 Hua Yang
Hua Yang Yu Fang2
Yu Fang2- 1The First Hospital of Hunan University of Chinese Medicine, Changsha, China
- 2Department of Pathology, The First Hospital of Hunan University of Chinese Medicine, Changsha, China
- 3Medical College, Hunan University of Chinese Medicine, Changsha, China
- 4Medical Department, The First Hospital of Hunan University of Chinese Medicine, Changsha, China
Background: Breast cancer continues to be a leading cause of cancer-related deaths among women worldwide. Despite advancements in diagnostics and therapies, challenges such as metastasis, recurrence, and resistance remain prevalent. Recently, research has shifted from traditional genomic analyses to the study of epigenetic regulation, which includes DNA methylation, histone modifications, and non-coding RNAs. Given the rapid expansion of literature in this field, a systematic overview of its evolution and emerging trends is necessary.
Methods: We performed a comprehensive bibliometric analysis of 5,271 articles on breast cancer epigenetics, sourced from the Web of Science Core Collection, covering the years 2000 to 2024. Utilizing tools like CiteSpace and VOSviewer, with support from RStudio, Pajek, and HisCite, we analyzed co-citation networks, keyword co-occurrence, and burst detection. This analysis included visualizations of collaboration among authors, institutions, and countries. Metrics such as modularity, silhouette scores, and betweenness centrality were used to ensure analytical rigor and to identify thematic evolution and emerging research frontiers.
Results: From 2000 to 2018, the number of annual publications increased steadily, with citation peaks occurring in 2021. The United States led in research output and influence, followed by China. Leading institutions included Johns Hopkins University and the University of Texas MD Anderson Cancer Center. Keyword and co-citation analyses revealed four research phases: (1) early studies focused on promoter hypermethylation of tumor suppressor genes like RASSF1A; (2) an in-depth investigation of molecular mechanisms, including epithelial-mesenchymal transition and chromatin remodeling; (3) translational research involving HDAC inhibitors and DNA methylation biomarkers; and (4) recent advancements in multi-omics integration, synthetic lethality, and the study of epigenetics in the tumor microenvironment. Emerging research directions include the targeted removal of epigenetic memory, metabolism-epigenetics networks, and single-cell epigenomic profiling.
Conclusion: This bibliometric study outlines the trajectory of research in breast cancer epigenetics, highlighting its evolution from basic methylation studies to advanced therapeutic exploration. Future research should focus on targeting epigenetic memory to combat drug resistance and recurrence, developing synthetic lethality strategies, and employing single-cell technologies for dynamic epigenetic mapping. These findings provide a strategic roadmap for researchers and policymakers navigating the evolving landscape of breast cancer epigenetics.
1 Background
Breast cancer is the most prevalent malignancy affecting women globally. In 2022, there were approximately 2.3 million new cases and 670,000 deaths, as reported (1). There is significant heterogeneity in clinical outcomes and therapeutic responses across different molecular subtypes. Despite advances in early detection and targeted therapies, challenges such as drug resistance, metastasis, and recurrence persist, indicating that breast cancer is the leading cause of cancer-related deaths among women. Although traditional genomic approaches can identify mutations like BRCA1/2 and PIK3CA, they fail to fully account for the dynamic adaptability of tumor cells or the role of non-genetic factors on disease progression (2). This knowledge gap has redirected research focus toward epigenetic regulation—a reversible and heritable mechanism that modulates gene expression without altering DNA sequences—positioning it at the forefront of oncological research.
Epigenetic mechanisms—such as DNA methylation, histone modifications, chromatin remodeling, and non-coding RNAs—play a crucial role in breast carcinogenesis (3). For instance, hypermethylation of promoter regions in tumor suppressor genes such as RASSF1A and BRCA1 occurs in 40%–60% of breast cancers cases, correlating with advanced stages and poor prognosis (4). Conversely, global hypomethylation in triple-negative breast cancer (TNBC) leads to genomic instability and facilitates immune evasion. Histone deacetylases (HDACs) and methyltransferases, such as EZH2, frequently exhibit dysregulation, reshaping the tumor microenvironment (TME) and maintaining stemness (5). The clinical relevance of these findings is highlighted by FDA-approved epigenetic therapies, including HDAC inhibitors like Entinostat and DNMT inhibitors (6).
Over the past two decades, the field has experienced exponential growth, with pioneering studies such as The Cancer Genome Atlas (TCGA) pan-cancer analyses linking epigenetic subtypes to therapeutic vulnerabilities (7). However, the rapid surge in research outputs has fragmented the research landscape, obscuring evolutionary trends, collaborative networks, and emerging frontiers. Bibliometric analysis, a quantitative method for evaluating publication patterns, authorship contributions, and keyword dynamics, provides a systematic approach to map this complex domain. Since its introduction by Pritchard in 1969 (8), bibliometrics has evolved into various computational tools like VOSviewer and CiteSpace, which analyze publications and co-cited references to identify research shifts and trends.
To date, there has been no comprehensive bibliometric assessment of breast cancer epigenetics, which limits the strategic prioritization of research efforts. This study addresses this gap by analyzing 5,271 publications from 2000 to 2024. We utilize co-citation networks, keyword clustering, and burst detection to: (1) describe the temporal evolution of the field; (2) identify influential contributors and institutions; (3) characterize thematic shifts from locus-specific methylation to pan-cancer epigenetic reprogramming; and (4) forecast future directions including synthetic lethality strategies and single-cell epigenomic profiling. By integrating multidimensional bibliographic data, this work provides a roadmap for researchers, clinicians, and policymakers to understand the transformative potential of epigenetics in breast oncology.
2 Methods
2.1 Data source and search strategy
The bibliometric analysis was conducted via the Web of Science Core Collection (WoSCC), a well-recognized and frequently used database for bibliometric research, due to its comprehensive overview of essential data such as publications, citations, authors, references, and keywords. The search strategy utilized was as follows: TS=(“Epigenet*” OR “Epigenesis, Genetic” OR “Epigenetic Processes”) AND TS=(“Breast Cancer” OR “Breast Neoplasm*” OR “Mammary Cancer” OR “Breast Tumor*” OR “Breast Malignan*” OR “Breast Lesion*”) AND DT=(Article). Literature published between January 1, 2000, and December 31, 2024, was retrieved, and the data was confined to the Science Citation Index-Expanded (SCIE) database. Additionally, documents categorized as “retracted paper” “proceedings paper,” and “book chapter” were excluded. The retrieval process was conducted on a single day (January 1, 2025) to minimize potential confounding bias from daily database updates. To ensure the authenticity and reliability of the research data, two trained investigators independently collected the data, and a third colleague intervened in discussions only when divergent opinions needed resolution. Ultimately, 5,271 documents were obtained after manual reference screening, and full records along with cited references were downloaded in plain text for further analysis. A detailed flowchart of the study procedures is provided in Figure 1.
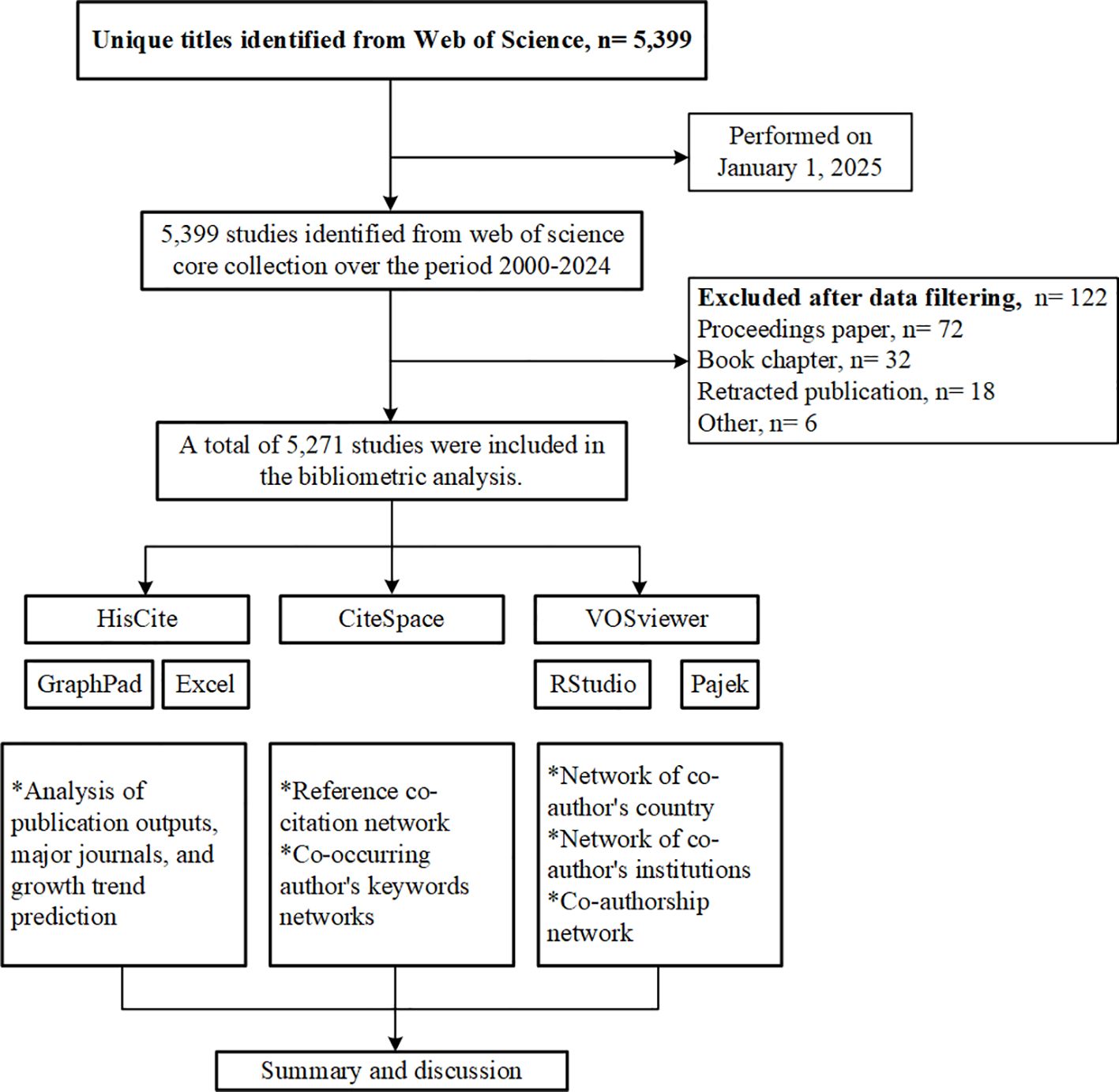
Figure 1. Flow chart of the bibliometric study.
