 Darja Smigoc Schweiger1
Darja Smigoc Schweiger1 Katja Goricar2Tinka Hovnik3Andrijana Mendez4Natasa Bratina1,5Jernej Brecelj5,6Blanka Vidan-Jeras4
Katja Goricar2Tinka Hovnik3Andrijana Mendez4Natasa Bratina1,5Jernej Brecelj5,6Blanka Vidan-Jeras4 Tadej Battelino1,5
Tadej Battelino1,5 Vita Dolzan2*
Vita Dolzan2*- 1Department of Pediatric Endocrinology, Diabetes and Metabolic Diseases, University Children's Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 2Pharmacogenetics Laboratory, Faculty of Medicine, Institute of Biochemistry, University of Ljubljana, Ljubljana, Slovenia
- 3Unit of Special Laboratory Diagnostics, University Children's Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia
- 4Tissue Typing Centre, Blood Transfusion Center of Slovenia, Ljubljana, Slovenia
- 5Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
- 6Department of Gastroenterology, Hepatology and Nutrition, University Children's Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia
Genetic polymorphisms in genes coding for inflammasome components nucleotide-binding oligomerization domain leucine rich repeat and pyrin domain-containing protein 3 (NLRP3) and caspase recruitment domain-containing protein 8 (CARD8) have been associated with autoinflammatory and autoimmune diseases. On the other hand several studies suggested that NLRP3 inflammasome contributes to maintenance of gastrointestinal immune homeostasis and that activation of NLRP3 is regulated by protein tyrosine phosphatase non-receptor 22 (PTPN22). PTPN22 polymorphism was implicated in the risk for various autoimmune diseases including type 1 diabetes (T1D) but not for celiac disease (CD). The aim of our study was to evaluate the role of inflammasome related polymorphisms in subjects with either T1D or CD as well as in subjects affected by both diseases. We examined PTPN22 rs2476601 (p.Arg620Trp), NLRP3 rs35829419 (p.Gln705Lys), and CARD8 rs2043211 (p.Cys10Ter) in 66 subjects with coexisting T1D and CD, 65 subjects with T1D who did not develop CD, 67 subjects diagnosed only with CD and 127 healthy unrelated Slovenian individuals. All results were adjusted for clinical characteristic and human leukocyte antigen (HLA) risk. PTPN22 rs2476601 allele was significantly more frequent among subjects with T1D (Padj = 0.001) and less frequent in subjects with CD (Padj = 0.039) when compared to controls. In patients with coexisting T1D and CD this variant was significantly less frequent compared to T1D group (Padj = 0.010). Protective effect on CD development in individuals with T1D was observed only within the low risk HLA group. On the other hand, we found no association of NLRP3 rs35829419 and CARD8 rs2043211 with the development of T1D, CD or both diseases together. In conclusion PTPN22 rs2476601polymorphism was significantly associated with the risk of developing T1D in Slovenian population, while no associations of proinflammatory NLRP3 and CARD8 polymorphisms with T1D and CD were observed. Interestingly, the same PTPN22 variant protected from CD. We hypothesize that this effect may be mediated through the NLRP3 inflammasome activation.
Introduction
Type 1 diabetes (T1D) is characterized by chronic autoimmune mediated destruction of pancreatic β-cells, leading to symptomatic partial, or in most cases, absolute insulin deficiency requiring lifelong administration of exogenous insulin (1). Celiac disease (CD) is a chronic immune-mediated disorder characterized by the presence of variable combination of clinical manifestations ranging from enteropathy to a systemic affliction of different organ systems and appearance of autoimmune antibodies elicited by ingestion of gluten and related prolamines (2). Both multifactorial diseases have a strong autoimmune and genetic component and often segregate together (3). Because of common association of T1D with CD, screening for CD is recommended in all children with T1D (1, 2). In individuals with T1D positive serum anti-tissue transglutaminase (tTG) antibodies may spontaneously normalize even on a gluten-containing diet (4). Therefore, duodenal biopsy is still required in asymptomatic individuals with T1D to confirm the diagnosis of CD (2). The majority of patients develop CD after the onset of T1D and the risk is significantly higher in children with T1D diagnosed before 5 years of age (5). In addition to CD autoimmune thyroid disease (ATD) is frequently seen in individuals with T1D (6). Having T1D and ATD has been reported to be a risk factor for the development of CD (5). On the other hand children affected by CD were at increased risk of subsequent hypothyroidism (7).
Shared susceptibility alleles in the Human leukocyte antigen (HLA) region encoding HLA-DQ2 and DQ8 molecules which account for much of the etiology of T1D and CD contribute to the coexistence of both diseases. The risk is further modified by the HLA genotype, which is the combination of HLA alleles inherited from both parents (8, 9). In our previous study, DR3-DQ2/DR4-DQ8 and DR3-DQ2/DR3-DQ2 genotypes were found to be highly predisposing for developing both diseases together (10). However, disease co-occurrence is greater that could be accounted for HLA-DR-DQ genotypes. In the prospective birth cohort study persistent islet autoantibodies were associated with increasing risk of subsequent development tTG antibodies, suggesting that T1D autoimmunity is a risk factor for subsequent CD (11). Cumulative genetic risk associated with both T1D and CD increases substantially when combined with the effect of genetic polymorphisms outside the HLA region (12). Among non-HLA factors shared pathophysiological mechanisms may involve many genes of the innate immune signaling, which is the first line of defense against invading microorganisms not only by directly eliminating the pathogen but also through further shaping of the adaptive immunity (13). It is hypothesized that the environment increasingly conducive to T1D development may potentiate autoimmunity by dysregulating innate immune processes in genetically susceptible individuals (14, 15). The inflammasomes are highly conserved pattern-recognition receptors expressed by cells of the innate immune system and some tissue cells. These cells recognize microbial pathogens as well as cellular stress and damage, leading to production of pro-inflammatory cytokines interleukin (IL)-1β and IL-18 and triggering a process of inflammation-related cell death (16). The best characterized is the NLRP3 inflammasome, the protein complex including nucleotide-binding oligomerization domain leucine rich repeat and pyrin domain-containing protein 3 (NLRP3) protein, adapter protein apoptosis-associated speck-like protein and procaspase-1. Interactions among these proteins tightly regulate inflammasome function in order to ensure immune activity only when appropriate (17). NLRP3 rs35829419 (p.Gln705Lys) is a gain-of-function polymorphism associated with pro-inflammatory phenotype (18, 19). Caspase recruitment domain-containing protein 8 (CARD8) negatively regulates NLRP3 inflammasome through its binding with NLRP3 protein. CARD8 rs2043211 (p.Cys10Ter) results in non-functional protein and leads to loss of CARD-8 inhibition of caspase-1 (20). Aberrant NLRP3 inflammasome activation has been linked to various immune disorders (21) and can also contribute to the onset and progression of metabolic diseases (22, 23). It was shown that NLRP3 played an important role in the immune pathogenesis of T1D development in NOD mice (24). It seems that NRLP3 also plays a regulatory and reparative role in the maintenance of immune tolerance and intestinal epithelial barrier integrity (25).
