 Ashish Shrestha
Ashish Shrestha Yangchan Li
Yangchan Li Lixia Huang
Lixia Huang Shaoli Li
Shaoli Li Yanbin Zhou
Yanbin Zhou Jincui Gu
Jincui Gu Ziying Lin
Ziying Lin- 1Department of Respiratory and Critical Care Medicine, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 2Department of Radiation, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
Background: KEAP1 and STK11 are frequently mutated in NSCLC, and are associated with compromised response to immunotherapy, the underlying mechanism of which is not fully understood.
Methods: To assess the impact of KEAP1/STK11 mutations on immune profiles, we analyzed RNA-seq data from the TCGA lung cancer cohort and the GSE72094 cohort. Differential expression, pathway enrichment, and correlation analyses were performed to elucidate the underlying mechanisms. Key findings were further validated using a single-cell RNA-seq dataset. Additionally, the prognostic significance of these mutations in immunotherapy was evaluated using immune checkpoint inhibitor (ICI) cohorts from our medical center and published studies.
Results: We observed the simultaneous upregulation of pathways involved in oxidoreductase activity and down-regulation of interferon signaling pathways by mutation of KEAP1 or STK11, and developed a redox signature driven by KEAP1/STK11 mutations. Redox score exhibited negative correlation with expression of STING/MDA5, which function as sensors of dsDNA/dsRNA and activate downstream interferon signaling. Redox score and STING/MDA5 expression manifested the exact opposite impact on the infiltrating level of most immune cells. Analysis of single cell RNA sequencing dataset indicated that redox phenotype specifically impacted expressional level of STING/MDA5 in cancer cells but other cell types within tumor immune microenvironment. Prognostic significance of redox signature was validated in immunotherapy cohorts of lung cancer and melanoma, which all indicated a significant worse outcome associated with higher redox score.
Conclusions: Collectively, we associated the redox status mediated by loss-function mutations of KEAP1 or STK11 to immune evasion and immunotherapeutic resistance by suppressing STING/MDA5 expression and interferon signaling of cancer cells. Our findings link redox homeostasis to STING/MDA5 expression and tumor immunogenicity, raising the possibility that targeting this axis could represent a future strategy to enhance ICI efficacy.
Introduction
Non-small cell lung cancer (NSCLC), which constitutes the majority of lung cancer cases, continues to pose a significant challenge in oncology due to its aggressive nature and limited treatment options (1). The advent of immunotherapy has revolutionized cancer treatment, offering new hope for patients with advanced NSCLC (2–5). However, the response to immune checkpoint inhibitors varies widely, and understanding the underlying mechanisms of resistance is essential for improving therapeutic outcomes.
One approach to address immunotherapy resistance is to define tumor-intrinsic genetic mutations that modulate the tumor immune microenvironment (TIME) and therapy response. Among the myriad of genetic alterations that drive lung carcinogenesis, mutations in the genes like KEAP1 (Kelch-like ECH-associated protein 1) and STK11 (serine/threonine kinase 11) have emerged as significant players in influencing immune surveillance and therapeutic response to immune check-point inhibitors (ICIs) (6, 7). KEAP1 is mutated in approximately 20% of lung adenocarcinomas and squamous cell carcinomas, as identified by cancer genome sequencing studies (8). It is the third most frequently mutated gene in NSCLC, encodes a protein crucial for the ubiquitination and proteasomal degradation of Nuclear factor erythroid 2-related factor 2 (NRF2), and works as a critical component during the anti-oxidant response (9–11). Its role extends beyond cellular homeostasis, as alterations in KEAP1 function have been implicated in modulating immunogenicity and restricting the efficacy of immunotherapeutic interventions (7, 12, 13). STK11, also known as LKB1 (Liver kinase B1), whose loss-function mutation occurs in approximately 10% of NSCLC, is a tumor suppressor gene that plays a critical role in cellular processes such as metabolic reprogramming, cell polarity, and proliferation (14–17). In recent studies, STK11 loss has also been associated with reduced immune-infiltration and poor response to immunotherapy (18, 19). However, the mechanism by which KEAP1 or STK11 mutations impeding tumor immunogenicity and immunotherapeutic response remains elusive.
The crosstalk between KEAP1 and STK11 in the context of metabolic reprogramming and therapeutic resistance has been reported previously (20). For instance, loss of LKB1 upregulates the KEAP1-NRF2 pathway, driving glutamine dependence and resistance to radiotherapy and ferroptosis (21, 22). Whether metabolic alteration is also the common mechanism shared by KEAP1 and STK11 in immune remodeling is yet to be evaluated. Specifically, mutations in KEAP1 and STK11 are both known to be involved in modulating cellular redox homeostasis. A growing body of evidence suggests that the redox phenotype of tumors, characterized by alterations in oxidative stress response pathways, can significantly influence the immunological landscape (23–25). In particular, tumors with a high redox phenotype have been associated with a T cell-exclusion microenvironment, which is generally less responsive to immunotherapeutic interventions (26). Whether alteration of redox homeostasis is the key player of immune suppression among tumors with mutant KEAP1 or STK11 and how it interacts with TIME is yet to be explored.
This article aims to dissect the complex relationship between KEAP1/STK11 mutations, the redox homeostasis, and immune remodeling in NSCLC. Our results showed that tumors with mutant KEAP1/STK11 exhibited upregulation of oxidoreductase activity and repression of interferon signaling. KEAP1/STK11 mutations or redox phenotype are associated with downregulation of genes involved in dsDNA/dsRNA sensing like STING and MDA5, which leads to repression of downstream interferon signaling and immune exclusion. Redox signature was predictive of immunotherapeutic outcomes in NSCLC and other cancer type.
Methods
Patients’ cohort
Discovery cohorts
To explore the correlation between KEAP1/STK11 mutations and outcomes of immunotherapy in NSCLC, we retrospectively assembled a cohort of 185 consecutive patients in our medical center who had received immunotherapy treatment and undergone molecular profiling between March 2010 and April 2023 (as detailed in Supplementary Table 1). Clinical data including therapeutic regiment, line of treatment, progression free survival (PFS), overall survival (OS), response to immunotherapy as accessed by Response Evaluation Criteria in Solid Tumors (RECIST) standard, mutation status of KEAP1 and STK11 were retrieved from medical records. Additionally, we incorporated an independent cohort from the Memorial Sloan Kettering Cancer Center and MD Anderson Cancer Center (MSKCC/MDACC cohort) (27), comprising patients with advanced NSCLC who had undergone Programmed Death- (Ligand) 1 (PD-(L)1) checkpoint blockade and comprehensive genomic profiling of their tumors (n=179). Clinical data, including treatment regimen, therapeutic outcomes, OS, PFS, and the mutation status of KEAP1 and STK11, were sourced from the cBioportal database (https://www.cbioportal.org/).
In order to assess the influence of KEAP1/STK11 mutations on immune profiles, we included NSCLC patients (n=1144) with available whole-exome sequencing data and genomic mutational profiles from The Cancer Genome Atlas (TCGA). Clinical information, RNA sequencing data and mutational status of KEAP1/STK11 were retrieved from the cBioportal database, which can be accessed through the following link: https://www.cbioportal.org/study/summary?id=nsclc_tcga_broad_2016. Additionally, we acquired another dataset, GSE72094, comprising 441 lung adenocarcinoma tumors that had been profiled using microarray-based gene expression assays and included information on STK11 mutations. This dataset was sourced from the Gene Expression Omnibus (GEO) under the accession number GSE72094. A comprehensive summary of the details for TCGA cohort and GSE72094 cohort were shown in Supplementary Tables 2 and 3 respectively.
Single-cell RNA sequencing cohort
A published dataset (28) of single-cell RNA sequencing (scRNA-seq), which included treatment-naïve samples from 42 patients diagnosed with advanced NSCLC, was repurposed in our study as scRNA-seq cohort. Raw data of RNA sequencing, which involved 88794 single cells, was obtained from the GEO database under the accession number GSE148071. Processed data of scRNA-seq were presented in Supplementary Table 4.
Immunotherapy cohorts
In order to confirm the predictive significance of the redox signature for immunotherapy outcomes, we methodically gathered pre-treatment transcriptomic data and clinical information from two immune checkpoint inhibitor (ICI) cohorts: the Ravi lung cancer cohort (29), the Gide melanoma cohort (30). All patients within these cohorts were treated with anti-PD-1 therapy, specifically nivolumab or pembrolizumab. For the Ravi lung cancer cohort, we exclusively included cases where ICIs were administered as a first-line treatment. We collected clinical data such as PFS, OS, and the clinical response to immunotherapy, which was evaluated using the RECIST standards. A comprehensive summary of the details for all ICI cohorts is presented in Supplementary Tables 5 and 6.
Assessment of immune infiltration based on RNA-seq data
Infiltrating levels of 20 leukocyte subtypes were quantified based on the expression profiles of their corresponding leukocyte signatures, which were derived from previously published research (31). The enrichment scores for each leukocyte signature were determined using the single-sample gene set enrichment analysis (ssGSEA) method, as implemented by the R-packages (GSEABase and GSVA) (32). The gene signatures for the 20 leukocyte subtypes are detailed in the Supplementary Table 7.
Differential expression analysis and pathway analysis
Genes that were differentially expressed between two comparator groups—mutant KEAP1 versus wild-type KEAP1, and mutant STK11 versus wild-type STK11—were identified using differential expression analysis facilitated by the DESeq2 package (version 1.26.0) within the R software environment (version 3.6.3). Genes were classified as differentially expressed (DEGs) if they exhibited a log2(fold change) greater than 1 and a P-value less than 0.05. The gene lists of DEGs that were consistently up-regulated in tumors with mutant KEAP1 and STK11 across all datasets (TCGA-luad, TCGA-lusc, GSE72094) were subjected to Gene Ontology (GO) analysis. This analysis was conducted using the ‘msigdbr’ package (version 7.5.1) and the clusterProfiler package (version 4.12.6) within the R software environment (version 4.4.1). Conversely, DEGs that were down-regulated in both mutant KEAP1 and STK11 tumors were analyzed using Reactome pathway analysis, which was implemented with the ReactomePA R package (version 1.48.0).