2.2 Data analysis and visualization
CiteSpace (version 6.4.R1 Advanced) and VOSviewer (version 1.6.17) were the primary tools used, supplemented by additional data analysis and visualization software, including RStudio (version 2024.09.1), Pajek (version 5.19, 64-bit), HisCite (version 12.03.17), GraphPad Prism (version 10.1.2), and Microsoft Excel 2021, to complete the research. These tools facilitated key bibliometric analyses, such as co-citation analysis of references, co-occurrence analysis of keywords, and collaboration network analysis of authors, institutions, and countries. Co-citation analysis, a bibliometric method that examines the frequency with which two documents are cited together in subsequent publications, is a powerful approach for identifying influential literature and integrating cross-disciplinary research ideas (9). Co-occurrence refers to the simultaneous appearance of two keywords within the same publications. Co-authorship illustrates collaborative contributions between research entities and serves as an indicator of cooperation among authors, institutions, or countries (10). To examine recent research trends, the study period focused on the last five years (2020–2024) and the most recent year (2024), with all analyses repeated for these specific time frames.
CiteSpace, developed by Professor Chaomei Chen, is a primary software tool for analyzing medical research trends (11). In this study, CiteSpace was employed to conduct clustering, timeline, and burst analyses of co-cited references and co-occurring keywords, along with constructing co-citation networks. In order to achieve better graphical presentation, we clicked on “all in one” and “optimize layout” to separate each cluster and assign different colors (based on keywords), then clicked “find clusters” to obtain the individual clusters. The clustering results were evaluated via structural metrics such as modularity, which assesses network modularization, and silhouette scores, which measure the homogeneity and quality of the clusters. The labels and results were meticulously re-examined to determine whether adjustments were necessary. It is widely acknowledged that a Q value of ≥ 0.3 indicates significant modularity in the network, with higher values reflecting enhanced clustering performance (12, 13). Similarly, an S value of ≥ 0.5 denotes reasonable clustering quality, with values approaching 1 indicating improved network homogeneity (14).
A citation burst refers to a sudden and significant increase in the number of citations for a specific publication within a defined period (15). Clusters with numerous nodes exhibiting strong citation bursts may indicate emerging trends in current or future research. Betweenness centrality measures the extent to which a node acts as a bridge on the shortest paths connecting other node pairs, highlighting its role in the network’s structure. The analysis parameters were as follows: (1) the time frame was from 2000 to 2024; (2) the time slicing interval was one year; and (3) the g-index, an extension of Hirsch’s h-index, was used to evaluate the global citation performance of the article set (16). The quantity of exhibited nodes was determined by the g-index. Due to fluctuations in citation volumes over time frames, the k-values for the g-index were 25 for 2000–2024, 50 for 2020–2024, and 100 for 2024; (4) all other parameters were left at their normal settings.
VOSviewer is a sophisticated tool for viewing and mapping diverse network data, and is proficient at processing extensive datasets and generating high-quality network maps (17). This study employed VOSviewer to generate collaboration network maps for countries, institutions, and authors. Before constructing the national collaboration network map, we first assigned the geographical data of the involved countries in RStudio, primarily using the “sp”, “rgdal”, and “tidyverse” packages. Then, we used VOSviewer for visualization. Detailed methods can be found at https://github.com/like-firework/Progress-in-Epigenetic-Research-of-Breast-Cancer. The research of institutional and author partnerships utilized VOSviewer to construct the network, which was further modified with Pajek. Citation data for the papers were evaluated with HisCite software to identify highly cited articles, and bar charts were created via GraphPad. Excel was used for data storage during the investigation.
3 Results
3.1 Analysis of co‐cited references: publications, most cited papers, and research clusters
3.1.1 Analysis of publications
As of December 31, 2024, a total of 5,271 articles have been published within this field, indicating significant research activity. Figure 2A illustrates the annual trends in both publication and citation counts. Our analysis reveals a substantial increase in both metrics. The lowest publication output occurred in 2000 with 20 articles, whereas the peak was in 2018 with 371 articles. The number of citations were lowest in 2000, totaling 17, and reached a maximum in 2021, with 21,749 citations. The sustained high number of published papers over the past decade highlights the popularity and maturation of this research field.
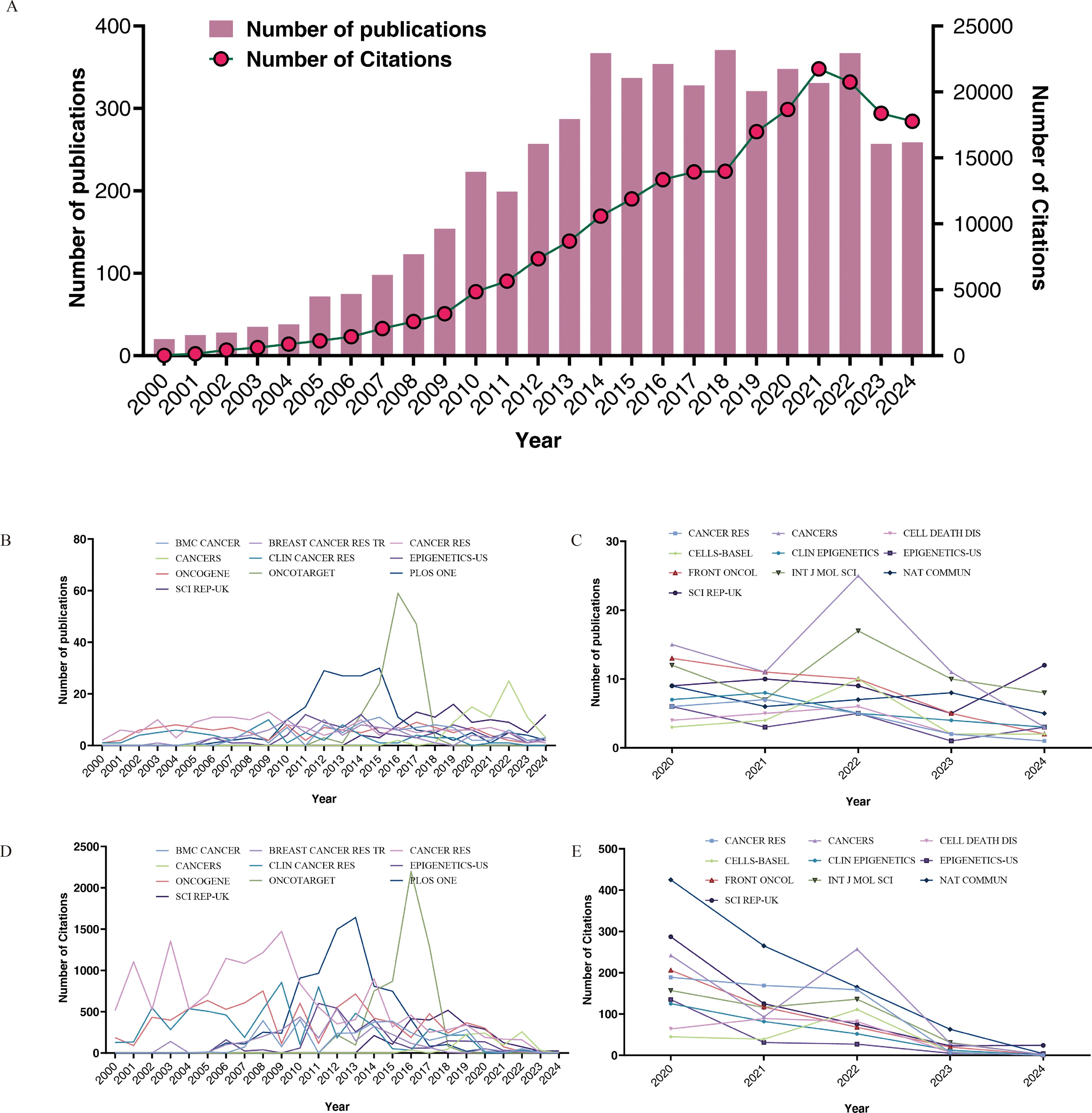
Figure 2. Annual number of publications and citations from 2000 to 2024 (A), top 10 journals by publication volume for the time period 2000-2024 (B) and 2020-2024 (C), top 10 cited journals for the time period 2000-2024 (D) and 2020-2024 (E).
Table 1 presents the journals with the highest number of published papers and their respective citation frequencies. We identified the top 15 journals based on publications for the periods 2000–2024 and 2020–2024. Notably, PLOS ONE has the highest volume of published articles; however, each article in CANCER RES receives, on average, 2.1 times more citations than those in PLOS ONE, indicating that the papers published in this journal have a higher long-term impact in the field. Journals such as ONCOTARGET, ONCOGENE, and SCI REP-UK also demonstrated a considerable number of publications. Over the past five years, the number of relevant publications in journals such as CANCERS, INT J MOL SCI, and SCI REP-UK has increased. Based on standardized citations (referring to the total number of citations a journal has received divided by the total number of articles it has published), we believe that during the period from 2000 to 2024, articles published in CANCER RES and CLIN CANCER RES exhibited relatively high impact. In the past five years, the leading journals have been SEMIN CANCER BIOL and NAT COMMUN. Furthermore, we examined the publication counts and citation trends of the top 10 journals from both 2000–2024 and 2020–2024, as illustrated in Figures 2B–E.