Tyrosine phosphorylation is an important regulatory mechanism that prevents aberrant inflammasome activation. A recent study has shown that protein tyrosine phosphatase non-receptor 22 (PTPN22) interacts with and dephosphorylates NLRP3 upon proinflammatory insults, allowing robust NLRP3 activation and IL-1β secretion (26). The PTPN22 rs2476601 (p.Arg620Trp) polymorphism has been linked to several autoimmune diseases (27). The association between PTPN22 rs2476601 and T1D was initially identified by Bottini et al. in two independent populations (28). Interestingly, the results of comprehensive meta-analysis showed no or weak association of PTPN22 rs2476601 with CD (29, 30) and some autoimmune diseases primarily affecting the gastrointestinal system (31).
Previous studies have shown conflicting and heterogeneous findings with regard to the role of NLRP3 and PTPN22 polymorphisms in either T1D or CD onset. However, T1D and CD frequently occur in the same individual, pointing to shared mechanisms. The aim of our study was to investigate the role of PTPN22 rs2476601 (p.Arg620Trp), NLRP3 rs35829419 (p.Gln705Lys), and CARD8 rs2043211 (p.Cys10Ter) in subjects with T1D who did not develop CD, in subjects with CD who did not develop T1D and in subjects who developed both diseases. Furthermore, the role of these polymorphisms was assessed while also taking into account the presence of high or low risk HLA genotype.
Materials and Methods
Study Design and Subjects
In total 325 individuals, who were previously HLA typed (10), were included in our study. Patients were stratified into three groups: 66 with coexisting T1D and CD (T1D+CD), 65 with T1D who did not develop CD after minimum of 10 years of follow up and 67 diagnosed only with CD. A group of 127 healthy unrelated Slovenian individuals served as a control group. In all subjects CD was diagnosed according to the relevant European Society for Pediatric Gastroenterology Hepatology and Nutrition (ESPGHAN) criteria (2, 32). In individuals with T1D serological screening for CD started at the onset of T1D and continued yearly. Study included 862 children from 0 to 17.99 years of age at diagnosis, referred to the University Children's Hospital Ljubljana, Slovenia at the onset of T1D, from 1995 to 2009. Screening for celiac disease was based on the detection of endomysial immunoglobulin A (IgA) autoantibodies (EMA) measured by indirect immunofluorescence until 2000. Afterwards tTG measured by enzyme linked immunosorbent assay were used for screening. Total serum IgA was used to exclude IgA deficiency. In IgA-deficient individuals IgG-specific anti-bodies (tTG and/or EMA IgG) were detected. Since individuals with T1D may have fluctuating levels of CD-specific antibodies, the definitive diagnosis of CD was established by an intestinal biopsy sample showing a Marsh score of 2 or higher in 69 participants. This study included 66 participants for whom DNA samples were available. Most of the subjects were diagnosed with CD after T1D diagnosis (designated as T1D first), while 22 (33.8%) had CD before T1D onset or were diagnosed with CD at the time of T1D diagnosis as the result of the screening (designated as CD first).
PTPN22, NLRP3, and CARD8 Genotyping
For the detection of PTPN22 rs2476601 (c.1858T>C) polymorphism we used TaqMan SNP Genotyping assay (ID:C_16021387_20; Applied Biosystems, Foster City, CA, US). NLRP3 rs35829419 (c.2113C>A, p.Gln705Lys) and CARD8 rs2043211 (c.304A>T; p.Phe102Ile, p.Cys10Ter) were genotyped using a fluorescence-based competitive allele-specific (KASPar) assay according to the manufacturer's instructions (KBiosciences, Herts, UK). Genotyping was repeated in 20% of samples to check for genotyping reliability.
HLA DRB1, DQB1, and DQA1 Typing
The DRB1, DQB1, and DQA1 genotyping was performed as previously described (10). We performed PCR-sequence-specific primer (PCR-SSP) typing (Olerup SSP, Stockholm, Sweden), PCR-reverse sequence-specific oligonucleotide probe (PCR-rSSO) typing assay (LabType SSO, One Lambda, Kanoga Park, CA, USA) with Luminex technology (LABSCAN TM 100, Luminex corporation, Austin, TX, USA) and PCR-sequencing based typing (PCR-SBT) (Conexio's SBT Resolver™, Conexio-Genomics, Fremantle, Western Australia).