Development of redox signature and scoring pipeline
Differential expression analysis was performed to identify genes upregulated by mutant KEAP1 in the TCGA-LUAD and TCGA-LUSC cohorts, as well as those upregulated by mutant STK11 in the TCGA-LUAD and GSE72094 cohorts. From these analyses, we identified 148 overlapping genes common to all four upregulated gene sets. These genes were subsequently subjected to GO pathway enrichment analysis, which revealed that the top 8 enriched pathways were associated with oxidoreductase activity and antioxidant functions. Further refinement of the 148-gene pool identified 26 genes (Supplementary Table 7) implicated in these 8 pathways, which we designated as the redox signature. To quantify the enrichment of this redox signature in individual tumors, we applied the single-sample Gene Set Enrichment Analysis (ssGSEA) algorithm. This method ranks genes based on expression and calculates a normalized enrichment score (NES) for each sample, eliminating the need for batch adjustment. The ssGSEA analysis was performed using the normalized gene expression matrix and an unweighted redox gene list as input, implemented via the R packages GSEABase and GSVA (see Supplementary Material for R code).
Single-cell sequencing analysis
Pre-processed RNA sequencing data of single cells derived from 42 treatment naïve NSCLC tumors were obtained from the public available dataset GSE148071. The Seurat package (version 4.4.1) was employed to create the object, filtering out cells of poor quality based on the following criteria: lower than 200 or higher than 5000 expressed genes, and cells with >20% mitochondrial content. Expression matrices were normalized by function NormalizeData and ScaleData. The top 2000 variable genes, identified by the FindVariableFeatures function, were used for principal components analysis. The first 30 principal components and resolution 0.6 were used with FindClusters function to generate 30 cell clusters, which was assigned to 8 major cell types based on the expression pattern of the following canonical markers: Endothelial cells (PECAM1, FLT1, VWF), cancer cells (EPCAM, KRT19, SOX2, EGFR), Alveolar cells (CLDN18, AQP4, SFTPC), Fibroblasts (COL1A1, COL1A2, DCN), T cells (CD2, CD3D, TRAC, NKG7), B cells (CD79A, IGHG3, IGHA2), Myeloid cells (CD68, CD14, LYZ), Neutrophils (CSF3R, FCGR3B). All these cluster markers were decided based on reference to previous publication (28, 33) and CellMarker dataset (http://bio-bigdata.hrbmu.edu.cn/CellMarker/). Bulk expression profiles of each tumor were inferred based on the average expression profiles of all the cells derived from the same tumor. AverageExpression function was used to generated averaged expression profiles of each cellular subset.
Statistical analysis
Statistical analyses were performed using R v4.4.1 (https://www.r-project.org) or SPSS software (v26). Comparison of enrichment scores or gene expression level between two groups was analyzed by two-sided Wilcoxon tests. Spearman correlation analysis was applied to evaluate correlation between two continuous parameters. Distribution of categorical data between two groups were analyzed using the chi-square test. Kaplan-Meier curve was used to estimate median OS and PFS, with statistical difference between two groups accessed by log-rank test. Univariate and multivariate Cox regression analyses were performed to assess the prognostic significance of multiple variables. Similarly, univariate and multivariate logistic regression analyses were conducted to evaluate the influence of different variables on the clinical response to immune checkpoint inhibitors (ICIs). A two-sided p-value of less than 0.05 was considered statistically significant. The study design and key statistical analyses were summarized in a flow diagram, as illustrated in Figure 1.
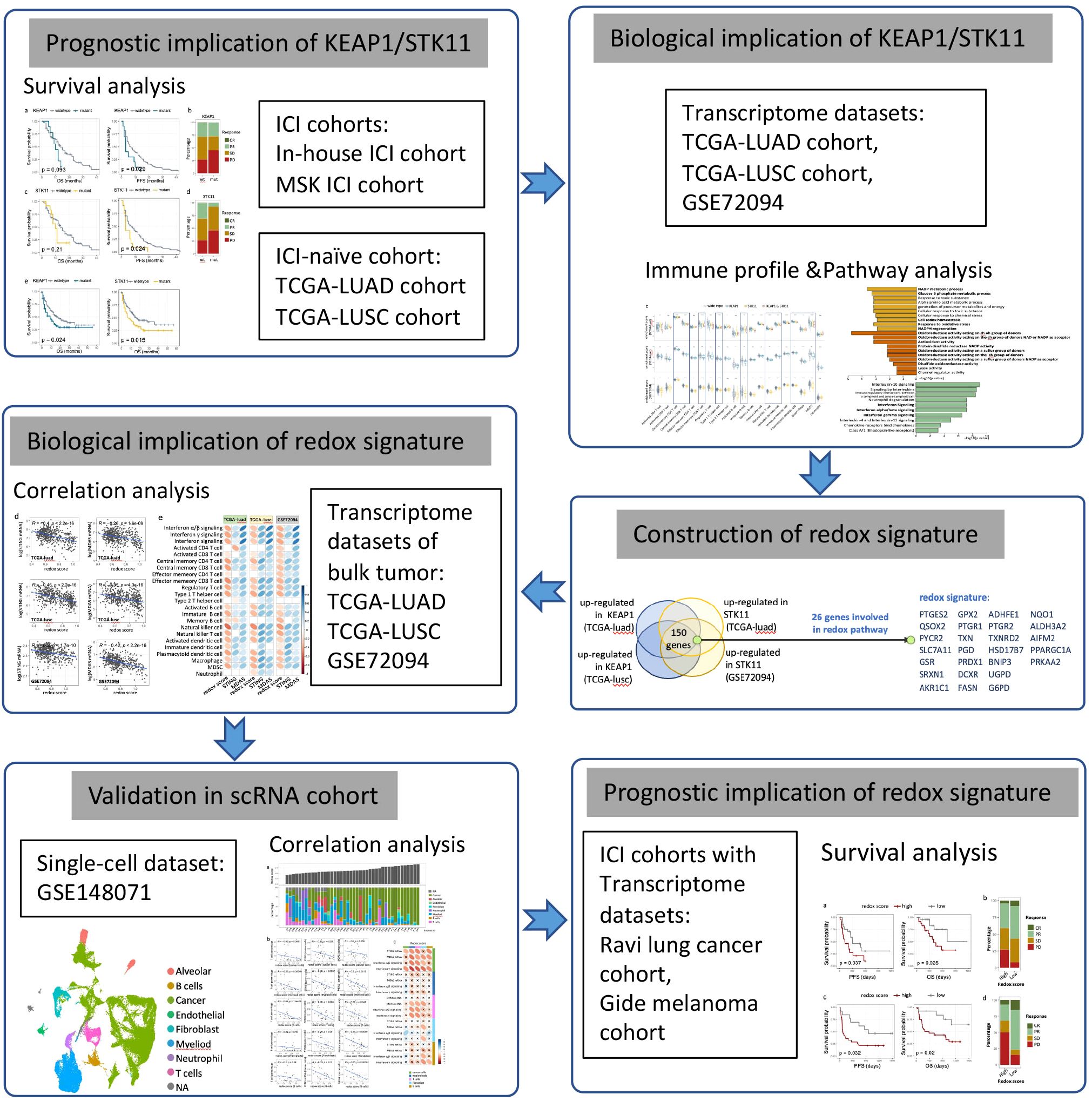
Figure 1. Flow diagram demonstrating the outlook of the study.
Results
KEAP1/STK11 mutations diminishes immunotherapy efficacy in NSCLC
Mutations in KEAP1 and STK11 had been frequently reported to diminish immunotherapy efficacy (6, 7). An in-house cohort comprising 185 patients of advanced NSCLC treated with PD-1/PD-L1 immunotherapy alone or in combination with chemotherapy was deployed to verify the prognostic impact of mutant KEAP1/STK11. In line with previous studies (6, 7), we found that patients harboring mutant KEAP1 or STK11 had significant inferior PFS as compared to those with wild-type KEAP1/STK11 (Figures 2a, c). The proportion of patients achieving complete response (CR) or partial response (PR) was markedly lower in patients with mutant KEAP1/STK11 as compared to those with wild-type variants (Figures 2b, d). Although not statistically significant in our cohort, patients with KEAP1/STK11 mutations tended to have reduced OS (Figures 2a, c). Analysis of an additional immunotherapy cohort, the MSK lung cancer cohort, corroborated our findings, demonstrating that mutations in KEAP1 or STK11 were linked to unfavorable outcomes, as evidenced by significantly poorer OS (Figure 2e). It is noteworthy that KEAP1 and STK11 did not affect survival in NSCLC patients who underwent surgical treatment (Supplementary Figures 1a, b), suggesting that their prognostic significance is specific to the context of immunotherapy.