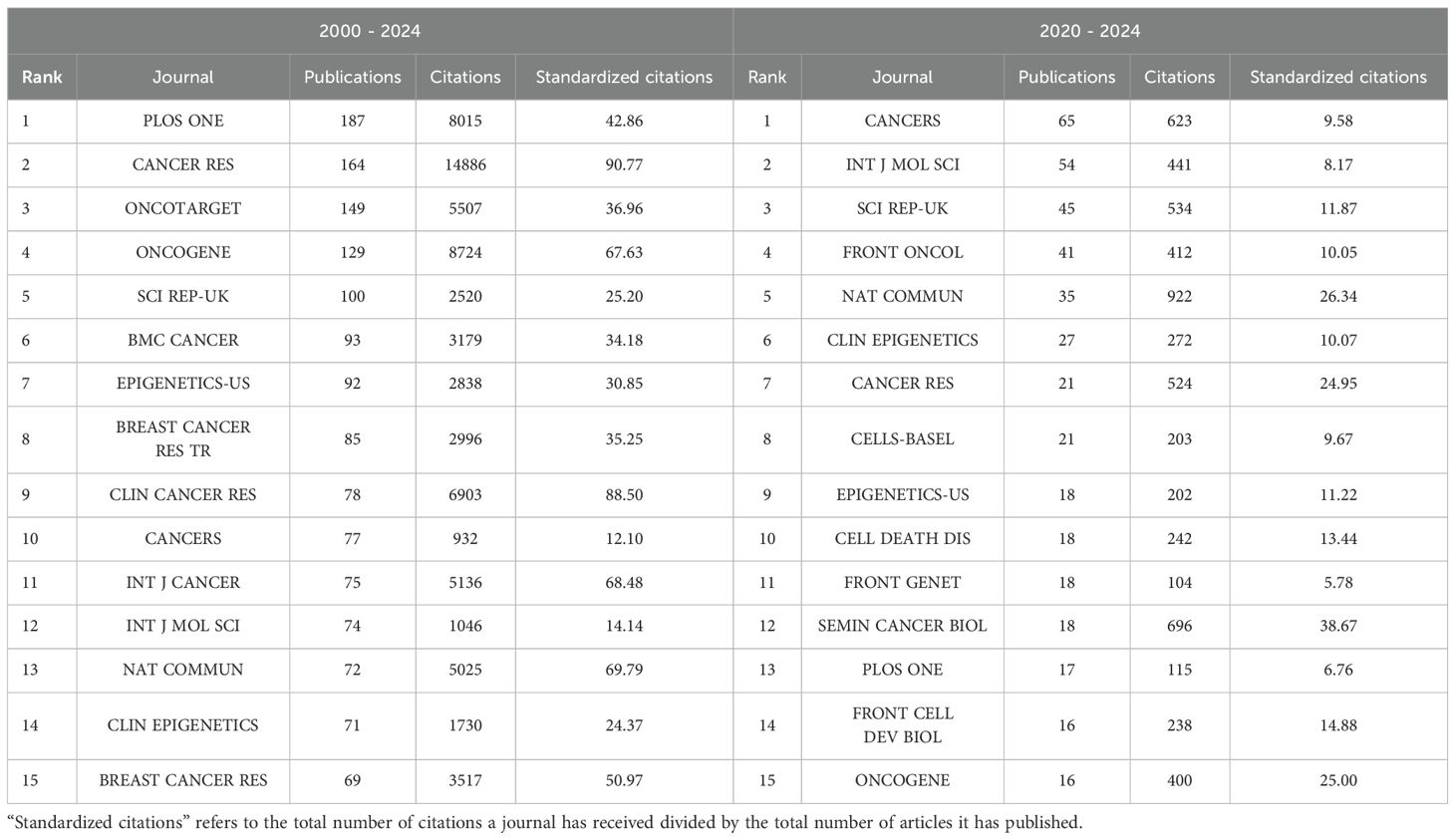
Table 1. Top 15 journals for publications.
3.1.2 The most cited papers
We have identified the 10 most referenced works in both the literature and the network from 2000 to 2024, as detailed in Tables 2 and 3. The most cited paper is by Koboldt et al., with 9,254 citations in the literature and 270 in the network. This study provides a comprehensive molecular characterization of breast cancer subtypes, emphasizing mutations in TP53, PIK3CA, and GATA3 (18). The research of Horvath et al. follows, with 4,045 citations, introducing a universal age predictor on the basis of DNA methylation and identifying age acceleration in cancer tissues (19). The third most cited study is that of Irizarry et al., which was cited 1,670 times and shows that DNA methylation changes in colon cancer occur mainly at CpG island shores, affecting gene expression (20). Additionally, in the network, Burbee et al.’s and Fackler et al.’s studies, with 64 and 56 citations respectively, explored the role of RASSF1A and other genes in breast cancer epigenetics (4, 21).
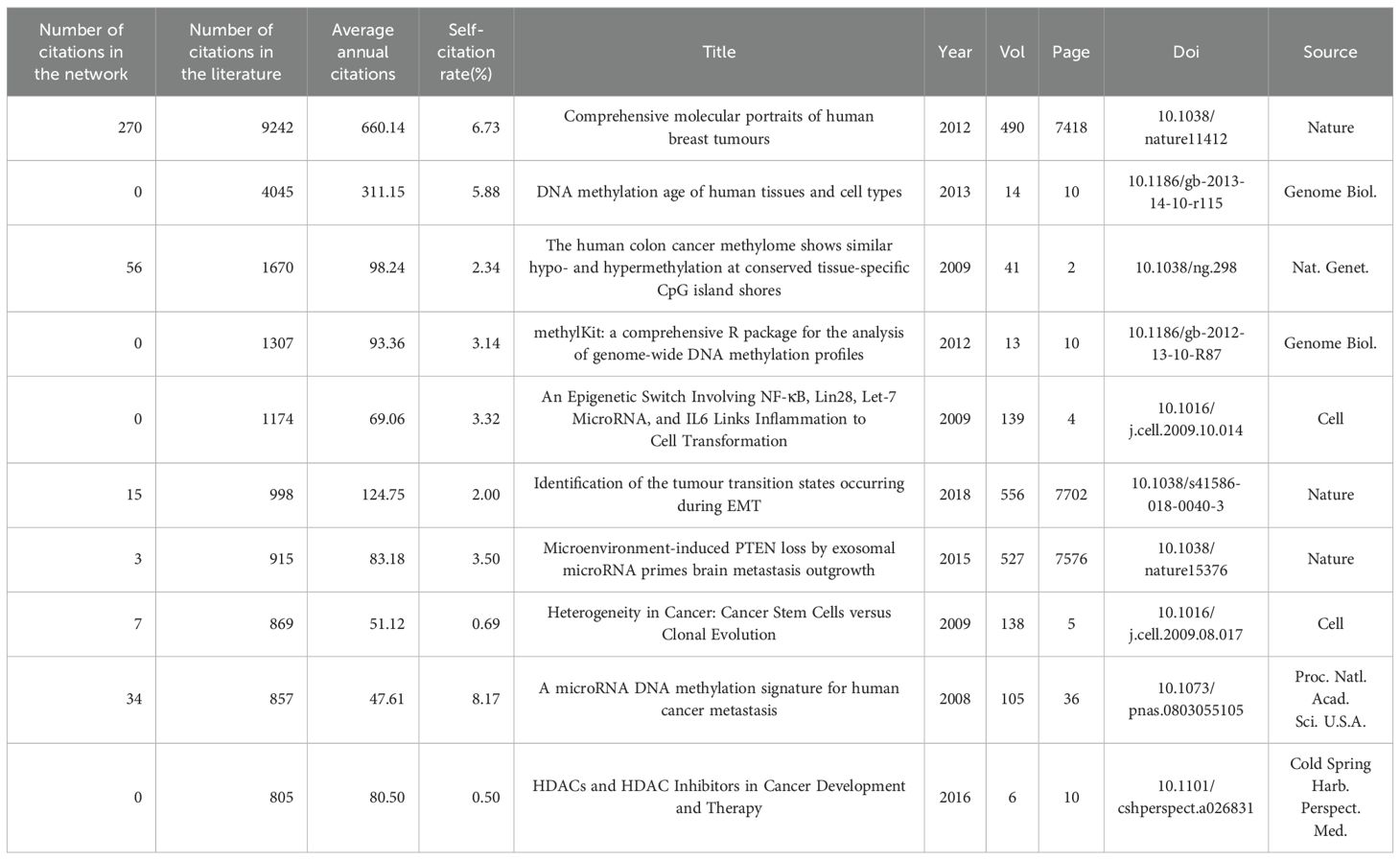
Table 2. The top 10 most frequently cited papers in the literature between 2000 and 2024.
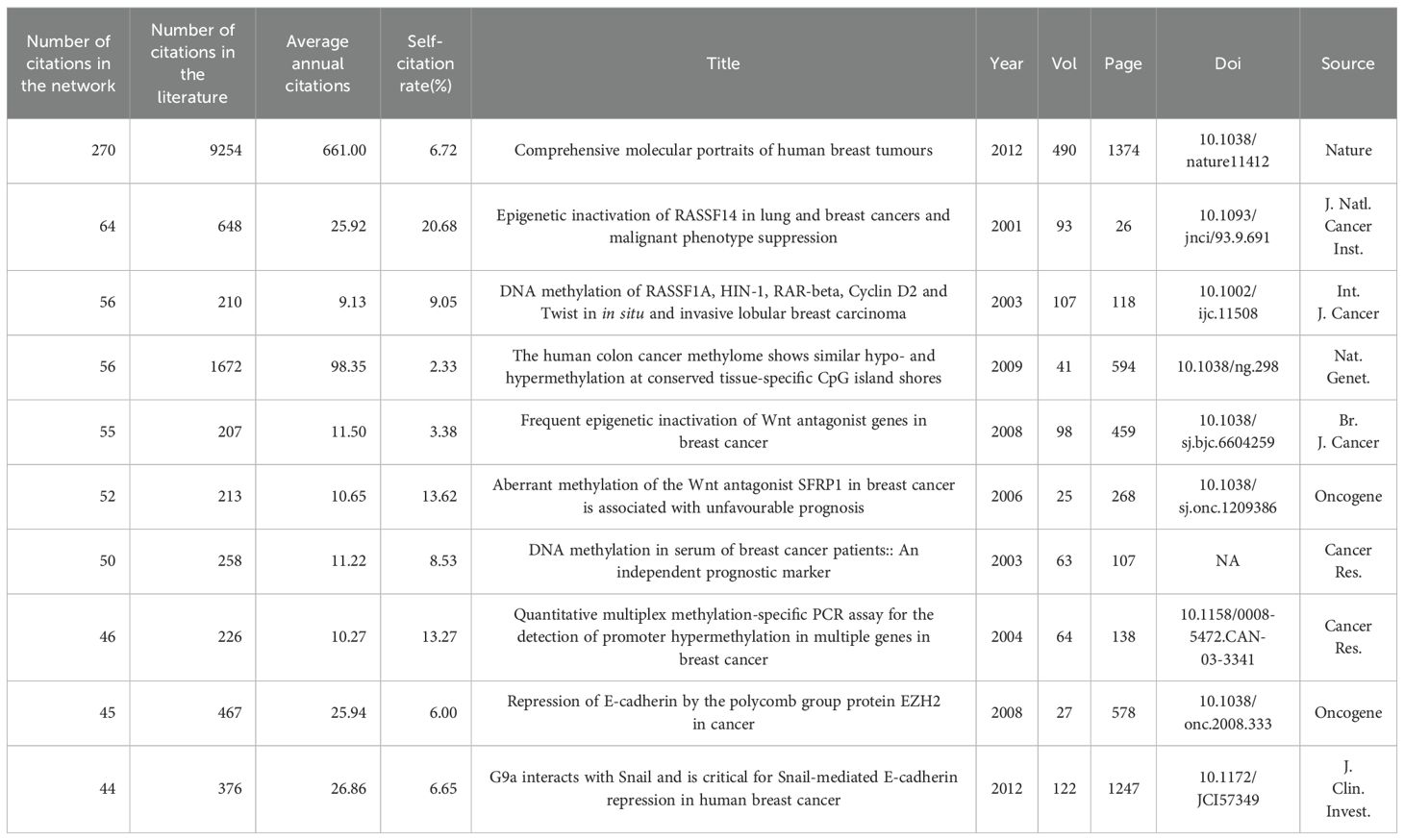
Table 3. The top 10 most frequently cited papers in the network between 2000 and 2024.