Statistical Analysis
HLA alleles were deduced into three loci haplotypes (DRB1-DQA1-DQB1) as described earlier (10). DRB1*03:01-DQA1*05:01-DQB1*02:01 haplotype was abbreviated as DR3-DQ2, DRB1*07:01–DQA1*02:01-DQB1*02:02 as DR7-DQ2 and DRB1*11/12–DQA1*05:05–DQB1*03:01 as DR5-DQ7. DRB1*04:01-DQA1*03:01-DQB1*03:02, DRB1*04:02-DQA1*03:01-DQB1*03:02, DRB1*04:04-DQA1* 03:01-DQB1*03:02, DRB1*04:05-DQA1*03:01-DQB1*03:02, and DRB1*04:08-DQA1*03:01-DQB1*03:04 haplotypes were considered as DR4-DQ8. X was defined as non-DR3-DQ2 and non-DR4-DQ8 haplotype. HLA-DRB1-DQA1-DQB1/DRB1-DQA1-DQB1 genotypes (combination of two DRB1-DQA1-DQB1 haplotypes) were grouped into two HLA-risk categories encoded as low-risk (LR) and high-risk (HR) categorical variables by means of the known risk associated with T1D and CD in the Slovenian population. The heterozygous genotype DR3-DQ2/DR4-DQ8 and genotype DR4-DQ8/ DR4-DQ8 were defined as HR for T1D (8, 33, 34). Since the HLA influence on CD susceptibility showed a dose effect, DR3-DQ2/ DR3-DQ2 and DR3-DQ2/ DR7-DQ2, which are DQB1*02 homozygous combinations were classified as high risk for CD (10). DR3-DQ2/X and DR4-DQ8/X single dose combinations, DRB1*07-DQA1*02:01-DQB1*02:02/DRB1*11/12-DQA1*05:05-DQB1*03:01 haplotype combination with DQ2 alleles carried in trans as well as non-DQ2 and non-DQ8 (X/X) haplotype combinations were considered as low risk.
For the description of continuous and categorical variables, median with interquartile range and frequencies were used, respectively. Standard chi square test was used to compare distribution of categorical clinical characteristics between different patient groups and to assess deviation from Hardy-Weinberg equilibrium. Nonparametric Mann-Whitney and Kruskal-Wallis tests were used to compare continuous clinical characteristics between different patient groups. Logistic regression was used for comparison of genotype frequencies, calculating odds ratio (OR) and 95% confidence interval (CI), as well as for the analysis of interaction between polymorphisms. Two-tailed Fisher's exact test was used for comparisons of the frequencies of HLA deduced genotypes. The level of statistical significance was set at 0.05. All statistical analyses were performed using IBM SPSS Statistics 19.0 (IBM Corporation, Armonk, NY, USA).
Results
Clinical Characteristics of Subjects With T1D, CD, and T1D+CD
Clinical characteristics of subjects with T1D, CD, or both including HLA-risk categories are shown in Table 1. Subjects with CD were the youngest at the time of the first diagnosis, but the difference did not reach statistical significance when all three groups were compared (P = 0.072, Supplementary Figure 1). Antithyroid antibodies were observed in 18 individuals with T1D (27.7%) and in 16 individuals (24.2%) with coexisting T1D and CD with no significant difference between the two groups. Three individuals (4.5%) were diagnosed with ATD in the CD only group (Table 1).
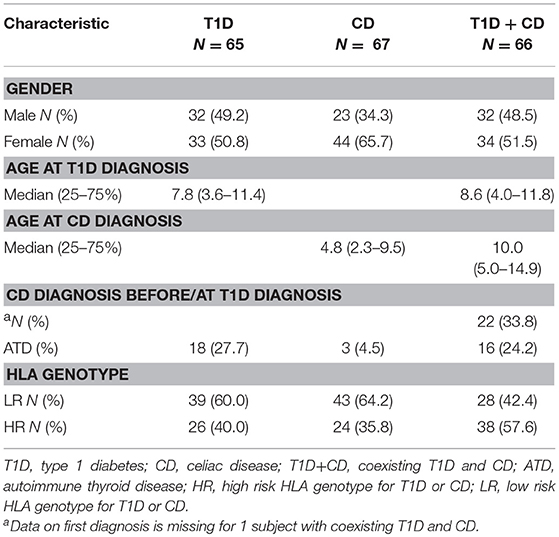
Table 1. Characteristics of subject with T1D, CD, and T1D+CD.
HLA-DRB1-DQA1-DQB1 Deduced Genotype Frequencies in Individuals With Coexisting T1D and CD
The distribution of HLA genotypes differed significantly among the groups (Supplementary Table 1). Two high-risk genotypes DR3-DQ2/DR4-DQ8 (p < 0.001) and DR3-DQ2/DR3-DQ2 (p < 0.001) were associated with an increased risk for developing the combination of both diseases. On the other hand low risk genotypes had neutral effect on co-occurrence. When individuals with T1D who later developed CD were compared to those with T1D alone, genotype DR3-DQ2/DR3-DQ2 (OR = 7.10, p = 0.002) conferred an increased risk for CD and DR4-DQ8/X (OR = 0.19, p = 0.001) was negatively associated. Comparisons of individuals in whom CD diagnosis proceeded T1D with CD only group revealed that DR3-DQ2/DR4-DQ8 genotype was associated with an increased risk for developing T1D (OR = 9.96, p = 0.002).
PTPN22, NLRP3, and CARD8 Genotype Frequencies Distribution
Genotype frequencies were in agreement with HWE in the control group (P = 0.413 for PTPN22 rs2476601, P = 0.461 for NLRP3 rs35829419, and P = 0.616 for CARD8 rs2043211) as well as in the entire study group (P = 0.823 for PTPN22 rs2476601, P = 0.138 for CARD8 rs2043211, and P = 0.607 for NLRP3 rs35829419). Minor allele frequencies were 14.2% for PTPN22 rs35829419, 38.2% for CARD8 rs2043211, and 6.6% for NLRP3 rs2476601 (Table 2).
