Abstract
Cancer treatment currently includes a variety of approaches. Chemotherapy, targeted therapy, and immunotherapy are combined based on cancer characteristics to develop personalized treatment plans. However, drug resistance can hinder the progress of treatment over time. Methylglyoxal (MG) is a metabolite with hormesis, exhibiting both pro-tumor and anti-tumor actions depending on its concentration during cancer progression. The MG-related metabolic pathway is being explored in the development of anti-cancer drugs, focusing on reducing MG stress or exploiting its cytotoxic effects to inhibit cancer progression. This article investigates the dual role of MG in cancer, emphasizing its effects on cell metabolism and tumor progression. It proposes MG capture therapy for the pre-cancerous stage and MG toxicity therapy for the cancer stage, contributing to the development of precise and individualized cancer treatments.
InSR, Insulin Receptor; NK cell, natural killer cell.
1 Introduction
According to data from the International Agency for Research on Cancer (IARC), the incidence and mortality of cancer remain remarkably high. Nearly 20 million new cases and approximately 10 million deaths were reported in 2022; this upward trend is projected to persist (1). Despite significant advances in precision medicine and immunotherapy in recent years, cancer remains a substantial global health challenge (2). Critical risk factors, including smoking, obesity, radiation, and infection, can disrupt normal cellular metabolism, leading to an imbalance between metabolite generation and detoxification, which consequently promotes cancer development. At present, the development of traditional cytotoxic drugs has slowed down (3). At the same time, due to the complexity of cancer cell metabolism and compensatory mechanisms, the efficacy of strategies exclusively targeting tumor cell building block synthesis remains limited (4). Therefore, there is an urgent need to explore new therapeutic targets in order to overcome the limitations of conventional treatments and to enhance the clinical efficacy of cancer therapy.
An important metabolic feature of cancer is the Warburg effect, whereby cancer cells rely predominantly on glycolysis for energy even under normoxic conditions. This metabolic reprogramming not only fulfills the energy demands required for the rapid proliferation of cancer cells but also results in the accumulation of numerous toxic by-products (e.g., methylglyoxal [MG], L-2-hydroxyglutaric acid, and glutaryl coenzyme A) (4–6). MG, a highly reactive dicarbonyl compound produced by glycolysis, has been shown to be strongly associated with a variety of metabolic disorders, including cancer, particularly among high-risk groups such as patients with diabetes mellitus, obesity, or advanced age. Persistent accumulation of MG significantly increases cancer risk (7–9). The abnormal accumulation of MG induces genotoxicity, cellular dysfunction, and subsequent disease by forming advanced glycation end products (AGEs) through glycation modifications of biological macromolecules, including DNA and proteins; this phenomenon is termed “MG stress” (10). Additionally, AGEs further exacerbate inflammation and oxidative stress by activating the receptor for advanced glycation end-products (RAGE), thus promoting tumor growth, invasion, and metastasis (11). In recent years, increasing evidence has indicated that the dysregulation of MG metabolism, particularly abnormal changes in expression levels of the key MG detoxification enzyme glyoxalase 1 (GLO1), is closely associated with the development of various malignant tumors (for detailed mechanisms, see Section 5.2.1).
It is worth noting that the effect of MG exhibits a prominent dose-dependent dual effect known as hormesis: at low concentrations, MG can induce malignant transformation of normal cells and promote carcinogenesis; however, at higher concentrations, it exerts significant cytotoxicity, directly inducing apoptosis in cancer cells and inhibiting tumor growth (12). This unique dose-dependent feature provides important clinical insights into cancer treatment, emphasizing the potential therapeutic value of targeting the MG metabolic pathway. On the one hand, for high-risk groups such as individuals with metabolic syndrome, advanced age or diabetes, lowering MG levels using MG scavengers or GLO1 agonists is expected to be an effective cancer prevention strategy; on the other hand, for cancer cells with high glycolytic activity and high GLO1 expression, targeting the MG metabolic pathway to selectively elevate intracellular MG levels beyond the cytotoxic threshold can significantly enhance the sensitivity of cancer cells to chemotherapy, radiotherapy, and even immunotherapy. This strategy not only provides a therapeutic approach for targeting cancer-specific metabolic alterations but also holds promise for overcoming drug resistance and toxic side effects associated with traditional therapies.
Therefore, an in-depth understanding of the dual mechanism of MG metabolism in cancer progression, as well as clarification of the precise applications of targeting MG metabolic pathways in cancer prevention and treatment, is of substantial clinical significance and research importance. The aim of this review is to summarize the latest advances in research regarding MG metabolism and cancer development, to propose specific directions for future investigation and potential clinical strategies, and to provide a novel theoretical basis and practical guidance for precision cancer therapy.
2 The biological processes of MG
2.1 Formation of MG
MG is a highly reactive dicarbonyl compound with cytotoxic properties and has the molecular formula C3H4O2. It was first identified in red blood cells. Several studies have reported on the concentration of MG under physiological conditions, but significant discrepancies exist across the studies. For instance, Rabbani et al. reported the concentration range of MG in mammalian cells as 1–4 mM using stable isotope dilution liquid chromatography-tandem mass spectrometry (SID-LC-MS/MS), whereas the plasma MG level in healthy individuals was measured at 132 ± 63 nM (13). In contrast, plasma MG concentrations in healthy individuals measured by Scheijen et al. were higher, reaching 212 ± 8 nM (14). This significant discrepancy in measurements may stem from variations in assay methodology, sample preparation, instrument sensitivity, and study sample size. Specifically, the higher concentrations reported by Scheijen et al. may be attributed to their relatively small sample sizes or insufficient elimination of interfering factors during the experimental procedure, whereas the study by Rabbani et al. may reflect a more rigorous sample pretreatment and analytical approach. However, few studies in the literature systematically compare different assays, highlighting the urgent need for larger, standardized studies to clarify baseline MG levels across various tissues and physiological states. Furthermore, clarifying the reasons for differences in measurements is essential to elucidate the pathologic links between MG levels and disease such as diabetes and cancer.
The major pathways of MG production include glycolysis (9), glycated protein degradation, lipid, carbohydrate, and protein metabolic processes (15, 16) (Figure 1). Additionally, dietary intake constitutes one of the primary sources of MG in the human body (17). Consequently, plasma MG concentrations are generally higher in diabetic patients than in healthy individuals, with a reported mean value of 277 ± 9 nM (14). Under normal circumstances, the formation of MG accounts for only 0.1 - 0.4% of the glucose flux in glycolysis (18), yet its glycation activity is 10,000 - 50,000 times that of glucose and is regarded as the most potent glycating agent (19). Abnormal accumulation of MG is capable of triggering MG stress, which has been linked to the pathology of various diseases, including aging, diabetic complications, and cancer (10).
Figure 1
2.2 Detoxification of MG
MG, as a cytotoxic metabolite, can cause severe cellular damage if accumulated in excess. In response to this potential threat, specialized intracellular detoxification mechanisms exist to prevent MG accumulation. Detoxification of MG mainly depends on the glyoxalase system. This system was first discovered by Dakin-Dudley and Neuberg et al. in 1913 (20) and consists of GLO1, GLO2, and glutathione (GSH), which are widely present in the cytoplasm of mammalian cells, with GLO2 is also partially distributed in mitochondria (21). The detoxification process of the glyoxalase system depends on GSH: MG first reacts nonenzymatically with GSH to produce a hemisulfide acetal intermediate, which is subsequently converted to S-D-lactoylglutathione catalyzed by GLO1; immediately thereafter, S-D-lactoylglutathione is hydrolyzed to the nontoxic D-lactic acid catalyzed by GLO2 and releases GSH to reenter the next cycle. Ordinarily, the glyoxalase system metabolizes 99% of MG, safeguarding the proteome and genome from damage (22). Therefore, the glyoxalase system plays a vital role throughout the processes of cell development, maturation, aging, and death, and is essential for maintaining normal biological functions (23). Notably, aldehyde dehydrogenases (ALDHs) and aldehyde-keto reductases (AKRs) can convert MG to pyruvate or hydroxyacetone, respectively, undertaking a small amount of MG detoxification (24, 25).
2.3 The glycation reaction of MG
Glycation is a common non-enzymatic biochemical reaction in which the carbonyl group on a reducing sugar molecule reacts with the amino group of a protein, nucleic acid, or lipid to form AGEs, a process also known as the Maillard reaction (26). MG, an important precursor of AGEs, exerts strong pro-oxidative and pro-inflammatory effects, inducing protein and DNA dysfunction through glycation modifications, thereby triggering pathological responses such as cellular dysfunction (7, 10). MG modifies DNA through non-enzymatic reactions, with deoxyguanosine (dG) being the most susceptible nucleotide. Common MG-DNA adducts, including CEdG and MGdG, were identified using SID-LC-MS/MS,. These MG-DNA adducts contribute to genomic instability, exhibit mutagenic potential, and are associated with malignant transformation (27). MG also interacts with proteins by modifying amino acid residues, primarily arginine and lysine, thereby altering their structure and function. The protein adducts induced by MG include MG-H1, AP, and CEL (28, 29). Specific arginine residues, such as Arg-114, Arg-186 and Arg-218, have been identified as key sites for MG modification (30). Amino acid modification by MG significantly alters the structure and function of proteins, which in turn affects cell signaling, including the induction of apoptosis and the regulation of epithelial-mesenchymal transition (EMT) processes. This alteration is closely associated with various pathological processes such as aging, diabetes, cardiovascular disease, and cancer (31, 32). Notably, Aglago et al. measured and analyzed plasma AGEs in 1,378 patients with primary colorectal cancer using ultra-high-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). Their findings revealed a strong association between methylglyoxal-derived AGEs (CEL, MG-H1) and the risk of colorectal cancer (33). The aforementioned studies suggest that MG-mediated glycation plays a crucial role in the pathological process of cancer.
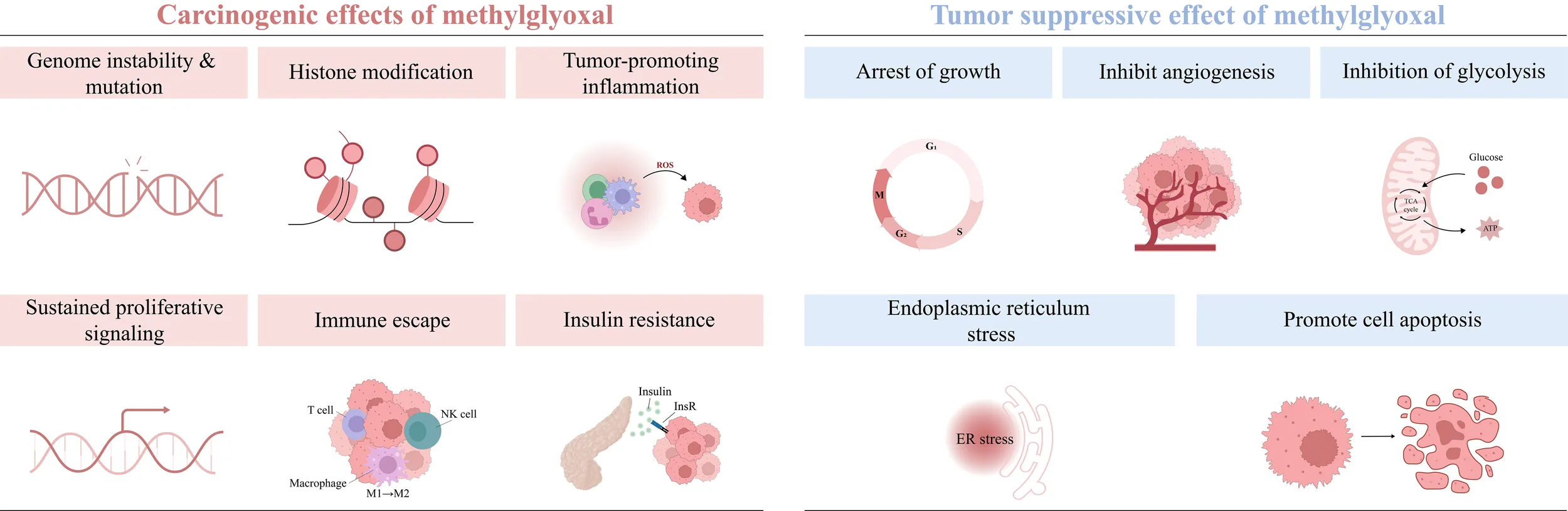
3 MG mediates the occurrence and development of tumors
Tumorigenesis and progression are driven by on complex interactions between cancer cells and their microenvironment. The tumor microenvironment (TME) consists of immune cells, cancer-associated fibroblasts (CAFs), the vascular system, the extracellular matrix (ECM), and a metabolic microenvironment characterized by inflammation and oxidative stress. MG, a highly reactive metabolite, significantly impacts cancer cell signaling and the TME by modifying nucleic acids, proteins, and lipids, thereby promoting tumor development. (Figure 2) (Table 1). Early studies have shown that continuous subcutaneous injection of MG (0.2 mL/dose, 10 mg/mL twice a week for 10 weeks) induced tumor formation in rats, with a tumorigenicity rate of 22.22% (34). In colorectal cancer (CRC) and anaplastic thyroid cancer (ATC) tissues, MG adduct levels adducts are positively correlated with tumor malignancy and metabolic activity, and the oncogenic effects of MG can be effectively reversed by MG scavengers or GLO1 agonists (32, 35). Epidemiological studies further support the association between MG accumulation and increased cancer risk, particularly in populations with metabolic abnormalities such as advanced age, obesity, and hyperglycemia (7, 36, 37).