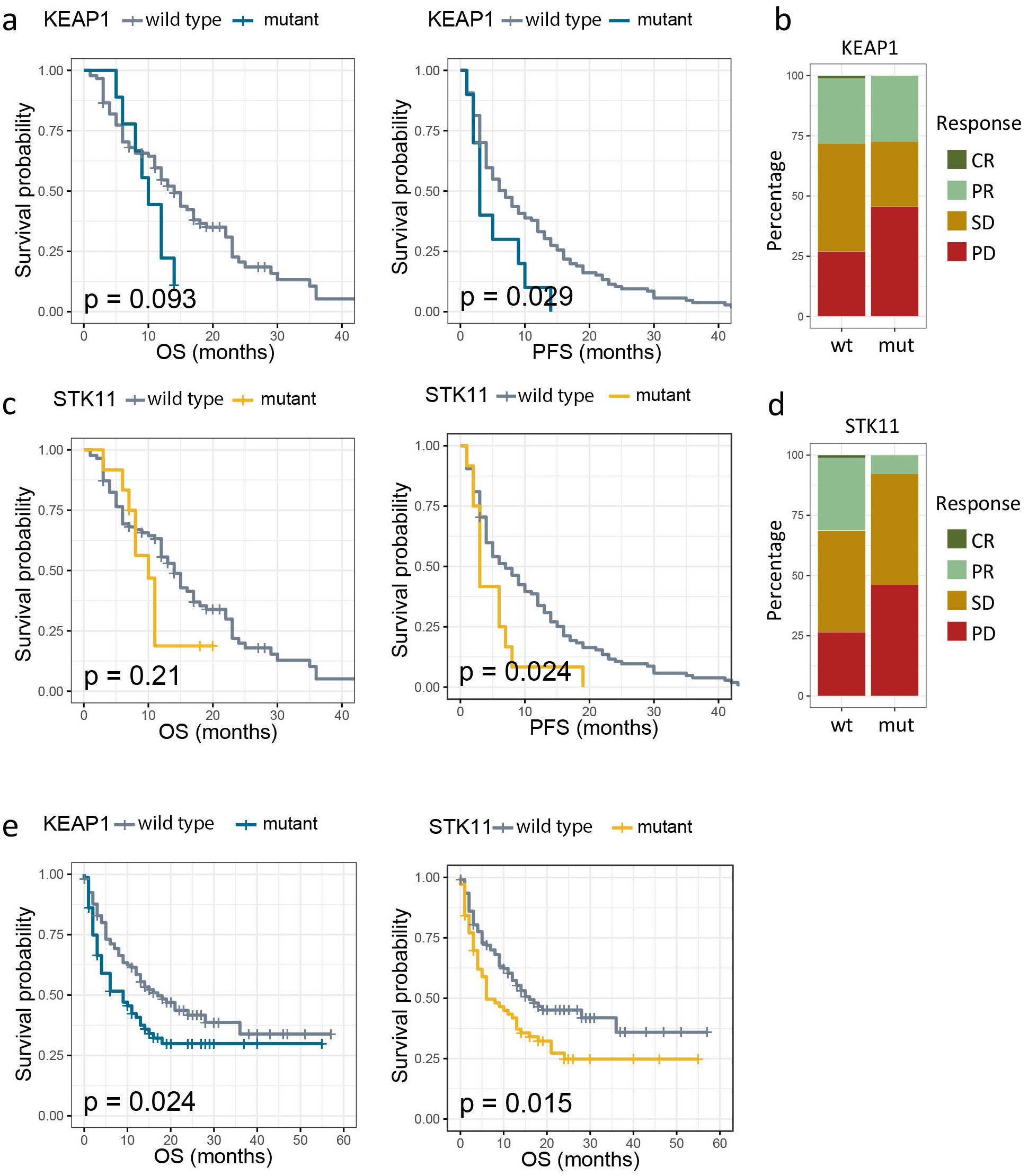
Figure 2. Association between mutation status of KEAP1/STK11 and therapeutic outcomes to immunotherapy. (a, c). Kaplan–Meier survival curves of progression-free survival (PFS) and overall survival (OS) between patients with wild-type and mutant KEAP1 (a), as well as between patients with wild-type and mutant STK11 (c) in our own immunotherapy cohort; (b, d) Bar charts showing the distribution of patients with complete response (CR), partial response (PR), stable disease (SD) or progression disease (PD) between patients with wild-type and mutant KEAP1 (a), as well as between patients with wild-type and mutant STK11 (c) in our own immunotherapy cohort; (e) Kaplan–Meier survival curves of OS by mutational status of KEAP1 or STK11 among NSCLC patients from MSK immunotherapy cohort.
NSCLC with mutant KEAP1 or STK11 exhibited immunosuppressive microenvironment in a similar pattern
We subsequently investigated the influence of KEAP1/STK11 mutations on established biomarkers of immunotherapeutic response, such as PD-L1 expression and tumor mutation burden (TMB). In our analysis of the TCGA lung cancer dataset, where protein expression levels were accessible, we found no significant differences in PD-L1 expression levels between tumors harboring wild-type and mutant KEAP1/STK11, both in adenocarcinoma (TCGA-LUAD) and squamous cell carcinoma (TCGA-LUSC) (Figure 3a). Furthermore, TMB was not affected by STK11 mutations; and in fact, it was even elevated in tumors with mutant KEAP1 according to our analysis of the MSK lung cancer dataset (Figure 3b). These observations suggest that the poor response to immunotherapy in patients with mutant KEAP1/STK11 is not due to reduced PD-L1 expression or TMB levels, but rather may be attributed to alterations in the immune microenvironment.
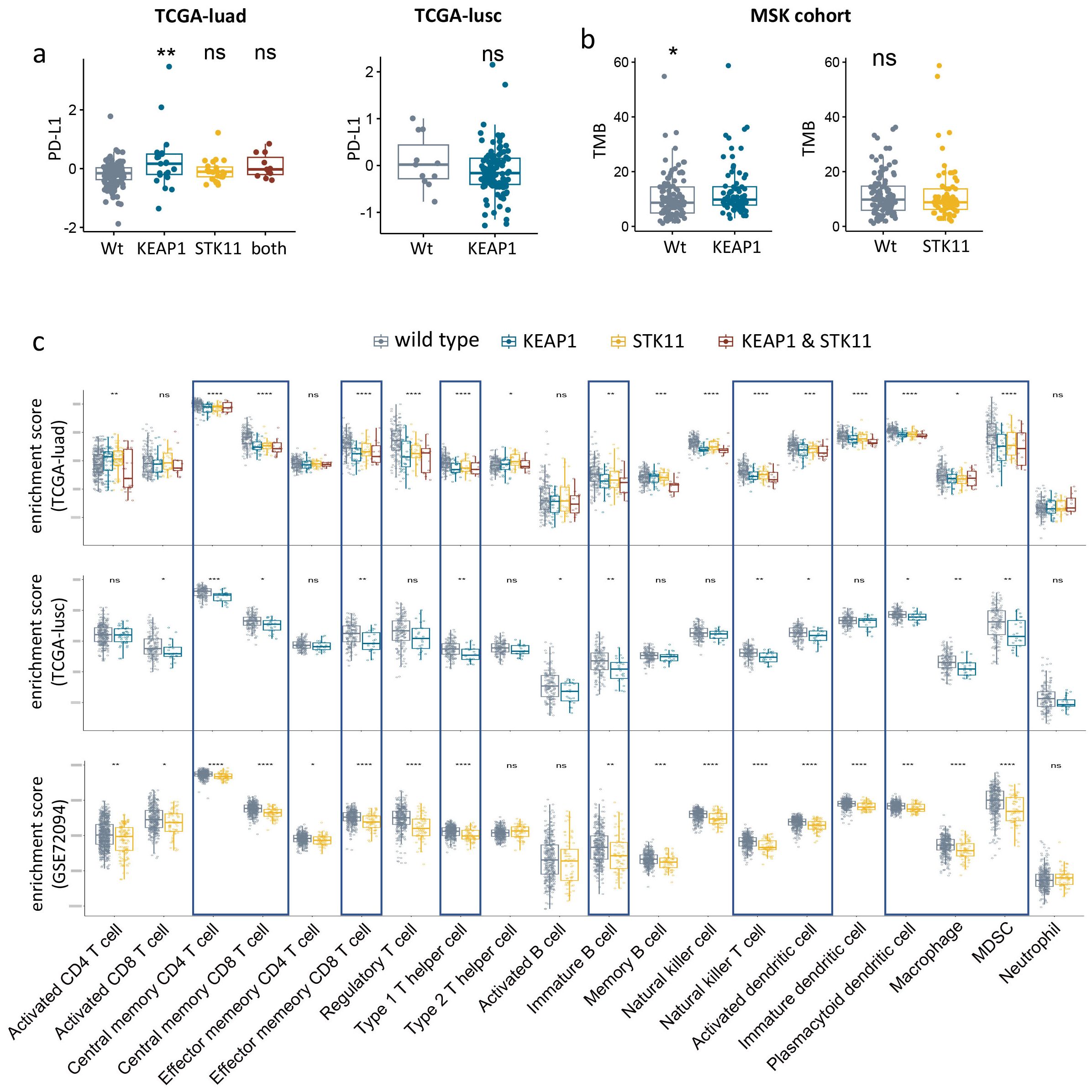
Figure 3. Immunologic consequences of KEAP1/STK11 mutations. (a) Boxplots showing PD-L1 protein expression level in NSCLC with mutant or wild-type KEAP1/STK11. Analysis was performed in two cohorts (TCGA-luad, TCGA-lusc). (b) Boxplots showing TMB level of KEAP1/STK11 mutant or wild-type tumors from MSK immunotherapy cohort. (c) Boxplots showing infiltrating abundance of 20 immune cells among tumors with mutant or wild-type KEAP1/STK11. Analysis was performed in three cohorts (TCGA-luad, TCGA-lusc, GSE72094). (a, c) As for TCGA-luad cohort, tumors were categorized into four groups based on mutational status of KEAP1 and STK11. Analysis only involved mutational status of KEAP1 or STK11 in TCGA-lusc cohort and GSE72094 cohort respectively, owing to insufficiency or unavailability of relevant data. (a–c) Data are presented as median with quartiles. Wilcoxon tests was used to determined significance in difference between two groups. Kruskal -Wallis was performed for multiple comparison. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To access the impact of KEAP1/STK11 mutations on immune landscape of lung cancer, we utilized two bulk RNA-seq datasets: TCGA-lung cancer and GSE72094, for our analysis. The infiltration levels of 20 different leukocyte populations were deduced from the enrichment scores of their respective gene signatures (as detailed in Supplementary Table 7). Figure 3c illustrates that lung tumors with mutations in KEAP1 or STK11 displayed an immunosuppressive phenotype, characterized by a significant reduction in the infiltration of a broad spectrum of leukocytes. These included central memory CD4 T cells, central memory CD8 T cells, effector memory CD8 T cells, type 2 T helper cells, immature B cells, natural killer T cells, activated dendritic cells, plasmacytoid dendritic cells, macrophages, and myeloid-derived suppressor cells (MDSCs). Due to the limited number of tumors with detecs. Table TK11 mutations (only three), the analysis of STK11 status in the TCGA-LUSC cohort was not conducted. Similarly, the assessment of KEAP1 status in the GSE72094 cohort was not possible due to the absence of pertinent data. It’s intriguing to notice that the impact of mutant KEAP1 and STK11 on the infiltration levels of various leukocytes was similar, which indicated KEAP1 and STK11might employ analogous mechanisms in immune regulation.