In the past five years, many studies, including Becker et al.’s 2020 study, have focused on cancer-associated fibroblasts and their epigenetic and metabolic roles in breast cancer under hypoxic conditions (22). Research by Corr et al.’s 2020 research demonstrated how myocardial infarction might accelerate the progression of breast cancer by reprogramming immune-suppressive monocytes through epigenetic mechanisms (23). In 2021, Chen et al. systematically analyzed RNA adenosine modifications in colorectal cancer and developed a WM_Score model for predicting patient survival and TME features (24). Collectively, these works underscore the crucial role of epigenetic regulation in cancer progression and TME dynamics.
Furthermore, we assessed the impact of publications from 2000–2024 and 2020–2024 by evaluating citation bursts (Additional file 2: Supplementary Table S1). The timeline is depicted with a segmented blue line indicating each year, while a red line denotes the duration of citation bursts. Notably, the strongest and most recent citation bursts are from Sung et al.’s “Global Cancer Statistics 2020,” (25) Waks and Winer’s “Breast Cancer Treatment: A Review,” (26) and Loibl et al.’s “Breast Cancer.” (27).
3.1.3 Cluster of research
We constructed cluster-based co-citation networks for the periods 2000–2024, 2020–2024, and 2024. All three networks demonstrated well-structured and reliable configurations, with the 2000–2024 network attaining a modularity (Q) of 0.7311 and a silhouette score (S) of 0.8866, the 2020–2024 network achieving Q = 0.691 and S = 0.8481, and the 2024 network reaching Q = 0.6597 and S = 0.8634. Each reference was depicted as a single node, with node size reflecting co-citation frequency. The citation rings were color-coded in chronological order, and their thickness was proportional to the number of citations within the corresponding time frame. The arrows indicate dependency links among clusters. Detailed descriptions of the major clusters of co-cited references can be found in Additional File 3: Supplementary Table S2, and the visualization of each network is shown in Additional File 1: Supplementary Figures S1 and S2. Additionally, landscape visualization depicts the start, duration, and cessation of each cluster, where peak heights represent cluster activity levels.
In the co-citation network from 2000 to 2024, we identified 16 distinct clusters. Clusters were numerically designated by size, with the largest as #0 and the smallest as #15. Each cluster was characterized by qualitative measures, including label, size, silhouette score, and the average year of co-cited references. Landscape visualization succinctly illustrates the evolution of research themes in this field since 2000 (Figure 3). The silhouette scores of the identified clusters are all greater than 0.8, indicating that the members within each cluster exhibit good homogeneity. Based on the analysis of these clusters, we suggest that one of the earliest identified epigenetic alterations in breast cancer is the loss of gene expression resulting from promoter hypermethylation.
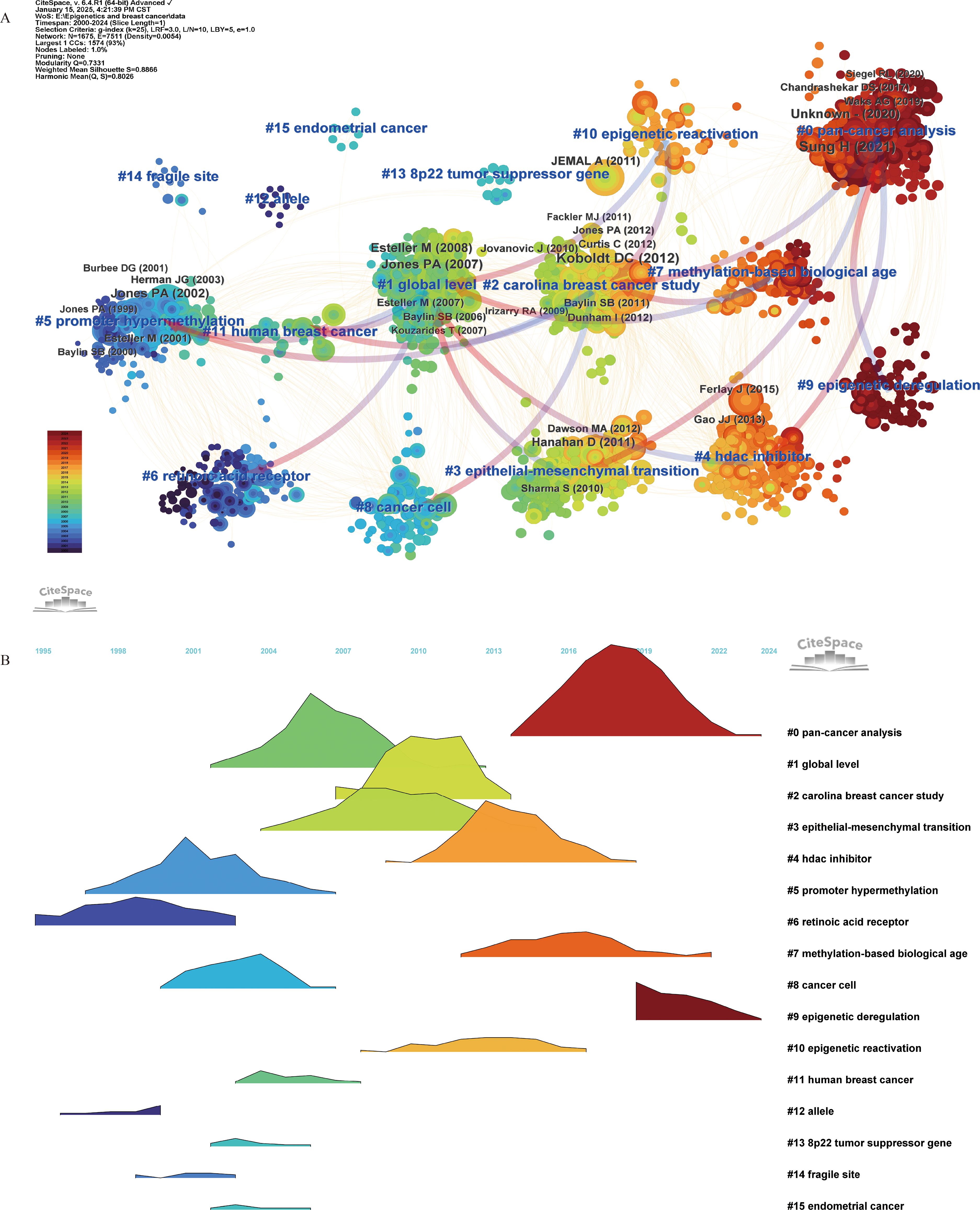
Figure 3. Co-citation reference (2000–2024) network and clustering visualization of hotspots, and citation relationships between 16 clusters (A). Landscape Visualization of clusters from 2000 to 2024 (B). Each node represents a co-cited reference, and the size of the node is proportional to its co-citation frequency. The tree rings around each node indicate the citation burst period. The gradient from blue to red corresponds to the time span from 2000 to 2024, with dark blue representing 2000 and dark red representing 2024. Blue labels denote the cluster number and name, where cluster #0 is the smallest and cluster #15 is the largest. The arrows indicate dependency links among clusters.
This hypothesis is substantiated by five early clusters: #12 “allele”, #6 “retinoic acid receptor”, #14 “fragile site”, #5 “promoter hypermethylation”, and #13 “8p22 tumor suppressor gene”. Research subsequently deepen, with studies like #1 “global level” and #2 “Carolina breast cancer study” focusing on metastasis and cancer progression, particularly #3 epithelial-mesenchymal transition. Over the past decade, emphasis has been placed on epigenetic regulatory mechanisms, including #10 “epigenetic reactivation” and #9 “epigenetic deregulation”; predictive measures like #7 “methylation-based biological age”; and treatment modalities exemplified by #4 “HDAC inhibitor”. Notably, pan-cancer analysis has gained prominence in recent years, exemplified by cluster #0 pan-cancer. Detailed information for each cluster can be found in Additional File 3: Supplementary Table S2 A.
In addition, we analyzed the co-cited references published over the past 5 years (2020–2024) and the most recent year (2024) (Figure 4) to understand the latest research trends, detailed information can be found in Additional File 3: Supplementary Table S2, sections B and C. Consistent with our previous analyses, scholars over the past five years have primarily focused on exploring epigenetic regulatory mechanisms and therapeutic strategies. In the 2020–2024 network, biomarker screening has emerged as a major research focus, as reflected in cluster #0 “emerging cancer hallmark” and cluster #4 “prognostic marker”. Moreover, #8 “tumor heterogeneity”, #11 “oxidative phosphorylation”, and #12 “microbial mechanism” represent important directions in pathogenesis research. The network highlights #2 “synthetic lethality strategies” and #3 “epigenetic restoration” as emerging epigenetic intervention strategies. In the 2024 network, #0 “epigenetic memory”, #5 “dormant breast cancer cell”, and chemotherapy resistance in breast cancer—illustrated by #1 “cisplatin response” and #6 “therapy resistance”—are considered major research directions.