Figure 2
Table 1
| In vitro | ||||
|---|---|---|---|---|
| Cancer Categories | Cell Types | Processing approach | Laboratory Findings | References |
| ATC | AL62 | MG: 5uM | Migration and invasion capacity↑ | (32) |
| SqCLC | SW1573 | IHC | ▲ AP-Hsp27↑. Caspase↓ ▲ Evade cell apoptosis | (72) |
| NSCLC | NCI-H231 | IHC | (71) | |
| PC | T3M4 | The Gemcitabine Provocation Test | ▲ Glycolysis (GLUT1 and LDH) and MG stress (AP)↑ ▲ Acquiring gemcitabine resistance | (95) |
| MIA-PaCa-2 | MG: 200uM; CEL: 100ug/mL | ▲ YAP↑ ▲ cell proliferation↑ | (75) | |
| BC | MDA-MB-231; MDA-MB-468 | shGLO1; MG: 300/500uM | ▲ MEK/ERK/SMAD1↑ ▲ Migration and invasion capacity↑ | (77) |
| MCF-7 | MG-BSA-AGEs: 50/100 ug/ml | ▲ p-ERK1/2, p-CREB1↑ ▲ Proliferation and migration ability↑ | (78) | |
| MDA-MB-231 | MG-BSA-AGEs: 50/100 ug/ml | ▲ MMP9, p-ERK1/2, p-STAT3↑ ▲ Proliferation, migration, and invasive ability↑ | (79) | |
| MDA-MB-231; MDA-MB-468 | MG: 300uM | ▲ YAP, vimentin↑. E-cadherin↓ ▲ Proliferation and migration ability↑ | (74) | |
| CC | SW480 | AGEs: 100ug/ml | ▲ PI3K/Akt↑ ▲ Cell proliferation, invasion, and EMT↑ | (76) |
| CRC tissue | IHC | The high expression of AP and MG-H1 is positively associated with the malignancy and metabolic activity of the tumor. | (35) | |
| CRC | KRAS^WT-LIM121 | shGLO1 | ▲ MG stress, Akt↑ ▲ Acquiring resistance to cetuximab | (96) |
| CC | HCT116 | AGEs: 50-400mg/l | ▲ ChREBP↑ ▲ Cell proliferation (max: 200mg/l)↑ | (94) |
| HCC | HepG2 | |||
| In vivo | ||||
| Cancer Categories | Model | Processing approach | Laboratory Findings | References |
| BC | mice/chicken embryo membranes | shGLO1-MD-MBA-231 | ▲ Nuclear accumulation of YAP↑ ▲ Tumorigenic and metastatic capacity↑ | (74) |
| mice | shGLO1-MD-MBA-231 | Pulmonary tumor burden↑ | (77) | |
| PC | KC-Mito mice | Carnosine (FL-926-16): 30mg/kg/day, Oral | Mice untreated with carnosine exhibited a higher incidence of pancreatic cancer. | (75) |
The pro-cancer manifestations of MG/AGEs in vivo and in vitro.
SqCLC, squamous cell lung cancer; NSCLC, non-small cell lung cancer; PC, pancreatic carcinoma; BC, breast cancer; HCC, hepatocellular carcinoma; CC, colon cancer; IHC, immunohistochemistry.
↑: High expression/promotion; ↓: Low expression/inhibition.
3.1 Gene mutagenesis
Cancer development typically involves activation of proto-oncogenes and inactivation of tumor suppressor genes, with these alterations are closely associated with DNA damage and mutation accumulation. In addition to traditional oncogenic factors such as radiation, inflammation, and aging, MGs also exhibits significant genotoxicity (38, 39). MG modifies approximately 1% of DNA nucleotides, increasing the frequency of base mismatches, particularly at the G:C site (40, 41), and inducing various forms of DNA damage, such as sister chromatid exchanges (42), DNA adduct formation (43), and strand breaks (27). Additionally, MG induces the degradation of the tumor suppressor protein BRCA2, weakening the cell’s DNA repair capacity and further increases mutation accumulation (44, 45). Despite the cell’s defense mechanisms against DNA glycation, the long-term accumulation of MG eventually exceeds the cell’s repair capacity, causing a significant increase in mutational load. Therefore, studying MG-mediated DNA modification can help to elucidate the mechanism of mutation, which is particularly critical for cancer prevention in individuals with advanced age or metabolic disorders.
3.2 Histone modification
Histone modifications play a central role in maintaining chromosome structure and regulating gene expression, serving as an important modality for epigenetic regulation (46). MG primarily modifies arginine and lysine residues in the N-terminal tail of histones, affecting their structure and function, decreasing nucleosome stability, leading to chromatin loosening, and thus altering gene transcriptional activity (47). Simultaneously, MG modifications can also alter the electrically charged properties of histones, thereby weakening their binding strength to DNA, affecting the DNA-binding efficiency of transcription factors, and activating or inhibiting the expression of genes associated with carcinogenesis (48). Additionally, MG modifications may reduce the interaction efficiency of histones with DNA damage repair proteins, leading to DNA damage accumulation and further increasing the risk of tumorigenesis (49). Moreover, MG modifications may have synergistic or competitive effects with other histone modifications, such as acetylation and methylation, to co-regulate the expression of tumor-associated genes (49, 50). Studies have shown that high levels of histone MG modifications in breast cancer cells are associated with abnormal cell proliferation (51). Additionally, MG-specific modifications of histones may trigger autoimmune responses, making MG-modified histone autoantibodies promising novel immune markers for early diagnosis and prognostic assessment of cancer (52). Thus, MG profoundly impacts epigenetic regulation through histone glycation modification, offering a new dimension of understanding in tumorigenesis and development.
3.3 The cross-linking of MG stress, oxidative stress and chronic inflammation
One of the key features of MG stress is the activation of RAGE, which triggers a series of signaling cascades that drive chronic inflammation and oxidative stress, thereby promoting tumor progression (53).
3.3.1 MG stress and oxidative stress
Imbalance in redox homeostasis is one of the hallmarks of cancer cells and can lead to malignant tumor progression and drug resistance (54). MG activates various transcription factors, such as MAPK, NF-κB, and p-21, through the AGEs/RAGE pathway (55), and promotes NADPH oxidase activation, causing ROS accumulation and oxidative stress (56–58). Furthermore, MG further increases ROS production through RAGE-independent pathways, such as mitochondrial damage (59), inhibition of antioxidant enzymes (60), and non-enzymatic reactions (57), exacerbating oxidative damage and mutation risk. Notably, oxidative stress can drive a shift in cellular metabolic patterns from oxidative phosphorylation (OXPHOS) to glycolysis, a process known as the Warburg effect. For example, ROS cause a decrease in OXPHOS efficiency by affecting mitochondrial DNA (mtDNA), electron transport chain (ETC) complexes (e.g., complexes I and III), and membrane lipids, which prompts the cell to shift to glycolysis to maintain energy metabolism (61). It has been shown in CAF that accumulation of ROS leads to mitochondrial dysfunction and a shift toward glycolytic metabolism, thus creating a nutrient-rich microenvironment to meet the metabolic demands of cancer cells (62). Meanwhile, oxidative stress prompts isolated cancer cells to aggregate and form cell clusters, inducing microenvironmental hypoxia, thereby activating hypoxia-inducible factor 1-α (HIF-1α)-mediated glycolysis (63). The increase in glycolytic activity is accompanied by an increase in MG production, thus forming an oxidative stress-MG stress feedback loop. However, there is no direct evidence to support the establishment of the “oxidative stress-glycolysis-MG stress” loop, but it provides an important direction for future research.
3.3.2 MG stress and chronic inflammation
The role of chronic inflammation in cancer progression is well-established (64). Circulating AGEs levels have been found to correlate with the inflammatory marker C-reactive protein (65). Activation of relevant transcriptional regulators (NF-κb, STAT3, HIF-1α) by the MG/AGEs/RAGE axis promotes increased secretion of inflammation-associated factors (e.g., TNF-α, IL-1, IL-6) (11, 65), as well as the recruitment of inflammatory cells, thereby shaping an inflammatory microenvironment that promotes tumor growth (66). Additionally, foodborne MG also promotes the release of pro-inflammatory factors such as IL-8 and IL-6 (67). In turn, chronic inflammation can also inhibit GLO1 activity, leading to further accumulation of MG, thus creating a vicious cycle that promotes cancer development, thus forming a closed loop. The link between chronic inflammation and cancer driven by MG/RAGE signaling has been discussed in detail (11) and will not be revisited here.
The crosstalk between chronic inflammation and oxidative stress has been well-established (68). In conclusion, the crosstalk between MG stress, oxidative stress and chronic inflammation has become a reality, and MG acts as a bridge to construct the microenvironment of tumorigenesis and development.
3.4 Cellular proliferation and apoptosis
One of the important mechanisms by which MG promotes tumor growth is through the regulation of signaling pathways involved in cell proliferation and apoptosis. MG directly modifies the Cys(77) site of Akt, increasing Akt phosphorylation, and promote cell cycle progression and cell proliferation (69, 70). Additionally, MG inhibits cytochrome c-mediated caspase activation and blocks apoptosis by modifying Hsp27. Studies have shown that MG modification of Hsp27 significantly reduces the sensitivity of tumor cells to chemotherapeutic drugs, a phenomenon closely associated with the activation of the Akt pathway (71–73). Furthermore, MG can inhibit the activity of large tumor suppressor 1 (LATS1) and promote the translocation of YAP to the nucleus by modifying Hsp90, accelerating the proliferation and metastasis of breast cancer cells (74). In a diabetes-associated pancreatic cancer model, MG significantly promotes the development and invasion of KrasG12D/+ pancreatic cancer through activation of ERK1/2 signaling and YAP nuclear translocation (75). In ATC cells, MG similarly promotes TGF-β1 secretion and activates the FAK signaling pathway, thereby enhancing their invasive phenotype (32).
AGEs formed by MG also regulate the proliferation and migration of tumor cells through the RAGE signaling pathway. For example, AGEs can activate the PI3K/Akt signaling pathway, inhibit apoptosis of colon cancer cells, and promote their proliferation and metastasis (76). In breast cancer cells, low-dose AGEs activate ERK1/2 and CREB1 to enhance cancer cell proliferation and migration (77, 78), and similar RAGE-dependent pro-proliferative and migratory effects were observed in the highly invasive breast cancer MDA-MB-231 cell line (79).
In summary, MG plays a key role in cancer cell survival and proliferation by regulating multiple signaling pathways, including Akt, Hsp27, and YAP, and modulating cell proliferation and anti-apoptotic signaling through the AGEs/RAGE pathway.