KEAP1/STK11 mutations confer redox phenotype and suppression of IFN signaling in lung cancer
To investigate the shared biological processes influenced by KEAP1 and STK11 mutations, we focused on the 151 differentially expressed genes that were consistently up-regulated by these mutations across all cohorts. These genes were subjected to Gene Ontology (GO) pathway analysis. Figure 3a displays the top 10 GO biological pathways (GOBP) and top 10 GO molecular functions (GOMF) that were concurrently up-regulated by mutant KEAP1 and STK11. Notably, the majority of the up-regulated pathways were associated with oxidoreductase activity, suggesting that a redox phenotype is the predominant feature in tumors with mutant KEAP1 or STK11. Among the 148 overlapping up-regulated genes, 25 were identified as being involved in redox activity and were selected to form a redox signature for further investigation (Figure 4a). Spearman correlation analysis revealed that all 26 genes within the redox signature were significantly and negatively correlated with the infiltration levels of most leukocytes, particularly those that were markedly reduced in tumors with mutant KEAP1 or STK11 (Supplementary Figure 2a). This correlation suggests that the redox phenotype may be a critical factor driving immune evasion in tumors harboring KEAP1 or STK11 mutations.
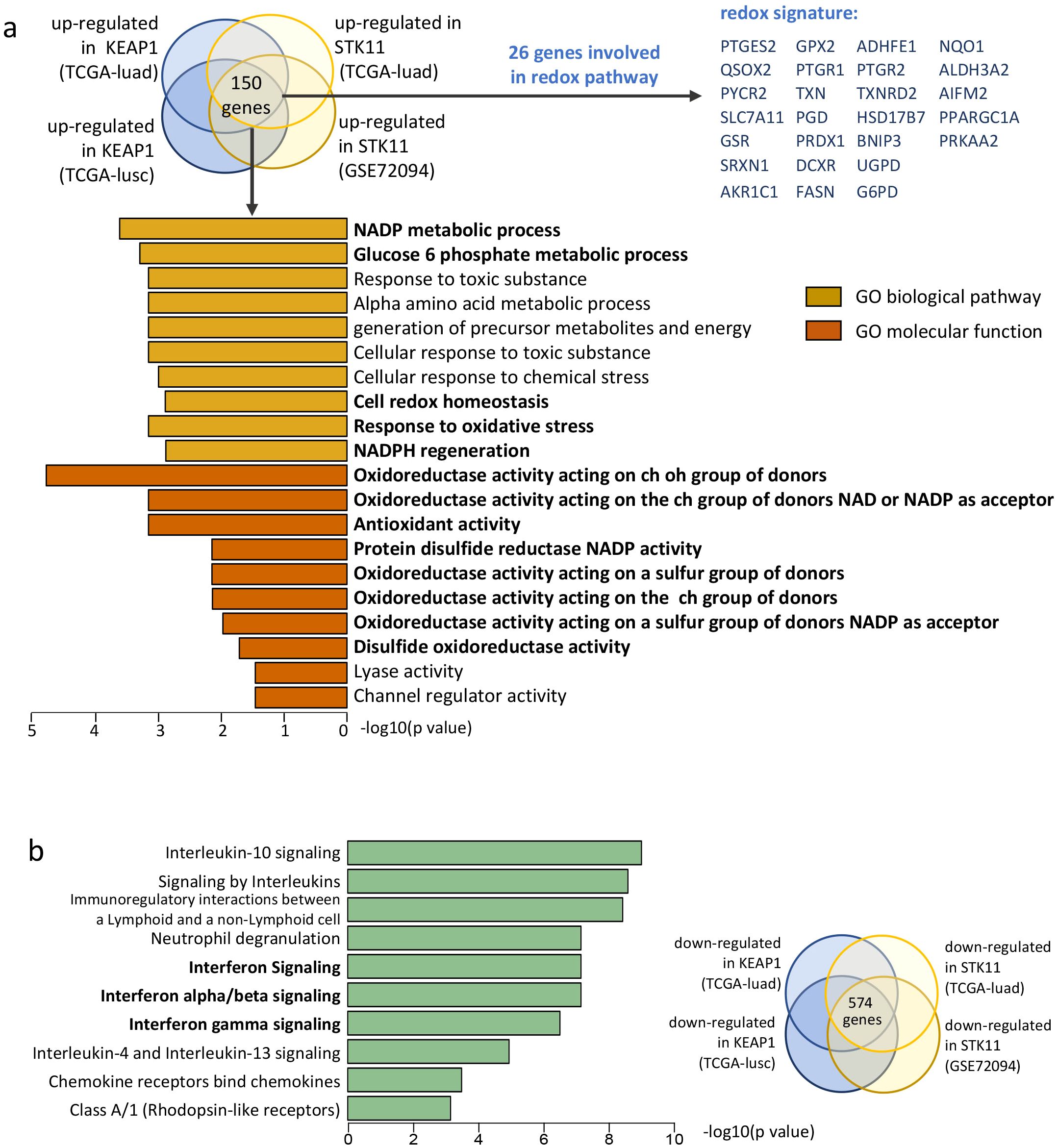
Figure 4. Pathway analysis reveals enrichment of redox pathways and down-regulation of IFN signaling in NSCLC with KEAP1/STK11 mutations. (a) Gene signature enrichment analysis (GSEA) was performed for the overlapped up-regulated genes based on Gene Ontology (GO) categories. Diagram on the top showing the generation of overlapped genes up-regulated by KEAP1 and STK11 among different cohorts. A total 150 overlapped genes identified, among which 26 genes were identified to be involved in redox biological process. Bar plot showing the top 10 most significantly enriched pathways for the GO-biological pathway category and GO-molecular function category respectively. (b) Gene signature enrichment analysis (GSEA) was performed for the overlapped down-regulated based on Reactome categories. Diagram on the right showing the generation of overlapped genes down-regulated by KEAP1 and STK11 among different cohorts, with a total 574 overlapped genes identified. Bar plot showing the top 10 most significantly enriched pathways.
We calculated the enrichment score of the redox signature for each tumor using the ssGSEA method. As depicted in Supplementary Figure 2b, the redox score did not demonstrate prognostic relevance among NSCLC patients who underwent surgical treatment. Specifically, there was no observed difference in OS or PFS between patients with high and low redox scores within the TCGA cohorts (TCGA-LUAD and TCGA-LUSC). We proceeded to assess the correlation between the redox score and various clinicopathological characteristics, including histology, gender, smoking history, and TNM stage. The redox score was significantly elevated in tumors originating from squamous cell carcinoma as compared to adenocarcinoma, and also in tumors from male patients as compared to female patients (Supplementary Figure 2c). However, neither smoking history nor TNM staging had a significant influence on the redox status of NSCLC tumors (Supplementary Figure 2c).
To elucidate the mechanism through which the redox phenotype facilitates immune evasion in NSCLC with mutations in KEAP1 or STK11, we conducted a further analysis to identify pathways enriched in genes that are down-regulated by these mutations. We identified a total of 574 down-regulated genes that were common across all cohorts. These genes were then subjected to Reactome pathway enrichment analysis. Notably, three of the top 10 enriched pathways were associated with the activation of interferon signaling, including the pathways for interferon α/β signaling and interferon γ signaling (Figure 4b).
Redox phenotype mediates immune exclusion by repressing STING/MDA5 expression and interferon signaling
Cell-autonomous interferon responses are typically regulated by pathways involving in sensing double-stranded DNA (dsDNA), double-stranded RNA (dsRNA), or single-stranded RNA (ssRNA) (Figure 5a) (34, 35). As redox imbalance had been associated with DNA damage (36), we first evaluated signaling of DNA repair in tumors with KEAP1/STK11 mutations. Of note, biological pathways of DNA repair or double-strand break repair were unaltered by mutations in either KEAP1 or STK11 (Supplementary Figure 3). We then proceeded to examine the differential expression of genes along the signaling axes of dsDNA/dsRNA/ssRNA sensing in tumors with wild-type versus mutant KEAP1/STK11. Figure 5b and Supplementary Figures 4a–c reveal that mRNA expression levels of STING (Stimulator of Interferon Genes), MDA5 (Melanoma Differentiation-Associated protein 5) and RIG-I (Retinoic acid-inducible gene I) are significantly downregulated in tumors with mutant KEAP1 or STK11 across all cohorts. We also noted a reduction in CGAS (Cyclic guanosine monophosphate-adenosine monophosphate synthase) expression due to KEAP1/STK11 mutations in the TCGA-LUSC and GSE72094 cohorts, but not in the TCGA-LUAD cohort. Similarly, IFI16 (Interferon Gamma Inducible Protein 16) expression was downregulated by KEAP1/STK11 mutations in the TCGA-LUAD and GSE72094 cohorts, yet this effect was not observed in the TCGA-LUSC cohort.
