 Jan Halbritter
Jan Halbritter Anna Seidel1
Anna Seidel1 Luise Müller
Luise Müller Ria Schönauer
Ria Schönauer Bernd Hoppe
Bernd Hoppe- 1Division of Nephrology, Department of Internal Medicine, University of Leipzig, Leipzig, Germany
- 2Division of Pediatric Nephrology, University Children’s Hospital, Bonn, Germany
Kidney stone disease is an increasingly prevalent condition with remarkable clinical heterogeneity, with regards to stone composition, age of manifestation, rate of recurrence, and impairment of kidney function. Calcium-based kidney stones account for the vast majority of cases, but their etiology is poorly understood, notably their genetic drivers. As recent studies indicate, hereditary conditions are most likely underestimated in prevalence, and new disease genes are constantly being identified. As a consequence, there is an urgent need of a more efficient documentation and collection of cases with underlying hereditary conditions, to better understand shared phenotypic presentation and common molecular mechanisms. By implementation of a centralized patient registry on hereditary kidney stone disease in Germany, we aim to help closing the vast knowledge gap on genetics of kidney stone disease. In this context, clinical registries are indispensable for several reasons: first, delineating better phenotype–genotype associations will allow more precise patient stratification in future clinical research studies. Second, identifying new disease genes and new mechanisms will further reduce the rate of unknown nephrolithiasis/nephrocalcinosis etiology; and third, deciphering new molecular targets will pave the way to develop drugs for recurrence prevention in severely affected families.
Incidence and prevalence of kidney stone disease continues to rise in the general population. With a lifetime prevalence of up to 10%, nephrolithiasis (NL) and nephrocalcinosis (NC) are therefore major health burdens, especially in the Western World (1). NL and NC are associated with significant morbidity and progression to chronic kidney disease due to recurrence, repetitive surgical/endoscopic intervention, and concomitant inflammation. On a simplified level, kidney stone formation results from an imbalance of urinary inhibitors (e.g., citrate, magnesium, uromoduline, and pyrophosphate) and promoters (e.g., oxalate, calcium, phosphate, urate, and cystine) of crystallization, exceeding supersaturation with consecutive aggregation, nucleation, and stone growth at Randall’s plaque (Figure 1). This imbalance can be due to altered enteral and/or renal handling of either promotors or inhibitors, such as enteral malsecretion of oxalate or renal malreabsorption of calcium (Figure 1).
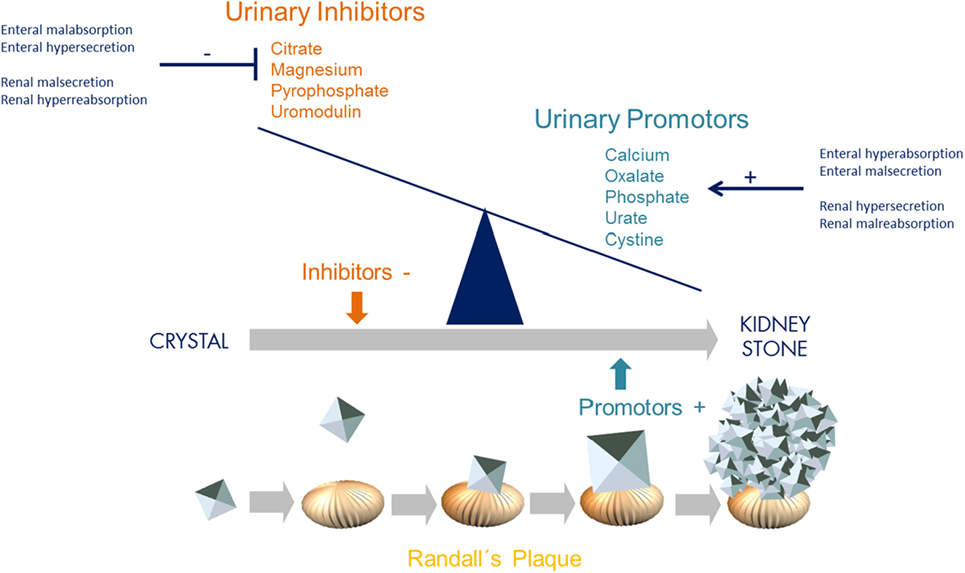
Figure 1. Imbalance of urinary inhibitors and promotors of crystallization leading to kidney stone formation. Concentration of urinary inhibitors and promotors is influenced and controlled by both intestinal and renal transporters (GI–kidney axis). These transporters and exchangers, such as SLC26A1, are responsible for secretion and absorption. With regards to oxalate, enteral hyperabsorption but malsecretion, and/or renal hypersecretion but malabsorption leads to urinary oxalate levels exceeding supersaturation and thereby promoting crystallization and consecutive stone formation [adapted from Ref. (2)].