3.5 Immune escape
The immune system plays a key role in the prevention and treatment of cancer. However, MG can impair the immune response and facilitate immune escape. Natural killer (NK) cells, as important anti-tumor immune cells, are required to kill tumor cells by forming cleavage synaptic clefts to contact tumor cells. However, MG modification of NK cell proteins may interfere with this contact, affecting the recognition and adhesion of NK cells to tumor cells and inhibiting their immunocidal effects on tumor cells without affecting the cells’ own activity (80). Myeloid-derived suppressor cells (MDSCs) are commonly recruited to the tumor microenvironment (81), where they transfer self-produced MG to T cells in a cell-contact-dependent manner, depleting intracellular L-arginine and inducing T-cell paralysis (82). MG also inhibits the expression of CD83, a maturation marker on the surface of dendritic cells (DCs), impeding DC maturation and T cell activation capacity (83); meanwhile, MG induces oxidative damage and apoptosis in human monocytes (84). Furthermore, MG promotes macrophage polarization toward an immunosuppressive M2 phenotype (85, 86), and the above suggests that MG drives cancer cell immune escape by affecting the immune cell’s own function and activity. Notably, MG can also affect the expression of tumor cell-associated immune proteins, thereby interfering with anti-tumor immune effects. For example, MG treatment increased the expression of CD24 protein in breast cancer cells to evade phagocytosis by macrophages (77, 87). Therefore, in the therapeutic strategy, the MG burden is reduced by the MG scavenger N-acetylcysteine (NAC) (88) and genistein attenuates MG stress damage to immune cells (84), which restores anti-tumor immunity and reduces the risk of immune escape.
3.6 Insulin resistance
Hyperinsulinemia, a hallmark of insulin resistance (IR), creates a tumor-friendly environment by promoting cellular proliferation (89). Obesity and metabolic syndrome significantly increase MG accumulation, further exacerbating IR and promotes type 2 diabetes (T2D) (90). The exact mechanism of MG-induced IR is still unclear and may involve the following aspects: disrupting the normal conduction of the insulin signaling pathway (91); decreasing the stability of the insulin molecule itself (92); and directly decreasing the sensitivity of insulin-targeted tissues. These mechanisms interact to form a vicious circle, further exacerbating metabolic abnormalities and ultimately promoting the formation of the tumor microenvironment, suggesting that the MG metabolic pathway is an important potential target for cancer prevention and treatment.
Furthermore, MG/AGEs promote the proliferation, invasion, and migration of tumor cells by cross-linking the extracellular matrix (93) and modifying metabolism (94). To summarize, MG constructs a cancer-promoting microenvironment through multilevel modifications; in-depth study of MG metabolic abnormalities will provide important theoretical and clinical insights for cancer prevention and treatment.
4 MG inhibits the occurrence and development of tumors
In recent years, researchers have identified the dual role of MG in tumors: at lower doses, it induces the malignant transformation of normal cells, resulting in carcinogenesis and invasion; at higher doses, it directly induces cell apoptosis and inhibits tumor growth, a phenomenon known as hormesis (12). MG exerts cytotoxic effects on cancer cells through multiple mechanisms, including inducing cell growth arrest, activating apoptosis/necrosis signals, and inhibiting glycolysis (Figure 3) (Table 2).
Figure 3
Table 2
| In vitro | ||||
|---|---|---|---|---|
| Cancer Categories | Cell Types | Processing approach | Laboratory Findings | References |
| Leukemia | HL60 | MG: 33-524uM IC50 = 238uM | ▲ Inhibit DNA and protein synthesis. ▲ Induce cell growth arrest; ▲ High concentrations induce cell apoptosis. | (31, 101) |
| Jukat | MG: 0.25-0.5mM | ▲ JNK, caspase-3↑ ▲ Cell apoptosis↑ | (114) | |
| CML | Blood | MG: 1/2mM | ▲ Mitochondrial complex I, glycolysis, ATP↓ ▲ Cell viability↓ | (123) |
| EAC | EAC cell | MG: 2/5mM | ▲ Glycolysis, mitochondrial respiratory, ATP↓ ▲ Tumor cell death↑ | (121, 122) |
| MG: 0.2/0.4mM | ▲ DNA and protein synthesis↓ ▲ Cell proliferation↓ | (100) | ||
| EAC | MG: 2/5mM | Glycolysis, mitochondrial respiratory↓ | (120–123) | |
| Leukemia | MG: 1/2mM | |||
| CESC | ||||
| PC | PC-3 | MG: 1-5mM | ▲ Cyclin D1, CDK2, CDK4↓ ▲ Glycolysis, Cell proliferation↓ ▲ Cell cycle arrest (G1), cell apoptosis↑ | (124) |
| LNCaP | MG: 1mM | ▲ NF-kB, Bcl-2, Bcl-XL↓. Bax↑ ▲ Cell apoptosis↑ | (109) | |
| GBM | U87MG/T98G | MG: 25uM | ▲ ATP↓ ▲ Cell cycle arrest (G0), cell apoptosis↑ | (102) |
| BC | MDA-MB-231/4T1 | MG: IC50: 0.6mM/1.0mM | ▲ ROS, caspase, Bax↑ ▲ Bcl-2, p-AKT, nuclear translocation of NF-κB↓ ▲ Cell proliferation↓ Cell apoptosis↑ | (110) |
| MCF-7 | MG: 24h IC50 = 2.7mM 48h IC50 = 1.4mM | Induce mitochondrial functional disorder, suppress the proliferation of tumor cells. | (132) | |
| MCF-7 | MG-BSA-AGEs: 200ug/ml | ▲ caspase-3↑ ▲ Cell proliferation↓ Cell apoptosis↑ | (78) | |
| MCF-7/MDA-MB-231 | MG: 0.4/0.8uM; shGLO1 | ▲ p-JNK, p-p38↑ MMP-9, Bcl-2↓ ▲ Cell proliferation, migration, and invasion↓ ▲ Cell apoptosis↑ | (133) | |
| Osteosarcoma | U266 | MG: 5/10uM+ Cisplatin: 3ug/ml | ▲ ROS, PKCδ↑ ▲ Strengthen cisplatin-induced cell apoptosis. | (113) |
| HCC | Huh-7/HepG2 | MG: 1uM | ▲ Facilitate the nuclear translocation of p53; ▲ Attenuate the migratory, invasive and adhesive capabilities of cancer cells. | (116) |
| RC | CT26 | MG: 200uM + RT 6Gy | ▲ ER stress, cGAS-STING↑ ▲ ROS, Caspase-3, Bax↑ Bcl-2↓ ▲ Enhanced immune cell infiltration within the tumor (CD8+ T cells, NK cells) ▲ Cell apoptosis↑ Tumor volume↓ | (131) |
| CRC | SW480/SW620/DLD-1/HCT-15 | shGLO1 | ▲ STAT1, p53, Bax↑ c-Myc, Bcl-2↓ ▲ Cell apoptosis↑ ▲ Cell proliferation and migration↓ | (134) |
| NSCLC | 3553T3/LGSP | MG: 200uM | ▲ MG-H1↑ ▲ Cell viability↓ | (135) |
| In vivo | ||||
| Cancer Categories | Model | Processing approach | Laboratory Findings | References |
| YOSHIDA ASCITES HEPATOMA | AH-130 mice | MG:12.5,25,50,100mg/kg Intraperitoneal | ▲ Cell proliferation↓ ▲ Cell cycle arrest (S)↑ ▲ Survival rate and survival period of tumor-bearing rats↑ | (99) |
| BC | 4T1 mice | MG:15, 35mg/kg tail vein injection | ▲ Cell apoptosis↑ ▲ Tumor growth↓ | (110) |
| CRC | shGLO1-SW620mice | ▲ Tumor growth↓ | (134) | |
The tumor-suppressive manifestations of MG in vivo and in vitro.
CML, chronic myelocytic leukemia; EAC, Ehrlich ascites carcinoma; CESC, cervical squamous cell carcinoma; GBM, glioblastoma; RC, rectal cancer.
↑: High expression/promotion; ↓: Low expression/inhibition.
4.1 Growth arrest
The anti-cancer potential of MG was first proposed in the 1960s (97). Early studies have shown that intraperitoneal injection of MG in mice inhibits the growth of ascites sarcoma 180 cells but has limited effect on established solid tumors (98). Tessitore et al. found that MG interfered with cell cycle progression, significantly inhibiting the proliferation of rat Yoshida ascites tumor (AH-130) cells, and prolonged the survival of the animals (99). Subsequent studies further confirmed that MG interfered with the growth of Ehrlich ascites tumor cells by inhibiting DNA and protein synthesis (100). MG-induced DNA-DNA polymerase cross-linking severely impedes DNA replication and is one of the key mechanisms for its growth inhibitory effects (43). In vitro studies by Thornalley et al. also found that MG induced G1 phase cell cycle arrest in HL60 leukemia cells (IC50 = 238µM) and further triggered apoptosis (DNA fragmentation) at higher concentrations (524 µM) (31, 101). Similarly, Paul-Samojedny et al. demonstrated that MG induced G0 phase arrest and apoptosis in human glioblastoma cells (102). Notably, the toxicity of MG to tumor cells was positively correlated with the rate of cell proliferation and negatively correlated with the degree of differentiation. These studies suggest that one of the key potentials of MG in tumor therapy lies in its ability to inhibit cell cycle progression.
4.2 Apoptosis
MG accumulation in cancer cells, especially when GLO1 is inhibited, significantly promotes apoptosis in tumor cells (103). The mechanism of MG-induced apoptosis involves DNA damage, oxidative stress, and the activation of apoptotic signaling pathways.
MG-induced DNA damage plays a central role in initiating apoptosis.MG induces DNA strand breaks through the formation of adducts (e.g., MGdG) and cross-linking structures with DNA, activating the intracellular DNA damage response, and triggering apoptosis by cell cycle arrest when the damage exceeds the cellular ability to repair it (27, 104). The presence of a GLO1 inhibitor significantly enhances the MG-induced DNA damage effects, thereby accelerating apoptosis (27).
Apart from DNA damage, MG-induced apoptosis involves oxidative stress and impairment of mitochondrial function. The apoptosis induced by MG involves the activation of the MAPK pathway and caspase-3 (105). MG can increase the level of ROS or modify mitochondrial proteins, such as the mitochondrial permeability transition pore (PTP), damage the membrane potential, and trigger the mitochondrial-dependent apoptotic pathway (105–107). For example, in L929 fibrosarcoma cells, MG-induced apoptosis was dependent on ROS accumulation (108). Similarly, in triple-negative breast cancer (TNBC) cells and prostate cancer cells, MG promotes activation of the mitochondrial apoptotic pathway by upregulating Bax, downregulating Bcl-2, and inhibiting NF-κB activity (109, 110). Notably, Laga et al. observed that MG inhibits NF-κB activation by directly suppressing the binding of NF-κB p65 to DNA (111). Furthermore, MG amplifies oxidative stress damage and enhances the sensitivity of cancer cells to chemotherapeutic drugs (e.g., cisplatin) by activating multiple signaling pathways, including protein kinase Cδ (PKCδ), JNK, and p38 MAPK (112–114). Notably, cancer cells with weaker antioxidant capacity showed greater sensitivity to MG (115). Moreover, MG-induced p53 nuclear translocation has also been shown to significantly inhibit migration and invasion in hepatocellular carcinoma cells (116). Intracellular calcium overload further exacerbates MG-induced cellular homeostasis disruption and promotes apoptosis (117).
4.3 Inhibition of angiogenesis
MG inhibits angiogenesis by disrupting the key molecular interactions required for the formation of new blood vessels in tumors. Specifically, MG modifies the transcriptional coactivator p300 and reduces its ability to bind to HIF-1α, thus limiting the function of HIF-1α to promote angiogenesis under hypoxic conditions and inhibiting nutrient supply to the tumor (118). Furthermore, the modification of extracellular matrix (e.g., type IV collagen) by MG can directly damage endothelial cells, further preventing angiogenesis and even triggering endothelial cell denudation and apoptosis (anoikis), limiting tumor progression (119).
