical pathway – PCP signaling occurs through WNT-Fz receptor interaction without the involvement of LRP5/6 co-receptors. This results in activation of the DVL protein, which in turn activates a small GTPase such as RAC (63). Activated RAC further stimulates JNK activation (64). DVL also forms a complex with DSH associated activator of morphogenesis 1 (DAAM1), which results in the activation of other GTPases: RHO and, subsequently, Rho-associated kinase (ROCK) and myosin (63). The actin remodeling process associated with cell polarization and motility is controlled by signals emanating from both RAC and RHO activation (64). Non-canonical WNT ligands such as WNT5A also interact with ROR family orphan receptor tyrosine kinases such as ROR2 to activate JNK, RHOA, etc. This has been shown to antagonize the canonical WNT signaling pathway by inhibiting the transcriptional activation potential of the β-catenin/TCF complex, thus reducing the expression of Cyclin D1 (65, 66).
The second branch of the non-canonical pathway – WNT/Ca2+ signaling is characterized by WNT-Fz-induced phospholipase C (PLC) activation that increases cytoplasmic Ca2+ levels. Several Ca2+-responsive enzymes such as protein kinase C (PKC), calcineurin, and calcium/calmodulin-dependent protein kinase II (CaMKII) are activated upon sensing the intracellular Ca2+ flux (67). CaMKII further activates the transcription factors nuclear factor of activated T cells (NFAT), TGF-β activated kinase (TAK1), and Nemo-like kinase (NLK), all of which result in decrease in the levels of intracellular cGMP, which antagonizes the canonical WNT signaling (68, 69). Moreover, TAK1 activates NLK, which in turn phosphorylates TCF. This prevents the β-catenin-TCF complex from binding DNA, thereby inhibiting gene transcription (70). The WNT/Ca2+ signaling regulates various developmental processes such as cytoskeletal rearrangements, cellular adhesion, dorsoventral patterning, and tissue separation in embryos (71).
The non-canonical WNT signaling pathway also has the potential to inhibit canonical WNT signaling by promoting the proteasomal degradation of β-catenin through an alternative E3 ubiquitin ligase complex containing APC, Ebi, and Siah1 or Siah2 (72, 73). This does not involve GSK3β or β-TrCP and neither requires activation of CaMKII or NFAT (74). Thus, the antagonism of canonical WNT signaling by non-canonical WNTs involves multiple mechanisms that may or may not involve calcium (71).
Coactivators Regulating the Output of the WNT Signaling Pathways
The canonical WNT signaling pathway is dependent on β-catenin, whereas the non-canonical WNT signaling pathway is independent of β-catenin (75). Once in the nucleus, β-catenin interacts with members of the TCF/LEF family of transcription factors. β-catenin can also interact with a variety of other transcription factors like FOXO, HIF1, Sox family members, nuclear receptors, etc. Thus, nuclear β-catenin can perform divergent functions, which adds complexity to the interpretation of canonical WNT signaling (76). Transcriptional coactivators like cAMP response element-binding protein (CREB)-binding protein (CBP) or its closely related homolog, p300, are key regulators of RNA polymerase II-mediated transcription (77). β-catenin can recruit either CBP or p300 along with other components of the basal transcriptional machinery to generate a transcriptionally active complex, which leads to the expression of a variety of downstream target genes (78). Differential utilization of these coactivators by β-catenin can lead to different cellular outputs (75). Canonical WNT signaling pathway mediated by WNT3A stabilizes β-catenin and leads to its association with the coactivator CBP for transcribing genes related to self-renewal, potency, and proliferation (77). However, the non-canonical WNT signaling pathway mediated by WNT5A induces the activation of various kinases such as PKC, CaMKII, SIK, AMPK, and MAPK (5, 8, 79). PKC further phosphorylates Ser89 of p300, which increases its affinity for β-catenin (77). The association of coactivator p300 with β-catenin drives the gene expression program from a proliferative state to a differentiative state; that also governs planar cell polarity, convergent extension, and cytoskeletal reorganization (80). Thus, coordinated integration between both canonical and non-canonical WNT signaling pathways is extremely crucial for regulating cell proliferation with differentiation and adhesion (75). Canonical and non-canonical WNT signaling pathways are, hence, highly dynamic, coupled, and not mutually exclusive with cross-talk occurring between them, which is dependent on the type of cell, tissue, and specific stage of development (77).
Kinases Regulating the Activity of β-Catenin
Alternative signal transduction pathways from various growth factor receptors and ion channels can activate a myriad of kinases, which also have the potential to phosphorylate either CBP or p300, thereby controlling differential utilization of coactivators by β-catenin. Moreover, these kinases can phosphorylate β-catenin directly (77), thereby playing key roles in regulating the localization, expression, and function of β-catenin (81). We will be discussing some of those kinases in the following sections.
Cell Cycle-Associated Kinases
Aurora kinase A (AURKA) is required for centrosome function and spindle assembly during mitosis that, once activated, phosphorylates Polo-like kinase 1 (PLK1) at Thr210 in presence of the co-factor Bora (82–85). PLK1 is the master regulator of the cell cycle, playing an important role in M-phase progression (86). PLK1 in turn phosphorylates several substrates, FOXM1 being one of them. FOXM1 then transcribes target genes involved in cell cycle progression, cell proliferation, genomic stability, chemoresistance, and DNA damage repair (87–89). All components of the AURKA-PLK1-FOXM1 axis appear to be hyperactivated due to multiple events associated with a BCR-ABL fusion protein that promotes resistance to tyrosine kinase inhibitors in chronic myeloid leukemia (CML) (90–94). The AURKA-PLK1-FOXM1 axis interacts with β-catenin, which supports the resistance of BCR-ABL+ leukemic stem cells to tyrosine kinase inhibitors (86, 95–97). PLK1 physically interacts with β-catenin and phosphorylates it on Ser718 in the M phase of the cell cycle, indicating an important M-phase specific function of β-catenin such as the regulation of centrosomes (86). Deregulated PLK1 expression has been reported in many cancers, which in turn could alter β-catenin regulation in the M phase, thus leading to abnormal cell cycle control and chromosome instability (79, 86, 98). Moreover, PLK1 regulates the activity of another cell cycle regulatory kinase, NIMA-related protein kinase 2 (Nek2). Nek2 phosphorylates Thr41, Ser37, and Ser33 at the N-terminus of β-catenin along with some five additional sites; these are the amino acid residues phosphorylated by GSK3β as well. This inhibits the interaction of β-catenin with β-TRCP, thereby stabilizing it. Nek2 regulates centrosome disjunction/splitting; thus, β-catenin stabilized by Nek2 accumulates at centrosomes in mitosis, regulating the centrosome cycle (81). AURKA sequesters AXIN from the destruction complex, while FOXM1 associates with β-catenin, thereby enabling its nuclear import and recruitment to the TCF/LEF transcription complex to support leukemic cell proliferation and survival (99). So, all 3 components of the AURKA-PLK1-FOXM1 axis regulate β-catenin transcriptional activity. Inhibition of these components ultimately results in the dephosphorylation of FOXM1, causing the release of β-catenin from its binding, which leads to its cytoplasmic relocation and degradation. The proliferation and survival of BCR-ABL+ cells are thus blocked by the inhibition of the β-catenin-mediated transcriptional activity (100).
Protein Kinase C (PKC)
Once activated, β-catenin is translocated to the nucleus. Its stability is regulated by the E3 ubiquitin ligase tripartite motif-containing protein 33 (TRIM33), which is independent of GSK3β and β-TRCP (101). However, this regulation is dependent on the PKC family member PKCδ, which is activated upon prolonged WNT stimulation. PKCδ phosphorylates β-catenin at Ser715, thereby facilitating its interaction with TRIM33. β-catenin is then targeted for degradation, shutting off the WNT pathway. Apart from inhibiting the canonical WNT pathway mediated by WNT3a, TRIM33 can inhibit EGF-induced β-catenin transactivation (101). TRIM33 is thus known to act as a tumor suppressor in various cancers including clear cell renal cell carcinoma (102), chronic myelomonocytic leukemia (103), hepatocellular carcinoma (104), and pancreatic cancer (105). In fact, PKC is a family of 10 protein serine/threonine kinases that are encoded by 9 genes (106–109). PKC family members are divided into three subfamilies: classical (PKCα, PKCβ1, PKCβ2, and PKCγ), novel (PKCδ, PKCϵ, PKCη, and PKCθ) and atypical (PKCζ and PKCι) (108, 109). Classical and novel PKC isoforms display dependency on second messengers for activation. For example, classical PKC isoforms are diacylglycerol (DAG) and Ca2+-responsive while novel PKC isoforms are dependent on DAG. While several PKC family members have been implicated in tumorigenesis, PKCδ acts as a tumor suppressor, as its main function is to induce apoptosis, apart from regulating β-catenin degradation (101, 108–114). Another PKC family member that serves as a tumor suppressor in intestinal cancer is the atypical PKCζ that induces Ser45 phosphorylation of β-catenin, which is independent of CK1α and is thus important for GSK3β-mediated phosphorylation (115). The classical PKC isoform PKCα induces phosphorylation of β-catenin at N-terminal serine residues, which in turn results in enhanced proteasomal degradation of β-catenin and a reduction of transcriptional regulations (116). Furthermore, PKCα phosphorylates ROR1 (RORα) at Ser35, which can eventually limit the transcriptional regulation of β-catenin (117). Collectively, these studies suggest that several PKC isoforms play important roles in the regulation of WNT/β-catenin signaling.
Protein Kinase A (PKA)
The cyclic AMP (cAMP)-dependent protein kinase, protein kinase A (PKA), has diverse cellular functions including cell proliferation, differentiation, cell cycle regulation, and apoptosis. The cAMP/PKA pathway plays a highly complex and cell-specific role in regulating cell growth, as it stimulates growth for some cell types while inhibiting others (118, 119). PKA can even have contrasting effects of promoting or inhibiting cell proliferation in the same cell type, such as the vascular smooth muscle cells, depending on the agonist that stimulates its activity (120, 121). The presenilin1 complex in Alzheimer’s disease contains presenilin1, GSK3β, the catalytic subunit of PKA and β-catenin. PKA induces Ser45 phosphorylation on β-catenin, thereby enhancing GSK3β-dependent phosphorylation of β-catenin and its subsequent proteasomal degradation, which is independent of the WNT-controlled AXIN complex (122). However, in contrast, activation of PKA has also been shown to increase β-catenin accumulation in both the cytosol and nucleus of HEK293 cells after stimulation with prostaglandin E2 (PGE2) (123). PKA phosphorylates β-catenin on Ser552 and Ser675 that stabilize β-catenin by inhibiting its ubiquitination without affecting the formation of the destruction complex and GSK3β-dependent phosphorylation (123–125). PKA-induced phosphorylation of β-catenin at Ser675 promotes TCF/LEF transactivation and binding to its transcriptional coactivator CREB-binding protein (CBP) (124). Thus, the phosphorylation of β-catenin by PKA at different serine residues determines the outcome of β-catenin regulation.