The underlying etiology of NL is thought to be multifactorial with an environmental, notable dietary, hormonal, and genetic component. In twin studies, the heritability of kidney stones has been estimated at 56% (3), and up to two-thirds of hypercalciuric stone formers have relatives with NL (4). Although calcium-containing kidney stones account for more than 80% of all, the genetic basis of such stones remains largely unknown (5). Except for variants in CLDN14, TRPV5, SLC34A1, ALPL, CASR, and UMOD, genome-wide association studies have yet to yield substantial genetic factors (6–8). However, risk alleles have been identified within genes that were also found to transmit the disease on a Mendelian basis, such as CASR, SLC34A1, and SLC2A9 (9, 10). To date, more than 30 single genes with an Online Mendelian Inheritance in Man-defined phenotype have been identified to be implicated in NL/NC, if mutated (Table 1).
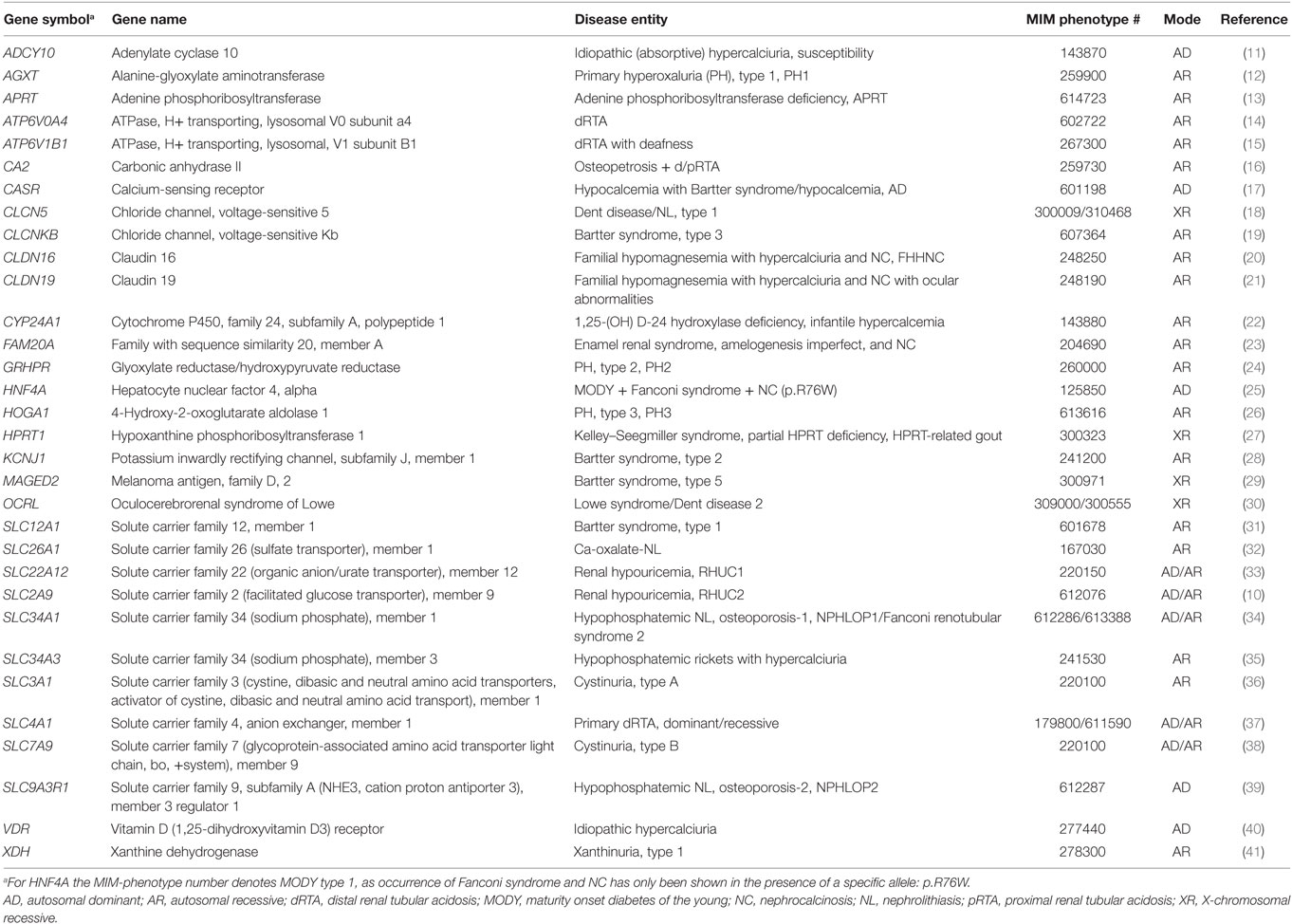
Table 1. Genes, known to cause monogenic forms of NL/NC.
Modes of inheritance in monogenic forms include autosomal-dominant, autosomal-recessive, and X-linked transmission. Interestingly, in several of these genes, both recessive and dominant modes of inheritance have been reported: SLC7A9, SLC34A1, SLC34A3, SLC2A9, SLC22A12, and SLC4A1. While most of the syndromic and severe congenital disorders exhibit a recessive inheritance pattern (Bartter, Lowe, Dent, FHHNC, and distal renal tubular acidosis with sensorineural deafness), milder conditions are rather associated with mutations in dominant genes. The majority of encoded proteins constitute renal solute transporters (e.g., SLC34A1, SLC34A3, and SLC9A3R1), but also chloride channels (CLCN5), tight-junction proteins (e.g., CLDN16/CLDN19), and metabolizing enzymes (e.g., AGXT, APRT, and CYP24A1) have been found defective in patients with NL/NC. Hence, the underlying defect is mostly located in the tubular system of the kidney itself and can therefore be attributed as tubulopathy. Conversely, a priori extrarenal conditions, as in primary hyperoxaluria (PH) where dysfunction of liver enzymes (AGXT, GRHPR, and