4.4 Inhibition of glycolysis
MG blocks tumor cell energy metabolism by directly inhibiting key enzymes of glycolysis (e.g., GAPDH) and interfering with the function of mitochondrial respiratory chain complex I, leading to energy depletion in cancer cells and triggering apoptosis (120–123). For example, in prostate cancer PC-3 cells, MG leads to cell cycle arrest (G1 phase) and significantly inhibits glycolysis, inducing cancer cell death (124).
4.5 Immune activation
MG plays a complex role in the tumor immune microenvironment, both by promoting immune escape and, in some cases, by stimulating the body’s anti-tumor immune response. For example, MG treatment in mouse tumor models enhances macrophage phagocytosis and increases splenic CD8⁺ and CD4⁺ T-cell counts, thereby elevating immune cytotoxicity (125, 126). Moreover, MG derivative MG-H1 down-regulates PD-L1 expression in tumor cells in metastatic prostate cancer (mPCa), whereas high expression of GLO1 limits the accumulation of MG-H1 and maintains the expression of PD-L1, which inhibits CD8⁺ T cell function and promotes immune escape in mPCa. This mechanism may explain the resistance of mPCa to immune checkpoint inhibitors (e.g., atezolizumab) (127). Thus, inhibition of GLO1 may enhance the efficacy of immunotherapy against PD-L1 high-expressing tumors, providing a potential therapeutic strategy. In conjunction with Section 3.5, it is clear that MG and its derivatives exhibit a complex dual role in tumor immunotherapy, both driving tumor immune escape and enhancing immune response in specific contexts, with the reasons for this difference still needing to be examined. Therefore, a deeper understanding of their specific mechanisms is crucial for optimizing immunotherapeutic strategies in future studies.
4.6 Endoplasmic reticulum stress
MG can modify proteins to make them lose their original structure and function, resulting in the accumulation of misfolded proteins and inducing endoplasmic reticulum (ER) stress response. Glycated proteins lose their normal functions and serve as targets for proteasomal degradation (128). However, covalent modification of the 20S proteasome by MG reduces its degradation activity, leading to persistent activation of the unfolded protein response (UPR), which ultimately triggers apoptosis and inflammatory responses (129, 130). Moreover, MG enhances DNA damage with ROS-induced endoplasmic reticulum stress, which in turn amplifies cGAS-STING pathway activation, leading to greater CD8+T cell and NK cell infiltration in the tumor microenvironment, thereby increasing the radiosensitivity of rectal cancer (131). Therefore, MG may be a potential sensitizer and immunomodulator for tumor radiotherapy.
5 Targeted therapies for MG
In recent years, targeting MG metabolism by developing MG scavengers and GLO1 modulators has emerged as a promising strategy for cancer therapy. Given the dual role of MG in tumor progression, the rational design of therapeutic strategies must take into account the patient’s metabolic status, disease stage, and tumor type. Targeting high-risk groups with elevated MG levels (e.g., the elderly, obese, and diabetic patients) using MG scavengers or lifestyle modifications to reduce chronic MG stress may be effective in reducing cancer risk. Conversely, inhibition of the MG detoxification pathway may be a proven treatment for tumor cells that have developed into cancer and show increased MG detoxification ability.
5.1 MG capturing therapy in the pre-cancerous stage - reducing the risk of cancer
Studies have indicated that MG scavengers such as carnosine can effectively neutralize reactive dicarbonyls and reduce MG stress response (136). In breast cancer cells and ATC cells, MG scavengers (e.g., carnosine, aminoguanidine) as well as GLO1 agonists (e.g., resveratrol) have been shown to reverse the invasive phenotype of cells induced by MG stress (32, 77). In animal models, carnosine has also been found to suppress cancer progression resulting from hyperglycemic conditions, such as pancreatic cancer and breast cancer (74, 75). Furthermore, MG stress is a key driver of drug resistance in cancer cells. For instance, in KRAS-mutant colorectal cancer cells, high glycolytic flux leads to MG accumulation, which activates the Akt survival pathway via the PI3K/mTOR2 axis and Hsp27 regulation, thereby mediating resistance to cetuximab. Specifically, MG-modified Hsp27 is more prone to forming stable functional oligomers with an extended half-life, preventing Akt dephosphorylation and degradation, thereby promoting drug resistance (73). Similar mechanisms are seen in gemcitabine-resistant pancreatic cancer cells, and MG scavengers (metformin, carnosine) are effective in reversing these resistances (95, 96). This suggests that MG scavengers and GLO1 agonists are not only indicated for cancer prevention but may also improve the sensitivity of chemotherapy, especially in high-risk groups with metabolic abnormalities (Table 3).
Table 3
| MG scavenge | Cancer Categories | Model | Dosage | Laboratory Findings | References |
|---|---|---|---|---|---|
| Metformin/aminoguanidine | PC | T3M4 | 10mM | HSF1, HSP27, HSP90↓ Suppress the proliferation of gemcitabine-resistant cells. | (95) |
| Carnosine | MIA PaCa-2 | MG:200uM+Carnosine: 20 mM | ▲ YAP↓ ▲ Cell proliferation↓ | (75) | |
| Carnosine/aminoguanidine | BC | shGLO1-MDA-MB-231/468;MCF7 | 10mM | Decrease the migratory and invasive capabilities induced by MG stress. | (77) |
| Carnosine | shGLO1-MDA-MB-231 mice | 100mg/kg Intraperitoneal | Pulmonary tumor burden and Metastatic foci↓ | ||
| Carnosine | CRC | KRAS^G12V- SW48 mice | Carnosine: 100 mg/kg+ Cetuximab: 0.5mg Intraperitoneal | ▲ PARP, caspase-3↑ ▲ Cell apoptosis↑ Tumor growth↓ | (96) |
| KRAS^G12V- SW48 | Carnosine: 100mM+ Cetuximab:30ug/ml | ||||
| Aminoguanidine/resveratrol | ATC | AL62/8505C | AG: 1mM/ Resveratrol: 50uM | Attenuate the migratory and invasive capacity of cells. | (32) |
The therapeutic role of MG scavengers in cancer.
↑: High expression/promotion; ↓: Low expression/inhibition.
Notably, diet and lifestyle can have a significant impact on the levels of MG and its derived AGEs. Both high-fat and high-sugar diets, as well as smoking, increase the production of AGEs (7, 65). Specific cooking methods (e.g., grilling, deep-frying) also increase the level of AGEs in food (137). Various dietary strategies have been proposed to reduce the load of MG/AGEs. For example, dietary intake of quercetin and genistein was effective in reducing MG levels in plasma and tissues in mice (138, 139). A randomized controlled study that included 540 subjects showed that following a Mediterranean diet based on vegetables, fruits, fish, whole grains, legumes, and olive oil was effective in reducing serum MG and AGEs levels in humans (140). Therefore, the intake of MG/AGEs can be reduced through the following dietary interventions: (I) Reducing the intake of exogenous MG/AGEs, such as avoiding high temperature processing (barbecuing, frying, baking), recommending low temperature cooking methods such as steaming, boiling, stewing, and reducing the intake of foods high in sugar and fat (such as processed foods, red meat, refined carbohydrates); (II) Increasing the intake of MG scavengers and antioxidants, such as chicken, beef and fish rich in carnosine to directly remove MG and inhibit the generation of AGEs (141); Antioxidant foods such as vegetables, fruits, beans and olive oil, which are rich in polyphenols and dietary fiber, can effectively reduce the oxidative stress and inflammatory response induced by MG (142), all of which contribute to reducing the risk of cancer and improving the prognosis of cancer patients.
5.2 MG toxic therapy in the cancer stages - enhancing treatment sensitivity
5.2.1 GLO1 overexpression—protective adaptive mechanisms in cancer cells
Similar to Darwinian evolutionary theory, highly glycolysis-dependent cancer cells have evolved adaptive protective mechanisms under the selective pressure of MG, the most critical of which is the high expression of GLO1 (143). GLO1 overexpression endows cancer cells with stronger MG detoxification ability, prevents them from MG-induced apoptosis, and promotes tumor invasion and development of drug resistance (9, 144) (a discussion of the toxic killing effects of MG on cancer cells can be found in Section 4). Currently, high GLO1 expression has been confirmed in various malignant tumors, including gastric cancer, pancreatic cancer, endometrial cancer, hepatocellular carcinoma, and prostate cancer, and is closely associated with tumor invasion, metastasis, and chemotherapy resistance (143, 145–147). In breast cancer, lung cancer, bladder cancer, and colon cancer, GLO1 gene amplification was significantly associated with poor survival outcomes, suggesting that GLO1 could be used as a biomarker for poor prognosis (148, 149). At the same time, the expression of GLO1 showed variability in different types of the same tumor. For example, proteomic studies of breast cancer revealed that overexpression of GLO1 was significantly associated with the grading of breast cancer (150). In bladder cancer, superficial bladder cancer (SBC) exhibited higher GLO1 activity than invasive bladder cancer (IBC) (151). This finding suggests that GLO1 can be used as a marker to differentiate tumor grading and typing, which in turn improves accuracy and reliability in clinical diagnosis. Therefore, GLO1 is not only suitable as a predictive marker for tumor progression and therapeutic resistance, but also has potential clinical applications: by quantitatively detecting the expression level of GLO1 in cancer tissues or circulating tumor cells, the response or resistance risk of patients to conventional chemotherapeutic drugs can be assessed before clinical treatment, and then optimize personalized treatment plans. In addition, GLO1 may also be an important therapeutic target by inhibiting GLO1 activity and inducing MG toxicity to selectively kill cancer cells, thus providing a new strategy for the treatment of drug-resistant cancers. (Table 4).
Table 4
| GLO1 inhibitor | Cancer Categories | Model | Dosage | Laboratory Findings | References |
|---|---|---|---|---|---|
| BBGC | Leukemia | HL60 | IC50 = 4.23 ± 0.01uM | Suppress cell growth, inhibit DNA synthesis, and induce cell apoptosis. | (103) |
| MOL/CML | UK711/ K562 | 12.5uM | ▲ caspase↑, Cell apoptosis↑ ▲ Augment the therapeutic sensitivity of drug-resistant cells. | (144) | |
| CML | HA-K562/KCL22 | 21.6uM/40.7uM | Cell apoptosis↑ | (154) | |
| HA-K562 mice | 10mg/kg Intraperitoneal | The survival duration of mice↑ | |||
| NB | SH-SY5Y | 10uM | ▲ MG, JNK, p38 MAPK, caspase-3↑ ▲ Cell viability↓ | (166) | |
| CML | HA-CML mice | 10mg/kg, gavage | ▲ Cell apoptosis↑ ▲ The survival duration of mice↑ | (154) | |
| EP | AMOL | THP-1 | IC50 = 1.4 ± 0.2mM | ▲ LDH, ATP↓ ▲ Cell necrosis | (155) |
| Curcumin | BC | MCF-7 | 24h IC50 = 24.5uM 48h IC50 = 11.4uM | ▲ Mitochondrial dysfunction↑ ▲ Cell proliferation↓ | (132) |
| OT55 | HCC | SMMC-7721 | 1.56-50uM | MG, Cell apoptosis↑ | (164) |
| mice | 10mg/kg, Intraperitoneal | ▲ MG, STAT1, p53, Bax↑ ▲ c-Myc, Bcl-2↓ ▲ Cell apoptosis↑ Tumor growth↓ |
The therapeutic effects of GLO1 inhibitors in cancer.
↑: High expression/promotion; ↓: Low expression/inhibition.
5.2.2 Targeting GLO1 therapy—new strategies to improve treatment sensitivity
Therapies targeting GLO1 offer a novel strategy for cancer therapy. Preliminary studies have shown that targeting GLO1 by small molecule inhibitors such as S-p-bromobenzylglutathione cyclopentyl diester (BBGC) significantly inhibits the growth and induces apoptosis of leukemia and other tumor cells, while reversing the resistance of cancer cells to chemotherapeutic drugs (103, 144, 152–154). Furthermore, the competitive GLO1 inhibitor ethyl pyruvate (EP) showed specific antitumor activity in leukemia cell lines, mainly by depleting cancer cell ATP reserves without impairing normal cell function (155). The anticancer effects of methotrexate and troglitazone are also partially dependent on GLO1 activity inhibition, further highlighting the potential of GLO1 as a predictor of treatment response (156, 157).