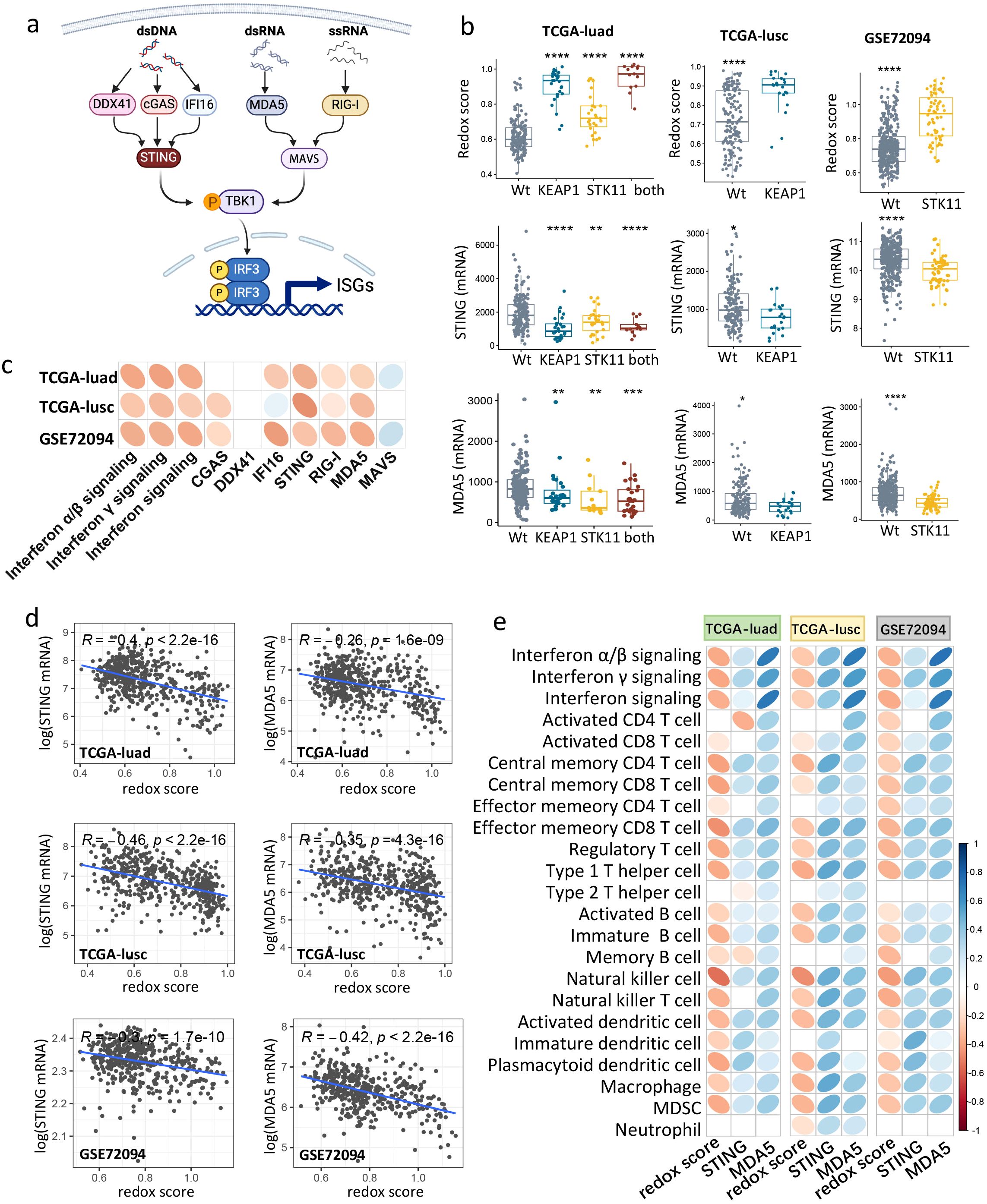
Figure 5. Redox scoring negatively correlated with STING/MDA5 expression, IFN signaling and immune infiltration in NSCLC. (a) Schematic of dsDNA, dsRNA, ssRNA sensing pathways that induce IFN signaling. (b) Boxplots showing STING/MDA5 mRNA expression and redox score among tumors with mutant or wild-type KEAP1/STK11 in three cohorts (TCGA-luad, TCGA-lusc, GSE72094). As for TCGA-luad cohort, tumors were categorized into four groups based on mutational status of KEAP1 and STK11. Analysis only involved mutational status of KEAP1 or STK11 in TCGA-lusc cohort and GSE72094 cohort respectively, owing to insufficiency or unavailability of relevant data. Data are presented as median with quartiles, and Wilcoxon tests was used to determined significance in difference between wild-type group and other groups. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (c) Correlation matrix depicting correlation of redox score with IFN signaling and genes involved in dsDNA/dsRNA/ssRNA sensing in three datasets (TCGA-luad, TCGA-lusc and GSE72094). (d) Scatter plot showing the correlation between redox score and mRNA expression of STING/MDA5 in three datasets (TCGA-luad, TCGA-lusc and GSE72094). Correlation coefficients (r value) and P value of Spearman Correlation were shown. (e) Correlation matrix depicting correlation of redox score and mRNA expression of STING with infiltrating level of 20 immune cells in three datasets (TCGA-luad, TCGA-lusc and GSE72094). (c, e) Spearman Correlation analysis was performed, with blue ellipse obliquely upward representing positive correlation, and red ellipse obliquely downward representing negative correlation. The flatness of ellipse and the depth of the color represent the magnitude of the correlation (r value). Ellipse was presented only for those with significant correlation (P value < 0.05).
We next determined the correlation between the redox phenotype and IFN signaling, as well as genes involved in dsDNA/dsRNA/ssRNA sensing. As illustrated in Figure 5c, the redox score was found to be negatively correlated with the enrichment scores of all interferon signaling pathways. Notably, the expression levels of genes involved in dsDNA/dsRNA/ssRNA sensing, specifically STING, MDA5 and RIG-I, were negatively correlated with the redox score across all datasets (Figures 5c–d). CGAS and IFI16 also showed a negative correlation with the redox score, but this was only observed in certain cohorts (Figure 5c).
We then assessed the correlation between genes involved in dsDNA/dsRNA/ssRNA sensing and the immune profiles of NSCLC tumors using Spearman correlation analysis. As shown in Supplementary Figure 5d, mRNA expression of dsDNA sensors (STING, CGAS) or dsRNA/ssRNA sensors (MDA5, RIG-I) were significantly and positively correlated with the infiltrating abundance of the majority of immune cells across all cohorts. The positive correlation between IFI16 expression and immune infiltration was observed exclusively in the adenocarcinoma cohorts (GSE72094 and TCGA-LUAD) (Supplementary Figure 5d). Collectively, these findings suggest that KEAP1/STK11 mutations are associated with the downregulation of genes involved in dsDNA/RNA sensing, particularly STING and MDA5, which may be a key driver of immune evasion.
Given that STING and MDA5 are the downstream components in dsDNA/dsRNA sensing and trigger the activation of IFN signaling, their suppression could be a pivotal mechanism by which the redox phenotype drives immune evasion in lung cancer. Specifically, we observed an inverse relationship between KEAP1/STK11 mutations and changes in redox scores and STING/MDA5 expression levels across all cohorts (Figure 5b). Correlation analysis further indicated that the redox score and STING/MDA5 expression had opposite effects on the infiltration levels of nearly all immune cells across all cohorts (Figure 5e). It is noteworthy that the immune cells that were significantly reduced in tumors with KEAP1/STK11 mutations were also the ones that showed significant positive and negative correlations with STING/MDA5 expression and redox scores, respectively (Figure 5e). Collectively, our findings suggested that the redox phenotype, driven by KEAP1/STK11 mutations, promotes immune evasion by downregulating genes involved in dsDNA/dsRNA sensing especially STING and MDA5, and thus suppresses the downstream interferon signaling pathway.
scRNA-seq analysis reveals redox phenotype specifically impacts STING/MDA5 expression of cancer cells
To identify the predominant cellular subtypes contributing to the redox status, we utilized single-cell RNA sequencing (scRNA-seq) data from 42 NSCLC tumors for further analysis. Cell clusters were classified into T cells, B cells, myeloid cells, neutrophils, fibroblasts, endothelial cells, alveolar cells, and cancer cells based on the expression of canonical marker genes (Supplementary Figures 5a–b). Notably, most redox-associated genes were found to be highly expressed across various cell types (Supplementary Figure 4c), although a slightly higher redox score was observed in cancer cells compared to other cell types (Supplementary Figure 5d).
We then assessed the impact of the redox phenotype on the immune composition within the TIME. As depicted in Figure 6a, the percentage of various immune cells decreased with an increase in the redox score of bulk tumors. To determine redox status of which cellular subtype plays a major role in shaping the immunosuppressive microenvironment, we calculated the redox score for five major cell types (cancer cells, myeloid cells, T cells, fibroblasts, and B cells) based on the averaged expression profiles for each tumor. Figure 5b shows that the redox scores of both myeloid cells and T cells were significantly negatively correlated with the T cell percentage. The redox scores of cancer cells, fibroblasts, and B cells also exhibited a negative, albeit not statistically significant correlation with the T cell percentage.