HOGA1) cause oxalate accumulation with secondary renal affection, are conceivable causes of NL/NC. Although each disease phenotype is thought to represent a relatively rare entity, single-gene causes may account for a significant number of patients by their broad genetic heterogeneity (42). Apart from genetic heterogeneity, there is also an allelic variation, where truncating variants rather result in a loss of function and missense variants (hypomorphs) may cause rather subtle defects, which can be clinically overseen, especially in adult stone formers. Another recently appreciated phenomenon is about gene dosage effects in several of the aforementioned kidney stone genes. In SLC34A3 for instance, encoding one of the main phosphate transporters in the proximal tubule (NaPiIIc), it was shown that heterozygous individuals can no longer be merely regarded as healthy carriers, as they display renal calcifications and/or bone manifestation significantly more frequent than wild-type individuals; but still to a lesser degree than biallelic (homozygous and compound heterozygous) individuals (43). Similar observations were reported for families with mutations in CYP24A1 (44). The contribution of monogenic disorders to the overall prevalence of kidney stone disease has not been studied comprehensively in the past. Especially, genetic evidence based on broad screenings of a multitude of causative genes in large patient cohorts is lacking. Comprehensive genetic testing has been too costly and inefficient in the past. For most individuals with NL/NC, mutation analysis for a causative genetic defect has therefore not been accessible, despite the fact that knowledge of the molecular cause of NL/NC may have important consequences for prognosis, prophylaxis and/or treatment. Only rough estimates have been derived from clinical observation studies: based on a huge data collection of stone composition analysis, it was concluded that monogenic causes do not exceed 9.6% in children and 1.6% in adults (45). In the last decade, however, this situation has begun to change, with the advent of high-throughput sequencing techniques.