Receptor Tyrosine Kinases (RTKs)
Receptor tyrosine kinase (RTK) consists of a family of around 60 mammalian protein tyrosine kinases (106, 107). The RTK epidermal growth factor receptor (EGFR) activates AKT that directly phosphorylates β-catenin on Ser552, which increases the cytosolic and nuclear β-catenin levels and transcriptional regulation, thereby promoting tumor cell invasion (126). Furthermore, the proliferation of PTEN-deficient intestinal stem cells that initiate intestinal polyposis is driven by AKT activation where AKT phosphorylates β-catenin on Ser552 (127). Ultraviolet (UV) irradiation activates EGFR in keratinocytes, resulting in the phosphorylation of β-catenin at Tyr654, which is responsible for its dissociation from the E-cadherin/β-catenin/α-catenin complex (128). Furthermore, UV-induced EGFR activation allows for the nuclear translocation of β-catenin and transcriptional activation through interaction with TCF4 (128). Fibroblast growth factor-2 (FGF-2) activates MAP kinase signaling in osteoblasts, which in turn phosphorylates β-catenin. The phosphorylation is mediated by MEKK2 (MAP3K2) at Ser675 that stabilizes β-catenin and increases its activity by recruiting the deubiquitinase USP15 (129). In colorectal cancer, a loss-of-function mutation in APC is very common, stabilizing β-catenin due to the lack of destruction complex. However, 60% of colorectal cancer patients carry mutations in KRAS and BRAF genes that result in uncontrolled MAPK signaling. Oncogenic activation of KRAS/BRAF/MEK signaling increases the transcriptional activities of β-catenin/TCF4 and c-MYC promoter and increases the mRNA levels of c-Myc, AXIN2, and Lef1 (130). Another MAPK, p38γ, phosphorylates β-catenin at Ser605 (131). FGFR2, FGFR3, EGFR, and TRKA increase the cytosolic β-catenin concentration by dissociating β-catenin from cadherin complex through direct phosphorylation at Tyr142 (132). The RTK MET interacts with β-catenin in hepatocytes, colon cancer, and breast cancer cell lines; this association occurs at the region of cell-cell contact (133, 134). The association is constitutive but can be abrogated by hepatocyte growth factor (HGF) stimulation. HGF induces tyrosine phosphorylation of β-catenin at Tyr654 and Tyr670, resulting in its dissociation from MET (135). Hepatocyte growth factor-like protein (HGFL) induced RON activation, which resulted in tyrosine phosphorylation of β-catenin at Tyr654 and Tyr670, inducing its nuclear accumulation and transcriptional activation in breast cancer (136). Type-1 insulin-like growth factor (IGF-1) causes the nuclear translocation of β-catenin in the context of IGF-1R signaling, which leads to the activation of β-catenin target genes such as c-MYC and cyclin (137–141). This is brought about by the direct binding of sequences between amino acid residues 695 and 781 in the C-terminus of β-catenin to sequences located between amino acid residue 600 and the C-terminus of insulin receptor substrate-1 (IRS-1), in both the nucleus and the cytoplasm (142). The PTB domain of IRS-1 then translocates the β-catenin-IRS-1 complex to the nucleus (143). β-catenin binding to IRS-1 with its C-terminus may thus prevent phosphorylation at its N-terminus by GSK3β, which primes β-catenin for ubiquitination and degradation (8, 144). So, IRS-1, a docking protein for both IGF-1 and insulin receptors, can regulate the subcellular localization and activity of β-catenin in cells that are responsive to the mitogenic action of IGF-1 (142, 145).
Janus Kinase 3 (JAK3)
Adherens junctions are multiprotein complexes in cell-cell junctions that connect neighboring cells to maintain the epithelial tissue structure (146). β-catenin is an adherens junctions-associated protein that links the cytoplasmic domain of cadherins to the α-catenin-associated actin cytoskeleton and has been implicated in adherens junctions remodeling (16). Janus kinase 3 (JAK3) is a non-receptor tyrosine kinase that transmits intracellular signals through interactions with the γ chain of several cytokine receptors upon stimulation with cytokines (147). It associates with β-catenin in adherens junctions and phosphorylates Tyr30, Tyr64, and Tyr86 (148). However, JAK3-mediated phosphorylation is required prior to the Tyr654 phosphorylation of β-catenin. Phosphorylation on those sites by JAK3 suppressed EGF-induced epithelial-mesenchymal transition (EMT) and, rather, induced epithelial barrier functions by adherens junctions localization of phosphorylated β-catenin via its association with α-catenin. Moreover, a reverse effect was seen with increased EMT and compromised epithelial barrier functions upon the loss of JAK3-mediated β-catenin phosphorylation, which even abrogated the localization of β-catenin in the adherens junctions (148).
p21-Activated Kinase (PAK)
p21-activated kinase (PAK) is a family of six protein serine/threonine kinases that acts mainly as the effector proteins for the Rho GTPases CDC42 and RAC (149). PAK-family proteins regulate various cellular processes including cell proliferation and cell survival, cell motility, cytoskeletal reorganization, oncogenic transformation, and gene transcription (149, 150). JNK2 and PAK1 both are the downstream effectors of RAC1. More advanced colon cancer samples often contained K-Ras mutation, which elevated two signaling branches: K-Ras/RAC1/JNK2 cascade and K-Ras/RAC1/PAK1 cascade (151–154). Thus, JNK2-mediated phosphorylation of β-catenin at Ser191 and Ser605 increased, which led to its nuclear translocation (151, 152). Additionally, RAC1 induces β-catenin Ser191 and Ser605 phosphorylation, which is mediated by JNK2; phosphorylation on those sites is required for the nuclear localization of β-catenin (151, 155). Apart from RAC1, the EGF and IGF signaling pathways may induce PAK1 expression (156–158). Another kinase, PAK4, shuttles between the nucleus and cytoplasm and interacts with β-catenin in both cellular compartments (159). It was observed that both PAK1 and PAK4 mediated phosphorylation of β-catenin on Ser675 stabilizes it by inhibiting its degradation and thus promotes TCF/LEF transcriptional activity (159, 160). Therefore, one downstream effector of RAC1 mediates the nuclear translocation of β-catenin, while the other downstream effector promotes its transcriptional activity.
Tyrosine Kinases
Several tyrosine phosphorylation sites have been identified on β-catenin and have been shown to play important roles. In the presence of a disrupted cellular contact and active TGF-β1, E-cadherin and the epithelial integrin α3β1 associate with TGF-β1 receptors, where Tyr654 in β-catenin gets phosphorylated by the epithelial integrin in primary alveolar epithelial cells (AECs) and the phosphorylated β-catenin forms a complex with phospho-SMAD2, resulting in EMT initiation in idiopathic pulmonary fibrosis (IPF) (161, 162). This epithelial integrin-mediated cross-talk between WNT and TGF-β1 signaling pathways, which is required for pulmonary fibrogenesis and EMT, is done with the help of TGF-β1mediated activation of SRC family kinases (163). In a hypoxic condition, reactive oxygen species (ROS) activates SRC that phosphorylates β-catenin on Tyr654. This tyrosine-phosphorylated β-catenin gets complexed with SRC and HIF1α in primary human lung adenocarcinomas and lung tumor cell lines to promote the transcriptional activity of HIF1α and hypoxia-induced EMT (164). Another substrate identified for SRC in β-catenin is Tyr86 (165). An SFK FYN induces tyrosine phosphorylation of β-catenin at Tyr142 and disrupts its association with α-catenin (37). Other tyrosine kinases such as FER, FES, and c-MET also participate in the phosphorylation of Tyr142 (37). This disrupts the binding of α-catenin to β-catenin and, instead, favors the binding of β-catenin to the nuclear transporter B-cell lymphoma 9 (Bcl9), which acts as a co-activator in WNT signaling (166). Protein tyrosine kinase (PTK6) is a distant SRC family member. It is regulated by C-terminal tyrosine phosphorylation, which is similar to SRC but lacks an N-terminal myristoylation site and, therefore, cannot localize to the membrane-like other SRCs. PTK6 interacts with both the cytoplasmic and nuclear β-catenin and directly phosphorylates β-catenin at several tyrosine residues including Tyr64, Tyr142, Tyr331, and Tyr333 (167). In the nucleus, PTK6 inhibits β-catenin function, which was shown to be independent of tyrosine phosphorylation, suggesting a possible kinase-independent role of PTK6 in the transcriptional regulation of β-catenin function (167).
BCR-ABL Fusion Protein
The BCR-ABL fusion protein, which is frequently found in chronic myeloid leukemia (CML), physically interacts with β-catenin and phosphorylates it at Tyr86 and Tyr654 (168). Phosphorylation of these tyrosine sites by BCR-ABL prevents the association of β-catenin with AXIN/GSK3β complex; thereby, serine/threonine phosphorylation is prevented. This subsequently increases the cytosolic and nuclear accumulation of β-catenin that is required for self-renewal of BCR-ABL-positive CML cells (169). This process can be reversed by tyrosine phosphatase SHP1, where SHP1-mediated dephosphorylation of Tyr86 and Tyr654 restores GSK3β-dependent serine/threonine phosphorylation and, thereby, β-catenin degradation (170). In another mechanism, BCR-ABL tyrosine kinase controls β-catenin mediated transcriptional activity indirectly, without physical interaction. Chibby1 (CBY1) is a small protein that represses β-catenin transcriptional activation; it interacts with the C-terminal activation domain of β-catenin that hinders its binding with the TCF/LEF transcription factors (171). The scaffolding protein 14-3-3 drives the nuclear export of CBY1 and β-catenin by forming a stable tripartite complex with them (172, 173). However, BCR-ABL fusion protein downregulates CBY1 at the transcriptional level by promoter hypermethylation and at the post-transcriptional level by directing CBY1 toward proteasome-dependent degradation through SUMOylation (171–173). This retains β-catenin in the nucleus and sustains its activation, which is required for the proliferation of CML cells (174).