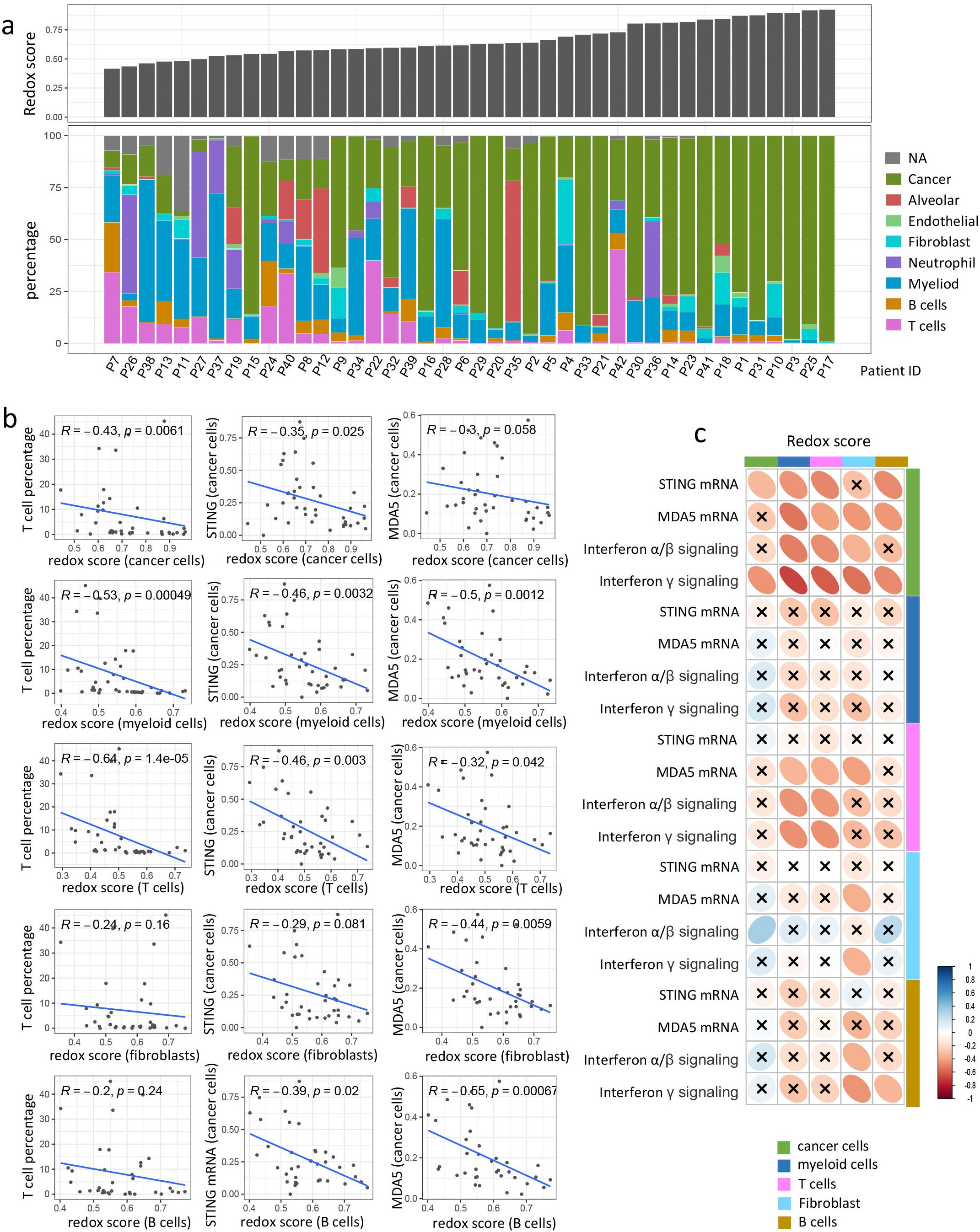
Figure 6. scRNA analysis reveals redox phenotype down-regulates STING/MDA5 expression in cancer cells and facilitates immune exclusion. (a) Barplots showing bulk redox score and proportion of different cellular components in each tumor. Tumors were arranged according to bulk redox score in ascending order. (b) Scatter plot showing the correlation of T cell percentage or STING/MDA5 mRNA expression in cancer cells with redox score of different cellular components. Correlation coefficients (r value) and P value of Spearman Correlation were shown. (c) Correlation matrix showing correlation of redox score with STING/MDA5 mRNA expression or IFN signaling (Interferon α/β signaling and Interferon γ signaling) across different cell types. Spearman Correlation analysis was performed, with blue ellipse obliquely upward representing positive correlation, and red ellipse obliquely downward representing negative correlation. The flatness of ellipse and the depth of the color represent the magnitude of the correlation (r value). The cross mark represents the failure of the significance test (P value > 0.05).
We further explored the changes in expressional level of genes involved in dsDNA/dsRNA/ssRNA sensing and the enrichment of downstream interferon signaling pathway across different cell types in response to the redox status of various cell types. It is intriguing to observe that STING/MDA5 expression in cancer cells showed a negative correlation with the redox scores of nearly all major cellular subtypes, including cancer cells, myeloid cells, T cells, fibroblasts, and B cells (Figures 6b–c). MDA5 expression in T cells were also negatively correlated with redox score of myeloid cells, T cells and fibroblast. However, the STING/MDA5 expression of other cell types exhibited no significant correlation with redox score of any cell types (Figure 6c). As for other genes involved in dsDNA/dsRNA/ssRNA sensing like CGAS, RIG-I, MAVS, DDX41, and IFI16, whose expression in either cancer cells or other cell types, were not significantly impacted by redox status of any cell types (Supplementary Figure 5e). Additionally, interferon α/β signaling and interferon γ signaling in cancer cells were negatively influenced by the redox status of nearly all cellular subtypes (Figure 6c). Interferon signaling in T cells also negatively correlated with the redox scores of T cells and myeloid cells (Figure 6c). Nevertheless, no significant correlation was observed between interferon signaling and redox scores in most other cell types (Figure 6c). Collectively, these findings suggest that the redox phenotype mediates immune exclusion primarily by suppressing STING/MDA5 expression and interferon signaling in cancer cells.
Redox signature predicts response to immunotherapy
To ascertain the prognostic relevance of redox signatures in the context of immunotherapy, we assessed the relationship between redox scores and the efficacy of PD-1/PD-L1 checkpoint inhibitors in two distinct patient cohorts: the Ravi lung cancer cohort, the Gide melanoma cohort. In the Ravi lung cancer cohort, we enrolled 48 patients with advanced NSCLC who were treated with PD-L1 blockade as their first-line therapy. As depicted in Figure 7a, patients with lower redox scores exhibited significantly extended PFS and OS compared to those with higher redox scores. Specifically, the median PFS was 529.4 days for the low redox group versus 221.8 days for the high redox group (p = 0.037), and the median OS was 843.7 days for the low redox group versus 491.2 days for the high redox group (p = 0.025). Cox regression analysis, adjusting for PD-L1 expression (protein level), smoking status, gender, age, histology (adenocarcinoma vs. squamous), and co-mutations (KRAS, TP53, KEAP1, STK11), further confirmed redox status as the only significant predictor of reduced OS (HR 3.60 [1.54 -8.41], P = 0.00306) and PFS (HR 3.12 [1.09-8.94], P = 0.0337) in univariate analysis but not multivariate analysis (Supplementary Tables 8.1, 8.2). In terms of response rates, 55% of patients with low redox scores achieved a complete response (CR) or partial response (PR), contrasted with 37% in the high redox group (Figure 7b). However, logistic regression analysis indicated that none of the above-mentioned variables, including redox status, were significant predictors of clinical response (Supplementary Table 8.3).
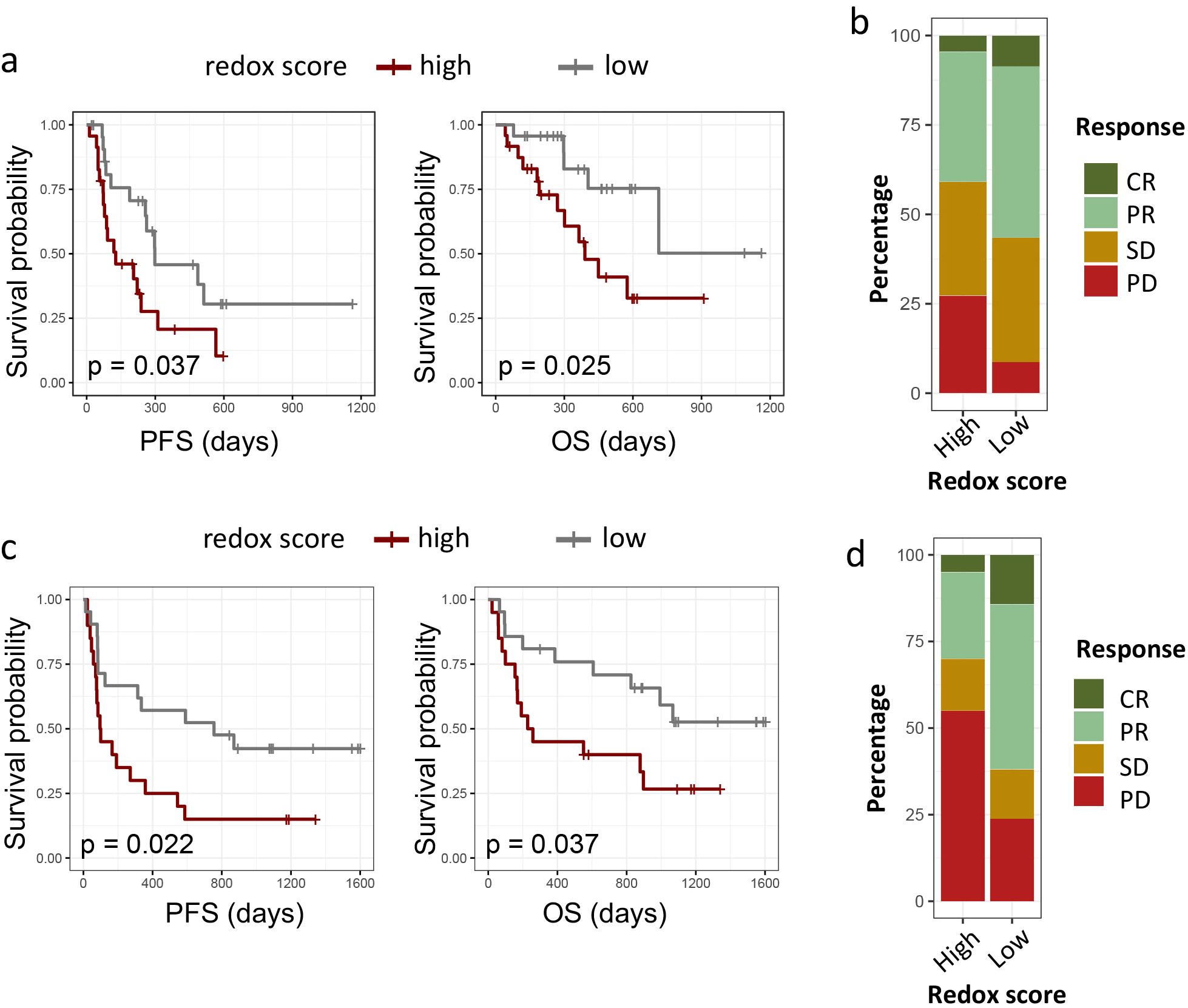
Figure 7. Impact of redox phenotypes on response to ICIs in multiple cancers. (a, c). Kaplan–Meier survival curves of PFS and OS between high redox group and low redox group in Ravi lung cancer cohort (a) and Gide melanoma cohort (c). (b, d) 100-percent bar plots showing the distribution of patients with complete response (CR), partial response (PR) stable disease (SD) or progression disease (PD) between high redox group and low redox group in Ravi lung cancer cohort (b) and Gide melanoma cohort (d). Tumors were categorized into high and low redox group with medium redox score as cut-off value for both cohorts.