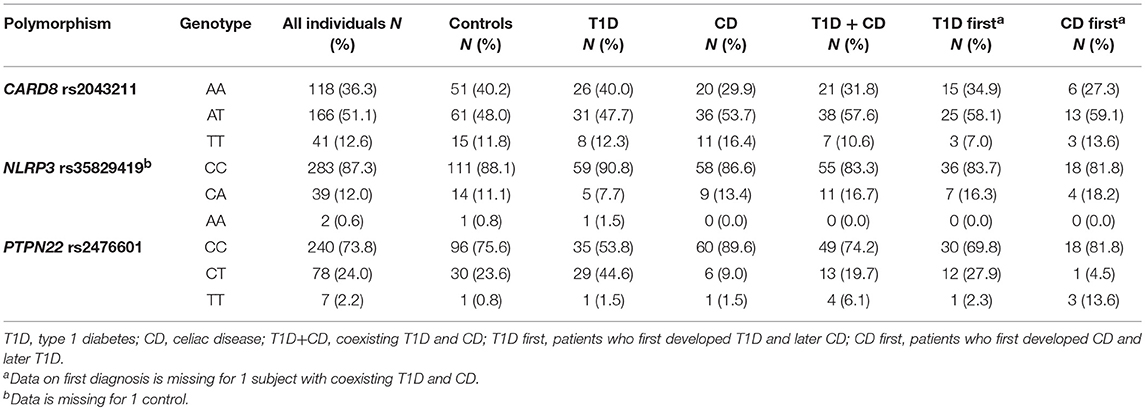
Table 2. Genotype frequencies of CARD8, NLRP3, and PTPN22 polymorphisms.
Association Between PTPN22, NLRP3, and CARD8 Genotype Frequencies and Risk of T1D, CD, and T1D+CD
Proportion of carriers of at least one polymorphic PTPN22 rs2476601allele was significantly higher among subjects withT1D (OR = 2.65, 95% CI = 1.41–5.00, P = 0.003) and the difference remained statistically significant after adjustment for gender (OR = 3.02, 95% CI = 1.54–5.93, P = 0.001). On the other hand, polymorphic PTPN22 rs2476601allele was significantly less common in subjects with CD when compared to controls (OR = 0.36, 95% CI = 0.15–0.87, P = 0.024), the difference remained significant after adjustment for gender (OR = 0.39, 95% CI = 0.16–0.96, P = 0.039). There were no significant differences in genotype frequencies for NLRP3 rs35829419 and CARD8 rs2043211 between cases and controls, not even after adjustment for clinical parameters (Table 3).

Table 3. Association between CARD8, NLRP3, and PTPN22 genotype frequencies and risk of T1D, CD and T1D+CD.
Comparison of PTPN22, NLRP3, and CARD8 Genotype Frequencies of Subjects With T1D, CD, and T1D+CD
Proportion of carriers of at least one polymorphic PTPN22 rs2476601allele in subjects with coexisting T1D and CD was significantly lower compared to T1D (OR = 0.41, 95% CI = 0.19–0.85, P = 0.016) and the difference remained significant after adjustment for age at diagnosis, gender and HLA genotype (OR = 0.36, 95% CI = 0.17–0.78, P = 0.010). Proportion of carriers of at least one polymorphic PTPN22 rs2476601 allele in subjects with coexisting T1D and CD was significantly higher compared to CD (OR = 2.97, 95% CI = 1.14–7.75, P = 0.026), however the difference was no longer significant after adjustment for age at diagnosis, gender and HLA genotype (OR = 2.42, 95% CI = 0.82–7.12, P = 0.109). NLRP3 rs35829419 and CARD8 rs2043211 genotype frequencies did not differ significantly among subjects with T1D, CD, or coexisting diseases (Table 4). No significant differences in genotype frequencies were observed when subjects with coexisting T1D and CD were stratified according to the order of the disease onset (T1D first and CD first groups) and compared to patients with T1D only and CD only as well as when compared between themselves (Table 4).
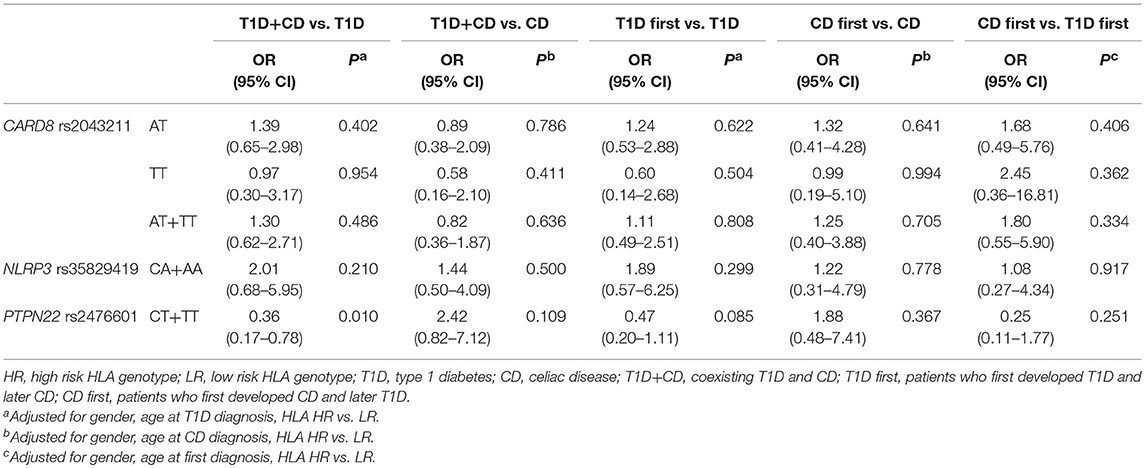
Table 4. Comparison of CARD8, NLRP3, and PTPN22 genotype frequencies of subjects with T1D, CD, and T1D+CD.