Casein Kinase 2 (CK2)
Casein kinase 2 (CK2) is ubiquitously expressed in the nucleus and cytoplasm of eukaryotic cells (175). CK2 phosphorylates many transcription factors, tumor suppressors, and proto-oncoproteins involved in cancer, thereby regulating a multitude of cellular processes. One such important function is the regulation of protein stability, thus contributing to cell proliferation and transformation (176). It also plays an important role in embryonic development. CK2 phosphorylates β-catenin at Thr393, which leads to its stabilization and increases its contributions in transcriptional regulations (176). This is probably mediated by decreased affinity to AXIN in the destruction complex (177). Another study identified Ser29, Thr102, and Thr112 in β-catenin as CK2 phosphorylation sites that were required for interaction with α-catenin. CK2 phosphorylation of these residues was also important for degradation of β-catenin, as pre-phosphorylation of β-catenin by CK2 stabilizes its binding to components of the destruction complex, AXIN, and GSK3β, further enhancing the activity of GSK3β. Thus, the cytoplasmic turnover of β-catenin is controlled by the combined action of CK2 and GSK3β (178). Therefore, CK2 appears to have dual roles in β-catenin regulation: one for canonical WNT signaling and the other for cell adhesion (166).
Protein Kinase D1 (PKD1)
Protein kinase D1 (PKD1) lies downstream of the signaling pathways initiated by diacylglycerol and PKC. Diacylglycerol dictates the intracellular localization of PKD1, while PKC activates it by phosphorylation (179). PKD1 served as a tumor suppressor in advanced prostate cancer, where its expression was downregulated; later, it was found that it interacted with E-cadherin (180, 181). PKD1 interacts with β-catenin and phosphorylates it on Thr112 and Thr120 that inhibit the nuclear localization of β-catenin, thereby decreasing its transcriptional activity. This was probably because these threonine phosphorylations on β-catenin increased its interaction with α-catenin and E-cadherin, thereby linking the cytoskeleton. This suggests that Thr120 phosphorylation is critical for cell-cell adhesion (182, 183). However, it has been suggested that PKD1 expression is transcriptionally repressed by β-catenin, indicating the involvement of a negative auto-regulatory loop (184).
In summary, β-catenin activity is tightly controlled by a series of tyrosine and serine/threonine kinases (summarized in Table 1). Phosphorylation on a specific β-catenin residue or several residues can affect its stability, localization, and interaction with other partners (Figure 5). For example, phosphorylation by tyrosine or serine/threonine kinases in the N-terminal region (except for Tyr86 and Ser191) tags β-catenin for degradation or inhibits its nuclear translocation. On the other hand, phosphorylation of the C-terminal region increases the stability of β-catenin as well as its nuclear translocation (except for Ser718). Nevertheless, different regions of β-catenin display affinity to different regulatory proteins probably due to phosphorylation modifications (8). Since a deregulated β-catenin function can contribute to malignancy, kinases involved in the regulation of β-catenin stability, localization, and function can be attractive targets for therapeutic implications. Pharmacological inhibitors against BCR-ABL, EGFR, and MET have been developed for several cancers (185–187) and probably can be repurposed to regulate β-catenin activity. However, inhibition of kinases that promote β-catenin degradation or inhibits its function can result in transcriptional activation of WNT/β-catenin target genes.
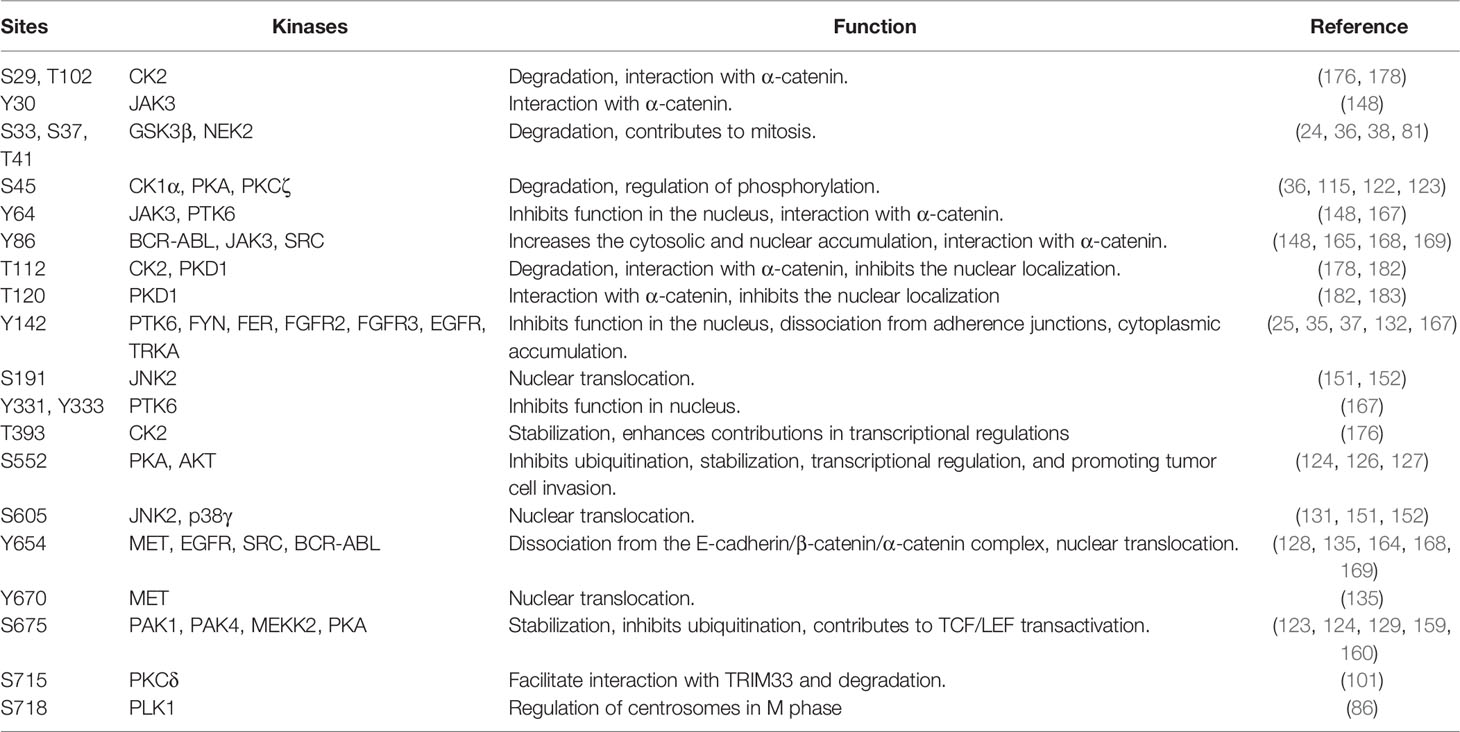
Table 1 Kinases regulating β-catenin activity.
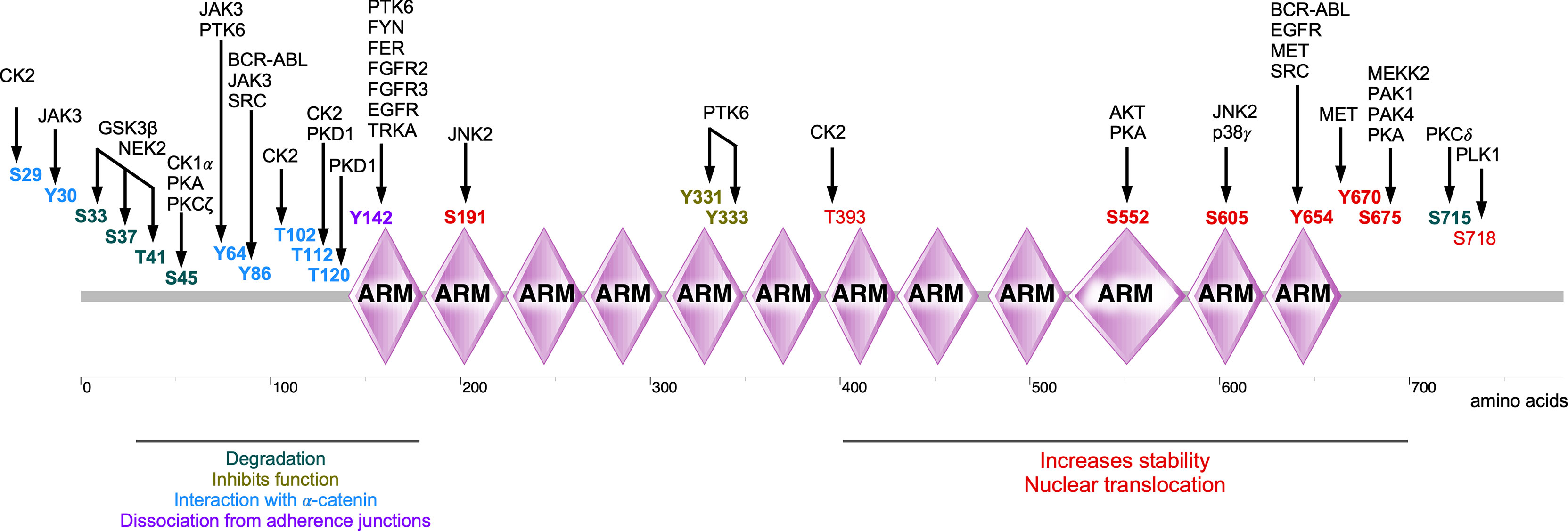
Figure 5 Phosphorylations sites of β-catenin. Several kinases are involved in the phosphorylation of β-catenin, thereby regulating its stability, localization, and activity of β-catenin.
Conclusion
Like other cellular signaling events, WNT/β-catenin signaling is tightly controlled by protein phosphorylation and dephosphorylation. For example, in classical WNT/β-catenin signaling, the formation and activity of the destruction complex are phosphorylation-dependent, which further regulates the stability of β-catenin in a phosphorylation-dependent manner (8). Nevertheless, as discussed above, several kinases are involved in the regulation of this evolutionarily conserved pathway modulating cellular adhesion, polarity, motility, migration, proliferation, and differentiation (188–190).