In breast cancer models, MG induction or GLO1 inhibition can achieve significant anticancer effects by activating the MAPK signaling pathway and decreasing the expression of anti-apoptotic proteins (e.g., Bcl-2, MMP-9) (133). Of interest, GLO1 inhibitors have been shown to reverse the sensitivity of tamoxifen-resistant breast cancer cells, revealing that GLO1 may be a biomarker for predicting the efficacy of endocrine therapies and a target for overcoming treatment resistance (158). Curcumin, as a natural inhibitor of GLO1 activity, inhibits the proliferation of breast cancer cells by inducing mitochondrial dysfunction (132). Moreover, non-pharmacological interventions, like peritumoral electroacupuncture, have also been demonstrated to increase tumor sensitivity to chemotherapeutic agents by inhibiting GLO1, further strengthening the potential of GLO1 as a targeted therapeutic indicator (159).
In lung cancer, inhibition of GLO1 expression activity leads to MG accumulation, which can significantly inhibit tumor growth and induce cell apoptosis (135, 160). Similarly, GLO1 inhibitors have been shown to limit tumor progression and increase the sensitivity of conventional anticancer therapeutic agents in colorectal cancer (134, 161), melanoma, hepatocellular carcinoma (147, 162), and gastric cancer (163). For instance, 3,3’-[3-(5-chloro-2–35 hydroxyphenyl)-3-oxopropane-1,1-diyl]bis(4-hydroxycoumarin) (OT-55) inhibits the GLO1 activity, induces MG accumulation and triggers apoptosis of liver cancer cells (164). Other GLO1 inhibitors, such as BBGC, TER 117, and shikonin, have also shown synergistic chemotherapy sensitization effects when combined with anticancer drugs (165–167).
In summary, targeted GLO1 inhibition therapy can effectively improve the sensitivity of tumor treatment by selectively utilizing the accumulation of MG toxicity in cancer cells, especially showing outstanding value in the treatment of drug-resistant tumors. Meanwhile, as a potential biomarker of prognosis and treatment response, GLO1 can assist clinical decision-making and optimize treatment options, thereby promoting the further development of precision medicine in oncology.
6 Conclusion and perspective
The dual role of MG in cancer reveals its intricate and diverse functions in cellular metabolism and tumor progression. On the one hand, as a toxic by-product of glycolysis, high concentrations of MG exhibit significant anti-tumor effects by inducing apoptosis, DNA damage, energy metabolism disruption, and endoplasmic reticulum stress. On the other hand, at lower concentrations or under long-term chronic exposure conditions, MG promotes cancer cell proliferation, invasion, and metastasis through glycation modification, AGEs-RAGE signaling pathway, and immune microenvironment remodeling, driving tumor progression. This apparent dose-dependent “double-edged sword” characteristic, i.e., hormesis, opens up new research ideas and therapeutic possibilities in the field of tumor metabolism and tumor microenvironment research.
It is important to note that differences in the susceptibility of different cancer cells and tumor subtypes to MG effects may depend on differences in intracellular MG cumulative load, MG detoxification ability (e.g., GLO1 activity), and intracellular redox homeostasis. However, the specific threshold concentration of MG has not yet been clarified, and how different exposure frequencies and durations affect the survival, apoptosis, or transformation potential of tumor cells remains an important question to be addressed. In addition, the performance of MG-regulated dose thresholds in vivo tumor models and clinical patients has not been systematically investigated. Therefore, future studies need to further clarify the following key issues: (I) Determine the safety and toxicity dose thresholds of MG in different cancer types, and clarify at what concentration and exposure time the effect of MG shifts from cancer-promoting to cancer-suppressing, which would provide a precise biological basis for the dose-dependent effect of MG; (II) Systematically assess the variability of GLO1 activity and MG metabolic load in different tumor cell types and tumor microenvironments, and to gain a deeper understanding of how cellular stress induced by MG exposure affects the survival, apoptosis, and invasive capacity of cancer cells, thus providing theoretical support for individualized therapy; (III) Explore the synergistic mechanisms and combined application strategies of MG modulation with existing clinical therapies (e.g., chemotherapy, radiotherapy, immunotherapy), especially in the context of therapeutic drug resistance to clarify the specific mechanisms and clinical significance of MG in overcoming drug resistance; (IV) Elucidate the exact mechanism of MG stress, oxidative stress and chronic inflammation cross-linking network in the construction of tumor microenvironment, and to identify the key nodes of MG-related signaling pathways in the dynamic evolution of the tumor microenvironment, so as to develop effective intervention strategies.
In terms of clinical application, targeting the MG metabolic pathway provides a dual therapeutic strategy: first, for tumors with high glycolytic activity, by inhibiting GLO1 activity to selectively elevate MG levels in cancer cells and enhance the sensitivity of tumor cells to therapy; and second, for high-risk populations (e.g., diabetes, obesity, and the elderly), by reducing MG stress through MG scavengers or GLO1 agonists to prevent cancer progression. However, how to accurately control MG levels to maximize efficacy and minimize toxicity in the course of clinical treatment and cancer prevention remains an important challenge.
In conclusion, the dose-dependent threshold of MG effects, individual variability, and its synergism with cancer therapeutics must be clarified in future cellular, animal model, and clinical studies. This not only has important theoretical significance for understanding the role of MG in tumor biology, but also provides a broad prospect for the development of precise and individualized cancer therapies based on the MG metabolic pathway.
Statements
Author contributions
ZAW: Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing – original draft. SJL: Software, Investigation, Writing – original draft. MHZ: Data curation, Formal analysis, Writing – original draft. ML: Funding acquisition, Methodology, Supervision, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was financially supported by the National Natural Science Foundation of China (82274423).
Acknowledgments
First of all, I would like to give my heartfelt thanks to all the people who have ever helped me in this paper. My sincere and hearty thanks and appreciations go firstly to my supervisor, Dr. Min Liu, whose suggestions and encouragement have given me much insight into these cancer studies. It has been a great privilege and joy to study under his guidance and supervision. I am also extremely grateful to all my friends and classmates who have kindly provided me assistance and companionship in the course of preparing this paper. Additionally, I would like to express my heartfelt gratitude to my family and my wife, Yingxue Zhang, for their selfless love and unwavering support throughout. Finally, I am really grateful to all those who devote much time to reading this thesis and give me much advice, which will benefit me in my later study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
BrayFLaversanneMSungHFerlayJSiegelRLSoerjomataramIet al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74(3):229–63. doi: 10.3322/caac.21834
2
SiegelRLMillerKDJemalA. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
3
ScottECBainesACGongYMooreRPamukGESaberHet al. Trends in the approval of cancer therapies by the FDA in the twenty-first century. Nat Rev Drug Discovery. (2023) 22:625–40. doi: 10.1038/s41573-023-00723-4
4
LeeNSpearsMECarlisleAEKimD. Endogenous toxic metabolites and implications in cancer therapy. Oncogene. (2020) 39:5709–20. doi: 10.1038/s41388-020-01395-9
5
WarburgOWindFNegeleinE. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
6
GatenbyRAGilliesRJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. (2004) 4:891–9. doi: 10.1038/nrc1478
7
LinJ-AWuC-HLuC-CHsiaS-MYenG-C. Glycative stress from advanced glycation end products (AGEs) and dicarbonyls: an emerging biological factor in cancer onset and progression. Mol Nutr Food Res. (2016) 60:1850–64. doi: 10.1002/mnfr.201500759
8
Kold-ChristensenRJohannsenM. Methylglyoxal metabolism and aging-related disease: moving from correlation toward causation. Trends Endocrinol Metab TEM. (2020) 31:81–92. doi: 10.1016/j.tem.2019.10.003
9
ThornalleyPJRabbaniN. Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev Biol. (2011) 22:318–25. doi: 10.1016/j.semcdb.2011.02.006
10
SchalkwijkCGStehouwerCDA. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev. (2020) 100:407–61. doi: 10.1152/physrev.00001.2019
11
RiehlANémethJAngelPHessJ. The receptor RAGE: bridging inflammation and cancer. Cell Commun Signal CCS. (2009) 7:12. doi: 10.1186/1478-811X-7-12
12
AlfaroukKOAlqahtaniSSAlshahraniSMorgensternJSupuranCTReshkinSJ. The possible role of methylglyoxal metabolism in cancer. J Enzyme Inhib Med Chem. (2021) 36:2010–5. doi: 10.1080/14756366.2021.1972994
13
RabbaniNThornalleyPJ. Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat Protoc. (2014) 9:1969–79. doi: 10.1038/nprot.2014.129
14
ScheijenJLJMSchalkwijkCG. Quantification of glyoxal, methylglyoxal and 3-deoxyglucosone in blood and plasma by ultra performance liquid chromatography tandem mass spectrometry: evaluation of blood specimen. Clin Chem Lab Med. (2014) 52:85–91. doi: 10.1515/cclm-2012-0878
15
KalaposMP. Where does plasma methylglyoxal originate from? Diabetes Res Clin Pract. (2013) 99:260–71. doi: 10.1016/j.diabres.2012.11.003
16
LylesGAChalmersJ. The metabolism of aminoacetone to methylglyoxal by semicarbazide-sensitive amine oxidase in human umbilical artery. Biochem Pharmacol. (1992) 43:1409–14. doi: 10.1016/0006-2952(92)90196-p
17
DegenJHellwigMHenleT. 1,2-dicarbonyl compounds in commonly consumed foods. J Agric Food Chem. (2012) 60:7071–9. doi: 10.1021/jf301306g
18
ThornalleyPJ. Glyoxalase I–structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. (2003) 31:1343–8. doi: 10.1042/bst0311343
19
ShinoharaMThornalleyPJGiardinoIBeisswengerPThorpeSROnoratoJet al. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. (1998) 101:1142–7. doi: 10.1172/JCI119885
20
CastorLRGLocatelliKAXimenesVF. Pro-oxidant activity of apocynin radical. Free Radic Biol Med. (2010) 48:1636–43. doi: 10.1016/j.freeradbiomed.2010.03.010
21
HopkinsFGMorganEJ. On the distribution of glyoxalase and glutathione. Biochem J. (1945) 39:320–4. doi: 10.1042/bj0390320
22
RabbaniNThornalleyPJ. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem Soc Trans. (2014) 42:425–32. doi: 10.1042/BST20140018
23
McLellanACThornalleyPJ. Glyoxalase activity in human red blood cells fractioned by age. Mech Ageing Dev. (1989) 48:63–71. doi: 10.1016/0047-6374(89)90026-2
24
BabaSPBarskiOAAhmedYO’TooleTEConklinDJBhatnagarAet al. Reductive metabolism of AGE precursors: A metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes. (2009) 58:2486–97. doi: 10.2337/db09-0375
25
RabbaniNThornalleyPJ. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. (2012) 42:1133–42. doi: 10.1007/s00726-010-0783-0
26
ShenC-YLuC-HWuC-HLiK-JKuoY-MHsiehS-Cet al. The development of maillard reaction, and advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with AGE-related diseases. Mol Basel Switz. (2020) 25:5591. doi: 10.3390/molecules25235591
27
ThornalleyPJWarisSFlemingTSantariusTLarkinSJWinklhofer-RoobBMet al. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. (2010) 38:5432–42. doi: 10.1093/nar/gkq306
28
SchleicherEDWagnerENerlichAG. Increased accumulation of the glycoxidation product N(Epsilon)-(Carboxymethyl)Lysine in human tissues in diabetes and aging. J Clin Invest. (1997) 99:457–68. doi: 10.1172/JCI119180
29
MaessenDEMStehouwerCDASchalkwijkCG. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin Sci Lond Engl 1979. (2015) 128:839–61. doi: 10.1042/CS20140683
30
AhmedNThornalleyPJ. Peptide mapping of human serum albumin modified minimally by methylglyoxal in vitro and in vivo. Ann N Y Acad Sci. (2005) 1043:260–6. doi: 10.1196/annals.1333.031
31
KangYEdwardsLGThornalleyPJ. Effect of methylglyoxal on human leukaemia 60 cell growth: modification of DNA G1 growth arrest and induction of apoptosis. Leuk Res. (1996) 20:397–405. doi: 10.1016/0145-2126(95)00162-x
32
AntognelliCMorettiSFrosiniRPuxedduESidoniATalesaVN. Methylglyoxal acts as a tumor-promoting factor in anaplastic thyroid cancer. Cells. (2019) 8:547. doi: 10.3390/cells8060547
33
AglagoEKSchalkwijkCGFreislingHFedirkoVHughesDJJiaoLet al. Plasma concentrations of advanced glycation end-products and colorectal cancer risk in the EPIC study. Carcinogenesis. (2021) 42:705–13. doi: 10.1093/carcin/bgab026
34
NagaoMFujitaYWakabayashiKNukayaHKosugeTSugimuraT. Mutagens in coffee and other beverages. Environ Health Perspect. (1986) 67:89–91. doi: 10.1289/ehp.866789
35
ChiavarinaBNokinM-JBellierJDurieuxFBletardNShererFet al. Methylglyoxal-mediated stress correlates with high metabolic activity and promotes tumor growth in colorectal cancer. Int J Mol Sci. (2017) 18:213. doi: 10.3390/ijms18010213
36
BorugianMJShepsSBWhittemoreASWuAHPotterJDGallagherRP. Carbohydrates and colorectal cancer risk among Chinese in North America. Cancer Epidemiol biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. (2002) 11:187–93.
