 Gilles Maussion
Gilles Maussion Cecilia Rocha
Cecilia Rocha Geneviève Bernard
Geneviève Bernard Lenore K. Beitel
Lenore K. Beitel Thomas M. Durcan
Thomas M. Durcan- 1Department of Neurology and Neurosurgery, Montreal Neurological Institute and Hospital, McGill University, Montreal, QC, Canada
- 2Departments of Neurology and Neurosurgery, Pediatrics and Human Genetics, McGill University, Montreal, QC, Canada
- 3Division of Medical Genetics, Department of Internal Medicine, McGill University Health Center, Montreal, QC, Canada
- 4Child Health and Human Development Program, Research Institute of the McGill University Health Center, Montreal, QC, Canada
- 5MyeliNeuroGene Laboratory, Research Institute of the McGill University Health Center, Montreal, QC, Canada
Autism Spectrum Disorders (ASDs) is a multigenic and multifactorial neurodevelopmental group of disorders diagnosed in early childhood, leading to deficits in social interaction, verbal and non-verbal communication and characterized by restricted and repetitive behaviors and interests. To date, genetic, descriptive and mechanistic aspects of the ASDs have been investigated using mouse models and post-mortem brain tissue. More recently, the technology to generate stem cells from patients' samples has brought a new avenue for modeling ASD through 2D and 3D neuronal models that are derived from a patient's own cells, with the goal of building new therapeutic strategies for treating ASDs. This review analyses how studies performed on mouse models and human samples can complement each other, advancing our current knowledge into the pathophysiology of the ASDs. Regardless of the genetic and phenotypic heterogeneities of ASDs, convergent information regarding the molecular and cellular mechanisms involved in these disorders can be extracted from these models. Thus, considering the complexities of these disorders, patient-derived models have immense potential to elucidate molecular deregulations that contributed to the different autistic phenotypes. Through these direct investigations with the human in vitro models, they offer the potential for opening new therapeutic avenues that can be translated into the clinic.
Introduction: Genetics of Autism
Autism is a complex, multigenic and multifactorial neurodevelopmental disorder that was originally defined based on three clinical criteria (i) abnormalities in social interactions, (ii) deficits in verbal and non-verbal communication and (iii) the presence of stereotypical and repetitive behaviors (1). As the genetic causes of autism have been investigated, the literature now refers more to Autism Spectrum Disorders (ASDs) (2) rather than autism, originally defined by Kanner. ASDs can be classified in two main categories: non-syndromic and syndromic based on a lack of or association to clinical manifestations outside of the autistic features, respectively. The non-syndromic ASDs are diagnosed based on structured interviews (3, 4) during childhood, according to specific clinical criteria. They are divided between those caused by specific gene mutations (5), deletions, disruptions (6, 7) or copy number variations (8) leading to abnormal gene dosage. Idiopathic forms of autism, whose genetic bases remain unclear represent the majority of ASDs' cases. Syndromic forms of autism such as Fragile X (9) or Rett syndromes (10) along with Tuberous sclerosis (11) and others (see Table 1) are characterized by a specific set of clinical features as well as the autistic triad. They are often monogenic, genetically well characterized and account for 10–20% of ASDs' cases (18). Examples of specific set of clinical features that can be seen in the syndromic ASDs include facial dysmorphisms, epilepsy, intellectual disability, and systemic manifestations. These features are not observed in non-syndromic forms of autism (2).