High-Throughput Mutation Analysis in Patients with NL/NC
To investigate patients with kidney stone disease for the presence of pathogenic mutations in known disease genes, we established a gene panel based on microfluidic multiplex-PCR and consecutive NextGen sequencing (Fluidigm™/NGS) (46, 47).
In a “pilot-study,” we consecutively recruited 268 genetically unresolved individuals from typical kidney stone clinics; 102 pediatric and 166 adult probands. As a result, we identified 50 deleterious variants in 14 out of 30 analyzed genes, leading to a molecular diagnosis in 15% of all cases. In the pediatric subgroup, we detected a causative mutation in 21%, while among adults, deleterious variants were present in 11% (Figure 2A) (48). Mutations in the cystinuria-gene SLC7A9 were found most frequently in the adult cohort (Figure 2B). Two follow-up studies were able to confirm these results. First, in an exclusively pediatric cohort of 143 NL/NC patients, 17% of cases were explained by mutations in 14 different genes (49). Second, in a cohort of 51 families with age of NL/NC manifestation before 25 years, targeted WES was used to detect a genetic cause in almost 30% (50). Not surprisingly, recessive mutations were more frequently found among neonates and in cases of congenital disease, whereas dominant conditions usually manifested later in life. These data indicate that genetic kidney stone disease is an underdiagnosed condition, despite the fact that the molecular diagnosis will potentially influence prognosis, prophylaxis, and/or treatment. A limitation worth mentioning, however, is a potential selection bias due to recruitment from specialist kidney stone clinics in all of the three aforementioned studies.
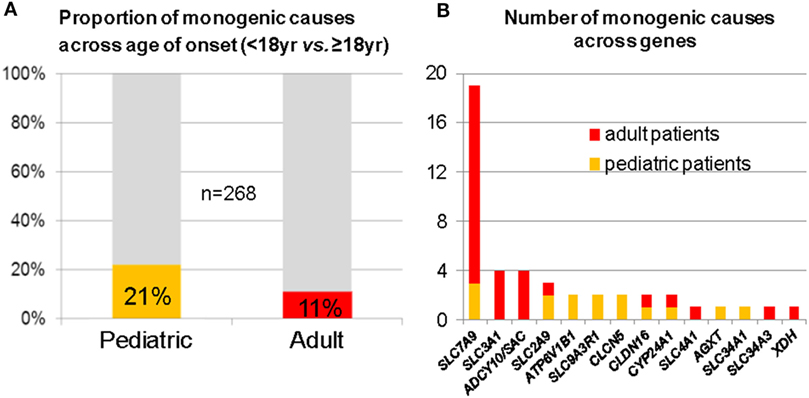
Figure 2. Mutation analysis of 30 known monogenic nephrolithiasis (NL)/nephrocalcinosis (NC) genes in 268 patients with NL/NC. (A) Fraction of monogenic causes in pediatric and adult subcohort. (B) Number of monogenic causes across genes (red denotes adults; orange denotes pediatric patients). Of note, SLC7A9 was found most frequently mutated, especially in young adults.
Identification of Novel Human Disease Genes by Candidate-Gene Approach
High-throughput mutation analysis is also used to screen for pathogenic variants in various candidate genes. One of the most interesting recent findings was the discovery of human mutations in SLC26A1 (32). Since the first description of Ca-oxalate (CaOx) kidney stone formation and NC in Slc26a1 (Sat1)-knockout mice by Dawson et al. in 2010, SLC26A1 has been a bona fide NL-candidate gene (51). SLC26A1 encodes an anion exchanger expressed at the basolateral membrane of proximal renal tubules, ileum, and jejunum. Consequently, by using a candidate-gene approach, pathogenic variants were identified in humans with a history of early onset CaOx-NL, namely, two unrelated individuals with biallelic missense variants (32). Functionally, pathogenicity of the identified variants was demonstrated in vitro, leading to intracellular mis-trafficking and impaired transport activity (32). Defective SLC26A1 therefore constitutes a new cause of CaOx-NL and should be considered when testing individuals for causes of recurrent CaOx-stone formation.