Multiplicative Interaction
When comparing subjects that were first diagnosed with T1D and then developed CD (T1D first) to patients with only T1D, we observed a multiplicative interaction between PTPN22 and HLA (OR = 7.19, 95% CI = 1.03–50.29, P = 0.047) (Table 5). Subjects with LR HLA and PTPN22 CC genotypes were the reference category. Carriers of at least one polymorphic PTPN22 allele were significantly less frequent among subjects withT1D first compared to T1D only within HLA LR group (P = 0.015), but not in the HLA HR group. Among carriers of at least one polymorphic PTPN22 allele, T1D first were more likely HLA HR than T1D (P = 0.014). A similar trend was observed when comparing group of subjects with both T1D and CD (T1D first + CD first) to subjects with only T1D or CD, but it did not reach statistical significance. No multiplicative interaction was observed between NLRP3 rs35829419 and CARD8 rs2043211 (Supplementary Table 2).
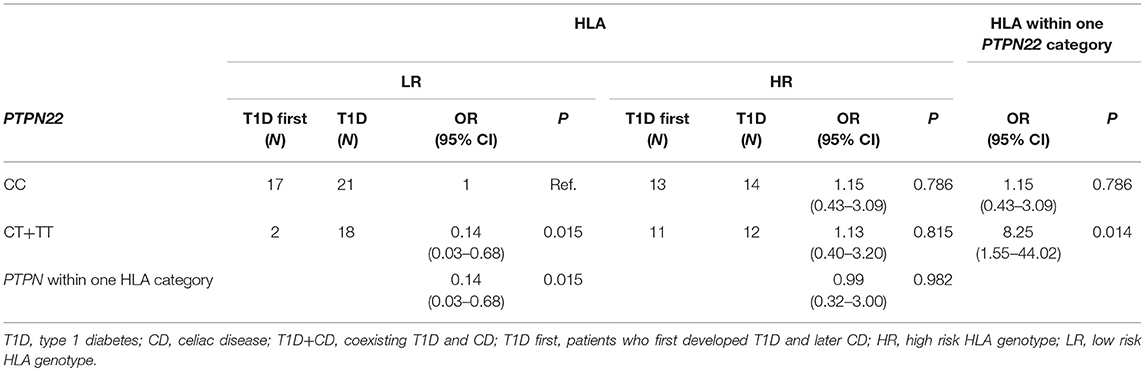
Table 5. Interaction between PTPN22 rs2476601 and HLA (comparing T1D first to T1D).
Discussion
The present study investigated the role of PTPN22 and inflammasome polymorphisms in the risk for development of T1D and CD as separate or concurrent diseases. Our results suggest a dual role of the PTPN22 polymorphism in the development of autoimmune diseases: while rs2476601 polymorphism was associated with increased risk for T1D, it was protective against development of CD. On the other hand, NLRP3 and CARD8 polymorphisms were not associated with the risk of either T1D or CD.
Previous studies reported associations of PTPN22 and NLRP3 polymorphisms with several autoimmune diseases suggesting their general role in the etiology of autoimmunity, however these risk alleles are not shared among all autoimmune disease (31, 35). NOD mice models showed an important role of aberrant NLRP3 activation in the immune pathogenesis of T1D (24). In preclinical studies NLRP3 deficient animals have been shown to have defective epithelial barrier function and increased intestinal inflammation (36). In human studies of CD, peripheral blood mononuclear cells and monocytes responded to gliadin fraction by a robust secretion of IL-1β and IL-1α and a slightly elevated production of IL-18 (37). In contrast with these functional studies, our results did not confirm the role of functional NLRP3 rs35829419 and CARD8 rs2043211 polymorphisms that promote NLRP3 inflammasome activation in the risk of either T1D or CD. Conflicting results were also reported in the literature, as the minor NLRP3 rs35829419 A allele was reported to be associated with CD in Brazilian population (38), however in Italian patients this allele appeared to have a protective role against the development of CD (39).
Our finding that PTPN22 polymorphism has a dual role in the development of autoimmune diseases is of particular interest as recent studies showed that NLRP3 activation is negatively regulated by tyrosine phosphorylation (40). The presence of autoimmunity associated PTPN22 variant in mice (PTPN22-619W) resulted in dephosphorylation and activation of NLRP3, while loss of PTPN22 resulted in decreased NLRP3 mediated IL-1β secretion. PTPN22 affected NLRP3 activation only in the lamina propria, resulting in only minor changes of IL-18 activation, which is mainly produced by intestinal epithelial cells. Furthermore, loss of PTPN22 resulted in aggravated intestinal inflammation (26). Of note PTPN22 c.1858C>T variant leading to p.Arg620Trp substitution (rs2476601) showed association pattern with target tissue specificity being less involved in pathogenesis of autoimmune diseases triggered by environmental factors primarily acting on target tissues such as gut mucosa (31). Supporting the notion of negligible effect of PTPN22 rs2476601 on CD development, no statistically significant association of this polymorphism with CD was found in a north European (41, 42) and Spanish population (29). Nevertheless, we observed an apparent protective effect of PTPN22 rs2476601 polymorphism on CD development, suggesting a role of PTPN22 in facilitating inflammasome activation in intestinal lamina propria.
Previous studies have consistently associated PTPN22 rs2476601 with the T1D onset in several populations (43). A definite effect of this polymorphism on the appearance of autoantibodies and progression to clinical T1D was observed in the DIPP study cohort, where the carriers of the PTPN22 rs2476601 polymorphism showed enhanced appearance of T1D associated autoantibodies when exposed to cow milk in early infancy (44). Consistent with these reports we observed significantly higher frequency of carriers of at least one rs2476601 T allele among subjects withT1D without associated CD as compared to the control group supporting a role of this polymorphism in disease susceptibility in the Slovenian population. Given a role of PTPN22 in pathogenesis of the T1D and lack of association to CD we also investigated the role of PTPN22 in T1D with accompanying CD. However, unlike to the individuals with isolated T1D, PTPN22 rs2476601 was not associated with increased risk for T1D with accompanying CD. These results suggest that PTPN22 rs2476601 may not influence T1D pathophysiological mechanisms in all cases, while previous reports on the association of this variant and APSIII onset suggest common pathogenic pathways (45). The discrepancies observed among studies may in part be explained by considerable differences of PTPN22 rs2476601 frequencies in populations of European origin (31). Minor allele frequency for PTPN22 rs2476601 in our population was 14.2%, resembling more closely those from northern European populations, which are among the highest in Europe (46).