Figure 4. Co-citation network of references with corresponding clusters obtained with CiteSpace for the time period 2020-2024 (A) and 2024 (B). Each node represents a co-cited reference, and the size of the node is proportional to its co-citation frequency. The tree rings around each node indicate the citation burst period. Blue labels indicate the cluster number and name, where Cluster #0 is the smallest and Cluster #15 is the largest. In Panel (A), the gradient from red to blue represents the time span from 2020 to 2024, with dark red indicating 2020 and dark blue indicating 2024. Arrows denote the dependency relationships among clusters. In Panel (B), the gradient from purple to red corresponds to clusters ranging from smallest to largest, with purple representing the smallest cluster (#11) and red indicating the largest cluster (#15).
3.2 Analysis of co‐occurrence of keywords
The primary goal of constructing a co-occurrence network of keywords and conducting burst analysis is to offer a comprehensive overview of the current research landscape and predict the evolution of research hotspots over time. Within this network, each node represents a frequently co-occurring keyword, with node size reflecting the frequency of occurrence and the color of the annual ring indicating the time of appearance.
Between 2000 and 2024, key terms in the network include breast cancer, growth, DNA methylation, metastasis, proliferation, epigenetics, gene expression, prognosis, BRCA1, tumor suppressor, and epigenetic regulation. The connections among these keywords are depicted via VOSviewer in Figure 5A. The three keywords exhibiting the highest burst strengths are tumor suppressor genes, promoter hypermethylation, and CpG islands, with their evolutionary trajectories shown in Figure 5B.

Figure 5. Visualization of the co-occurring network of keywords from 2000 to 2024 (A), citation burst analysis of keywords between 2000 and 2024 (B), and timeline visualization of the network (C) for the time period 2020-2024. In Panel (A), each node represents a keyword. The larger the node, the greater the number of associated publications. Keywords belonging to the same category are shown in the same color. In panel B, red indicates the duration of a burst, indicating that keywords are frequently quoted, while green indicates keywords that are not frequently quoted. In Panel (C), each node represents the time when a keyword first appeared. The larger the node, the more frequently the keyword appeared. The rings surrounding the nodes indicate the duration over which the keyword continued to appear. Purple lines represent the earliest occurrence (2020), while red lines represent the most recent (2024). The cluster labels are generated by the CiteSpace software based on the timeline displayed on the right side of the figure.
Keyword changes over the past five years serve as crucial indicators of emerging research hotspots. Therefore, a clustering analysis of keywords from 2020 to 2024 was performed, resulting in eight clusters (Q=0.335, S=0.6549, Figure 5C): #0 cancer stem cells, #1 activation, #2 breast cancer, #3 DNA methylation, #4 oxidative stress, #5 neoadjuvant chemotherapy, #6 prevention, #7 heterogeneity, and #8 mutations. Detailed information on each cluster is available in Additional File 4: Supplementary Table S3 A. In the burst term analysis, histone H3, microenvironment, and liquid biopsy emerged as the latest hotspot terms, as detailed in Additional File 4: Supplementary Table S3 B.
3.3 Analysis of the cooperation network across authors, institutions and countries
Using VOSviewer, we constructed co-occurrence networks for authors, institutions, and countries (Figure 6). Within the author co-occurrence network, 22 major collaboration networks were identified. By employing the average citation score (citations per paper) as the primary metric of impact, Horvath Steve, Yao Jun, Hung Mien-Chie, Zhang Lin, and Brown Myles were identified as the most influential authors, with Yao Jun and Zhang Lin in the same cluster. Among the 16 major institutional collaboration networks, Johns Hopkins University (101 publications), University of Texas MD Anderson Cancer Center (98 publications), NCI (93 publications), Chinese Academy of Sciences (85 publications), and Shanghai Jiao Tong University (77 publications) had the highest number of publications. Johns Hopkins University (9,365 citations), Harvard University (7,892 citations), and the University of Texas MD Anderson Cancer Center (7,648 citations) received the most citations. Furthermore, the University of Texas MD Anderson Cancer Center presented the most interinstitutional connections. Additionally, the countries with the highest number of publications were the United States (2,072 publications), China (1,258 publications), the UK (345 publications), Germany (334 publications), Italy (292 publications), and Canada (240 publications). International collaborations predominantly occurred between neighboring countries, with the United States facilitating the most connections. In addition, detailed information on publications by authors, institutions, and countries can be found in Additional File 4: Supplementary Tables S4-S6, respectively.
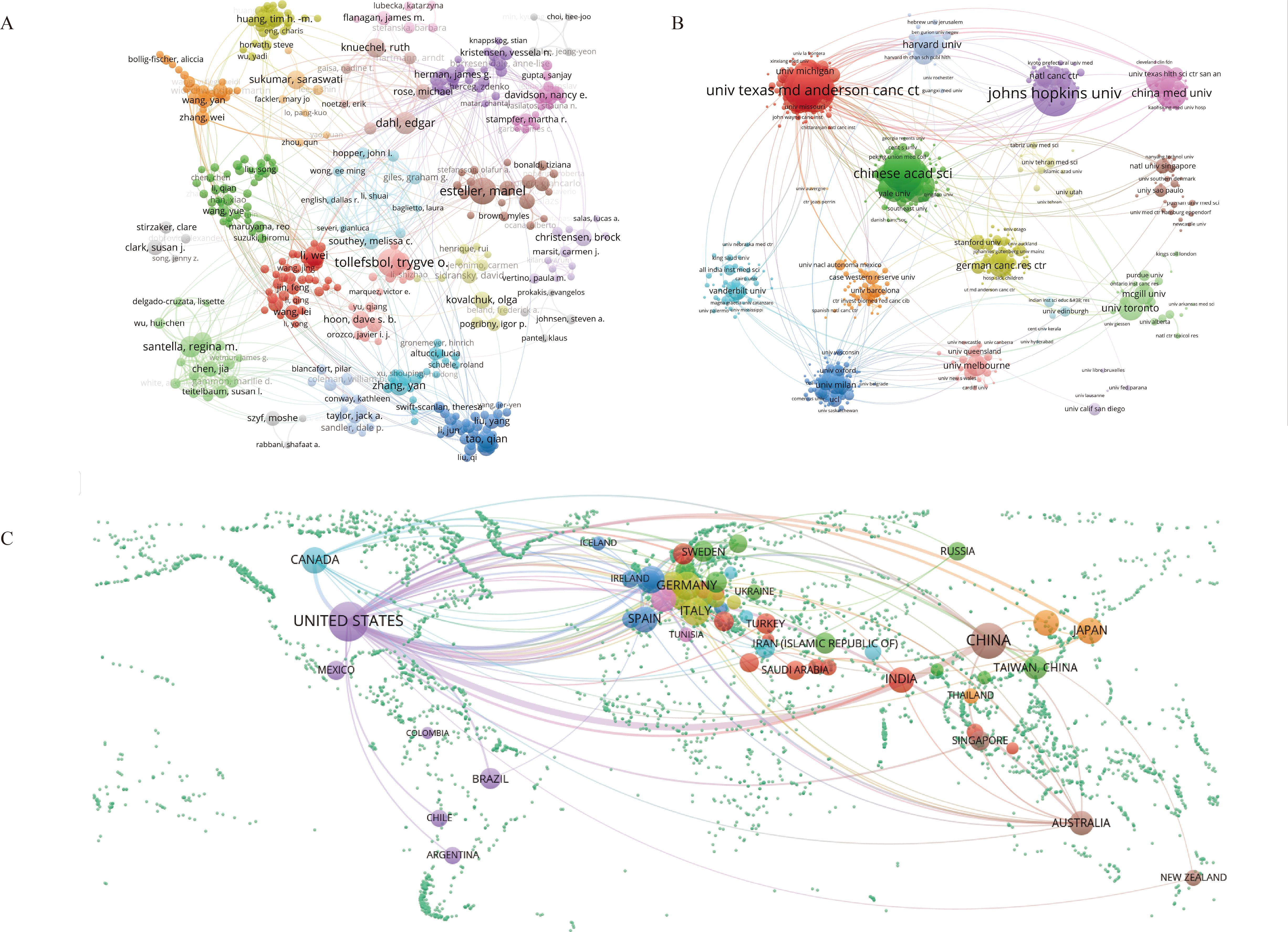
Figure 6. Visualization of the co-occurring network of authors (A), institutions (B), and countries (C) from 2000 to 2024. In Panels (A-C), the nodes represent authors, institutions, and countries, respectively. Larger nodes indicate a greater number of associated publications. Elements belonging to the same category are displayed in the same color.
4 Discussion
4.1 Summary of the main findings
The bibliometric analysis revealed a continuing increase in annual publications from 2000 (n = 20) to 2018 (n = 371), with citation counts peaking at 21,749 in 2021. This trend underscores the progressive development of epigenetic research on breast cancer and advances in related analysis techniques. Geographically, the United States published the most papers, contributing 2,072 articles (39.3% of the total), followed by China with 1,258 articles (23.9% of the total). When it comes to the perspective of research impact, countries such as Pakistan, Finland, and Russia achieved higher citation counts despite having published fewer papers. At the institutional level, Johns Hopkins University leads in paper production (101 papers), whereas the University of Texas MD Anderson Cancer Center stands out for its impact, with 7,648 citations and the highest interinstitutional connectivity (degree centrality = 0.43). Notably, Chinese institutions such as the Chinese Academy of Sciences and Shanghai Jiao Tong University are emerging. In terms of journal publications, PLOS ONE and CANCER RESEARCH have the most publications, but CANCER RESEARCH is cited 2.1 times more often per article than PLOS ONE. Additionally, Horvath Steve, Yao Jun, and Hung Mien-Chie are notable for publishing the most papers in this field. They have made significant contributions to the study of epigenetic age acceleration and breast cancer risk, as well as to the regulation of key epigenetic factors (28–30).