Much is already known about WNT/β-catenin signaling and the roles of its components in various cancers. However, there are still several possible missing links that need to be studied in the regulation of WNT/β-catenin. For instance, aurora family kinases seem to be involved in the stabilization of β-catenin (191). However, the exact mechanism of how β-catenin stabilization is mediated by aurora family kinases remains to be determined. Although WNT/β-catenin signaling is highly simplified in the figures depicted in this review, the interaction between various signaling proteins as described above makes it highly complicated. Both the canonical and non-canonical pathways involving β-catenin intersect at many levels, which adds several layers of complexity. Thus, attempts should be made to precisely define and dissect these pathways, so that they can be better targeted as therapies in the future.
Author Contributions
KS and JK outlined the content, reviewed the literature, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Kungliga Fysiografiska Sällskapet i Lund (KS), the Crafoord Foundation (JK), Magnus Bergvalls Stiftelse (JK), the Swedish Cancer Society (JK), and the Swedish Childhood Cancer Foundation (JK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Perugorria MJ, Olaizola P, Labiano I, Esparza-Baquer A, Marzioni M, Marin JJG, et al. Wnt-Beta-Catenin Signalling in Liver Development, Health and Disease. Nat Rev Gastroenterol Hepatol (2019) 16(2):121–36. doi: 10.1038/s41575-018-0075-9
2. Parsons MJ, Tammela T, Dow LE. WNT as a Driver and Dependency in Cancer. Cancer Discov (2021) 11(10):2413–29. doi: 10.1158/2159-8290.CD-21-0190
3. Kleeman SO, Leedham SJ. Not All Wnt Activation Is Equal: Ligand-Dependent Versus Ligand-Independent Wnt Activation in Colorectal Cancer. Cancers (Basel) (2020) 12(11):3355. doi: 10.3390/cancers12113355
4. Vallee A, Lecarpentier Y, Vallee JN. The Key Role of the WNT/beta-Catenin Pathway in Metabolic Reprogramming in Cancers Under Normoxic Conditions. Cancers (Basel) (2021) 13(21):5557. doi: 10.3390/cancers13215557
5. Bell IJ, Horn MS, Van Raay TJ. Bridging the Gap Between Non-Canonical and Canonical Wnt Signaling Through Vangl2. Semin Cell Dev Biol (2021). doi: 10.1016/j.semcdb.2021.10.004
6. Lojk J, Marc J. Roles of Non-Canonical Wnt Signalling Pathways in Bone Biology. Int J Mol Sci (2021) 22(19):10840. doi: 10.3390/ijms221910840
7. Trejo-Solis C, Escamilla-Ramirez A, Jimenez-Farfan D, Castillo-Rodriguez RA, Flores-Najera A, Cruz-Salgado A. Crosstalk of the Wnt/beta-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells. Pharmaceuticals (Basel) (2021) 14(9):871. doi: 10.3390/ph14090871
8. Yu F, Yu C, Li F, Zuo Y, Wang Y, Yao L, et al. Wnt/beta-Catenin Signaling in Cancers and Targeted Therapies. Signal Transduct Target Ther (2021) 6(1):307. doi: 10.1038/s41392-021-00701-5
9. Nusse R, Clevers H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell (2017) 169(6):985–99. doi: 10.1016/j.cell.2017.05.016
10. Hiremath IS, Goel A, Warrier S, Kumar AP, Sethi G, Garg M. The Multidimensional Role of the Wnt/beta-Catenin Signaling Pathway in Human Malignancies. J Cell Physiol (2021) 273:199–238. doi: 10.1002/jcp.30561
11. Taank Y, Agnihotri N. Understanding the Regulation of Beta-Catenin Expression and Activity in Colorectal Cancer Carcinogenesis: Beyond Destruction Complex. Clin Transl Oncol (2021) 23(12):2448–59. doi: 10.1007/s12094-021-02686-7
12. Mege RM, Ishiyama N. Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harb Perspect Biol (2017) 9(5):a028738. doi: 10.1101/cshperspect.a028738
13. Menck K, Heinrichs S, Baden C, Bleckmann A. The WNT/ROR Pathway in Cancer: From Signaling to Therapeutic Intervention. Cells (2021) 10(1):142. doi: 10.3390/cells10010142
14. Söderholm S, Cantù C. The WNT/beta-Catenin Dependent Transcription: A Tissue-Specific Business. WIREs Mech Dis (2021) 13(3):e1511. doi: 10.1002/wsbm.1511
15. Zhao B, Xue B. Self-Regulation of Functional Pathways by Motifs Inside the Disordered Tails of Beta-Catenin. BMC Genomics (2016) 179Suppl 5):484. doi: 10.1186/s12864-016-2825-9
16. Xing Y, Takemaru K, Liu J, Berndt JD, Zheng JJ, Moon RT, et al. Crystal Structure of a Full-Length Beta-Catenin. Structure (2008) 16(3):478–87. doi: 10.1016/j.str.2007.12.021
17. Valenta T, Hausmann G, Basler K. The Many Faces and Functions of Beta-Catenin. EMBO J (2012) 31(12):2714–36. doi: 10.1038/emboj.2012.150
18. Kumar R, Bashyam MD. Multiple Oncogenic Roles of Nuclear Beta-Catenin. J Biosci (2017) 42(4):695–707. doi: 10.1007/s12038-017-9710-9
19. Masuda T, Ishitani T. Context-Dependent Regulation of the Beta-Catenin Transcriptional Complex Supports Diverse Functions of Wnt/beta-Catenin Signaling. J Biochem (2017) 161(1):9–17. doi: 10.1093/jb/mvw072
20. Shapiro L, Weis WI. Structure and Biochemistry of Cadherins and Catenins. Cold Spring Harb Perspect Biol (2009) 1(3):a003053. doi: 10.1101/cshperspect.a003053
21. Yu W, Yang L, Li T, Zhang Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front Oncol (2019) 9:989. doi: 10.3389/fonc.2019.00989
22. Wang B, Li X, Liu L, Wang M. Beta-Catenin: Oncogenic Role and Therapeutic Target in Cervical Cancer. Biol Res (2020) 53(1):33. doi: 10.1186/s40659-020-00301-7
23. Vergara D, Stanca E, Guerra F, Priore P, Gaballo A, Franck J, et al. Beta-Catenin Knockdown Affects Mitochondrial Biogenesis and Lipid Metabolism in Breast Cancer Cells. Front Physiol (2017) 8:544. doi: 10.3389/fphys.2017.00544
24. Tominaga J, Fukunaga Y, Abelardo E, Nagafuchi A. Defining the Function of Beta-Catenin Tyrosine Phosphorylation in Cadherin-Mediated Cell-Cell Adhesion. Genes Cells (2008) 13(1):67–77. doi: 10.1111/j.1365-2443.2007.01149.x
25. Brembeck FH, Rosario M, Birchmeier W. Balancing Cell Adhesion and Wnt Signaling, the Key Role of Beta-Catenin. Curr Opin Genet Dev (2006) 16(1):51–9. doi: 10.1016/j.gde.2005.12.007
26. Spink KE, Polakis P, Weis WI. Structural Basis of the Axin-Adenomatous Polyposis Coli Interaction. EMBO J (2000) 19(10):2270–9. doi: 10.1093/emboj/19.10.2270
27. Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of Axin, a Wnt Signal Negative Regulator, by Glycogen Synthase Kinase-3beta Regulates Its Stability. J Biol Chem (1999) 274(16):10681–4. doi: 10.1074/jbc.274.16.10681
28. Xing Y, Clements WK, Kimelman D, Xu W. Crystal Structure of a Beta-Catenin/Axin Complex Suggests a Mechanism for the Beta-Catenin Destruction Complex. Genes Dev (2003) 17(22):2753–64. doi: 10.1101/gad.1142603
29. Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, et al. Axin-Mediated CKI Phosphorylation of Beta-Catenin at Ser 45: A Molecular Switch for the Wnt Pathway. Genes Dev (2002) 16(9):1066–76. doi: 10.1101/gad.230302
30. Strovel ET, Wu D, Sussman DJ. Protein Phosphatase 2Calpha Dephosphorylates Axin and Activates LEF-1-Dependent Transcription. J Biol Chem (2000) 275(4):2399–403. doi: 10.1074/jbc.275.4.2399
31. Stamos JL, Weis WI. The Beta-Catenin Destruction Complex. Cold Spring Harb Perspect Biol (2013) 5(1):a007898. doi: 10.1101/cshperspect.a007898
32. Jho E, Lomvardas S. Costantini F. A GSK3beta Phosphorylation Site in Axin Modulates Interaction With Beta-Catenin and Tcf-Mediated Gene Expression. Biochem Biophys Res Commun (1999) 266(1):28–35. doi: 10.1006/bbrc.1999.1760
33. Kim SE, Huang H, Zhao M, Zhang X, Zhang A, Semonov MV, et al. Wnt Stabilization of Beta-Catenin Reveals Principles for Morphogen Receptor-Scaffold Assemblies. Science (2013) 340(6134):867–70. doi: 10.1126/science.1232389
34. Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI. Mechanism of Phosphorylation-Dependent Binding of APC to Beta-Catenin and Its Role in Beta-Catenin Degradation. Mol Cell (2004) 15(4):511–21. doi: 10.1016/j.molcel.2004.08.010
35. Bustos VH, Ferrarese A, Venerando A, Marin O, Allende JE, Pinna LA. The First Armadillo Repeat Is Involved in the Recognition and Regulation of Beta-Catenin Phosphorylation by Protein Kinase CK1. Proc Natl Acad Sci USA (2006) 103(52):19725–30. doi: 10.1073/pnas.0609424104
36. Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, et al. Control of Beta-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell (2002) 108(6):837–47. doi: 10.1016/s0092-8674(02)00685-2