Since HLA remains, by far, the strongest predictor of T1D and CD risk all results in our study were adjusted for HLA risk and only odds ratios adjusted for given HLA risk category were reported. Previous studies have shown that the strength of association between PTPN22 rs2476601 and T1D is stronger in low-risk HLA subjects, compared with that in subjects caring high-risk HLA genotypes (47). In our study, the interaction effect of HLA-risk category on the association of PTPN22 rs2476601 was observed when individuals who had first T1D and then developed CD (T1D first group) were compared to individuals with T1D who did not develop CD. Protective effect on CD development was observed within the low risk HLA category but no significant difference was observed between groups within the HLA high risk category. More than half of patients who had coexisting T1D and CD carried the high risk HLA-DR3-DQ2/DR3-DQ2 or HLA-DR3-DQ2/DR4-DQ8 genotype. On the other hand, DR4-DQ8/X was protective for developing CD in individuals with T1D. If the results could be confirmed on a larger number of patients of different ethnic origin, assessing the presence of HLA-DQ2 and DQ8 genotypes in individuals negative for celiac disease-specific antibodies at the time of T1D diagnosis could be considered as an algorithm tool to optimize further screening frequency. Yearly screening for CD may be considered in T1D individuals with high risk HLA-DR3-DQ2/DR3-DQ2 or HLA-DR3-DQ2/DR4-DQ8 genotypes. On the other hand, individuals carrying DR4-DQ8/X genotype could be further genotyped for PTPN22 rs2476601 (c.1858T>C) polymorphism and if positive, less frequent screening for CD may be suggested.
The small sample size was a limitation of our study. The possibility of insufficient power to detect associations of genetic variants with small effect on disease susceptibility must be considered. Our results suggest opposite effects of PTPN22 variant on T1D and CD susceptibility and provide interesting information on incidence of this variant in subjects diagnosed with both autoimmune diseases together. To better understand the complex effect of genetic factors and their regulatory role in the pathogenesis of autoimmune diseases larger studies and functional studies are warranted. Due to the low prevalence of subjects with concurrently diagnosed T1D and CD the main advantage of our study was the use of longitudinal data (48) on the entire pediatric T1D population form the prospective Slovene childhood-onset T1D register (49) from 1995 to 2009, which allowed a longer follow up, up to 10 years for T1D only group.
In conclusion this study showed that PTPN22 rs2476601 polymorphism was significantly associated with the risk of developing T1D in Slovenian population, and of interest, the same variant protected from CD. We hypothesize that the underlying mechanism for this controversial effect could be explained considering that NLRP3 activation is involved in maintaining intestinal homeostasis. However, no association of proinflammatory NLRP3 and CARD8 polymorphisms with CD were observed. Interestingly, PTPN22 rs2476601 did not influence T1D susceptibility in individuals with coexisting CD, suggesting heterogeneity in the disease-predisposing effect of PTPN22 rs2476601 in T1D with regard to the presence of CD. Although individuals with coexisting T1D and CD represent only a small percentage of all T1D cases, acknowledging the diversity of genetic background may help to improve immune intervention strategies aimed at curing T1D.
Statement of Prior Presentation
Part of this study was presented as a poster with a title of “Polymorphism c1858t in the protein tyrosine phosphatase non-receptor type 22 in co-occurrence of type 1 diabetes and celiac disease” (Abstract Code: A-910-0012-00527) at the 43rd Annual Conference of the International Society for Pediatric and Adolescent Diabetes in Innsbruck, Austria, October 18–21, 2017.
Ethics Statement
The study protocol was approved by the Republic of Slovenia National Medical Ethics Committee (No. 98/09/12) and written informed consent was obtained from all participating individuals and/or their parents, when appropriate, prior to the enrolment.
Author Contributions
DS collected clinical data, interpreted data, and drafted the manuscript. KG carried out the NLRP3 and CARD molecular genetic studies, statistical analysis, and edited the manuscript. TH carried out the PTPN22 molecular genetic studies. AM and BV-J performed analysis of the HLA genetic studies. NB and JB were responsible for the clinical data and biological sample collection used in this study. VD and TB contributed to the study design and conception and edited the manuscript. All the authors collaborated in revising the manuscript and accepted the final version of the manuscript.
Funding
This work was supported in part by the Slovenian Research Agency, grants nos. P1-0170, J3-9663, J3-2412, J3-4116, and P3-0343.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Nina Bratanic, MD for collected clinical data and Ms. Jurka Ferran for expert technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2019.00063/full#supplementary-material
Abbreviations
T1D, type 1 diabetes; CD, celiac disease; T1D+CD, coexisting T1D and CD; HLA, human leukocyte antigen; NLRP3, nucleotide-binding oligomerization domain leucine rich repeat and pyrin domain-containing protein 3; CARD8, caspase recruitment domain-containing protein 8; PTPN22, protein tyrosine phosphatase non-receptor 22; LR, low-risk; HR, high-risk.