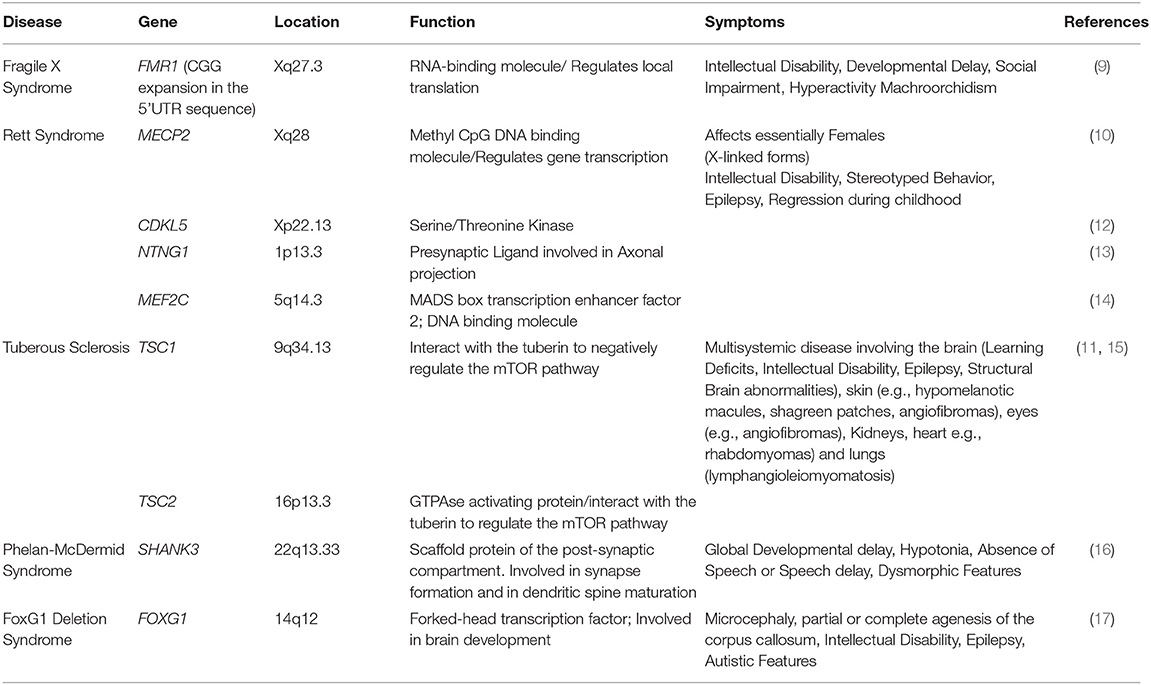
Table 1. Syndromic forms of autism that have been investigated in iPSC-derived studies.
Studies on syndromic forms of autism, as well as candidate gene approaches and genome-wide association studies, have led to the identification of more than 70 loci for ASDs in the human genome (18). Those loci are categorized into specific gene functions such as developmental programs (19), transcriptional (20), and translational regulation (21), cell signaling (22), gene imprinting (23), dendritic trafficking (24–26), and activity-dependent brain development and neurotransmission (27). Complementing these genetic studies, work on post-mortem brain tissue and mouse models have helped us better observe and understand changes that may occur in the context of ASDs at the cellular and molecular levels.
Over the past decade, technology has developed to derive induced pluripotent stem cells (iPSCs) from the somatic cells of patients with many different types of diseases. In the context of ASDs, this approach enables us to differentiate and grow human neurons from patients, within a dish, presenting a new in vitro tool to decipher how perturbations in specific genes or pathways may be either involved or the causative mechanisms in the development of ASDs, with the ultimate goal of developing new therapeutics. Multiple genetic susceptibility loci identified in ASDs strongly implies that these disorders are linked to genetic variants and risk factors across multiple genes that are best studied in human models. This review aims to discuss the strengths and limitations of mouse models, human post-mortem brain tissue and iPSCs in studying ASDs, and to summarize how these studies combined have contributed to advance our understanding into the molecular and cellular mechanisms that potentially cause ASDs.
Mouse Models in Autism Spectrum Disorders
Animal models have been a useful tool in understanding how genes linked to ASDs may contribute to the pathogenesis of these disorders. In order to study ASDs, a combination of genetic, chemically induced, and environmental models have been generated. In the genetic models, ASDs-associated genes have been inactivated to observe distinct phenotypes within the mice that can include altered gene expression, cell morphology, and behavioral or social deficits.
Syndromic form of autism that are genetically characterized such as Fragile X (FXS), Rett Syndrome, and Tuberous Sclerosis (Table 1) are studied in mouse models. At an adult stage, mice inactivated for Fmr1 (coding for the RNA-binding protein FMRP which acts as a translational repressor), present abnormal dendritic spine morphology, although one limitation of this model is that autistic traits often differ from one strain to another (28). Restoration of the abnormal protein synthesis in Fmr1 knockout mice by S6K1 (a translation regulator) deletion can stabilize neurological function (29). Fmr1 knockout leads to a disruption of synaptic protein interactions that notably involve metabotropic glutamate receptor subunit 5 (mGluR5) and Homer scaffold protein. Homer1a acts as a dominant negative isoform that prevents the normal interaction with mGluR5, and its deletion rescues, in H1a/Fmr1 double knockouts mice, mGluR5 signaling (30).