37. Piedra J, Miravet S, Castano J, Palmer HG, Heisterkamp N, Garcia de Herreros A, et al. P120 Catenin-Associated Fer and Fyn Tyrosine Kinases Regulate Beta-Catenin Tyr-142 Phosphorylation and Beta-Catenin-Alpha-Catenin Interaction. Mol Cell Biol (2003) 23(7):2287–97. doi: 10.1128/mcb.23.7.2287-2297.2003
38. Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a Beta-TrCP1-Skp1-Beta-Catenin Complex: Destruction Motif Binding and Lysine Specificity of the SCF(beta-TrCP1) Ubiquitin Ligase. Mol Cell (2003) 11(6):1445–56. doi: 10.1016/s1097-2765(03)00234-x
39. Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine Phosphorylation-Regulated Ubiquitination and Degradation of Beta-Catenin. J Biol Chem (1997) 272(40):24735–8. doi: 10.1074/jbc.272.40.24735
40. Niehrs C. The Complex World of WNT Receptor Signalling. Nat Rev Mol Cell Biol (2012) 13(12):767–79. doi: 10.1038/nrm3470
41. Tauriello DV, Jordens I, Kirchner K, Slootstra JW, Kruitwagen T, Bouwman BA, et al. Wnt/beta-Catenin Signaling Requires Interaction of the Dishevelled DEP Domain and C Terminus With a Discontinuous Motif in Frizzled. Proc Natl Acad Sci USA (2012) 109(14):E812–20. doi: 10.1073/pnas.1114802109
42. Anastas JN, Moon RT. WNT Signalling Pathways as Therapeutic Targets in Cancer. Nat Rev Cancer (2013) 13(1):11–26. doi: 10.1038/nrc3419
43. Bienz M. Signalosome Assembly by Domains Undergoing Dynamic Head-to-Tail Polymerization. Trends Biochem Sci (2014) 39(10):487–95. doi: 10.1016/j.tibs.2014.08.006
44. Fiedler M, Graeb M, Mieszczanek J, Rutherford TJ, Johnson CM, Bienz M. An Ancient Pygo-Dependent Wnt Enhanceosome Integrated by Chip/LDB-SSDP. Elife (2015) 4:e09073. doi: 10.7554/eLife.09073
45. Tolwinski NS, Wehrli M, Rives A, Erdeniz N, DiNardo S, Wieschaus E. Wg/Wnt Signal Can Be Transmitted Through Arrow/LRP5,6 and Axin Independently of Zw3/Gsk3beta Activity. Dev Cell (2003) 4(3):407–18. doi: 10.1016/s1534-5807(03)00063-7
46. Hou Y, Ding M, Wang C, Yang X, Ye T, Yu H. TRIM11 Promotes Lymphomas by Activating the Beta-Catenin Signaling and Axin1 Ubiquitination Degradation. Exp Cell Res (2020) 387(2):111750. doi: 10.1016/j.yexcr.2019.111750
47. Takemaru KI, Moon RT. The Transcriptional Coactivator CBP Interacts With Beta-Catenin to Activate Gene Expression. J Cell Biol (2000) 149(2):249–54. doi: 10.1083/jcb.149.2.249
48. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The Cyclin D1 Gene Is a Target of the Beta-Catenin/LEF-1 Pathway. Proc Natl Acad Sci USA (1999) 96(10):5522–7. doi: 10.1073/pnas.96.10.5522
49. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of C-MYC as a Target of the APC Pathway. Science (1998) 281(5382):1509–12. doi: 10.1126/science.281.5382.1509
50. Schell MJ, Yang M, Teer JK, Lo FY, Madan A, Coppola D, et al. A Multigene Mutation Classification of 468 Colorectal Cancers Reveals a Prognostic Role for APC. Nat Commun (2016) 7:11743. doi: 10.1038/ncomms11743
51. Ignatov A, Bischoff J, Ignatov T, Schwarzenau C, Krebs T, Kuester D, et al. APC Promoter Hypermethylation Is an Early Event in Endometrial Tumorigenesis. Cancer Sci (2010) 101(2):321–7. doi: 10.1111/j.1349-7006.2009.01397.x
52. Banno K, Yanokura M, Iida M, Masuda K, Aoki D. Carcinogenic Mechanisms of Endometrial Cancer: Involvement of Genetics and Epigenetics. J Obstet Gynaecol Res (2014) 40(8):1957–67. doi: 10.1111/jog.12442
53. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic Mutations of the Beta-Catenin Gene are Frequent in Mouse and Human Hepatocellular Carcinomas. Proc Natl Acad Sci USA (1998) 95(15):8847–51. doi: 10.1073/pnas.95.15.8847
54. Bigas A, Guillen Y, Schoch L, Arambilet D. Revisiting Beta-Catenin Signaling in T-Cell Development and T-Cell Acute Lymphoblastic Leukemia. Bioessays (2020) 42(2):e1900099. doi: 10.1002/bies.201900099
55. Wu R, Zhai Y, Fearon ER, Cho KR. Diverse Mechanisms of Beta-Catenin Deregulation in Ovarian Endometrioid Adenocarcinomas. Cancer Res (2001) 61(22):8247–55.
56. Gamallo C, Palacios J, Moreno G, Calvo de Mora J, Suarez A, Armas A. Beta-Catenin Expression Pattern in Stage I and II Ovarian Carcinomas : Relationship With Beta-Catenin Gene Mutations, Clinicopathological Features, and Clinical Outcome. Am J Pathol (1999) 155(2):527–36. doi: 10.1016/s0002-9440(10)65148-6
57. Nguyen VHL, Hough R, Bernaudo S, Peng C. Wnt/beta-Catenin Signalling in Ovarian Cancer: Insights Into Its Hyperactivation and Function in Tumorigenesis. J Ovarian Res (2019) 12(1):122. doi: 10.1186/s13048-019-0596-z
58. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
59. Veeman MT, Axelrod JD, Moon RT. A Second Canon. Functions and Mechanisms of Beta-Catenin-Independent Wnt Signaling. Dev Cell (2003) 5(3):367–77. doi: 10.1016/s1534-5807(03)00266-1
60. Keller R. Shaping the Vertebrate Body Plan by Polarized Embryonic Cell Movements. Science (2002) 298(5600):1950–4. doi: 10.1126/science.1079478
61. Mlodzik M. Planar Cell Polarization: Do the Same Mechanisms Regulate Drosophila Tissue Polarity and Vertebrate Gastrulation? Trends Genet (2002) 18(11):564–71. doi: 10.1016/s0168-9525(02)02770-1
62. Xiao Q, Chen Z, Jin X, Mao R, Chen Z. The Many Postures of Noncanonical Wnt Signaling in Development and Diseases. BioMed Pharmacother (2017) 93:359–69. doi: 10.1016/j.biopha.2017.06.061
63. Marlow F, Topczewski J, Sepich D, Solnica-Krezel L. Zebrafish Rho Kinase 2 Acts Downstream of Wnt11 to Mediate Cell Polarity and Effective Convergence and Extension Movements. Curr Biol (2002) 12(11):876–84. doi: 10.1016/s0960-9822(02)00864-3
64. Habas R, Dawid IB, He X. Coactivation of Rac and Rho by Wnt/Frizzled Signaling Is Required for Vertebrate Gastrulation. Genes Dev (2003) 17(2):295–309. doi: 10.1101/gad.1022203
65. Yan L, Du Q, Yao J, Liu R. ROR2 Inhibits the Proliferation of Gastric Carcinoma Cells via Activation of Non-Canonical Wnt Signaling. Exp Ther Med (2016) 12(6):4128–34. doi: 10.3892/etm.2016.3883
66. Yuan Y, Niu CC, Deng G, Li ZQ, Pan J, Zhao C, et al. The Wnt5a/Ror2 Noncanonical Signaling Pathway Inhibits Canonical Wnt Signaling in K562 Cells. Int J Mol Med (2011) 27(1):63–9. doi: 10.3892/ijmm.2010.560
67. De A. Wnt/Ca2+ Signaling Pathway: A Brief Overview. Acta Biochim Biophys Sin (Shanghai) (2011) 43(10):745–56. doi: 10.1093/abbs/gmr079
68. Saneyoshi T, Kume S, Amasaki Y, Mikoshiba K. The Wnt/calcium Pathway Activates NF-AT and Promotes Ventral Cell Fate in Xenopus Embryos. Nature (2002) 417(6886):295–9. doi: 10.1038/417295a
69. Wang HY, Malbon CC. Wnt Signaling, Ca2+, and Cyclic GMP: Visualizing Frizzled Functions. Science (2003) 300(5625):1529–30. doi: 10.1126/science.1085259
70. Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, et al. The TAK1-NLK-MAPK-Related Pathway Antagonizes Signalling Between Beta-Catenin and Transcription Factor TCF. Nature (1999) 399(6738):798–802. doi: 10.1038/21674
71. Kohn AD, Moon RT. Wnt and Calcium Signaling: Beta-Catenin-Independent Pathways. Cell Calcium (2005) 38(3-4):439–46. doi: 10.1016/j.ceca.2005.06.022
72. Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, et al. Siah-1 Mediates a Novel Beta-Catenin Degradation Pathway Linking P53 to the Adenomatous Polyposis Coli Protein. Mol Cell (2001) 7(5):927–36. doi: 10.1016/s1097-2765(01)00241-6
73. Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi Collaborate in a Novel Pathway for Beta-Catenin Degradation Linked to P53 Responses. Mol Cell (2001) 7(5):915–26. doi: 10.1016/s1097-2765(01)00242-8
74. Topol L, Jiang X, Choi H, Garrett-Beal L, Carolan PJ, Yang Y. Wnt-5a Inhibits the Canonical Wnt Pathway by Promoting GSK-3-Independent Beta-Catenin Degradation. J Cell Biol (2003) 162(5):899–908. doi: 10.1083/jcb.200303158
75. Lai KKY, Nguyen C, Lee KS, Lee A, Lin DP, Teo JL, et al. Convergence of Canonical and Non-Canonical Wnt Signal: Differential Kat3 Coactivator Usage. Curr Mol Pharmacol (2019) 12(3):167–83. doi: 10.2174/1874467212666190304121131
76. Le NH, Franken P, Fodde R. Tumour-Stroma Interactions in Colorectal Cancer: Converging on Beta-Catenin Activation and Cancer Stemness. Br J Cancer (2008) 98(12):1886–93. doi: 10.1038/sj.bjc.6604401
77. Teo JL, Kahn M. The Wnt Signaling Pathway in Cellular Proliferation and Differentiation: A Tale of Two Coactivators. Adv Drug Delivery Rev (2010) 62(12):1149–55. doi: 10.1016/j.addr.2010.09.012