4.2 Evolution of epigenetic research topics in breast cancer and key articles
Through clustering and burst analyses of co-cited references, combined with a landscape visualization of clusters from 2000 to 2024 (Figure 3), we have identified four distinct phases in the evolution of breast cancer epigenetics research hotspots: the initial exploration phase, the systematic elucidation of molecular mechanisms, the clinical translation phase, and the emergence of novel insights into breast cancer epigenetics.
4.2.1 The budding phase
During this period, research focused on the promoter hypermethylation of tumor suppressor genes, particularly in the chromosome 8p22 region. Key clusters include #5 promoter hypermethylation (Year = 2001), #12 allele (Year = 1998), #13 8p22 tumor suppressor gene (Year = 2001), and #14 fragile site (Year = 2001). A seminal paper by Burbee et al. (21) first demonstrated the hypermethylation of the RASSF1A gene promoter in breast cancer, highlighting epigenetic silencing as a mechanism for oncogene inactivation, and paving the way for further studies on promoter methylation in other genes (21). Müller et al. found that serum RASSF1A and APC DNA methylation are strongly associated with poor prognosis in breast cancer patients, serving as crucial indicators for patient prognosis assessment (31). Consistent with the findings of Fackler et al., methylation differences in the promoters of the RASSF1A, Cyclin D2, RAR-beta, and Hin-1 genes were detected between lobular carcinoma in situ and invasive lobular carcinoma of the breast (4). In 2018, a study in India also confirmed that hypermethylation of RASSF1A is correlated with a poor prognosis in patients with breast cancer (32). Additional corroboration was found for the correlation between promoter hypermethylation and breast cancer in genes such as TMS1, ACS, DSC3, and BRCA1 (33–35).
4.2.2 Emphasize an in-depth investigation of the molecular mechanisms
The focus during this phase shifted from individual gene methylation changes to comprehensive, systematic elucidation (cluster #1, global level, Year = 2006). Methodological advances have greatly driven progress. In 2004, Fackler et al. introduced a high-sensitivity quantitative multiplex methylation-specific PCR method, allowing the detection of promoter methylation levels across multiple genes with less tissue (36). This method was subsequently used for quantitative gene methylation analysis in breast tissues (37–39). The advent of sequencing technologies such as GS 20 further facilitated the systematic interpretation of breast cancer epigenetics. Gary and colleagues utilized ChIP-seq technology to analyze methylation profiles in breast cancer cell lines and primary human mammary epithelial cells, revealing the unique interplay of DNA methylation with histone modifications the H3K9me3 and H3K27me3 modifications (40). In 2012, Akalin et al. released the methylKit R package to help researchers quickly identify statistically significant DNA methylation sites or regions, easing data analysis, and enhancing research efficiency (41).
At this phase, the Carolina Breast Cancer Study (cluster #2, Year = 2010) had a significant impact as a long-term, population-based research project conducted by the University of North Carolina at Chapel Hill, substantially influencing breast cancer epidemiology, molecular biology, and genetic analysis (42–44). Furthermore, research has expanded from individual gene changes to mechanisms involved in EMT (cluster #3, Year = 2009), such as the discovery by Neha et al. that Sox4 regulates EMT via the epigenetic modifier Ezh2, promoting breast cancer (45).
4.2.3 Epigenetic therapy
Clinical application is the ultimate purpose and value of academic research, serving as the culmination of theoretical endeavors. This study revealed that the primary direction to improve breast cancer prognosis through targeted epigenetics is epigenetic reactivation (#10, Year = 2013), which involves the reactivation of silenced tumor suppressor genes. Among these, HDAC inhibitors (cluster #4, Year = 2013) are representative regulatory drugs that indirectly induce histone acetylation by disrupting HDAC activity, leading to the re-expression of regulatory genes in cancer cells and reversing malignant phenotypes (46). To date, more than 60 clinical studies have been registered in ClinicalTrials.gov for breast cancer treatments (47), including Vorinostat, Entinostat, and Panobinostat (48–50). However, due to their toxicity and risk of overdose, HDAC inhibitors are often combined with drugs like Tamoxifen and Paclitaxel, targeting multiple oncogenic signaling pathways to overcome drug resistance in advanced breast cancer (51–53). DNA methyltransferase inhibitors, such as azacitidine and decitabine, are also a class of epigenetic therapeutic drugs. During DNA replication, these agents bind to DNA methyltransferases, inhibiting gene methylation and subsequently reactivating silenced tumor suppressor genes. Paradoxically, recent research indicates that treatment with these inhibitors may also result in the de novo hypermethylation of specific CpG sites (54). This novel mechanism could influence both their therapeutic effects and side effects. Clinically, the U.S. Food and Drug Administration has approved azacitidine and decitabine for the treatment of myelodysplastic syndromes and certain leukemias (55). Although their efficacy as monotherapies for breast cancer remains limited, they have a certain inhibitory effect on tumor invasion and metastasis, particularly in triple-negative breast cancer (56). Recently, an epigenetic therapeutic drug targeting the histone methyltransferase EZH2 has also shown certain potential in the treatment of breast cancer (57).
4.2.4 New insights into epigenetics of breast cancer
Thanks to advancements in whole-genome projects and bioinformatics analysis methods, researchers can integrate multi-omics data (cluster #0, pan-cancer analysis, Year = 2018) to analyze subtype-specific epigenetic characteristics of breast cancer and the regulatory role of the TME (cluster #9, epigenetic deregulation, Year = 2020). Llorente et al. combined proteomics, transcriptomics, epigenomics, chromatin accessibility, and functional analysis, finding that MAF interacts directly with estrogen receptor α, forming a unique chromatin landscape conducive to metastasis. They also discovered that the histone demethylase KDM1A promotes epigenomic remodeling, further impacting breast cancer metastasis (58). In this cluster, Trnkova et al.’s analysis of epigenetic dysregulation in the TME (59), and Zhou et al.’s exploration of the epigenetic regulation of TNBC (60) have significant influence. Moreover, a new direction, methylation-based biological age (cluster #7, Year = 2016), has emerged, which uses mathematical modeling to construct an epigenetic clock for assessing cellular aging and cancer prognosis, as seen in Kresovich et al.’s predictive model (61, 62).
4.3 Future development trends
Keywords burst analysis identifies metabolism, EMT, TNBC, treatment resistance, invasion, prognosis, and the TME as persistent hot topics. Analyzing significant literature from the past 5 years, particularly 2024, suggests future trends in breast cancer epigenetics, with a focus on the following directions:
4.3.1 Targeted epigenetic memory clearance for treating drug resistance and recurrence
In this study, keywords such as “epigenetic memory,” “transcription factor,” “cisplatin response,” “dormant breast cancer cell,” and “therapy resistance” were identified in the 2024 clustering, suggesting that this is a potential trend. Studies have shown that genetic changes in gene expression or behavior induced by prior stimuli can be recorded by the organism as epigenetic memory (63). These epigenetic marks facilitate the untimely reactivation of dormant tumor cells, leading to tumor recurrence and treatment resistance (64–66). Bian et al. reported that radiotherapy leads to the epigenetic activation of thrombospondin 1(THBS1), increasing the difficulty of healing cancer wounds, and upon re-injury, the expression of THBS1 increases, affecting wound healing, illustrating the impact of epigenetic memory (67).
As for breast cancer, the HDAC inhibitor trichostatin A helps overcome the resistance of breast cancer cells to tamoxifen (68), which provides reliable evidence for this research direction. A clinical study indicates that treatment in the pre-neoadjuvant window with DNA methyltransferase inhibitor (decitabine, 15 mg/m² × 4 doses over 5 days) and T-cell immune checkpoint inhibition (pembrolizumab, 200 mg, administered 2 weeks apart) combined, is beneficial for the treatment of locally advanced HER2-negative breast cancer (69).
4.3.2 Improving the metabolic and epigenetic interaction network, and treating breast cancer through a synthetic lethal strategy
Currently, researchers generally believe that tumor occurrence results from interactive networks rather than single gene mutations. It has been proven that hypoxia induces epigenetic reprogramming of normal fibroblasts, thereby producing a pro-glycolytic transcriptome similar to that of tumor-associated fibroblasts, leading to the occurrence of breast cancer (22). Combining epigenetic drugs with other antineoplastic agents is a promising treatment strategy for advanced cancers (70, 71). The therapeutic effect of HDAC inhibitors combined with anticancer drugs, such as NSC-3852 combined with olaparib (72), romidepsin and ATR inhibitors (73), is significantly enhanced.
The principle of synthetic lethality, which refers to the loss of viability resulting from the disruption of two genes, which, individually, do not cause lethality This principle is primarily used to treat advanced ovarian and breast cancers associated with BRCA1 or BRCA2 gene mutations (74). Notably, the clinical success of poly (ADP-ribose) polymerase inhibitors (PARPi) has made synthetic lethality an attractive targeted therapeutic strategy; however, its limitation in treatment resistance has also gradually become apparent (75, 76).
In this study, “oxidative phosphorylation,” “synthetic lethality strategies,” and “epigenetic mechanism” were identified, indicating that these may be directions for future research.
4.3.3 Single-cell sequencing tracks the dynamic changes in epigenetic modifications in breast cancer across space and time
Single-cell sequencing technology utilizes optimized next-generation DNA sequencing (NGS) to detect the sequences of individual cells, allowing the acquisition of cell sequence differences in specific microenvironments to facilitate the study of functional differences (77). Scholars have employed single-cell sequencing technology to analyze epigenetic alterations in breast cancer. Fang et al. employed single-cell sequencing analysis and scRNA-seq data to define nine CSs from normal tissues and tamoxifen-treated recurrent tumors. They ultimately demonstrated that BMP7 plays an oncogenic role in tamoxifen-resistant breast cancer cells by modulating MAPK signaling pathways. Cancer-specific enhancers in male breast cancer have also been identified through single-cell sequencing technology (78). In addition, single-cell sequencing technology has been integrated with omics approaches to reanalyze and define the regulatory logic of breast cancer. This integration has identified the most probable cells of origin for subtype-specific breast tumors and implemented linear mixed-effects models to quantify the associations between regulatory elements and gene expression in malignant and normal cells (79).