Further studies highlighted the importance of synaptic transmission in FXS, connecting it also to impairment of the GABAergic system (31). Interestingly, the GABA receptor agonist THIP (gaboxadol) was shown to restore neuronal excitability in the Fmr1 knockout mice (32). A cytoplasmic polyadenylation element-binding protein (CPEB) also binds to mRNA controlling neuronal translation and modulating synaptic function. Frm1/Cpeb1 double knockout mice display an amelioration in morphological, electrophysiological and behavioral phenotypes associated with FXS showing the importance of translational homeostasis for neural function (33). Studies on animal models suggest that Fmr1 inactivation leads to translational deregulations which underlie abnormalities in excitatory and inhibitory neurotransmission.
Rett Syndrome (RTT) is caused, in the majority of cases, by loss of function mutations in the MECP2 gene (Table 1). Mouse models have helped to investigate the impact of Mecp2 mutations in RTT pathogenesis. Mecp2 was shown to be critical for GABAergic neuronal function in Rett Syndrome (RTT) as Viaat-Mecp2−/y mice lack Mecp2 in GABAergic neurons, causing RTT-like features that include the development of stereotypes, self-injury, compulsive behavior and progressive motor dysfunction (34). Activation of Mecp2 expression in knockout mice reverses neurological symptoms (35). Interestingly, a study with 7 weeks and 21 weeks female Mecp2+/− mice showed behavioral and motor deficits that were identified in distinct genetic backgrounds However, some phenotypes were also identified in only one genetic background (36). This study stresses the importance of age and strain background selection for RTT social behavior research using Mecp2+/− female mice.
Mutations in TSC1 and TSC2 are known to cause Tuberous Sclerosis (TSC) (Table 1). Actually, a functional interaction between those two signaling proteins is required for the activation of mTOR complex 2 (37). The mTOR pathway is an important regulator of mitophagy and autophagy (38), and its activation by IGF-1 or other small molecules, can promote reversion of the developmental alterations observed in TSC. Behavioral abnormalities in Tsc1+/− and Tsc2+/− are rescued by inhibition of mTORC1 with rapamycin treatment (39). A TSC mouse model with Tsc2 loss under a Purkinje cell promotor showed increased repetitive behavior in Tsc2f/-; Cre mice. These social behavioral deficits were also prevented with rapamycin treatment (40). These mouse models have shown that dysregulation of the mTOR signaling pathways, induced by Tsc1 and Tsc2 inactivation, results in a Tuberous Sclerosis-like phenotype.
Shank3 is a synaptic scaffolding protein and gene mutations have been associated with an ASD phenotype (41). Male heterozygous mice with deleted Shank3 display impairments in NMDA receptor signaling function, synaptic trafficking (42), hippocampal excitatory transmission, and motor learning although no social interaction deficit is observed (43). Another study reported that Histone deacetylase 2 (HDAC2) is upregulated in Shank3-deficient mice and HDAC2 knockdown in the prefrontal cortex rescues social deficits (44). Treatment with romidepsin, a histone deacetylase (HDAC) inhibitor, increases NMDAR transcription levels, restores NMDAR synaptic function, and alleviated social deficits in Shank3-deficient mouse (44). This model shows that mutations in the gene coding for a synaptic protein leads to autistic-like behavioral phenotypes in the mouse.
Taken together these models can reveal the individual mechanisms involved in syndromic forms of ASDs serving as a platform for proof-of-principle studies. Although knockout mice can model monogenic genetically-defined ASDs, idiopathic forms of autism, likely caused by multigenic risk factors, cannot be investigated in animal models. Furthermore, regulatory and coding regions from mouse and human genomes have acquired a more complex epigenetic organization or significant differences in the coding mRNAs having been submitted to evolutionary selective pressures. Most importantly, the diagnosis of ASDs still relies on assessing behavioral phenotypes that remain difficult to mimic in mouse models. Thus, studies in human samples are crucial for a better understanding of ASDs.