79. Park JE, Hymel D, Burke TR Jr, Lee KS. Current Progress and Future Perspectives in the Development of Anti-Polo-Like Kinase 1 Therapeutic Agents. F1000Res (2017) 6:1024. doi: 10.12688/f1000research.11398.1
80. Rieger ME, Zhou B, Solomon N, Sunohara M, Li C, Nguyen C, et al. P300/Beta-Catenin Interactions Regulate Adult Progenitor Cell Differentiation Downstream of WNT5a/Protein Kinase C (PKC). J Biol Chem (2016) 291(12):6569–82. doi: 10.1074/jbc.M115.706416
81. Mbom BC, Siemers KA, Ostrowski MA, Nelson WJ, Barth AI. Nek2 Phosphorylates and Stabilizes Beta-Catenin at Mitotic Centrosomes Downstream of Plk1. Mol Biol Cell (2014) 25(7):977–91. doi: 10.1091/mbc.E13-06-0349
82. Van Horn RD, Chu S, Fan L, Yin T, Du J, Beckmann R, et al. Cdk1 Activity Is Required for Mitotic Activation of Aurora A During G2/M Transition of Human Cells. J Biol Chem (2010) 285(28):21849–57. doi: 10.1074/jbc.M110.141010
83. Lindon C, Grant R, Min M. Ubiquitin-Mediated Degradation of Aurora Kinases. Front Oncol (2015) 5:307. doi: 10.3389/fonc.2015.00307
84. Cyphers S, Ruff EF, Behr JM, Chodera JD, Levinson NM. A Water-Mediated Allosteric Network Governs Activation of Aurora Kinase a. Nat Chem Biol (2017) 13(4):402–8. doi: 10.1038/nchembio.2296
85. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-Like Kinase-1 Is Activated by Aurora A to Promote Checkpoint Recovery. Nature (2008) 455(7209):119–23. doi: 10.1038/nature07185
86. Arai T, Haze K, Iimura-Morita Y, Machida T, Iida M, Tanaka K, et al. Identification of Beta-Catenin as a Novel Substrate of Polo-Like Kinase 1. Cell Cycle (2008) 7(22):3556–63. doi: 10.4161/cc.7.22.7072
87. Wierstra I. The Transcription Factor FOXM1 (Forkhead Box M1): Proliferation-Specific Expression, Transcription Factor Function, Target Genes, Mouse Models, and Normal Biological Roles. Adv Cancer Res (2013) 118:97–398. doi: 10.1016/B978-0-12-407173-5.00004-2
88. Asteriti IA, De Mattia F, Guarguaglini G. Cross-Talk Between AURKA and Plk1 in Mitotic Entry and Spindle Assembly. Front Oncol (2015) 5:283. doi: 10.3389/fonc.2015.00283
89. Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-Dependent Phosphorylation of FoxM1 Regulates a Transcriptional Programme Required for Mitotic Progression. Nat Cell Biol (2008) 10(9):1076–82. doi: 10.1038/ncb1767
90. Seke Etet PF, Vecchio L, Nwabo Kamdje AH. Signaling Pathways in Chronic Myeloid Leukemia and Leukemic Stem Cell Maintenance: Key Role of Stromal Microenvironment. Cell Signal (2012) 24(9):1883–8. doi: 10.1016/j.cellsig.2012.05.015
91. Yang J, Ikezoe T, Nishioka C, Udaka K, Yokoyama A. Bcr-Abl Activates AURKA and AURKB in Chronic Myeloid Leukemia Cells via AKT Signaling. Int J Cancer (2014) 134(5):1183–94. doi: 10.1002/ijc.28434
92. Zhang J, Yuan C, Wu J, Elsayed Z, Fu Z. Polo-Like Kinase 1-Mediated Phosphorylation of Forkhead Box Protein M1b Antagonizes Its SUMOylation and Facilitates its Mitotic Function. J Biol Chem (2015) 290(6):3708–19. doi: 10.1074/jbc.M114.634386
93. Marumoto T, Zhang D, Saya H. Aurora-A - A Guardian of Poles. Nat Rev Cancer (2005) 5(1):42–50. doi: 10.1038/nrc1526
94. Benada J, Burdova K, Lidak T, von Morgen P, Macurek L. Polo-Like Kinase 1 Inhibits DNA Damage Response During Mitosis. Cell Cycle (2015) 14(2):219–31. doi: 10.4161/15384101.2014.977067
95. Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, et al. Loss of Beta-Catenin Impairs the Renewal of Normal and CML Stem Cells In Vivo. Cancer Cell (2007) 12(6):528–41. doi: 10.1016/j.ccr.2007.11.003
96. Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, et al. FoxM1 Promotes Beta-Catenin Nuclear Localization and Controls Wnt Target-Gene Expression and Glioma Tumorigenesis. Cancer Cell (2011) 20(4):427–42. doi: 10.1016/j.ccr.2011.08.016
97. Xia Z, Wei P, Zhang H, Ding Z, Yang L, Huang Z, et al. AURKA Governs Self-Renewal Capacity in Glioma-Initiating Cells via Stabilization/Activation of Beta-Catenin/Wnt Signaling. Mol Cancer Res (2013) 11(9):1101–11. doi: 10.1158/1541-7786.MCR-13-0044
98. Iliaki S, Beyaert R, Afonina IS. Polo-Like Kinase 1 (PLK1) Signaling in Cancer and Beyond. Biochem Pharmacol (2021) 193:114747. doi: 10.1016/j.bcp.2021.114747
99. Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, et al. MELK-Dependent FOXM1 Phosphorylation Is Essential for Proliferation of Glioma Stem Cells. Stem Cells (2013) 31(6):1051–63. doi: 10.1002/stem.1358
100. Mancini M, De Santis S, Monaldi C, Bavaro L, Martelli M, Castagnetti F, et al. Hyper-Activation of Aurora Kinase a-Polo-Like Kinase 1-FOXM1 Axis Promotes Chronic Myeloid Leukemia Resistance to Tyrosine Kinase Inhibitors. J Exp Clin Cancer Res (2019) 38(1):216. doi: 10.1186/s13046-019-1197-9
101. Xue J, Chen Y, Wu Y, Wang Z, Zhou A, Zhang S, et al. Tumour Suppressor TRIM33 Targets Nuclear Beta-Catenin Degradation. Nat Commun (2015) 6:6156. doi: 10.1038/ncomms7156
102. Xu Y, Wu G, Zhang J, Li J, Ruan N, Zhang J, et al. TRIM33 Overexpression Inhibits the Progression of Clear Cell Renal Cell Carcinoma In Vivo and In Vitro. BioMed Res Int (2020) 2020:8409239. doi: 10.1155/2020/8409239
103. Aucagne R, Droin N, Paggetti J, Lagrange B, Largeot A, Hammann A, et al. Transcription Intermediary Factor 1gamma Is a Tumor Suppressor in Mouse and Human Chronic Myelomonocytic Leukemia. J Clin Invest (2011) 121(6):2361–70. doi: 10.1172/JCI45213
104. Herquel B, Ouararhni K, Khetchoumian K, Ignat M, Teletin M, Mark M, et al. Transcription Cofactors TRIM24, TRIM28, and TRIM33 Associate to Form Regulatory Complexes That Suppress Murine Hepatocellular Carcinoma. Proc Natl Acad Sci USA (2011) 108(20):8212–7. doi: 10.1073/pnas.1101544108
105. Vincent DF, Yan KP, Treilleux I, Gay F, Arfi V, Kaniewski B, et al. Inactivation of TIF1gamma Cooperates With Kras to Induce Cystic Tumors of the Pancreas. PloS Genet (2009) 5(7):e1000575. doi: 10.1371/journal.pgen.1000575
106. Kazi JU, Kabir NN, Soh JW. Bioinformatic Prediction and Analysis of Eukaryotic Protein Kinases in the Rat Genome. Gene (2008) 410(1):147–53. doi: 10.1016/j.gene.2007.12.003
107. Kabir NN, Kazi JU. Comparative Analysis of Human and Bovine Protein Kinases Reveals Unique Relationship and Functional Diversity. Genet Mol Biol (2011) 34(4):587–91. doi: 10.1590/S1415-47572011005000035
108. Kazi JU. The Mechanism of Protein Kinase C Regulation. Front Biol (2011) 6(4):328–36. doi: 10.1007/s11515-011-1070-5
109. Kazi JU, Kabir NN, Rönnstrand L. Protein Kinase C (PKC) as a Drug Target in Chronic Lymphocytic Leukemia. Med Oncol (2013) 30(4):757. doi: 10.1007/s12032-013-0757-7
110. Isakov N. Protein Kinase C (PKC) Isoforms in Cancer, Tumor Promotion and Tumor Suppression. Semin Cancer Biol (2018) 48:36–52. doi: 10.1016/j.semcancer.2017.04.012
111. Newton AC. Protein Kinase C as a Tumor Suppressor. Semin Cancer Biol (2018) 48:18–26. doi: 10.1016/j.semcancer.2017.04.017
112. Perletti GP, Marras E, Concari P, Piccinini F, Tashjian AH Jr. PKCdelta Acts as a Growth and Tumor Suppressor in Rat Colonic Epithelial Cells. Oncogene (1999) 18(5):1251–6. doi: 10.1038/sj.onc.1202408
113. Kazi JU, Soh JW. Isoform-Specific Translocation of PKC Isoforms in NIH3T3 Cells by TPA. Biochem Biophys Res Commun (2007) 364(2):231–7. doi: 10.1016/j.bbrc.2007.09.123
114. Kabir NN, Rönnstrand L, Kazi JU. Protein Kinase C Expression Is Deregulated in Chronic Lymphocytic Leukemia. Leuk Lymphoma (2013) 54(10):2288–90. doi: 10.3109/10428194.2013.769220
115. Llado V, Nakanishi Y, Duran A, Reina-Campos M, Shelton PM, Linares JF, et al. Repression of Intestinal Stem Cell Function and Tumorigenesis Through Direct Phosphorylation of Beta-Catenin and Yap by PKCzeta. Cell Rep (2015) 10(5):740–54. doi: 10.1016/j.celrep.2015.01.007
116. Gwak J, Cho M, Gong SJ, Won J, Kim DE, Kim EY, et al. Protein-Kinase-C-Mediated Beta-Catenin Phosphorylation Negatively Regulates the Wnt/beta-Catenin Pathway. J Cell Sci (2006) 119(Pt 22):4702–9. doi: 10.1242/jcs.03256
117. Lee JM, Kim IS, Kim H, Lee JS, Kim K, Yim HY, et al. RORalpha Attenuates Wnt/beta-Catenin Signaling by PKCalpha-Dependent Phosphorylation in Colon Cancer. Mol Cell (2010) 37(2):183–95. doi: 10.1016/j.molcel.2009.12.022
118. Stork PJ, Schmitt JM. Crosstalk Between cAMP and MAP Kinase Signaling in the Regulation of Cell Proliferation. Trends Cell Biol (2002) 12(6):258–66. doi: 10.1016/s0962-8924(02)02294-8