5 Limitations
This bibliometric analysis offers a comprehensive overview of epigenetic research in breast cancer; however, it presents several limitations. First, relying exclusively on the WoSCC database might lead to an incomplete literature compilation, introducing potential biases. Despite this limitation, the WoSCC is recognized as a globally authoritative source, featuring high-impact academic journals and citation indexes, which help maintain the study’s credibility. Second, since there is currently no reliable method for the batch processing of self-citations and for the identification of predatory journals, the research results may be subject to a certain degree of bias. Third, the method’s focus on keyword co-occurrence and citation networks might miss emerging research trends not yet highly cited. To compensate for these deficiencies in recent research evaluations, future researchers may consider integrating journal impact, author influence, and advanced artificial intelligence technologies to develop new comprehensive evaluation models.
6 Conclusion
This bibliometric analysis provides a comprehensive overview of the evolution of epigenetic research in breast cancer over the past two decades. This highlights the progression from single-site methylation studies to the systematic exploration of epigenetic pathways and therapies, such as HDAC inhibitors, showcasing the field’s maturation. Future research should focus on targeted epigenetic memory clearance to combat drug resistance and recurrence, enhancement of the metabolic and epigenetic interaction networks, and the application of synthetic lethal strategies in breast cancer treatment. Additionally, interdisciplinary collaborations and the integration of emerging technologies, such as single-cell sequencing, are recommended to facilitate dynamic monitoring of epigenetic changes in drug development.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
HY: Writing – original draft, Conceptualization, Writing – review & editing, Methodology. YF: Writing – review & editing, Methodology, Resources. HW: Supervision, Formal Analysis, Writing – original draft. TL: Investigation, Writing – original draft, Data curation. QC: Supervision, Writing – review & editing. HL: Supervision, Writing – review & editing, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. Funding was received for the research and/or publication of this article: National Natural Science Foundation of China (8220150800); Hunan Provincial Department of Education Excellent Youth Project (23B0341); Hunan Provincial Administration of Traditional Chinese Medicine Scientific Research Program (B2024051).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1619346/full#supplementary-material
References
1. Kim J, Harper A, McCormack V, Sung H, Houssami N, Morgan E, et al. Global patterns and trends in breast cancer incidence and mortality across 185 countries. Nat Med. (2025) 31:1154–62. doi: 10.1038/s41591-025-03502-3
2. Xiong X, Zheng L-W, Ding Y, Chen Y-F, Cai Y-W, Wang L-P, et al. Breast cancer: Pathogenesis and treatments. Signal Transduct Target Ther. (2025) 10:49. doi: 10.1038/s41392-024-02108-4
3. Thakur C, Qiu Y, Pawar A, and Chen F. Epigenetic regulation of breast cancer metastasis. Cancer Metastasis Rev. (2024) 43:597–619. doi: 10.1007/s10555-023-10146-7
4. Fackler MJ, McVeigh M, Evron E, Garrett E, Mehrotra J, Polyak K, et al. DNA methylation of RASSF1A, HIN-1, RAR-beta, cyclin D2 and twist in in situ and invasive lobular breast carcinoma. Int J Cancer. (2003) 107:970–5. doi: 10.1002/ijc.11508
5. Yomtoubian S, Lee SB, Verma A, Izzo F, Markowitz G, Choi H, et al. Inhibition of EZH2 catalytic activity selectively targets a metastatic subpopulation in triple-negative breast cancer. Cell Rep. (2020) 30:755–. doi: 10.1016/j.celrep.2019.12.056
6. Bates SE. Epigenetic therapies for cancer. N Engl J Med. (2020) 383:650–63. doi: 10.1056/NEJMra1805035
7. Cheng MW, Mitra M, and Coller HA. Pan-cancer landscape of epigenetic factor expression predicts tumor outcome. Commun Biol. (2023) 6:1–18. doi: 10.1038/s42003-023-05459-w
9. Trujillo CM and Long TM. Document co-citation analysis to enhance transdisciplinary research. Sci Adv. (2018) 4:e1701130. doi: 10.1126/sciadv.1701130
10. Manoj Kumar L and George RJ PSA. Bibliometric analysis for medical research. Indian J Psychol Med. (2023) 45:277–82. doi: 10.1177/02537176221103617
11. Chen C. Searching for intellectual turning points: progressive knowledge domain visualization. Proc Natl Acad Sci. (2004) 101:5303–10. doi: 10.1073/pnas.0307513100
12. Chen C. CiteSpace II: Detecting and visualizing emerging trends and transient patterns in scientific literature. J Am Soc Inf Sci Technol. (2006) 57:359–77. doi: 10.1002/asi.20317
13. Zhu W, Xiao Y, and Xie L. Visualization analysis of poisoning-related research based on CiteSpace. Front Public Health. (2025) 13:1592916. doi: 10.3389/fpubh.2025.1592916
14. Chen C and Song M. Visualizing a field of research: a methodology of systematic scientometric reviews. PloS One. (2019) 14:e0223994. doi: 10.1371/journal.pone.0223994
15. Amjad T, Shahid N, Daud A, and Khatoon A. Citation burst prediction in a bibliometric network. Scientometrics. (2022) 127:2773–90. doi: 10.1007/s11192-022-04344-3
16. Egghe L. Theory and practise of the g-index. Scientometrics. (2006) 69:131–52. doi: 10.1007/s11192-006-0144-7
17. Bukar UA, Sayeed MS, Razak SFA, Yogarayan S, Amodu OA, and Mahmood RAR. A method for analyzing text using VOSviewer. Methodsx. (2023) 11:102339. doi: 10.1016/j.mex.2023.102339
18. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
19. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. (2013) 14:R115. doi: 10.1186/gb-2013-14-10-r115
20. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. (2009) 41:178–86. doi: 10.1038/ng.298
21. Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and Malignant phenotype suppression. J Natl Cancer Inst. (2001) 93:691–9. doi: 10.1093/jnci/93.9.691
22. Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep. (2020) 31:107701. doi: 10.1016/j.celrep.2020.107701
23. Corr EM, Brown EJ, Albers KB, Yamaguchi N, Narke D, Schlegel M, et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat Med. (2020) 26:1452–8. doi: 10.1038/s41591-020-0964-7
24. Chen H, Yao J, Bao R, Dong Y, Zhang T, Du Y, et al. Cross-talk of four types of RNA modification writers defines tumor microenvironment and pharmacogenomic landscape in colorectal cancer. Mol Cancer. (2021) 20:29. doi: 10.1186/s12943-021-01322-w
25. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
26. Waks AG and Winer EP. Breast cancer treatment: a review. JAMA. (2019) 321:288. doi: 10.1001/jama.2018.19323
27. Loibl S, Poortmans P, Morrow M, Denkert C, and Curigliano G. Breast cancer. Lancet. (2021) 397:1750–69. doi: 10.1016/S0140-6736(20)32381-3
28. Horvath S and Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. (2018) 19:371–84. doi: 10.1038/s41576-018-0004-3
29. Thirumurthi U, Shen J, Xia W, LaBaff AM, Wei Y, Li C-W, et al. MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci Signaling. (2014) 7:ra71. doi: 10.1126/scisignal.2005076
30. Yamaguchi H, Du Y, Nakai K, Ding M, Chang S-S, Hsu JL, et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene. (2018) 37:208–17. doi: 10.1038/onc.2017.311
31. Müller HM, Widschwendter A, Fiegl H, Ivarsson L, Goebel G, Perkmann E, et al. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res. (2003) 63:7641–5.