37
GiovannucciEHarlanDMArcherMCBergenstalRMGapsturSMHabelLAet al. Diabetes and cancer: A consensus report. Diabetes Care. (2010) 33:1674–85. doi: 10.2337/dc10-0666
38
RobertsMJWondrakGTLaureanDCJacobsonMKJacobsonEL. DNA damage by carbonyl stress in human skin cells. Mutat Res. (2003) 522:45–56. doi: 10.1016/s0027-5107(02)00232-4
39
PapoulisAal-AbedYBucalaR. Identification of N2-(1-Carboxyethyl)Guanine (CEG) as a guanine advanced glycosylation end product. Biochemistry. (1995) 34:648–55. doi: 10.1021/bi00002a032
40
ThornalleyPJ. Protecting the genome: defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem Soc Trans. (2003) 31:1372–7. doi: 10.1042/bst0311372
41
YuanBCaoHJiangYHongHWangY. Efficient and accurate bypass of N2-(1-carboxyethyl)-2’-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc Natl Acad Sci U S A. (2008) 105:8679–84. doi: 10.1073/pnas.0711546105
42
FagginPBassiAMFinolloRBrambillaG. Induction of sister-chromatid exchanges in chinese hamster ovary cells by the biotic ketoaldehyde methylglyoxal. Mutat Res. (1985) 144:189–91. doi: 10.1016/0165-7992(85)90138-1
43
Murata-KamiyaNKamiyaH. Methylglyoxal, an endogenous aldehyde, crosslinks DNA polymerase and the substrate DNA. Nucleic Acids Res. (2001) 29:3433–8. doi: 10.1093/nar/29.16.3433
44
JiangP. A glycolytic metabolite that drives BRCA2 haploinsufficiency. Cell. (2024) 187:2124–6. doi: 10.1016/j.cell.2024.03.028
45
KongLRGuptaKWuAJPereraDIvanyi-NagyRAhmedSMet al. A glycolytic metabolite bypasses “Two-hit” Tumor suppression by BRCA2. Cell. (2024) 187:2269–87.e16. doi: 10.1016/j.cell.2024.03.006
46
ThompsonLLGuppyBJSawchukLDavieJRMcManusKJ. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. (2013) 32:363–76. doi: 10.1007/s10555-013-9434-8
47
KnörleinAXiaoYDavidY. Leveraging histone glycation for cancer diagnostics and therapeutics. Trends Cancer. (2023) 9:410–20. doi: 10.1016/j.trecan.2023.01.005
48
Millán-ZambranoGBurtonABannisterAJSchneiderR. Histone post-translational modifications — Cause and consequence of genome function. Nat Rev Genet. (2022) 23:563–80. doi: 10.1038/s41576-022-00468-7
49
KimJJLeeSYMillerKM. Preserving genome integrity and function: the DNA damage response and histone modifications. Crit Rev Biochem Mol Biol. (2019) 54:208–41. doi: 10.1080/10409238.2019.1620676
50
DubeGTiamiouABizetMBoumahdYGasmiICrakeRet al. Methylglyoxal: A novel upstream regulator of DNA methylation. J Exp Clin Cancer Res CR. (2023) 42:78. doi: 10.1186/s13046-023-02637-w
51
ZhengQOmansNDLeicherROsunsadeAAgustinusASFinkin-GronerEet al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat Commun. (2019) 10:1289. doi: 10.1038/s41467-019-09192-z
52
MirARUddinMKhanFAlamKAliA. Dicarbonyl induced structural perturbations make histone H1 highly immunogenic and generate an auto-immune response in cancer. PLoS One. (2015) 10:e0136197. doi: 10.1371/journal.pone.0136197
53
RabbaniNXueMThornalleyPJ. Methylglyoxal-induced dicarbonyl stress in aging and disease: first steps towards glyoxalase 1-based treatments. Clin Sci Lond Engl 1979. (2016) 130:1677–96. doi: 10.1042/CS20160025
54
WeinbergFChandelNS. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol Life Sci CMLS. (2009) 66:3663–73. doi: 10.1007/s00018-009-0099-y
55
LanderHMTaurasJMOgisteJSHoriOMossRASchmidtAM. Activation of the receptor for advanced glycation end products triggers a P21(Ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. (1997) 272:17810–4. doi: 10.1074/jbc.272.28.17810
56
HayyanMHashimMAAlNashefIM. Superoxide ion: generation and chemical implications. Chem Rev. (2016) 116:3029–85. doi: 10.1021/acs.chemrev.5b00407
57
NakayamaMSaitoKSatoENakayamaKTerawakiHItoSet al. Radical generation by the non-enzymatic reaction of methylglyoxal and hydrogen peroxide. Redox Rep Commun Free Radic Res. (2007) 12:125–33. doi: 10.1179/135100007X200182
58
WautierMPChappeyOCordaSSternDMSchmidtAMWautierJL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. (2001) 280:E685–694. doi: 10.1152/ajpendo.2001.280.5.E685
59
RoscaMGMustataTGKinterMTOzdemirAMKernTSSzwedaLIet al. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol. (2005) 289:F420–430. doi: 10.1152/ajprenal.00415.2004
60
MorganPEDeanRTDaviesMJ. Inactivation of cellular enzymes by carbonyls and protein-bound glycation/glycoxidation products. Arch Biochem Biophys. (2002) 403:259–69. doi: 10.1016/s0003-9861(02)00222-9
61
MurphyMP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
62
PavlidesSVeraIGandaraRSneddonSPestellRGMercierIet al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. (2012) 16:1264–84. doi: 10.1089/ars.2011.4243
63
LabuschagneCFCheungECBlagihJDomartM-CVousdenKH. Cell clustering promotes a metabolic switch that supports metastatic colonization. Cell Metab. (2019) 30:720–34.e5. doi: 10.1016/j.cmet.2019.07.014
64
BalkwillFMantovaniA. Inflammation and cancer: back to Virchow? Lancet Lond Engl. (2001) 357:539–45. doi: 10.1016/S0140-6736(00)04046-0
65
TurnerDP. Advanced glycation end-products: A biological consequence of lifestyle contributing to cancer disparity. Cancer Res. (2015) 75:1925–9. doi: 10.1158/0008-5472.CAN-15-0169
66
MiyataTInagiRIidaYSatoMYamadaNOdaOet al. Involvement of beta 2-microglobulin modified with advanced glycation end products in the pathogenesis of hemodialysis-associated amyloidosis. Induction of human monocyte chemotaxis and macrophage secretion of tumor necrosis factor-alpha and interleukin-1. J Clin Invest. (1994) 93:521–8. doi: 10.1172/JCI117002
67
KuntzSRudloffSEhlJBretzelRGKunzC. Food derived carbonyl compounds affect basal and stimulated secretion of interleukin-6 and -8 in caco-2 cells. Eur J Nutr. (2009) 48:499–503. doi: 10.1007/s00394-009-0035-9
68
ReuterSGuptaSCChaturvediMMAggarwalBB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. (2010) 49:1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006
69
ChangTWangROlsonDJHMousseauDDRossARSWuL. Modification of akt1 by methylglyoxal promotes the proliferation of vascular smooth muscle cells. FASEB J Off Publ Fed Am Soc Exp Biol. (2011) 25:1746–57. doi: 10.1096/fj.10-178053
70
JiaXChangTWilsonTWWuL. Methylglyoxal mediates adipocyte proliferation by increasing phosphorylation of Akt1. PLoS One. (2012) 7:e36610. doi: 10.1371/journal.pone.0036610
71
SakamotoHMashimaTYamamotoKTsuruoT. Modulation of heat-shock protein 27 (Hsp27) anti-apoptotic activity by methylglyoxal modification. J Biol Chem. (2002) 277:45770–5. doi: 10.1074/jbc.M207485200
72
van HeijstJWJNiessenHWMMustersRJvan HinsberghVWMHoekmanKSchalkwijkCG. Argpyrimidine-modified heat shock protein 27 in human non-small cell lung cancer: A possible mechanism for evasion of apoptosis. Cancer Lett. (2006) 241:309–19. doi: 10.1016/j.canlet.2005.10.042
73
MearowKMDodgeMERahimtulaMYegappanC. Stress-mediated signaling in PC12 cells - the role of the small heat shock protein, Hsp27, and Akt in protecting cells from heat stress and nerve growth factor withdrawal. J Neurochem. (2002) 83:452–62. doi: 10.1046/j.1471-4159.2002.01151.x
74
NokinM-JDurieuxFPeixotoPChiavarinaBPeulenOBlommeAet al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife. (2016) 5:e19375. doi: 10.7554/eLife.19375
75
MeniniSIacobiniCde LatouliereLManniIVitaleMPilozziEet al. Diabetes promotes invasive pancreatic cancer by increasing systemic and tumour carbonyl stress in KrasG12D/+ Mice. J Exp Clin Cancer Res CR. (2020) 39:152. doi: 10.1186/s13046-020-01665-0
76
LiangH. Advanced glycation end products induce proliferation, invasion and epithelial-mesenchymal transition of human SW480 colon cancer cells through the PI3K/AKT signaling pathway. Oncol Lett. (2020) 19:3215–22. doi: 10.3892/ol.2020.11413
77
NokinM-JBellierJDurieuxFPeulenORademakerGGabrielMet al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res BCR. (2019) 21:11. doi: 10.1186/s13058-018-1095-7
78
Matou-NasriSSharafHWangQAlmobadelNRabhanZAl-EidiHet al. Biological impact of advanced glycation endproducts on estrogen receptor-positive MCF-7 breast cancer cells. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:2808–20. doi: 10.1016/j.bbadis.2017.07.011
79
SharafHMatou-NasriSWangQRabhanZAl-EidiHAl AbdulrahmanAet al. Advanced glycation endproducts increase proliferation, migration and invasion of the breast cancer cell line MDA-MB-231. Biochim Biophys Acta. (2015) 1852:429–41. doi: 10.1016/j.bbadis.2014.12.009
80
RosenstockPBezoldVBorkKSchefflerJHorstkorteR. Glycation interferes with natural killer cell function. Mech Ageing Dev. (2019) 178:64–71. doi: 10.1016/j.mad.2019.01.006
81
TcyganovEMastioJChenEGabrilovichDI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol. (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
82
BaumannTDunkelASchmidCSchmittSHiltenspergerMLohrKet al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol. (2020) 21:555–66. doi: 10.1038/s41590-020-0666-9
83
PriceCLSharpPSNorthMERainbowSJKnightSC. Advanced glycation end products modulate the maturation and function of peripheral blood dendritic cells. Diabetes. (2004) 53:1452–8. doi: 10.2337/diabetes.53.6.1452
84
WuH-JChanW-H. Genistein protects methylglyoxal-induced oxidative DNA damage and cell injury in human mononuclear cells. Toxicol Vitro Int J Publ Assoc BIBRA. (2007) 21:335–42. doi: 10.1016/j.tiv.2006.09.002
85
MaHDingZXieYLiLLiDLouKet al. Methylglyoxal produced by tumor cells through formaldehyde-enhanced Warburg effect potentiated polarization of tumor-associated macrophages. Toxicol Appl Pharmacol. (2022) 438:115910. doi: 10.1016/j.taap.2022.115910