References
1. Craig ME, Jefferies C, Dabelea D, Balde N, Seth A, Donaghue KC. International Society for Pediatric and Adolescent Diabetes. ISPAD clinical practice consensus guidelines 2014. Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatr Diabetes. (2014) 15(Suppl. 20):4–17. doi: 10.1111/pedi.12186
2. Husby S, Koletzko S, Korponay-Szabó IR, Mearin ML, Phillips A, Shamir R, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr. (2012) 54:136–60. doi: 10.1097/MPG.0b013e31821a23d0
3. Cronin CC, Shanahan F. Insulin-dependent diabetes mellitus and coeliac disease. Lancet. (1997) 349:1096–7. doi: 10.1016/S0140-6736(96)09153-2
4. Waisbourd-Zinman O, Hojsak I, Rosenbach Y, Mozer-Glassberg Y, Shalitin S, Phillip M, et al. Spontaneous normalization of anti-tissue transglutaminase antibody levels is common in children with type 1 diabetes mellitus. Dig Dis Sci. (2012) 57:1314–20. doi: 10.1007/s10620-011-2016-0
5. Cerutti F, Bruno G, Chiarelli F, Lorini R, Meschi F, Sacchetti C. Younger age at onset and sex predict celiac disease in children and adolescents with type 1 diabetes: an Italian multicenter study. Diabetes Care. (2004) 27:1294–8. doi: 10.2337/diacare.27.6.1294
6. Shun CB, Donaghue KC, Phelan H, Twigg SM, Craig ME. Thyroid autoimmunity in type 1 diabetes: systematic review and meta-analysis. Diabet Med. (2014) 31:126–35. doi: 10.1111/dme.12318
7. Canova C, Pitter G, Ludvigsson JF, Romor P, Zanier L, Zanotti R, et al. Celiac disease and risk of autoimmune disorders: a population-based matched birth cohort study. J Pediatr. (2016) 174:146–52.e1. doi: 10.1016/j.jpeds.2016.02.058
8. Noble JA, Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diab Rep. (2011) 11:533–42. doi: 10.1007/s11892-011-0223-x
9. Ploski R, Ek JE, Thorsby, Sollid LM. On the HLA-DQ (alpha 1*0501, beta 1*0201)-associated susceptibility in celiac disease: a possible gene dosage effect of DQB1*0201. Tissue Antigens. (1993) 41:173–7. doi: 10.1111/j.1399-0039.1993.tb01998.x
10. Smigoc Schweiger D, Mendez A, Kunilo Jamnik S, Bratanic N, Bratina N, Battelino T, et al. High-risk genotypes HLA-DR3-DQ2/DR3-DQ2 and DR3-DQ2/DR4-DQ8 in co-occurrence of type 1 diabetes and celiac disease. Autoimmunity. (2016) 49:240–7. doi: 10.3109/08916934.2016.1164144
11. Hagopian W, Lee HS, Liu E, Rewers M, She JX, Ziegler AG, et al. Co-occurrence of type 1 diabetes and celiac disease autoimmunity. Pediatrics. (2017) 140:e20171305. doi: 10.1542/peds.2017-1305
12. Lempainen J, Laine AP, Hammais A, Toppari J, Simell O, Veijola R, et al. Non-HLA gene effects on the disease process of type 1 diabetes: from HLA susceptibility to overt disease. J Autoimmun. (2015) 61:45–53. doi: 10.1016/j.jaut.2015.05.005
13. Tai N, Wong FS, Wen L. The role of the innate immune system in destruction of pancreatic beta cells in NOD mice and humans with type I diabetes. J Autoimmun. (2016) 71:26–34. doi: 10.1016/j.jaut.2016.03.006
14. Cabrera SM, Henschel AM, Hessner MJ. Innate inflammation in type 1 diabetes. Transl Res. (2016) 167:14–27. doi: 10.1016/j.trsl.2015.04.011
15. Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR, Gill GV, et al. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet. (2004) 364:1699–700. doi: 10.1016/S0140-6736(04)17357-1
16. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. (2015) 6:262. doi: 10.3389/fphar.2015.00262
17. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. (2016) 13:148–59. doi: 10.1038/cmi.2015.95
18. Paramel GV, Sirsjö A, Fransén K. Role of genetic alterations in the NLRP3 and CARD8 genes in health and disease. Mediat Inflamm. (2015) 2015:846782. doi: 10.1155/2015/846782
19. Sahdo B, Fransén K, Asfaw Idosa B, Eriksson P, Söderquist B, Kelly A, et al. Cytokine profile in a cohort of healthy blood donors carrying polymorphisms in genes encoding the NLRP3 inflammasome. PLoS ONE. (2013) 8:e75457. doi: 10.1371/journal.pone.0075457
20. Ito S, Hara Y, Kubota T. CARD8 is a negative regulator for NLRP3 inflammasome, but mutant NLRP3 in cryopyrin-associated periodic syndromes escapes the restriction. Arthritis Res Ther. (2014) 16:R52. doi: 10.1186/ar4483
21. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res. (2015) 8:15–27. doi: 10.2147/JIR.S51250
22. Benetti E, Chiazza F, Patel NS, Collino M. The NLRP3 inflammasome as a novel player of the intercellular crosstalk in metabolic disorders. Mediat Inflamm. (2013) 2013:678627. doi: 10.1155/2013/678627
23. Klen J, Goričar K, Janež A, Dolžan V. NLRP3 inflammasome polymorphism and macrovascular complications in type 2 diabetes patients. J Diabetes Res. (2015) 2015:616747. doi: 10.1155/2015/616747
24. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci USA. (2015) 112:11318–23. doi: 10.1073/pnas.1513509112
25. Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and Non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol. (2017) 8:36. doi: 10.3389/fimmu.2017.00036
26. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest. (2016) 126:1783–800. doi: 10.1172/JCI83669
27. Fousteri G, Liossis SN, Battaglia M. Roles of the protein tyrosine phosphatase PTPN22 in immunity and autoimmunity. Clin Immunol. (2013) 149:556–65. doi: 10.1016/j.clim.2013.10.006
28. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. (2004) 36:337–8. doi: 10.1038/ng1323