Post-Mortem Brain Studies in ASDs
Although animal models have partially recapitulated some of the behaviors observed in patients and provided insights concerning gene expression and morphological deregulation potentially relevant to the ASDs, none of these systems are entirely modeling the complexity of ASDs. Studies on post-mortem brain tissue from patients with ASDs have enabled researchers to observe changes at the cellular and molecular levels that could be implicated as causative factors in the disease's manifestations.
The brain regions of interest for these post-mortem studies were chosen based on (i) their function, (ii) their potential implication in the disease and (iii) on imaging studies. As such, several morphological studies have been performed on the cerebellum, which controls balance, posture, and regulation of fine movements (45) as it is thought to be involved in the repetitive behaviors associated with ASDs (46). The frontal cortex has also received much attention, given its role in executive function (47), decision making (48) and working memory and attentional processes (49). Furthermore, the frontal cortex is interconnected with the limbic system (including the amygdala and hippocampus), important for learning and memory through the superior temporal sulcus, and forms a network involved in social perception and cognition (50).
Morphological studies of the cerebellum have shown it to be decreased in size accompanied by a decrease in the overall number of Purkinje cells in autistic patients relative to controls (51–53). Similarly, the structure of frontal cortex had been examined and decreased size of the minicolumns was observed in patients with idiopathic ASDs (54, 55). Interestingly, studies have reported in patients with ASDs, (i) increase of cortical thickness (56), (ii) increase of head circumference (57) as well as, more specifically in the prefrontal cortex, an increase in the number of neurons (58). These findings suggest that changes in cell number and synapses in brains from autistic patients, modify the cortical organization, connectivity and network efficiency (59, 60).
Other morphological studies were conducted on post-mortem brain tissue from patients diagnosed with syndromic ASDs, namely Fragile X and Rett Syndromes, two disorders that present with opposing morphological phenotypes. A decreased number of dendritic spines has been observed in the frontal cortex and in the CA1 regions of patient with Rett syndrome (25, 61) whereas, an increased number of elongated and immature dendritic spines was observed in brain sections from Fragile X patients (62). Studies performed on human post-mortem brain tissue have shown abnormalities in dendritic arborization that are specific for syndromic autisms (Supplementary Table 1).
Besides these morphological studies, genes expression have also been assessed in brain areas from patients diagnosed with ASDs, either based on candidate or whole genome approaches. From one of these studies, the expression of the RELN gene, which had been associated with autism (63) was observed to be decreased in the cerebellum and in the cortex of patients with ASDs (64). Interestingly, increased methylation of the RELN promoter was also observed in the frontal cortex and cerebellum of patients with ASDs. Furthermore, concomitantly with decreased number and size of the Purkinje cells, that are GABAergic neurons, decreased expression of GAD1 (65) and Parvalbumin mRNA (66), were observed in the frontal cortex and in the cerebellum, respectively, of ASDs patients. Based on a candidate gene approach, the expression of two autism-associated genes, SLC25A12 and MARK1 (24, 67), and BDNF (68, 69) were found to be increased in Brodmann Area 46 of patients with idiopathic ASDs autistic patients (24, 70, 71).
Whereas changes in the expression of genes associated with ASDs had suggested that deregulation in molecular mechanisms involved in the dendritic trafficking and the synaptic plasticity occurs in brain regions from ASD patients, high throughout analyses aimed at generating non-biased profiles of the whole transcriptome have also been conducted on cortical or cerebellar regions (72–75) Interestingly, these independent research projects have identified in brain regions of ASD patients, deregulations of biological processes that were expected regarding the functions of ASDs-associated genes whose expression changes had already been observed (see Table 2). The first study focused on the whole transcriptome in the temporal cortex of patients with idiopathic ASDs and provided evidence of increased expression of genes involved in the immune system as well as deregulation of genes involved in cell-cell communication and cell cycle (72). Two other studies which investigated expression profiles of coding genes in the BA9, BA41/42, and cerebellar vermis (73) or the non-coding transcriptome in frontal and temporal lobes (75) have shown, in the brain of patients with idiopathic ASDs, increased expression of genes involved in inflammatory processes as well as a decrease in the expression levels of neuronal genes including several involved in synapse functioning. Interestingly, Parikshak et al. have also observed deregulation of primate-specific long non-coding RNAs associated with autism (75). A fourth study revealed age-dependent differential expression profiles between controls and patients with idiopathic ASDs (74). In young patients, deregulated genes were involved in cell number, cortical patterning, and differentiation whereas deregulation observed in the older patients involved genes in signaling and repair pathways. Interestingly, a gene set enrichment analysis of the four studies mentioned above has provided evidence for a correlation between a decrease in expression of synaptic and mitochondrial genes within distinct brain regions from ASDs patients (86).