86
ZambreSBangarNMistryAKatarmalPKhanMSAhmedIet al. Aldosterone, methylglyoxal, and glycated albumin interaction with macrophage cells affects their viability, activation, and differentiation. ACS Omega. (2024) 9:11848–59. doi: 10.1021/acsomega.3c09420
87
BaumannPCremersNKroeseFOrendGChiquet-EhrismannRUedeTet al. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res. (2005) 65:10783–93. doi: 10.1158/0008-5472.CAN-05-0619
88
PriceCLHassiHOSAEnglishNRBlakemoreAIFStaggAJKnightSC. Methylglyoxal modulates immune responses: relevance to diabetes. J Cell Mol Med. (2010) 14:1806–15. doi: 10.1111/j.1582-4934.2009.00803.x
89
GallagherEJLeRoithD. Hyperinsulinaemia in cancer. Nat Rev Cancer. (2020) 20:629–44. doi: 10.1038/s41568-020-0295-5
90
MirandaERHausJM. Glyoxalase I is a novel target for the prevention of metabolic derangement. Pharmacol Ther. (2023) 250:108524. doi: 10.1016/j.pharmthera.2023.108524
91
NgS-PNomuraWTakahashiHInoueKKawadaTGotoTet al. Methylglyoxal induces multiple serine phosphorylation in insulin receptor substrate 1 via the TAK1-P38-mTORC1 signaling axis in adipocytes. Biochem J. (2022) 479:2279–96. doi: 10.1042/BCJ20220271
92
JiaXOlsonDJHRossARSWuL. Structural and functional changes in human insulin induced by methylglyoxal. FASEB J Off Publ Fed Am Soc Exp Biol. (2006) 20:1555–7. doi: 10.1096/fj.05-5478fje
93
Rodriguez-TejaMGronauJHBreitCZhangYZMinamidateACaleyMPet al. AGE-modified basement membrane cooperates with endo180 to promote epithelial cell invasiveness and decrease prostate cancer survival. J Pathol. (2015) 235:581–92. doi: 10.1002/path.4485
94
ChenHWuLLiYMengJLinNYangDet al. Advanced glycation end products increase carbohydrate responsive element binding protein expression and promote cancer cell proliferation. Mol Cell Endocrinol. (2014) 395:69–78. doi: 10.1016/j.mce.2014.07.021
95
CrakeRGasmiIDehayeJLardinoisFPeifferRMaloujahmoumNet al. Resistance to gemcitabine in pancreatic cancer is connected to methylglyoxal stress and heat shock response. Cells. (2023) 12:1414. doi: 10.3390/cells12101414
96
BellierJNokinM-JCaprasseMTiamiouABlommeAScheijenJLet al. Methylglyoxal scavengers resensitize KRAS-mutated colorectal tumors to cetuximab. Cell Rep. (2020) 30:1400–16.e6. doi: 10.1016/j.celrep.2020.01.012
97
FrenchFAFreedlanderBL. Carcinostatic action of polycarbonyl compounds and their derivatives. I. 3-ethoxy-2-ketobutyraldehyde and related compounds. Cancer Res. (1958) 18:172–5.
98
EgyüdLGSzent-GyörgyiA. Cancerostatic action of methylglyoxal. Science. (1968) 160:1140. doi: 10.1126/science.160.3832.1140
99
TessitoreLBonelliGCostelliPMateraLPileriABaccinoFMet al. Effect of two aliphatic aldehydes, methylglyoxal and 4-hydroxypentenal, on the growth of yoshida ascites hepatoma AH-130. Chem Biol Interact. (1989) 70:227–40. doi: 10.1016/0009-2797(89)90046-x
100
ReiffenKASchneiderFA. Comparative study on proliferation, macromolecular synthesis and energy metabolism of in vitro-grown Ehrlich ascites tumor cells in the presence of glucosone, galactosone and methylglyoxal. J Cancer Res Clin Oncol. (1984) 107:206–10. doi: 10.1007/BF01032608
101
AyoubFMAllenREThornalleyPJ. Inhibition of proliferation of human leukaemia 60 cells by methylglyoxal in vitro. Leuk Res. (1993) 17:397–401. doi: 10.1016/0145-2126(93)90094-2
102
Paul-SamojednyMŁasutBPudełkoAFila-DaniłowAKowalczykMSuchanek-RaifRet al. Methylglyoxal (MGO) inhibits proliferation and induces cell death of human glioblastoma multiforme T98G and U87MG cells. Biomed Pharmacother Biomed Pharmacother. (2016) 80:236–43. doi: 10.1016/j.biopha.2016.03.021
103
ThornalleyPJEdwardsLGKangYWyattCDaviesNLadanMJet al. Antitumour activity of S-p-bromobenzylglutathione cyclopentyl diester in vitro and in vivo. Inhibition of glyoxalase I and induction of apoptosis. Biochem Pharmacol. (1996) 51:1365–72. doi: 10.1016/0006-2952(96)00059-7
104
RabbaniNXueMWeickertMOThornalleyPJ. Multiple roles of glyoxalase 1-mediated suppression of methylglyoxal glycation in cancer biology-involvement in tumour suppression, tumour growth, multidrug resistance and target for chemotherapy. Semin Cancer Biol. (2018) 49:83–93. doi: 10.1016/j.semcancer.2017.05.006
105
ChanW-HWuH-JShiaoN-H. Metabolic targets for cancer therapy. J Cell Biochem. (2007) 100:1056–69. doi: 10.1002/jcb.21114
106
GalluzziLKeppOVander HeidenMGKroemerG. Metabolic targets for cancer therapy. Nat Rev Drug Discovery. (2013) 12:829–46. doi: 10.1038/nrd4145
107
SpeerOMorkunaite-HaimiSLiobikasJFranckMHensboLLinderMDet al. Rapid suppression of mitochondrial permeability transition by methylglyoxal. Role of reversible arginine modification. J Biol Chem. (2003) 278:34757–63. doi: 10.1074/jbc.M301990200
108
Van HerrewegheFMaoJChaplenFWRGrootenJGevaertKVandekerckhoveJet al. Tumor necrosis factor-induced modulation of glyoxalase I activities through phosphorylation by PKA results in cell death and is accompanied by the formation of a specific methylglyoxal-derived AGE. Proc Natl Acad Sci U S A. (2002) 99:949–54. doi: 10.1073/pnas.012432399
109
AntognelliCMezzasomaLFettucciariKTalesaVNA. Novel mechanism of methylglyoxal cytotoxicity in prostate cancer cells. Int J Biochem Cell Biol. (2013) 45:836–44. doi: 10.1016/j.biocel.2013.01.003
110
RoyAAhirMBhattacharyaSParidaPKAdhikaryAJanaKet al. Induction of mitochondrial apoptotic pathway in triple negative breast carcinoma cells by methylglyoxal via generation of reactive oxygen species. Mol Carcinog. (2017) 56:2086–103. doi: 10.1002/mc.22665
111
LagaMCottynAVan HerrewegheFVanden BergheWHaegemanGVan OostveldtPet al. Methylglyoxal suppresses TNF-alpha-induced NF-kappaB activation by inhibiting NF-kappaB DNA-binding. Biochem Pharmacol. (2007) 74:579–89. doi: 10.1016/j.bcp.2007.05.026
112
LiuB-FMiyataSHirotaYHigoSMiyazakiHFukunagaMet al. Methylglyoxal induces apoptosis through activation of P38 mitogen-activated protein kinase in rat mesangial cells. Kidney Int. (2003) 63:947–57. doi: 10.1046/j.1523-1755.2003.00829.x
113
GodboutJPPesaventoJHartmanMEMansonSRFreundGG. Methylglyoxal enhances cisplatin-induced cytotoxicity by activating protein kinase cdelta. J Biol Chem. (2002) 277:2554–61. doi: 10.1074/jbc.M100385200
114
DuJSuzukiHNagaseFAkhandAAYokoyamaTMiyataTet al. Methylglyoxal induces apoptosis in Jurkat leukemia T cells by activating C-jun N-terminal kinase. J Cell Biochem. (2000) 77:333–44. doi: 10.1002/(sici)1097-4644(20000501)77:2<333::aid-jcb15>3.0.co;2-q
115
AmicarelliFColafarinaSCattaniFCiminiADi IlioCCeruMPet al. Scavenging system efficiency is crucial for cell resistance to ROS-mediated methylglyoxal injury. Free Radic Biol Med. (2003) 35:856–71. doi: 10.1016/s0891-5849(03)00438-6
116
LoarcaLSassi-GahaSArtlettCM. Two α-dicarbonyls downregulate migration, invasion, and adhesion of liver cancer cells in a P53-dependent manner. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver. (2013) 45:938–46. doi: 10.1016/j.dld.2013.05.005
117
CookLJDaviesJYatesAPElliottACLovellJJouleJAet al. Effects of methylglyoxal on rat pancreatic beta-cells. Biochem Pharmacol. (1998) 55:1361–7. doi: 10.1016/s0006-2952(97)00619-9
118
ThangarajahHYaoDChangEIShiYJazayeriLVialINet al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc Natl Acad Sci U S A. (2009) 106:13505–10. doi: 10.1073/pnas.0906670106
119
DoblerDAhmedNSongLEboigbodinKEThornalleyPJ. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes. (2006) 55:1961–9. doi: 10.2337/db05-1634
120
R.MH.JD.SkR.S. Inhibition of respiration of tumor cells by methylglyoxal and protection of inhibition by lactaldehyde. Int J Cancer. (1991) 47(4):603–9. doi: 10.1002/ijc.2910470421
121
HalderJRayMRayS. Inhibition of glycolysis and mitochondrial respiration of Ehrlich ascites carcinoma cells by methylglyoxal. Int J Cancer. (1993) 54:443–9. doi: 10.1002/ijc.2910540315
122
RaySDuttaSHalderJRayM. Inhibition of electron flow through complex I of the mitochondrial respiratory chain of Ehrlich ascites carcinoma cells by methylglyoxal. Biochem J. (1994) 303:69–72. doi: 10.1042/bj3030069
123
BiswasSRayMMisraSDuttaDPRayS. Selective inhibition of mitochondrial respiration and glycolysis in human leukaemic leucocytes by methylglyoxal. Biochem J. (1997) 323:343–8. doi: 10.1042/bj3230343
124
MilanesaDMChoudhuryMSMallouhCTazakiHKonnoS. Methylglyoxal-induced apoptosis in human prostate carcinoma: potential modality for prostate cancer treatment. Eur Urol. (2000) 37:728–34. doi: 10.1159/000020226
125
PalABhattacharyaIBhattacharyaKMandalCRayM. Methylglyoxal induced activation of murine peritoneal macrophages and surface markers of T lymphocytes in sarcoma-180 bearing mice: involvement of MAP kinase, NF-kappa beta signal transduction pathway. Mol Immunol. (2009) 46:2039–44. doi: 10.1016/j.molimm.2009.03.014
126
BhattacharyyaNPalAPatraSHaldarAKRoySRayM. Activation of macrophages and lymphocytes by methylglyoxal against tumor cells in the host. Int Immunopharmacol. (2008) 8:1503–12. doi: 10.1016/j.intimp.2008.06.005
127
AntognelliCMandaranoMProsperiESidoniATalesaVN. Glyoxalase-1-dependent methylglyoxal depletion sustains PD-L1 expression in metastatic prostate cancer cells: A novel mechanism in cancer immunosurveillance escape and a potential novel target to overcome PD-L1 blockade resistance. Cancers. (2021) 13:2965. doi: 10.3390/cancers13122965
128
RabbaniNXueMThornalleyPJ. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj J. (2016) 33:513–25. doi: 10.1007/s10719-016-9705-z