29. Rueda B, Nunez C, Orozco G, Lopez-Nevot MA, de la Concha EG, Martin J, et al. C1858T functional variant of PTPN22 gene is not associated with celiac disease genetic predisposition. Hum Immunol. (2005) 66:848–52. doi: 10.1016/j.humimm.2005.04.008
30. Santin I, Castellanos-Rubio A, Aransay AM, Castano L, Vitoria JC, Bilbao JR. The functional R620W variant of the PTPN22 gene is associated with celiac disease. Tissue Antigens. (2008) 71:247–9. doi: 10.1111/j.1399-0039.2007.01002.x
31. Zheng J, Ibrahim S, Petersen F, Yu X. Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes Immun. (2012) 13:641–52. doi: 10.1038/gene.2012.46
32. Revised criteria for diagnosis of coeliac disease. Report of Working Group of European Society of Paediatric Gastroenterology and Nutrition. Arch Dis Child. (1990) 65:909–11. doi: 10.1136/adc.65.8.909
33. Petrone A, Battelino T, Krzisnik C, Bugawan T, Erlich H, Di Mario U, et al. Similar incidence of type 1 diabetes in two ethnically different populations (Italy and Slovenia) is sustained by similar HLA susceptible/protective haplotype frequencies. Tissue Antigens. (2002) 60:244–53. doi: 10.1034/j.1399-0039.2002.600306.x
34. Battelino T, Ursic-Bratina N, Dolzan V, Stopar-Obreza M, Pozzilli P, Krzisnik C, et al. The HLA-DRB, -DQB polymorphism and anti-insulin antibody response in Slovenian patients with type 1 diabetes. Eur J Immunogenet. (2003) 30:223–7. doi: 10.1046/j.1365-2370.2003.00395.x
35. Kanneganti TD. Inflammatory Bowel disease and the NLRP3 inflammasome. N Engl J Med. (2017) 377:694–6. doi: 10.1056/NEJMcibr1706536
36. Zakie MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. (2010) 32:379–91. doi: 10.1016/j.immuni.2010.03.003
37. Palová-Jelínková L, Dánová K, Drašarová H, Dvorák M, Funda DP, Fundová P, et al. Pepsin digest of wheat gliadin fraction increases production of IL-1β via TLR4/MyD88/TRIF/MAPK/NF-κB signaling pathway and an NLRP3 inflammasome activation. PLoS ONE. (2013) 8:e62426. doi: 10.1371/journal.pone.0062426
38. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity. (2010) 43:583–9. doi: 10.3109/08916930903540432
39. Pontillo A, Vendramin A, Catamo E, Fabris A, Crovella S. The missense variation Q705K in CIAS1/NALP3/NLRP3 gene and an NLRP1 haplotype are associated with celiac disease. Am J Gastroenterol. (2011) 106:539–44. doi: 10.1038/ajg.2010.474
40. Spalinger MR, Lang S, Gottier C, Dai X, Rawlings DJ, Chan AC, et al. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy. (2017) 13:1590–601. doi: 10.1080/15548627.2017.1341453
41. Viken MK, Amundsen SS, Kvien TK, Boberg KM, Gilboe IM, Lilleby V, et al. Association analysis of the 1858C>T polymorphism in the PTPN22 gene in juvenile idiopathic arthritis and other autoimmune diseases. Genes Immun. (2005) 6:271–3. doi: 10.1038/sj.gene.6364178
42. Zhernakova A, Eerligh P, Wijmenga C, Barrera P, Roep BO, Koeleman BP. Differential association of the PTPN22 coding variant with autoimmune diseases in a Dutch population. Genes Immun. (2005) 6:459–61. doi: 10.1038/sj.gene.6364220
43. Törn C, Hadley D, Lee HS, Hagopian W, Lernmark Å, Simell O, et al. Role of type 1 diabetes-associated SNPs on risk of autoantibody positivity in the TEDDY study. Diabetes. (2015) 64:1818–29. doi: 10.2337/db14-1497
44. Lempainen J, Vaarala O, Mäkelä M, Veijola R, Simell O, Knip M, et al. Interplaybetween PTPN22 C1858T polymorphism and cow's milk formula exposure in type 1 diabetes. J Autoimmun. (2009) 33:155–64. doi: 10.1016/j.jaut.2009.04.003
45. Tavares NA, Santos MM, Moura R, Araújo J, Guimarães RL, Crovella S, et al. Association of TNF-α, CTLA4, and PTPN22 polymorphisms with type 1 diabetes and other autoimmune diseases in Brazil. Genet Mol Res. (2015) 14:18936–44. doi: 10.4238/2015.December.28.42
46. Hermann R, Lipponen K, Kiviniemi M, Kakko T, Veijola R, Simell O, et al. Lymphoid tyrosine phosphatase (LYP/PTPN22) Arg620Trp variant regulates insulin autoimmunity and progression to type 1 diabetes. Diabetologia. (2006) 49:1198–208. doi: 10.1007/s00125-006-0225-4
47. Smyth DJ, Cooper JD, Howson JM, Walker NM, Plagnol V, Stevens H, et al. PTPN22 Trp620 explains the association of chromosome 1p13 with type 1 diabetes and shows a statistical interaction with HLA class II genotypes. Diabetes. (2008) 57:1730–7. doi: 10.2337/db07-1131
48. Day S, Jonker AH, Lau LPL, Hilgers RD, Irony I, Larsson K, et al. Recommendations for the design of small population clinical trials. Orphanet J Rare Dis. (2018) 13:195. doi: 10.1186/s13023-018-0931-2
Keywords: inflammasome, NLRP3, CARD8, PTPN22, type 1 diabetes, celiac disease
Citation: Smigoc Schweiger D, Goricar K, Hovnik T, Mendez A, Bratina N, Brecelj J, Vidan-Jeras B, Battelino T and Dolzan V (2019) Dual Role of PTPN22 but Not NLRP3 Inflammasome Polymorphisms in Type 1 Diabetes and Celiac Disease in Children. Front. Pediatr. 7:63. doi: 10.3389/fped.2019.00063
Received: 07 November 2018; Accepted: 18 February 2019;
Published: 12 March 2019.
Edited by:
Andrew S. Day, University of Otago, New ZealandReviewed by:
Gianfranco Meloni, University of Sassari, ItalyVrinda Bhardwaj, Children's Hospital of Los Angeles, United States
Copyright © 2019 Smigoc Schweiger, Goricar, Hovnik, Mendez, Bratina, Brecelj, Vidan-Jeras, Battelino and Dolzan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vita Dolzan, dml0YS5kb2x6YW5AbWYudW5pLWxqLnNp