New Clinical Patient Registry for Hereditary Kidney Stone Disease
Most epidemiological data on increasing prevalence in Western countries are derived from US databases. Although urgently needed, centralized European databases are not available at the time. As aforementioned genetic studies on prevalence of hereditary kidney stone disease were executed with small cohorts from specialized centers in both Europe and the US, a translation to the general situation in Europe is not valid. While in the US, the Rare Kidney Stone Consortium constitutes a platform that integrates and coordinates registry, basic science, and clinical research activities for rare conditions such as cystinuria, PH, APRT deficiency, Dent and Lowe disease, no comparable data collection on patients with hereditary kidney stone disease has been implemented neither in Europe nor in Germany today. In collaboration with the existing European PH registry, OxalEurope (Prof. Bernd Hoppe, University of Bonn), and through funding by Deutsche Forschungsgemeinschaft and Else Kröner-Fresenius Stiftung, we recently established a clinical patient “Registry for hereditary kidney stone disease” at the University of Leipzig. The registry is nationally supported by the German Societies of Adult Nephrology (DGfN) and Pediatric Nephrology (GPN). It is further enrolled at the German Clinical Trials Register (DRKS-ID: DRKS00012891). As a fundamental part of study recruitment, high-throughput mutation analysis for known and novel kidney stone genes is offered on a research basis for patients without an established molecular diagnosis but with a clinical picture that points to an underlying genetic susceptibility: e.g., early age of onset (<40 years), positive family history, indicative phenotypes such as NC, cystinuria, or RTA, and severely recurrent NL (>3×) (Table 2). While patients with an already established genetic diagnosis are generally enrolled, cases with secondary NL/NC causes, such as malignancy, sarcoidosis, and primary hyperparathyroidism, do not get included in genetic analysis. To actively enroll patients, a clinical center will usually need approval by the local Institutional Review Board; a process for which we offer our help and assistance by providing respective templates. Upon ethics approval, consent form and clinical data sheets (e.g., clinical questionnaire) can be downloaded from our registry website (http://www.mks-registry.net). To ensure thorough clinical phenotyping, we will be asking for substantial patient information such as ethnicity, consanguinity, family history, age of onset, recurrence (defined as every putatively new kidney stone event), daily fluid intake, surgical interventions, and extrarenal involvement among others. The documents can be filled in by the patient with the help of the enrolling physician. In addition, biochemical serum parameters, including creatinine, eGFR, PTH, vitamin D, electrolytes, uric acid, and urinalysis (pH, calcium, phosphate, magnesium, uric acid, citrate, oxalate, and cystine, preferably from 24-h urine, if not spot urine), as well as data on stone composition analysis will be requested upon enrollment. Taking into account that 24-h urine collection and stone composition analysis is not routinely performed at all institutions, we include these parameters upon availability. 2-yearly clinical follow-up visits of enrolled patients are desirable but not mandatory. After registration, recruiting clinical centers will be provided with a personalized login to enter patient data via our registry website (http://www.mks-registry.net). Alternatively, we offer entering the data electronically when sent to us on paper. Entered data will be stored on a secured server and can be accessed by participating clinical centers to view their own patient data. The following websites provide further information:
https://www.dgfn.eu/hereditaere-nierensteinleiden.html
https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012891

Table 2. Inclusion criteria for mutation analysis in clinical patient registry.
In summary, kidney stone disease is an increasingly prevalent condition which is clinically heterogeneous and poorly understood, notably its genetic drivers. As a series of recent studies indicated, monogenic conditions are most likely underestimated in prevalence. By implementation of a centralized patient registry on hereditary kidney stone disease, we will contribute to overcome, at least in part, the vast knowledge gap on genetics of kidney stone disease. In this context, clinical registries are valuable sources for several reasons: first, delineating better phenotype–genotype associations will be crucial for more precise patient stratification in future clinical research studies. Second, identifying new disease genes with new disease mechanisms will diminish the gap of unknown NL/NC etiology; and third, deciphering new molecular targets helps to pave the way for developing drugs of recurrence prevention in severely affected families.
Ethics Statement
This study was carried out in accordance with the recommendations of “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig” with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the “Ethikkommission an der Medizinischen Fakultät der Universität Leipzig.”