129
XueMIrshadZRabbaniNThornalleyPJ. Increased cellular protein modification by methylglyoxal activates endoplasmic reticulum-based sensors of the unfolded protein response. Redox Biol. (2024) 69:103025. doi: 10.1016/j.redox.2024.103025
130
QueisserMAYaoDGeislerSHammesH-PLochnitGSchleicherEDet al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. (2010) 59:670–8. doi: 10.2337/db08-1565
131
ZhouHWangLLinZJiangCChenXWangKet al. Methylglyoxal from gut microbes boosts radiosensitivity and radioimmunotherapy in rectal cancer by triggering endoplasmic reticulum stress and cGAS-STING activation. J Immunother Cancer. (2023) 11:e007840. doi: 10.1136/jitc-2023-007840
132
S.BF.CA.P.ClS.JlFE. Resveratrol, curcumin and piperine alter human glyoxalase 1 in MCF-7 breast cancer cells. Int J Mol Sci. (2020) 21(15):5244. doi: 10.3390/ijms21155244
133
GuoYZhangYYangXLuPYanXXiaoFet al. Effects of methylglyoxal and glyoxalase I inhibition on breast cancer cells proliferation, invasion, and apoptosis through modulation of MAPKs, MMP9, and Bcl-2. Cancer Biol Ther. (2016) 17:169–80. doi: 10.1080/15384047.2015.1121346
134
ChenYFangLZhangJLiGMaMLiCet al. Blockage of Glyoxalase I Inhibits Colorectal Tumorigenesis and Tumor Growth via Upregulation of STAT1, P53, and Bax and Downregulation of c-Myc and Bcl-2. Int J Mol Sci. (2017) 18:570. doi: 10.3390/ijms18030570
135
LuengoAAbbottKLDavidsonSMHosiosAMFaubertBChanSHet al. Reactive metabolite production is a targetable liability of glycolytic metabolism in lung cancer. Nat Commun. (2019) 10:5604. doi: 10.1038/s41467-019-13419-4
136
HipkissAR. Glycotoxins: dietary and metabolic origins; possible amelioration of neurotoxicity by Carnosine, with special reference to Parkinson’s disease. Neurotox Res. (2018) 34:164–72. doi: 10.1007/s12640-018-9867-5
137
UribarriJWoodruffSGoodmanSCaiWChenXPyzikRet al. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. (2010) 110:911–16.e12. doi: 10.1016/j.jada.2010.03.018
138
ZhaoYZhuYWangPSangS. Dietary genistein reduces methylglyoxal and advanced glycation end product accumulation in obese mice treated with high-fat diet. J Agric Food Chem. (2020) 68:7416–24. doi: 10.1021/acs.jafc.0c03286
139
ZhaoYTangYSangS. Dietary quercetin reduces plasma and tissue methylglyoxal and advanced glycation end products in healthy mice treated with methylglyoxal. J Nutr. (2021) 151:2601–9. doi: 10.1093/jn/nxab176
140
Gutierrez-MariscalFMPodadera-HerrerosAAlcalá-DiazJFCardeloMPArenas-de LarrivaAPde la Cruz-AresSet al. Reduction of circulating methylglyoxal levels by a mediterranean diet is associated with preserved kidney function in patients with type 2 diabetes and coronary heart disease: from the CORDIOPREV randomized controlled trial. Diabetes Metab. (2024) 50:101503. doi: 10.1016/j.diabet.2023.101503
141
HipkissAR. Glycation, ageing and carnosine: are carnivorous diets beneficial? Mech Ageing Dev. (2005) 126:1034–9. doi: 10.1016/j.mad.2005.05.002
142
GantenbeinKVKanaka-GantenbeinC. Mediterranean diet as an antioxidant: the impact on metabolic health and overall wellbeing. Nutrients. (2021) 13:1951. doi: 10.3390/nu13061951
143
DavidsonSDCherryJPChoudhuryMSTazakiHMallouhCKonnoS. Glyoxalase I activity in human prostate cancer: A potential marker and importance in chemotherapy. J Urol. (1999) 161:690–1. doi: 10.1016/S0022-5347(01)61996-7
144
SakamotoHMashimaTKizakiADanSHashimotoYNaitoMet al. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood. (2000) 95:3214–8. doi: 10.1182/blood.V95.10.3214
145
HosodaFAraiYOkadaNShimizuHMiyamotoMKitagawaNet al. Integrated genomic and functional analyses reveal glyoxalase I as a novel metabolic oncogene in human gastric cancer. Oncogene. (2015) 34:1196–206. doi: 10.1038/onc.2014.57
146
WangYKuramitsuYUenoTSuzukiNYoshinoSIizukaNet al. Glyoxalase I (GLO1) is up-regulated in pancreatic cancerous tissues compared with related non-cancerous tissues. Anticancer Res. (2012) 32:3219–22.
147
HuXYangXHeQChenQYuL. Glyoxalase 1 is up-regulated in hepatocellular carcinoma and is essential for HCC cell proliferation. Biotechnol Lett. (2014) 36:257–63. doi: 10.1007/s10529-013-1372-6
148
SantariusTBignellGRGreenmanCDWidaaSChenLMahoneyCLet al. GLO1-A novel amplified gene in human cancer. Genes Chromosomes Cancer. (2010) 49:711–25. doi: 10.1002/gcc.20784
149
AlhujailyMAbbasHXueMde la FuenteARabbaniNThornalleyPJ. Studies of glyoxalase 1-linked multidrug resistance reveal glycolysis-derived reactive metabolite, methylglyoxal, is a common contributor in cancer chemotherapy targeting the spliceosome. Front Oncol. (2021) 11:748698. doi: 10.3389/fonc.2021.748698
150
Fonseca-SánchezMARodríguez CuevasSMendoza-HernándezGBautista-PiñaVArechaga OcampoEHidalgo MirandaAet al. Breast cancer proteomics reveals a positive correlation between glyoxalase 1 expression and high tumor grade. Int J Oncol. (2012) 41:670–80. doi: 10.3892/ijo.2012.1478
151
MeariniERomaniRMeariniLAntognelliCZucchiABaroniTet al. Differing expression of enzymes of the glyoxalase system in superficial and invasive bladder carcinomas. Eur J Cancer Oxf Engl 1990. (2002) 38:1946–50. doi: 10.1016/s0959-8049(02)00236-8
152
VinceRDalugeSWaddWB. Studies on the inhibition of glyoxalase I by S-substituted glutathiones. J Med Chem. (1971) 14:402–4. doi: 10.1021/jm00287a006
153
LoTWThornalleyPJ. Inhibition of proliferation of human leukaemia 60 cells by diethyl esters of glyoxalase inhibitors in vitro. Biochem Pharmacol. (1992) 44:2357–63. doi: 10.1016/0006-2952(92)90680-h
154
TakeuchiMKimuraSKurodaJAshiharaEKawataniMOsadaHet al. Glyoxalase-I is a novel target against Bcr-Abl+ Leukemic cells acquiring stem-like characteristics in a hypoxic environment. Cell Death Differ. (2010) 17:1211–20. doi: 10.1038/cdd.2010.6
155
BirkenmeierGHemdanNYAKurzSBiglMPierohPDebebeTet al. Ethyl pyruvate combats human leukemia cells but spares normal blood cells. PLoS One. (2016) 11:e0161571. doi: 10.1371/journal.pone.0161571
156
BartyikKTuriSOroszFKargE. Methotrexate inhibits the glyoxalase system in vivo in children with acute lymphoid leukaemia. Eur J Cancer Oxf Engl 1990. (2004) 40:2287–92. doi: 10.1016/j.ejca.2004.06.024
157
DaviesGFRoeslerWJJuurlinkBHJHarknessTAA. Troglitazone overcomes doxorubicin-resistance in resistant K562 leukemia cells. Leuk Lymphoma. (2005) 46:1199–206. doi: 10.1080/10428190500102555
158
NassNBrömmeH-JHartigRKorkmazSSelSHircheFet al. Differential response to α-oxoaldehydes in tamoxifen resistant MCF-7 breast cancer cells. PLoS One. (2014) 9:e101473. doi: 10.1371/journal.pone.0101473
159
WanY-XQiX-WLianY-YLiuZ-YWangHQiuY-Qet al. Electroacupuncture facilitates vascular normalization by inhibiting glyoxalase1 in endothelial cells to attenuate glycolysis and angiogenesis in triple-negative breast cancer. Cancer Lett. (2024) 598:217094. doi: 10.1016/j.canlet.2024.217094
160
SakamotoHMashimaTSatoSHashimotoYYamoriTTsuruoT. Selective activation of apoptosis program by S-p-bromobenzylglutathione cyclopentyl diester in glyoxalase I-overexpressing human lung cancer cells. Clin Cancer Res Off J Am Assoc Cancer Res. (2001) 7:2513–8.
161
KimHSLeeM-S. STAT1 as a key modulator of cell death. Cell Signal. (2007) 19:454–65. doi: 10.1016/j.cellsig.2006.09.003
162
LvNHaoSLuoCAbukiwanAHaoYGaiFet al. miR-137 inhibits melanoma cell proliferation through downregulation of GLO1. Sci China Life Sci. (2018) 61:541–9. doi: 10.1007/s11427-017-9138-9
163
ChengW-LTsaiM-MTsaiC-YHuangY-HChenC-YChiH-Cet al. Glyoxalase-I is a novel prognosis factor associated with gastric cancer progression. PLoS One. (2012) 7:e34352. doi: 10.1371/journal.pone.0034352
164
TaïbiNAl-BalasQABekariNTalhiOAl JabalGABenaliYet al. Molecular docking and dynamic studies of a potential therapeutic target inhibiting glyoxalase system: metabolic action of the 3, 3 ‘- [3- (5-chloro-2-hydroxyphenyl) -3-oxopropane-1, 1-diyl] - bis-4-hydroxycoumarin leads overexpression of the intracellular level of methylglyoxal and induction of a pro-apoptotic phenomenon in a hepatocellular carcinoma model. Chem Biol Interact. (2021) 345:109511. doi: 10.1016/j.cbi.2021.109511
165
InoueMNakagawaYAzumaMAkahaneHChimoriRManoYet al. The PKM2 inhibitor shikonin enhances piceatannol-induced apoptosis of glyoxalase I-dependent cancer cells. Genes Cells Devoted Mol Cell Mech. (2024) 29:52–62. doi: 10.1111/gtc.13084
166
KuhlaBLüthH-JHaferburgDWeickMReichenbachAArendtTet al. Pathological effects of glyoxalase I inhibition in SH-SY5Y neuroblastoma cells. J Neurosci Res. (2006) 83:1591–600. doi: 10.1002/jnr.20838
167
JohanssonASRidderströmMMannervikB. The human glutathione transferase P1–1 specific inhibitor TER 117 designed for overcoming cytostatic-drug resistance is also a strong inhibitor of glyoxalase I. Mol Pharmacol. (2000) 57:619–24. doi: 10.1124/mol.57.3.619
Summary
Keywords
cancer, hormesis, methylglyoxal, glyoxalase 1, therapeutics
Citation
Wang Z, Liu S, Zhang M and Liu M (2025) Dual roles of methylglyoxal in cancer. Front. Oncol. 15:1557162. doi: 10.3389/fonc.2025.1557162
Received
08 January 2025
Accepted
04 April 2025
Published
25 April 2025
Volume
15 - 2025
Edited by
Leilei Wu, University of Chinese Academy of Sciences, China
Reviewed by
Jing-Sheng Cai, Peking University People’s Hospital, China
Yang Pan, Zhejiang Cancer Hospital, China
Updates
Copyright
© 2025 Wang, Liu, Zhang and Liu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Liu, liumin6958@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.