32. Yadav P, Masroor M, Nandi K, Kaza RCM, Jain SK, Khurana N, et al. Promoter methylation of BRCA1, DAPK1 and RASSF1A is associated with increased mortality among Indian women with breast cancer. Asian Pac J Cancer Prev: APJCP. (2018) 19:443–8. doi: 10.22034/APJCP.2018.19.2.443
33. Wei M, Grushko TA, Dignam J, Hagos F, Nanda R, Sveen L, et al. BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res. (2005) 65:10692–9. doi: 10.1158/0008-5472.CAN-05-1277
34. Oshiro MM, Kim CJ, Wozniak RJ, Junk DJ, Muñoz-Rodríguez JL, Burr JA, et al. Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res: BCR. (2005) 7:R669–680. doi: 10.1186/bcr1273
35. Levine JJ, Stimson-Crider KM, and Vertino PM. Effects of methylation on expression of TMS1/ASC in human breast cancer cells. Oncogene. (2003) 22:3475–88. doi: 10.1038/sj.onc.1206430
36. Fackler MJ, McVeigh M, Mehrotra J, Blum MA, Lange J, Lapides A, et al. Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res. (2004) 64:4442–52. doi: 10.1158/0008-5472.CAN-03-3341
37. Lee JJ, Ko E, Cho J, Park HY, Lee JE, Nam SJ, et al. Methylation and immunoexpression of p16(INK4a) tumor suppressor gene in primary breast cancer tissue and their quantitative p16(INK4a) hypermethylation in plasma by real-time PCR. Korean J Pathol. (2012) 46:554–61. doi: 10.4132/KoreanJPathol.2012.46.6.554
38. Elliott GO, Johnson IT, Scarll J, Dainty J, Williams EA, Garg D, et al. Quantitative profiling of CpG island methylation in human stool for colorectal cancer detection. Int J Colorectal Dis. (2013) 28:35–42. doi: 10.1007/s00384-012-1532-5
39. Locke I, Kote-Jarai Z, Fackler MJ, Bancroft E, Osin P, Nerurkar A, et al. Gene promoter hypermethylation in ductal lavage fluid from healthy BRCA gene mutation carriers and mutation-negative controls. Breast Cancer Res: BCR. (2007) 9:R20. doi: 10.1186/bcr1657
40. Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. (2012) 22:246–58. doi: 10.1101/gr.125872.111
41. Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. (2012) 13:R87. doi: 10.1186/gb-2012-13-10-R87
42. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the carolina breast cancer study. JAMA. (2006) 295:2492–502. doi: 10.1001/jama.295.21.2492
43. O’Brien KM, Cole SR, Tse C-K, Perou CM, Carey LA, Foulkes WD, et al. Intrinsic breast tumor subtypes, race, and long-term survival in the carolina breast cancer study. Clin Cancer Res: Off J Am Assoc Cancer Res. (2010) 16:6100–10. doi: 10.1158/1078-0432.CCR-10-1533
44. Conway K, Edmiston SN, May R, Kuan PF, Chu H, Bryant C, et al. DNA methylation profiling in the carolina breast cancer study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast Cancer Res: BCR. (2014) 16:450. doi: 10.1186/s13058-014-0450-6
45. Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling EzH2 expression and epigenetic reprogramming. Cancer Cell. (2013) 23:768–83. doi: 10.1016/j.ccr.2013.04.020
46. West AC and Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. (2014) 124:30–9. doi: 10.1172/JCI69738
47. Huang M, Zhang J, Yan C, Li X, Zhang J, and Ling R. Small molecule HDAC inhibitors: promising agents for breast cancer treatment. Bioorg Chem. (2019) 91:103184. doi: 10.1016/j.bioorg.2019.103184
48. Ma W, Sun J, Xu J, Luo Z, Diao D, Zhang Z, et al. Sensitizing triple negative breast cancer to tamoxifen chemotherapy via a redox-responsive vorinostat-containing polymeric prodrug nanocarrier. Theranostics. (2020) 10:2463–78. doi: 10.7150/thno.38973
49. Roussos Torres ET, Ho WJ, Danilova L, Tandurella JA, Leatherman J, Rafie C, et al. Entinostat, nivolumab and ipilimumab for women with advanced HER2-negative breast cancer: a phase ib trial. Nat Cancer. (2024) 5:866–79. doi: 10.1038/s43018-024-00729-w
50. Qin G, Li Y, Xu X, Wang X, Zhang K, Tang Y, et al. Panobinostat (LBH589) inhibits wnt/β-catenin signaling pathway via upregulating APCL expression in breast cancer. Cell Signalling. (2019) 59:62–75. doi: 10.1016/j.cellsig.2019.03.014
51. Raha P, Thomas S, Thurn KT, Park J, and Munster PN. Combined histone deacetylase inhibition and tamoxifen induces apoptosis in tamoxifen-resistant breast cancer models, by reversing bcl-2 overexpression. Breast Cancer Res: BCR. (2015) 17:26. doi: 10.1186/s13058-015-0533-z
52. Wang L, Li H, Ren Y, Zou S, Fang W, Jiang X, et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. (2016) 7:e2063. doi: 10.1038/cddis.2015.328
53. Hasan A, Khan NA, Uddin S, Khan AQ, and Steinhoff M. Deregulated transcription factors in the emerging cancer hallmarks. Semin Cancer Biol. (2024) 98:31–50. doi: 10.1016/j.semcancer.2023.12.001
54. Brueckner B and Lyko F. DNA methyltransferase inhibitors: old and new drugs for an epigenetic cancer therapy. Trends Pharmacol Sci. (2004) 25:551–4. doi: 10.1016/j.tips.2004.09.004
55. Thomas X. DNA methyltransferase inhibitors in acute myeloid leukemia: Discovery, design and first therapeutic experiences. Expert Opin Drug Discov. (2012) 7:1039–51. doi: 10.1517/17460441.2012.722618
56. Wong KK. DNMT1: A key drug target in triple-negative breast cancer. Semin Cancer Biol. (2021) 72:198–213. doi: 10.1016/j.semcancer.2020.05.010
57. SChade AE, Perurena N, Yang Y, Rodriguez CL, Krishnan A, Gardner A, et al. AKT and EZH2 inhibitors kill TNBCs by hijacking mechanisms of involution. Nature. (2024) 635:755–63. doi: 10.1038/s41586-024-08031-6
58. Llorente A, Blasco MT, Espuny I, Guiu M, Ballaré C, Blanco E, et al. MAF amplification licenses ERα through epigenetic remodelling to drive breast cancer metastasis. Nat Cell Biol. (2023) 25:1833–47. doi: 10.1038/s41556-023-01281-y
59. Trnkova L, Buocikova V, Mego M, Cumova A, Burikova M, Bohac M, et al. Epigenetic deregulation in breast cancer microenvironment: implications for tumor progression and therapeutic strategies. BioMed Pharmacother. (2024) 174:116559. doi: 10.1016/j.biopha.2024.116559
60. Zhou L and Yu C-W. Epigenetic modulations in triple-negative breast cancer: therapeutic implications for tumor microenvironment. Pharmacol Res. (2024) 204:107205. doi: 10.1016/j.phrs.2024.107205
61. Kresovich JK, Xu Z, O’Brien KM, Weinberg CR, Sandler DP, and Taylor JA. Epigenetic mortality predictors and incidence of breast cancer. Aging (Milano). (2019) 11:11975–87. doi: 10.18632/aging.102523
62. Kresovich JK, Xu Z, O’Brien KM, Weinberg CR, Sandler DP, and Taylor JA. Methylation-based biological age and breast cancer risk. JNCI: J Natl Cancer Inst. (2019) 111:1051–8. doi: 10.1093/jnci/djz020
63. D’Urso A and Brickner JH. Mechanisms of epigenetic memory. Trends Genet: TIG. (2014) 30:230–6. doi: 10.1016/j.tig.2014.04.004
64. Qiu J, Xu B, Ye D, Ren D, Wang S, Benci JL, et al. Cancer cells resistant to immune checkpoint blockade acquire interferon-associated epigenetic memory to sustain T cell dysfunction. Nat Cancer. (2023) 4:43–61. doi: 10.1038/s43018-022-00490-y
65. Garcia-Martinez L, Zhang Y, Nakata Y, Chan HL, and Morey L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat Commun. (2021) 12:1786. doi: 10.1038/s41467-021-22024-3
66. Rosano D, Sofyali E, Dhiman H, Ghirardi C, Ivanoiu D, Heide T, et al. Long-term multimodal recording reveals epigenetic adaptation routes in dormant breast cancer cells. Cancer Discov. (2024) 14:866–89. doi: 10.1158/2159-8290.CD-23-1161
67. Bian X, Piipponen M, Liu Z, Luo L, Geara J, Chen Y, et al. Epigenetic memory of radiotherapy in dermal fibroblasts impairs wound repair capacity in cancer survivors. Nat Commun. (2024) 15:9286. doi: 10.1038/s41467-024-53295-1
68. Fan J, Yin W-J, Lu J-S, Wang L, Wu J, Wu F-Y, et al. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J Cancer Res Clin Oncol. (2008) 134:883–90. doi: 10.1007/s00432-008-0354-x
69. Bear HD, Deng X, Bandyopadhyay D, Idowu M, Jenkins TM, Kmieciak M, et al. T-cell immune checkpoint inhibition plus hypomethylation for locally advanced HER2-negative breast cancer: A phase 2 neoadjuvant window trial of decitabine and pembrolizumab followed by standard neoadjuvant chemotherapy. J Immunother Cancer. (2025) 13:e010294. doi: 10.1136/jitc-2024-010294
70. Bamberg LV, Heigwer F, Wandmacher AM, Singh A, Betge J, Rindtorff N, et al. Targeting euchromatic histone lysine methyltransferases sensitizes colorectal cancer to histone deacetylase inhibitors. Int J Cancer. (2022) 151:1586–601. doi: 10.1002/ijc.34155
71. Das C, Bhattacharya A, Adhikari S, Mondal A, Mondal P, Adhikary S, et al. A prismatic view of the epigenetic-metabolic regulatory axis in breast cancer therapy resistance. Oncogene. (2024) 43:1727–41. doi: 10.1038/s41388-024-03054-9
72. Sasaki Y, Inouchi T, Kise C, Nakatsuka R, Inoue A, Masutani M, et al. NSC-3852 synergistically enhances the cytotoxicity of olaparib in oral squamous cell carcinoma. Biochem Biophys Res Commun. (2025) 744:151166. doi: 10.1016/j.bbrc.2024.151166
73. Kurz L, Miklyaeva A, Skowron MA, Overbeck N, Poschmann G, Becker T, et al. ARID1A regulates transcription and the epigenetic landscape via POLE and DMAP1 while ARID1A deficiency or pharmacological inhibition sensitizes germ cell tumor cells to ATR inhibition. Cancers. (2020) 12:905. doi: 10.3390/cancers12040905
74. Patel PS, Algouneh A, and Hakem R. Exploiting synthetic lethality to target BRCA1/2-deficient tumors: where we stand. Oncogene. (2021) 40:3001–14. doi: 10.1038/s41388-021-01744-2
75. Wu S, Yao X, Sun W, Jiang K, and Hao J. Exploration of poly (ADP-ribose) polymerase inhibitor resistance in the treatment of BRCA1/2-mutated cancer. Genes Chromosomes Cancer. (2024) 63:e23243. doi: 10.1002/gcc.23243
76. Ciccone MA, Adams CL, Bowen C, Thakur T, Ricker C, Culver JO, et al. Inhibition of poly(ADP-ribose) polymerase induces synthetic lethality in BRIP1 deficient ovarian epithelial cells. Gynecol Oncol. (2020) 159:869–76. doi: 10.1016/j.ygyno.2020.09.040
77. Eberwine J, Sul J-Y, Bartfai T, and Kim J. The promise of single-cell sequencing. Nat Methods. (2014) 11:25–7. doi: 10.1038/nmeth.2769
78. Kim H, Wisniewska K, Regner MJ, Thennavan A, Spanheimer PM, and Franco HL. Single-cell transcriptional and epigenetic profiles of male breast cancer nominate salient cancer-specific enhancers. Int J Mol Sci. (2023) 24:13053. doi: 10.3390/ijms241713053
Keywords: bibliometric study, epigenetics, breast cancer, visualization, bibliometric, citespace, VOSviewer
Citation: Yang H, Fang Y, Wang H, Lu T, Chen Q and Liu H (2025) Progress in epigenetic research of breast cancer: a bibliometric analysis since the 2000s. Front. Oncol. 15:1619346. doi: 10.3389/fonc.2025.1619346
Received: 28 April 2025; Accepted: 21 August 2025;
Published: 05 September 2025.
Edited by:
Maha Mohamed Saber-Ayad, University of Sharjah, United Arab EmiratesReviewed by:
Iros Barozzi, Medical University of Vienna, AustriaSungchan Gwark, Ewha Womans University Seoul Hospital, Republic of Korea
Dilek Cansu Gurer Er, Izmir Institute of Technology, Türkiye
Copyright © 2025 Yang, Fang, Wang, Lu, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Liu, NDcwNTI2NjE3QHFxLmNvbQ==