Author Contributions
JH conceived and wrote the manuscript. BH, AS, LM, and RS edited the manuscript and built up the registry’s infrastructure that is introduced to the reader.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The research is funded by project grants from DFG (HA 6908/2-1) and EKFS (2016_A52) to JH. This work was further supported by the Federal Ministry of Education and Research (BMBF), Germany, FKZ: 01EO1501 to JH.
References
1. Scales CD Jr., Smith AC, Hanley JM, Saigal CS. Prevalence of kidney stones in the United States. Eur Urol (2012) 62:160–5.
2. Pfau A, Knauf F. Update on nephrolithiasis: core curriculum 2016. Am J Kidney Dis (2016) 68:973–85. doi:10.1053/j.ajkd.2016.05.016
3. Goldfarb DS, Fischer ME, Keich Y, Goldberg J. A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int (2005) 67:1053–61. doi:10.1111/j.1523-1755.2005.00170.x
4. Stechman MJ, Loh NY, Thakker RV. Genetics of hypercalciuric nephrolithiasis: renal stone disease. Ann N Y Acad Sci (2007) 1116:461–84. doi:10.1196/annals.1402.030
5. Sayer JA. Progress in understanding the genetics of calcium-containing nephrolithiasis. J Am Soc Nephrol (2017) 28:748–59. doi:10.1681/ASN.2016050576
6. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet (2009) 41:926–30. doi:10.1038/ng.404
7. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet (2010) 6:e1001039. doi:10.1371/journal.pgen.1001039
8. Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjörnsson G, et al. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun (2015) 6:7975. doi:10.1038/ncomms8975
9. Döring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet (2008) 40:430–6. doi:10.1038/ng.107
10. Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet (2008) 83:744–51. doi:10.1016/j.ajhg.2008.11.001
11. Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, et al. Identification and characterization of a gene with base substitutions associated with the absorptive hypercalciuria phenotype and low spinal bone density. J Clin Endocrinol Metab (2002) 87:1476–85. doi:10.1210/jcem.87.4.8300
12. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Mistargeting of peroxisomal L-alanine:glyoxylate aminotransferase to mitochondria in primary hyperoxaluria patients depends upon activation of a cryptic mitochondrial targeting sequence by a point mutation. Proc Natl Acad Sci U S A (1991) 88:10900–4. doi:10.1073/pnas.88.23.10900
13. Hidaka Y, Palella TD, O’Toole TE, Tarlé SA, Kelley WN. Human adenine phosphoribosyltransferase. Identification of allelic mutations at the nucleotide level as a cause of complete deficiency of the enzyme. J Clin Invest (1987) 80:1409–15. doi:10.1172/JCI113219
14. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet (2000) 26:71–5. doi:10.1038/79208
15. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet (1999) 21:84–90. doi:10.1038/5022
16. Venta PJ, Welty RJ, Johnson TM, Sly WS, Tashian RE. Carbonic anhydrase II deficiency syndrome in a Belgian family is caused by a point mutation at an invariant histidine residue (107 His→Tyr): complete structure of the normal human CA II gene. Am J Hum Genet (1991) 49:1082–90.
17. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med (1996) 335:1115–22. doi:10.1056/NEJM199610103351505
18. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. A common molecular basis for three inherited kidney stone diseases. Nature (1996) 379:445–9. doi:10.1038/379445a0
19. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet (1997) 17:171–8. doi:10.1038/ng1097-171
20. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science (1999) 285:103–6. doi:10.1126/science.285.5424.103
21. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet (2006) 79:949–57. doi:10.1086/508617
22. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med (2011) 365:410–21. doi:10.1056/NEJMoa1103864
23. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, et al. Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron Physiol (2012) 122:1–6. doi:10.1159/000349989
24. Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet (1999) 8:2063–9. doi:10.1093/hmg/8.11.2063
25. Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al. The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a β cell phenotype. J Med Genet (2014) 51:165–9. doi:10.1136/jmedgenet-2013-102066
26. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet (2010) 87:392–9. doi:10.1016/j.ajhg.2010.07.023
27. Davidson BL, Tarlé SA, van Antwerp M, Gibbs DA, Watts RW, Kelley WN, et al. Identification of 17 independent mutations responsible for human hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. Am J Hum Genet (1991) 48:951–8.
28. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet (1996) 14:152–6. doi:10.1038/ng1096-152
29. Laghmani K, Beck BB, Yang S-S, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med (2016) 374:1853–63. doi:10.1056/NEJMoa1507629
30. Reilly DS, Lewis RA, Ledbetter DH, Nussbaum RL. Tightly linked flanking markers for the Lowe oculocerebrorenal syndrome, with application to carrier assessment. Am J Hum Genet (1988) 42:748–55.
31. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet (1996) 13:183–8. doi:10.1038/ng0696-183
32. Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, et al. Mutations in SLC26A1 cause nephrolithiasis. Am J Hum Genet (2016) 98:1228–34. doi:10.1016/j.ajhg.2016.03.026
33. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature (2002) 417:447–52. doi:10.1038/nature742
34. Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med (2002) 347:983–91. doi:10.1056/NEJMoa020028
35. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet (2006) 78:193–201. doi:10.1086/499410
36. Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, et al. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet (1994) 6:420–5. doi:10.1038/ng0494-420
37. Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest (1997) 100:1693–707. doi:10.1172/JCI119694
38. Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet (1999) 23:52–7. doi:10.1038/12652
39. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med (2008) 359:1128–35. doi:10.1056/NEJMoa0802836
40. Scott P, Ouimet D, Valiquette L, Guay G, Proulx Y, Trouvé ML, et al. Suggestive evidence for a susceptibility gene near the vitamin D receptor locus in idiopathic calcium stone formation. J Am Soc Nephrol (1999) 10:1007–13.
41. Ichida K, Amaya Y, Kamatani N, Nishino T, Hosoya T, Sakai O. Identification of two mutations in human xanthine dehydrogenase gene responsible for classical type I xanthinuria. J Clin Invest (1997) 99:2391–7. doi:10.1172/JCI119421
42. Hildebrandt F. Genetic kidney diseases. Lancet (2010) 375:1287–95. doi:10.1016/S0140-6736(10)60236-X
43. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol (2014) 25:2366–75. doi:10.1681/ASN.2013101085
44. O’Keeffe DT, Tebben PJ, Kumar R, Singh RJ, Wu Y, Wermers RA. Clinical and biochemical phenotypes of adults with monoallelic and biallelic CYP24A1 mutations: evidence of gene dose effect. Osteoporos Int (2016) 27:3121–5. doi:10.1007/s00198-016-3615-6
45. Jungers P, Joly D, Blanchard A, Courbebaisse M, Knebelmann B, Daudon M. Lithiases rénales héréditaires monogéniques. Récents acquis diagnostiques et thérapeutiques. Nephrol Ther (2008) 4:231–55. doi:10.1016/j.nephro.2007.12.005
46. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet (2012) 49:756–67. doi:10.1136/jmedgenet-2012-100973
47. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet (2013) 132:865–84. doi:10.1007/s00439-013-1297-0
48. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol (2015) 26:543–51. doi:10.1681/ASN.2014040388
49. Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clin J Am Soc Nephrol (2016) 11:664–72. doi:10.2215/CJN.07540715
50. Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int (2018) 93:204–13. doi:10.1016/j.kint.2017.06.025
Keywords: nephrolithiasis, hereditary, nephrocalcinosis, kidney stone disease, monogenic, registry
Citation: Halbritter J, Seidel A, Müller L, Schönauer R and Hoppe B (2018) Update on Hereditary Kidney Stone Disease and Introduction of a New Clinical Patient Registry in Germany. Front. Pediatr. 6:47. doi: 10.3389/fped.2018.00047
Received: 04 January 2018; Accepted: 19 February 2018;
Published: 07 March 2018
Edited by:
Max Christoph Liebau, Universitätsklinikum Köln, GermanyReviewed by:
Fatos Yalcinkaya, Ankara University, TurkeyGianpaolo De Filippo, Bicêtre Hospital, France
David J. Sas, Mayo Clinic, United States
Copyright: © 2018 Halbritter, Seidel, Müller, Schönauer and Hoppe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Halbritter, amFuLmhhbGJyaXR0ZXJAbWVkaXppbi51bmktbGVpcHppZy5kZQ==