To evaluate the broader applicability of redox signatures in predicting responses to immunotherapy, we examined the Gide melanoma cohort. The Gide melanoma cohort comprised 41 melanoma patients who underwent anti-PD-1 treatment. Similarly, we observed a marked improvement in PFS and OS for patients with lower redox scores: the median PFS was 967 days for the low redox group versus 402 days for the high redox group (p = 0.04), and the median OS was 1278 days for the low redox group versus 616 days for the high redox group (p = 0.038) (Figure 7c). Cox regression analysis was limited to redox level, PD-L1 expression (mRNA level), smoking status, gender, and age, as data on other parameters were unavailable. As shown in Supplementary Table 9, both high redox level (HR for PFS 3.60 [1.54 -8.41], P = 0.00306; HR for OS 2.68 [1.08-6.63], P = 0.0328) and low PD-L1 expression (HR for PFS 0.14 [0.06 -0.33], P < 0.001; HR for OS 0.16 [0.06-0.46], P < 0.001) were significant independent predictor of reduced OS and PFS. Additionally, patients with lower redox scores demonstrated a higher objective response rate (ORR), with 84% achieving CR/PR in the low redox group, compared to 32% in the high redox group (Figure 7d). Multivariate logistic regression analysis also confirmed redox level (OR 0.16 [0.02-0.8], P = 0.04) and low PD-L1 expression (OR 15.79 [3.10-130.96], P = 0.0027) as independent predictor of clinical response to immunotherapy (Supplementary Table 9.3).
Discussion
With this study, we demonstrated that NSCLC with KEAP1 or STK11 mutation manifested enhanced redox phenotype and diminished immune infiltration. Redox status is associated with inhibition of interferon signaling, which could be attributed to downregulation of genes involved in dsDNA/dsRNA sensing like STING and MDA5 in cancer cells. Redox score and STING/MDA5 expression exhibited the exact opposite correlation with infiltrating level of different immune cells. Our study suggested that KEAP1 and STK11 shared common mechanism in immune regulation, which is associated with enhancement of redox phenotype and the subsequent inhibition of STING/MDA5 expression and the downstream interferon signaling in cancer cells. We also developed a redox signature which may be helpful in predicting outcomes to ICI treatment in NSCLC and other cancers.
Repression of type I interferon signaling in tumors harboring KEAP1 or STK11 mutations has been reported in previous studies (37, 38), yet the underlying mechanism is not well characterized. Cell-autonomous type I interferon responses are typically regulated by dsDNA/dsRNA sensing pathways, whose activation can be driven by overproduction of dsDNA or dsRNA, or overexpression of genes along these signaling axes (34, 35). Previous study reported that KEAP1 mutation resulted in upregulation of BRCA1, which is an important DNA damage repair gene, and thus reduced production of dsDNA (39). Yet our finding suggested neither KEAP1 nor STK11 interfered with DNA repairing in NSCLC. Instead, several genes involved in dsDNA/dsRNA sensing like STING and MDA5 were significantly downregulated by mutation of KEAP1 or STK11. STING as an intracellular dsDNA sensor that activates the innate immune response (40), was observed among tumors with mutant KEAP1 or STK11 in recent studies (37, 39, 41). STING activation can lead to the production of type I interferons and other pro-inflammatory cytokines, which are crucial for the immune system’s recognition and elimination of cancer cells (42). MDA5 is a crucial cytosolic RNA sensor that plays a pivotal role in the innate immune response by detecting viral infections and activating antiviral defenses (43, 44). Beyond its antiviral functions, emerging research underscores MDA5’s significance in cancer immunity, with its suppression being linked to immune evasion (45). Our study suggested that MDA5 expression was also significantly suppressed by mutation in KEAP1 or STK11. The mechanism by which STING/MDA5 expression is altered by mutant KEAP1 or STK11 and their interaction with metabolic reprogramming remains unclear.
Redox homeostasis, as defined by the balance between reactive oxygen species (ROS) and antioxidants, has been intricately linked to the regulation of immune system. However, most of the previous studies centered on the direct impact of oxidative changes on biological functions of immune cells (46–51), but neglect their impact on the immunogenicity of tumor cells. Our study reveals a specific association between the that redox status and feature of immune evasion by specifically inhibiting STING/MDA5, characterized by the suppression of intrinsic STING/MDA5 expression and the interferon response specifically within tumor cells, a phenomenon not observed in other cell types of the TIME. Particularly, scRNA analysis showed that redox status of immune cells like macrophage, T cells etc. also correlated with reduced STING/MDA5 expression in tumor cells and a suppressive TIME. These findings suggested that antioxidants derived from different cell types all contributed to the development of redox phenotype, which concurrently suppressed tumor immunogenicity by inhibiting STING/MDA5 expression and interferon signaling of tumor cells. We recognize that the observed suppression of STING/MDA5 due to redox alterations is primarily based on transcriptome data analysis, and further validation through in vitro experiments is required in the future.
The underlying mechanism by which redox phenotype suppressed STING/MDA5 expression is yet to be explored in further studies. Previous studies have suggested a link between KEAP1-NRF2 pathway and STING suppression (52, 53). NRF2 is the master transcription factor that control the expression of a battery of genes involved in antioxidant response and detoxification processes (54). KEAP1 negatively regulates NRF2 by directly binding and leading to its proteasomal degradation (55, 56). Loss-of -function mutations in KEAP1 lead to constitute activation of NRF2 signaling (57). NRF2 has been reported as a negative regulator of STING (52, 53, 58), although the underlying mechanism remains a puzzle.
The most compelling clinical implication of our study lies in the potential for improved patient stratification and innovative trial design. The association between our redox signature and poor outcomes in the KEAP1/STK11 double-mutant subgroup highlights a patient population in urgent need of better therapeutic options. These patients may be prioritized for more aggressive monitoring and considered for alternative treatment strategies beyond first-line immunotherapy. Looking forward, our findings advocate for the design of biomarker-driven clinical trials that specifically enroll patients with this high-risk molecular profile. Such trials could evaluate novel combinations, such as immunotherapy with (e.g., targeted redox-balancing agents or STING agonists), using our signature or the mutational status as an enrichment biomarker. This approach is essential to breaking the cycle of poor outcomes in this refractory population.
Conclusions
In summary, our study elucidated the mechanism by which redox phenotype mediated immune evasion in NSCLC harboring STK11 or KEAP1 mutation. We first established a connection between redox phenotype and repression of pathways involved in dsDNA/dsRNA sensing, and clarify their association in suppressing immune infiltration. We findings also suggested that redox status predominantly suppressed STING/MDA5 expression in tumor cells but not among other cell types within TIME. Our self-developed redox signature also may serve as a predictive biomarker for ICI responsiveness.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics committee of the first affliated hospital of Sun Yat-sen University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
ZL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Project administration, Visualization, Supervision, Writing – original draft, Writing – review & editing. AS: Formal analysis, Methodology, Project administration, Writing – original draft, Writing – review & editing. JG: Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. LH: Methodology, Project administration, Writing – original draft, Writing – review & editing. SL: Data curation, Formal analysis, Methodology, Project administration, Writing – original draft. YL: Project administration, Funding acquisition, Writing – original draft, Writing – review & editing. YZ: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by grants from the Guangzhou Science and Technology Program Project (No. 2023A04J2210, No. 2023A04J2213).