119. Cao W, Li J, Hao Q, Vadgama JV, Wu Y. AMP-Activated Protein Kinase: A Potential Therapeutic Target for Triple-Negative Breast Cancer. Breast Cancer Res (2019) 21(1):29. doi: 10.1186/s13058-019-1107-2
120. Hogarth DK, Sandbo N, Taurin S, Kolenko V, Miano JM, Dulin NO. Dual Role of PKA in Phenotypic Modulation of Vascular Smooth Muscle Cells by Extracellular ATP. Am J Physiol Cell Physiol (2004) 287(2):C449–56. doi: 10.1152/ajpcell.00547.2003
121. Marino A, Hausenloy DJ, Andreadou I, Horman S, Bertrand L, Beauloye C. AMP-Activated Protein Kinase: A Remarkable Contributor to Preserve a Healthy Heart Against ROS Injury. Free Radic Biol Med (2021) 166:238–54. doi: 10.1016/j.freeradbiomed.2021.02.047
122. Kang DE, Soriano S, Xia X, Eberhart CG, De Strooper B, Zheng H, et al. Presenilin Couples the Paired Phosphorylation of Beta-Catenin Independent of Axin: Implications for Beta-Catenin Activation in Tumorigenesis. Cell (2002) 110(6):751–62. doi: 10.1016/s0092-8674(02)00970-4
123. Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of Beta-Catenin by Cyclic AMP-Dependent Protein Kinase Stabilizes Beta-Catenin Through Inhibition of Its Ubiquitination. Mol Cell Biol (2005) 25(20):9063–72. doi: 10.1128/MCB.25.20.9063-9072.2005
124. Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of Beta-Catenin by Cyclic AMP-Dependent Protein Kinase. J Biol Chem (2006) 281(15):9971–6. doi: 10.1074/jbc.M508778200
125. Li CC, Le K, Kato J, Moss J, Vaughan M. Enhancement of Beta-Catenin Activity by BIG1 Plus BIG2 via Arf Activation and cAMP Signals. Proc Natl Acad Sci USA (2016) 113(21):5946–51. doi: 10.1073/pnas.1601918113
126. Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, et al. Phosphorylation of Beta-Catenin by AKT Promotes Beta-Catenin Transcriptional Activity. J Biol Chem (2007) 282(15):11221–9. doi: 10.1074/jbc.M611871200
127. He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, et al. PTEN-Deficient Intestinal Stem Cells Initiate Intestinal Polyposis. Nat Genet (2007) 39(2):189–98. doi: 10.1038/ng1928
128. Jean C, Blanc A, Prade-Houdellier N, Ysebaert L, Hernandez-Pigeon H, Al Saati T, et al. Epidermal Growth Factor Receptor/Beta-Catenin/T-Cell Factor 4/Matrix Metalloproteinase 1: A New Pathway for Regulating Keratinocyte Invasiveness After UVA Irradiation. Cancer Res (2009) 69(8):3291–9. doi: 10.1158/0008-5472.CAN-08-1909
129. Greenblatt MB, Shin DY, Oh H, Lee KY, Zhai B, Gygi SP, et al. MEKK2 Mediates an Alternative Beta-Catenin Pathway That Promotes Bone Formation. Proc Natl Acad Sci USA (2016) 113(9):E1226–35. doi: 10.1073/pnas.1600813113
130. Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS Signalling Promotes the Wnt/beta-Catenin Pathway Through LRP6 in Colorectal Cancer. Oncogene (2015) 34(38):4914–27. doi: 10.1038/onc.2014.416
131. Yin N, Qi X, Tsai S, Lu Y, Basir Z, Oshima K, et al. P38gamma MAPK is Required for Inflammation-Associated Colon Tumorigenesis. Oncogene (2016) 35(8):1039–48. doi: 10.1038/onc.2015.158
132. Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, et al. Receptor Tyrosine Kinases Activate Canonical WNT/beta-Catenin Signaling via MAP Kinase/LRP6 Pathway and Direct Beta-Catenin Phosphorylation. PloS One (2012) 7(4):e35826. doi: 10.1371/journal.pone.0035826
133. Hiscox S, Jiang WG. Association of the HGF/SF Receptor, C-Met, With the Cell-Surface Adhesion Molecule, E-Cadherin, and Catenins in Human Tumor Cells. Biochem Biophys Res Commun (1999) 261(2):406–11. doi: 10.1006/bbrc.1999.1002
134. Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, et al. Hepatocyte Growth Factor Induces Wnt-Independent Nuclear Translocation of Beta-Catenin After Met-Beta-Catenin Dissociation in Hepatocytes. Cancer Res (2002) 62(7):2064–71.
135. Zeng G, Apte U, Micsenyi A, Bell A, Monga SP. Tyrosine Residues 654 and 670 in Beta-Catenin Are Crucial in Regulation of Met-Beta-Catenin Interactions. Exp Cell Res (2006) 312(18):3620–30. doi: 10.1016/j.yexcr.2006.08.003
136. Wagh PK, Gray JK, Zinser GM, Vasiliauskas J, James L, Monga SP, et al. Beta-Catenin Is Required for Ron Receptor-Induced Mammary Tumorigenesis. Oncogene (2011) 30(34):3694–704. doi: 10.1038/onc.2011.86
137. Guo Q, Kim A, Li B, Ransick A, Bugacov H, Chen X, et al. A Beta-Catenin-Driven Switch in TCF/LEF Transcription Factor Binding to DNA Target Sites Promotes Commitment of Mammalian Nephron Progenitor Cells. Elife (2021) 10:e64444. doi: 10.7554/eLife.64444
138. Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L. IGF-II Induces Rapid Beta-Catenin Relocation to the Nucleus During Epithelium to Mesenchyme Transition. Oncogene (2001) 20(36):4942–50. doi: 10.1038/sj.onc.1204660
139. Playford MP, Bicknell D, Bodmer WF, Macaulay VM. Insulin-Like Growth Factor 1 Regulates the Location, Stability, and Transcriptional Activity of Beta-Catenin. Proc Natl Acad Sci USA (2000) 97(22):12103–8. doi: 10.1073/pnas.210394297
140. Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M. Insulin-Like Growth Factor-1 Induces Survival and Growth of Biologically Early Melanoma Cells Through Both the Mitogen-Activated Protein Kinase and Beta-Catenin Pathways. Cancer Res (2001) 61(19):7318–24.
141. Reiss K, Valentinis B, Tu X, Xu SQ, Baserga R. Molecular Markers of IGF-I-Mediated Mitogenesis. Exp Cell Res (1998) 242(1):361–72. doi: 10.1006/excr.1998.4113
142. Chen J, Wu A, Sun H, Drakas R, Garofalo C, Cascio S, et al. Functional Significance of Type 1 Insulin-Like Growth Factor-Mediated Nuclear Translocation of the Insulin Receptor Substrate-1 and Beta-Catenin. J Biol Chem (2005) 280(33):29912–20. doi: 10.1074/jbc.M504516200
143. Prisco M, Santini F, Baffa R, Liu M, Drakas R, Wu A, et al. Nuclear Translocation of Insulin Receptor Substrate-1 by the Simian Virus 40 T Antigen and the Activated Type 1 Insulin-Like Growth Factor Receptor. J Biol Chem (2002) 277(35):32078–85. doi: 10.1074/jbc.M204658200
144. Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein Kinase I Phosphorylates and Destabilizes the Beta-Catenin Degradation Complex. Proc Natl Acad Sci USA (2002) 99(3):1182–7. doi: 10.1073/pnas.032468199
145. Gorgisen G, Gulacar IM, Ozes ON. The Role of Insulin Receptor Substrate (IRS) Proteins in Oncogenic Transformation. Cell Mol Biol (Noisy-le-grand) (2017) 63(1):1–5. doi: 10.14715/cmb/2017.63.1.1
146. Campbell HK, Maiers JL, DeMali KA. Interplay Between Tight Junctions & Adherens Junctions. Exp Cell Res (2017) 358(1):39–44. doi: 10.1016/j.yexcr.2017.03.061
147. Senkevitch E, Durum S. The Promise of Janus Kinase Inhibitors in the Treatment of Hematological Malignancies. Cytokine (2017) 98:33–41. doi: 10.1016/j.cyto.2016.10.012
148. Mishra J, Das JK, Kumar N. Janus Kinase 3 Regulates Adherens Junctions and Epithelial Mesenchymal Transition Through Beta-Catenin. J Biol Chem (2017) 292(40):16406–19. doi: 10.1074/jbc.M117.811802
149. Rane CK, Minden A. P21 Activated Kinase Signaling in Cancer. Semin Cancer Biol (2019) 54:40–9. doi: 10.1016/j.semcancer.2018.01.006
150. Wang K, Baldwin GS, Nikfarjam M, He H. P21-Activated Kinase Signalling in Pancreatic Cancer: New Insights Into Tumour Biology and Immune Modulation. World J Gastroenterol (2018) 24(33):3709–23. doi: 10.3748/wjg.v24.i33.3709
151. Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 Activation Controls Nuclear Localization of Beta-Catenin During Canonical Wnt Signaling. Cell (2008) 133(2):340–53. doi: 10.1016/j.cell.2008.01.052
152. Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, et al. A Two-Step Model for Colon Adenoma Initiation and Progression Caused by APC Loss. Cell (2009) 137(4):623–34. doi: 10.1016/j.cell.2009.02.037
153. Yao D, Li C, Rajoka MSR, He Z, Huang J, Wang J, et al. P21-Activated Kinase 1: Emerging Biological Functions and Potential Therapeutic Targets in Cancer. Theranostics (2020) 10(21):9741–66. doi: 10.7150/thno.46913
154. Nheu T, He H, Hirokawa Y, Walker F, Wood J, Maruta H. PAK Is Essential for RAS-Induced Upregulation of Cyclin D1 During the G1 to S Transition. Cell Cycle (2004) 3(1):71–4. doi: 10.4161/cc.3.1.593
155. Al Thawadi H, Abu-Kaoud N, Al Farsi H, Hoarau-Vechot J, Rafii S, Rafii A, et al. VE-Cadherin Cleavage by Ovarian Cancer Microparticles Induces Beta-Catenin Phosphorylation in Endothelial Cells. Oncotarget (2016) 7(5):5289–305. doi: 10.18632/oncotarget.6677