Acknowledgments
The authors used deepseek LLM Version K1 to assist in the initial drafting of the background section. The generated text was substantially revised, fact-checked, and refined by the authors, who take full responsibility for the entire work’s integrity and accuracy.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript. AI tool (Deepseek) was used during the initial writing process for language assistance and brainstorming.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1676797/full#supplementary-material
References
1. Hanna N, Johnson D, Temin S, Baker S Jr., Brahmer J, Ellis PM, et al. Systemic therapy for stage IV non-small-cell lung cancer: american society of clinical oncology clinical practice guideline update. J Clin Oncol. (2017) 35:3484–515. doi: 10.1200/JCO.2017.74.6065
2. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
3. Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. (2019) 393:1819–30. doi: 10.1016/S0140-6736(18)32409-7
4. Wang J, Lu S, Yu X, Hu Y, Zhao J, Sun M, et al. Tislelizumab plus chemotherapy versus chemotherapy alone as first-line treatment for advanced squamous non-small-cell lung cancer: final analysis of the randomized, phase III RATIONALE-307 trial. ESMO Open. (2024) 9:103727. doi: 10.1016/j.esmoop.2024.103727
5. Forde PM, Spicer JD, Provencio M, Mitsudomi T, Awad MM, Wang C, et al. Overall survival with neoadjuvant nivolumab plus chemotherapy in lung cancer. N Engl J Med. (2025) 393:741–52. doi: 10.1056/NEJMoa2502931
6. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. (2015) 5:860–77. doi: 10.1158/2159-8290.CD-14-1236
7. Ricciuti B, Arbour KC, Lin JJ, Vajdi A, Vokes N, Hong L, et al. Diminished efficacy of programmed death-(Ligand)1 inhibition in STK11- and KEAP1-mutant lung adenocarcinoma is affected by KRAS mutation status. J Thorac Oncol. (2022) 17:399–410. doi: 10.1016/j.jtho.2021.10.013
8. Li J, Shi D, Li S, Shi X, Liu Y, Zhang Y, et al. KEAP1 promotes anti-tumor immunity by inhibiting PD-L1 expression in NSCLC. Cell Death Dis. (2024) 15:175. doi: 10.1038/s41419-024-06563-3
9. Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, et al. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. (2014) 74:808–17. doi: 10.1158/0008-5472.CAN-13-1655
10. Kansanen E, Kuosmanen SM, Leinonen H, and Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. (2013) 1:45–9. doi: 10.1016/j.redox.2012.10.001
11. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. (2004) 24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004
12. Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S, Tull D, et al. Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. (2018) 27:935–43.e4. doi: 10.1016/j.cmet.2018.02.006
13. Zavitsanou AM, Pillai R, Hao Y, Wu WL, Bartnicki E, Karakousi T, et al. KEAP1 mutation in lung adenocarcinoma promotes immune evasion and immunotherapy resistance. Cell Rep. (2023) 42:113295. doi: 10.1016/j.celrep.2023.113295
14. Calles A, Sholl LM, Rodig SJ, Pelton AK, Hornick JL, Butaney M, et al. Immunohistochemical loss of LKB1 is a biomarker for more aggressive biology in KRAS-mutant lung adenocarcinoma. Clin Cancer Res. (2015) 21:2851–60. doi: 10.1158/1078-0432.CCR-14-3112
15. Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad Sci U S A. (2014) 111:2554–9. doi: 10.1073/pnas.1312570111
16. Gill RK, Yang SH, Meerzaman D, Mechanic LE, Bowman ED, Jeon HS, et al. Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene. (2011) 30:3784–91. doi: 10.1038/onc.2011.98
17. Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. (2007) 448:807–10. doi: 10.1038/nature06030
18. Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. (2016) 76:999–1008. doi: 10.1158/0008-5472.CAN-15-1439
19. Schabath MB, Welsh EA, Fulp WJ, Chen L, Teer JK, Thompson ZJ, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. (2016) 35:3209–16. doi: 10.1038/onc.2015.375
20. Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. (2019) 79:3251–67. doi: 10.1158/0008-5472.CAN-18-3527
21. Sitthideatphaiboon P, Galan-Cobo A, Negrao MV, Qu X, Poteete A, Zhang F, et al. STK11/LKB1 mutations in NSCLC are associated with KEAP1/NRF2-dependent radiotherapy resistance targetable by glutaminase inhibition. Clin Cancer Res. (2021) 27:1720–33. doi: 10.1158/1078-0432.CCR-20-2859
22. Wohlhieter CA, Richards AL, Uddin F, Hulton CH, Quintanal-Villalonga À, Martin A, et al. Concurrent mutations in STK11 and KEAP1 promote ferroptosis protection and SCD1 dependence in lung cancer. Cell Rep. (2020) 33:108444. doi: 10.1016/j.celrep.2020.108444
23. Mullen L, Mengozzi M, Hanschmann EM, Alberts B, and Ghezzi P. How the redox state regulates immunity. Free Radic Biol Med. (2020) 157:3–14. doi: 10.1016/j.freeradbiomed.2019.12.022
24. Boreel DF, Span PN, Heskamp S, Adema GJ, and Bussink J. Targeting oxidative phosphorylation to increase the efficacy of radio- and immune-combination therapy. Clin Cancer Res. (2021) 27:2970–8. doi: 10.1158/1078-0432.CCR-20-3913
25. Morris G, Gevezova M, Sarafian V, and Maes M. Redox regulation of the immune response. Cell Mol Immunol. (2022) 19:1079–101. doi: 10.1038/s41423-022-00902-0
26. Wei XW, Lu C, Zhang YC, Fan X, Xu CR, Chen ZH, et al. Redox(high) phenotype mediated by KEAP1/STK11/SMARCA4/NRF2 mutations diminishes tissue-resident memory CD8+ T cells and attenuates the efficacy of immunotherapy in lung adenocarcinoma. Oncoimmunology. (2024) 13:2340154. doi: 10.1080/2162402X.2024.2340154
27. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
28. Wu F, Fan J, He Y, Xiong A, Yu J, Li Y, et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun. (2021) 12:2540. doi: 10.1038/s41467-021-22801-0
29. Ravi A, Hellmann MD, Arniella MB, Holton M, Freeman SS, Naranbhai V, et al. Genomic and transcriptomic analysis of checkpoint blockade response in advanced non-small cell lung cancer. Nat Genet. (2023) 55:807–19. doi: 10.1038/s41588-023-01355-5
30. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell. (2019) 35:238–55.e6. doi: 10.1016/j.ccell.2019.01.003
31. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. (2015) 12:453–7. doi: 10.1038/nmeth.3337
32. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. (2009) 462:108–12. doi: 10.1038/nature08460
33. Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. (2020) 182:1232–51.e22. doi: 10.1016/j.cell.2020.07.017
34. Heidegger S, Stritzke F, Dahl S, Daßler-Plenker J, Joachim L, Buschmann D, et al. Targeting nucleic acid sensors in tumor cells to reprogram biogenesis and RNA cargo of extracellular vesicles for T cell-mediated cancer immunotherapy. Cell Rep Med. (2023) 4:101171. doi: 10.1016/j.xcrm.2023.101171
35. Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci Transl Med. (2020) 12:eaay9013. doi: 10.1126/scitranslmed.aay9013
36. Pezone A, Olivieri F, Napoli MV, Procopio A, Avvedimento EV, and Gabrielli A. Inflammation and DNA damage: cause, effect or both. Nat Rev Rheumatol. (2023) 19:200–11. doi: 10.1038/s41584-022-00905-1
37. Li A, Wang Y, Yu Z, Tan Z, He L, Fu S, et al. STK11/LKB1-deficient phenotype rather than mutation diminishes immunotherapy efficacy and represents STING/type I interferon/CD8(+) T-cell dysfunction in NSCLC. J Thorac Oncol. (2023) 18:1714–30. doi: 10.1016/j.jtho.2023.07.020
38. Sheban D and Merbl Y. EMSY stabilization in KEAP1-mutant lung cancer disrupts genome stability and type I interferon signaling. Cell Death Differ. (2023) 30:1397–9. doi: 10.1038/s41418-023-01150-z
39. Xiaodan S, Peiyan Z, Hui L, Yan L, and Ying C. NRF2 participates in the suppressive tumor immune microenvironment of KRAS/KEAP1 co-mutant non-small cell lung cancer by inhibiting the STING pathway. Genes Dis. (2023) 10:1727–30. doi: 10.1016/j.gendis.2022.10.009
40. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. (2011) 478:515–8. doi: 10.1038/nature10429
41. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. (2019) 9:34–45. doi: 10.1158/2159-8290.CD-18-0689
42. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
43. Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marié IJ, and Levy DE. PKR transduces MDA5-dependent signals for type I IFN induction. PloS Pathog. (2016) 12:e1005489. doi: 10.1371/journal.ppat.1005489
44. Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. (2010) 7:354–61. doi: 10.1016/j.chom.2010.04.007
45. Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. (2019) 565:43–8. doi: 10.1038/s41586-018-0768-9
46. Rashida Gnanaprakasam JN, Wu R, and Wang R. Metabolic reprogramming in modulating T cell reactive oxygen species generation and antioxidant capacity. Front Immunol. (2018) 9:1075. doi: 10.3389/fimmu.2018.01075
47. Zhang B, Liu SQ, Li C, Lykken E, Jiang S, Wong E, et al. MicroRNA-23a curbsxnecrosis during early T cell activation by enforcing intracellular reactive oxygen species equilibrium. Immunity. (2016) 44:568–81. doi: 10.1016/j.immuni.2016.01.007
48. Feng P, Yang Q, Luo L, Sun Y, Lv W, Wan S, et al. The kinase PDK1 regulates regulatory T cell survival via controlling redox homeostasis. Theranostics. (2021) 11:9503–18. doi: 10.7150/thno.63992
49. D’Angelo JA, Dehlink E, Platzer B, Dwyer P, Circu ML, Garay J, et al. The cystine/glutamate antiporter regulates dendritic cell differentiation and antigen presentation. J Immunol. (2010) 185:3217–26. doi: 10.4049/jimmunol.1001199
50. Zhou Y, Fei M, Zhang G, Liang WC, Lin W, Wu Y, et al. Blockade of the phagocytic receptor merTK on tumor-associated macrophages enhances P2X7R-dependent STING activation by tumor-derived cGAMP. Immunity. (2020) 52:357–73.e9. doi: 10.1016/j.immuni.2020.01.014
51. Ahn J, Xia T, Rabasa Capote A, Betancourt D, and Barber GN. Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell. (2018) 33:862–73.e5. doi: 10.1016/j.ccell.2018.03.027
52. Zou J, Zheng Z, Ye W, Jin M, Yang P, Little PJ, et al. Targeting the smooth muscle cell KEAP1-Nrf2-STING axis with pterostilbene attenuates abdominal aortic aneurysm. Phytomedicine. (2024) 130:155696. doi: 10.1016/j.phymed.2024.155696
53. Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun. (2018) 9:3506. doi: 10.1038/s41467-018-05861-7
54. Hayes JD and Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. (2014) 39:199–218. doi: 10.1016/j.tibs.2014.02.002
55. Kensler TW, Wakabayashi N, and Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. (2007) 47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
56. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote Malignancy. Proc Natl Acad Sci U S A. (2008) 105:13568–73. doi: 10.1073/pnas.0806268105
57. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PloS Med. (2006) 3:e420. doi: 10.1371/journal.pmed.0030420
Keywords: redox phenotype, KEAP1/STK11 mutations, immune microenvironment, STING, MDA5
Citation: Shrestha A, Li Y, Huang L, Li S, Zhou Y, Gu J and Lin Z (2025) Redox phenotype confers T cell-exclusion microenvironment and resistance to immunotherapy by suppressing STING/MDA5 expression and interferon signaling in lung cancers harboring KEAP1/STK11 mutations. Front. Oncol. 15:1676797. doi: 10.3389/fonc.2025.1676797
Received: 31 July 2025; Accepted: 31 October 2025;
Published: 25 November 2025.
Edited by:
Kai Zhang, Zhengzhou University, ChinaReviewed by:
Caterina De Rosa, University of Campania Luigi Vanvitelli, ItalyJeong-Ryeol Gong, Korea Advanced Institute of Science and Technology (KAIST), Republic of Korea
Copyright © 2025 Shrestha, Li, Huang, Li, Zhou, Gu and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanbin Zhou, emhvdXliQG1haWwuc3lzdS5lZHUuY24=; Jincui Gu, Z3VqY0BtYWlsLnN5c3UuZWR1LmNu; Ziying Lin, bGluenk2NkBtYWlsLnN5c3UuZWR1LmNu
†These authors have contributed equally to this work