156. Sun J, Khalid S, Rozakis-Adcock M, Fantus IG. Jin T. P-21-Activated Protein Kinase-1 Functions as a Linker Between Insulin and Wnt Signaling Pathways in the Intestine. Oncogene (2009) 28(35):3132–44. doi: 10.1038/onc.2009.167
157. Fu H, Zhang W, Yuan Q, Niu M, Zhou F, Qiu Q, et al. PAK1 Promotes the Proliferation and Inhibits Apoptosis of Human Spermatogonial Stem Cells via PDK1/KDR/ZNF367 and ERK1/2 and AKT Pathways. Mol Ther Nucleic Acids (2018) 12:769–86. doi: 10.1016/j.omtn.2018.06.006
158. Barbe C, Loumaye A, Lause P, Ritvos O, Thissen JP. P21-Activated Kinase 1 Is Permissive for the Skeletal Muscle Hypertrophy Induced by Myostatin Inhibition. Front Physiol (2021) 12:677746. doi: 10.3389/fphys.2021.677746
159. Li Y, Shao Y, Tong Y, Shen T, Zhang J, Li Y, et al. Nucleo-Cytoplasmic Shuttling of PAK4 Modulates Beta-Catenin Intracellular Translocation and Signaling. Biochim Biophys Acta (2012) 1823(2):465–75. doi: 10.1016/j.bbamcr.2011.11.013
160. Zhu G, Wang Y, Huang B, Liang J, Ding Y, Xu A, et al. A Rac1/PAK1 Cascade Controls Beta-Catenin Activation in Colon Cancer Cells. Oncogene (2012) 31(8):1001–12. doi: 10.1038/onc.2011.294
161. Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, et al. Epithelial Cell Alpha3beta1 Integrin Links Beta-Catenin and Smad Signaling to Promote Myofibroblast Formation and Pulmonary Fibrosis. J Clin Invest (2009) 119(1):213–24. doi: 10.1172/JCI36940
162. Xu L, Cui WH, Zhou WC, Li DL, Li LC, Zhao P, et al. Activation of Wnt/beta-Catenin Signalling Is Required for TGF-Beta/Smad2/3 Signalling During Myofibroblast Proliferation. J Cell Mol Med (2017) 21(8):1545–54. doi: 10.1111/jcmm.13085
163. Ulsamer A, Wei Y, Kim KK, Tan K, Wheeler S, Xi Y, et al. Axin Pathway Activity Regulates In Vivo Py654-Beta-Catenin Accumulation and Pulmonary Fibrosis. J Biol Chem (2012) 287(7):5164–72. doi: 10.1074/jbc.M111.322123
164. Xi Y, Wei Y, Sennino B, Ulsamer A, Kwan I, Brumwell AN, et al. Identification of Py654-Beta-Catenin as a Critical Co-Factor in Hypoxia-Inducible Factor-1alpha Signaling and Tumor Responses to Hypoxia. Oncogene (2013) 32(42):5048–57. doi: 10.1038/onc.2012.530
165. Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-Cadherin/Catenin Association by Tyrosine Phosphorylation. J Biol Chem (1999) 274(51):36734–40. doi: 10.1074/jbc.274.51.36734
166. Daugherty RL, Gottardi CJ. Phospho-Regulation of Beta-Catenin Adhesion and Signaling Functions. Physiol (Bethesda) (2007) 22:303–9. doi: 10.1152/physiol.00020.2007
167. Palka-Hamblin HL, Gierut JJ, Bie W, Brauer PM, Zheng Y, Asara JM, et al. Identification of Beta-Catenin as a Target of the Intracellular Tyrosine Kinase PTK6. J Cell Sci (2010) 123(Pt 2):236–45. doi: 10.1242/jcs.053264
168. Coluccia AM, Vacca A, Dunach M, Mologni L, Redaelli S, Bustos VH, et al. Bcr-Abl Stabilizes Beta-Catenin in Chronic Myeloid Leukemia Through Its Tyrosine Phosphorylation. EMBO J (2007) 26(5):1456–66. doi: 10.1038/sj.emboj.7601485
169. Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-Macrophage Progenitors as Candidate Leukemic Stem Cells in Blast-Crisis CML. N Engl J Med (2004) 351(7):657–67. doi: 10.1056/NEJMoa040258
170. Simoneau M, Coulombe G, Vandal G, Vezina A, Rivard N. SHP-1 Inhibits Beta-Catenin Function by Inducing Its Degradation and Interfering With Its Association With TATA-Binding Protein. Cell Signal (2011) 23(1):269–79. doi: 10.1016/j.cellsig.2010.09.011
171. Takemaru K, Yamaguchi S, Lee YS, Zhang Y, Carthew RW, Moon RT. Chibby, a Nuclear Beta-Catenin-Associated Antagonist of the Wnt/Wingless Pathway. Nature (2003) 422(6934):905–9. doi: 10.1038/nature01570
172. Li FQ, Mofunanya A, Harris K, Takemaru K. Chibby Cooperates With 14-3-3 to Regulate Beta-Catenin Subcellular Distribution and Signaling Activity. J Cell Biol (2008) 181(7):1141–54. doi: 10.1083/jcb.200709091
173. Takemaru K, Fischer V, Li FQ. Fine-Tuning of Nuclear-Catenin by Chibby and 14-3-3. Cell Cycle (2009) 8(2):210–3. doi: 10.4161/cc.8.2.7394
174. Mancini M, Soverini S, Gugliotta G, Santucci MA, Rosti G, Cavo M, et al. Chibby 1: A New Component of Beta-Catenin-Signaling in Chronic Myeloid Leukemia. Oncotarget (2017) 8(50):88244–50. doi: 10.18632/oncotarget.21166
175. Borgo C, D'Amore C, Sarno S, Salvi M, Ruzzene M. Protein Kinase CK2: A Potential Therapeutic Target for Diverse Human Diseases. Signal Transduct Target Ther (2021) 6(1):183. doi: 10.1038/s41392-021-00567-7
176. Song DH, Dominguez I, Mizuno J, Kaut M, Mohr SC, Seldin DC. CK2 Phosphorylation of the Armadillo Repeat Region of Beta-Catenin Potentiates Wnt Signaling. J Biol Chem (2003) 278(26):24018–25. doi: 10.1074/jbc.M212260200
177. Wu H, Symes K, Seldin DC, Dominguez I. Threonine 393 of Beta-Catenin Regulates Interaction With Axin. J Cell Biochem (2009) 108(1):52–63. doi: 10.1002/jcb.22260
178. Bek S, Kemler R. Protein Kinase CKII Regulates the Interaction of Beta-Catenin With Alpha-Catenin and Its Protein Stability. J Cell Sci (2002) 115(Pt 24):4743–53. doi: 10.1242/jcs.00154
179. Youssef I, Ricort JM. Deciphering the Role of Protein Kinase D1 (PKD1) in Cellular Proliferation. Mol Cancer Res (2019) 17(10):1961–74. doi: 10.1158/1541-7786.MCR-19-0125
180. Jaggi M, Rao PS, Smith DJ, Wheelock MJ, Johnson KR, Hemstreet GP, et al. E-Cadherin Phosphorylation by Protein Kinase D1/protein Kinase C{mu} Is Associated With Altered Cellular Aggregation and Motility in Prostate Cancer. Cancer Res (2005) 65(2):483–92. doi: 10.1158/0008-5472.483.65.2
181. Syed V, Mak P, Du C, Balaji KC. Beta-Catenin Mediates Alteration in Cell Proliferation, Motility and Invasion of Prostate Cancer Cells by Differential Expression of E-Cadherin and Protein Kinase D1. J Cell Biochem (2008) 104(1):82–95. doi: 10.1002/jcb.21603
182. Du C, Jaggi M, Zhang C, Balaji KC. Protein Kinase D1-Mediated Phosphorylation and Subcellular Localization of Beta-Catenin. Cancer Res (2009) 69(3):1117–24. doi: 10.1158/0008-5472.CAN-07-6270
183. Aberle H, Schwartz H, Hoschuetzky H, Kemler R. Single Amino Acid Substitutions in Proteins of the Armadillo Gene Family Abolish Their Binding to Alpha-Catenin. J Biol Chem (1996) 271(3):1520–6. doi: 10.1074/jbc.271.3.1520
184. Nickkholgh B, Sittadjody S, Rothberg MB, Fang X, Li K, Chou JW, et al. Beta-Catenin Represses Protein Kinase D1 Gene Expression by Non-Canonical Pathway Through MYC/MAX Transcription Complex in Prostate Cancer. Oncotarget (2017) 8(45):78811–24. doi: 10.18632/oncotarget.20229
185. Rossari F, Minutolo F, Orciuolo E. Past, Present, and Future of Bcr-Abl Inhibitors: From Chemical Development to Clinical Efficacy. J Hematol Oncol (2018) 11(1):84. doi: 10.1186/s13045-018-0624-2
186. Du X, Yang B, An Q, Assaraf YG, Cao X, Xia J. Acquired Resistance to Third-Generation EGFR-TKIs and Emerging Next-Generation EGFR Inhibitors. Innovation (N Y) (2021) 2(2):100103. doi: 10.1016/j.xinn.2021.100103
187. Wang Q, Yang S, Wang K, Sun SY. MET Inhibitors for Targeted Therapy of EGFR TKI-Resistant Lung Cancer. J Hematol Oncol (2019) 12(1):63. doi: 10.1186/s13045-019-0759-9
188. Hong Y. aPKC: The Kinase That Phosphorylates Cell Polarity. F1000Res (2018) 7:903. doi: 10.12688/f1000research.14427.1
189. Jones MC, Zha J, Humphries MJ. Connections Between the Cell Cycle, Cell Adhesion and the Cytoskeleton. Philos Trans R Soc Lond B Biol Sci (2019) 374(1779):20180227. doi: 10.1098/rstb.2018.0227
190. Chen Y, Chen Z, Tang Y, Xiao Q. The Involvement of Noncanonical Wnt Signaling in Cancers. BioMed Pharmacother (2021) 133:110946. doi: 10.1016/j.biopha.2020.110946
Keywords: β-catenin, CTNNB1, GSK3β, adherens junctions, AXIN, CK1, frizzled
Citation: Shah K and Kazi JU (2022) Phosphorylation-Dependent Regulation of WNT/Beta-Catenin Signaling. Front. Oncol. 12:858782. doi: 10.3389/fonc.2022.858782
Received: 20 January 2022; Accepted: 16 February 2022;
Published: 14 March 2022.
Edited by:
Simone Patergnani, University of Ferrara, ItalyReviewed by:
Wioletta Skronska-Wasek, Boehringer Ingelheim, GermanyClaudio Cantù, Linköping University Hospital, Sweden
Simon Söderholm, in collaboration with reviewer CC
Copyright © 2022 Shah and Kazi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julhash U. Kazi, a2F6aS51ZGRpbkBtZWQubHUuc2U=