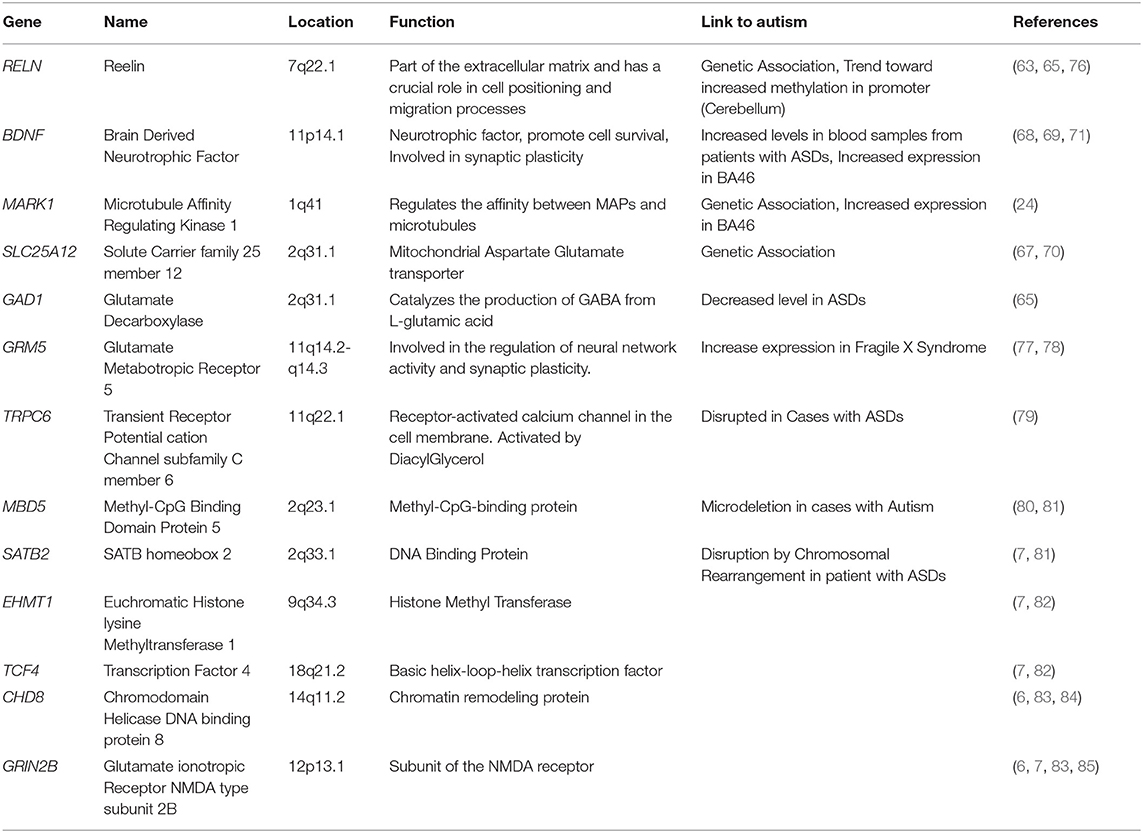
Table 2. Candidate genes of ASDs whose expression has either been assessed on post-mortem brain tissue and or in iPSC-derived cells.
These findings imply, that despite genetic and phenotypic heterogeneities, ASDs could be underpinned by deregulations of common molecular pathways and functions such as cell cycle, differentiation, mitochondrial function as well as synaptic plasticity and inflammation between brain regions; functions that had already been pointed out by genetics studies (Figure 1; Supplementary Table 1).
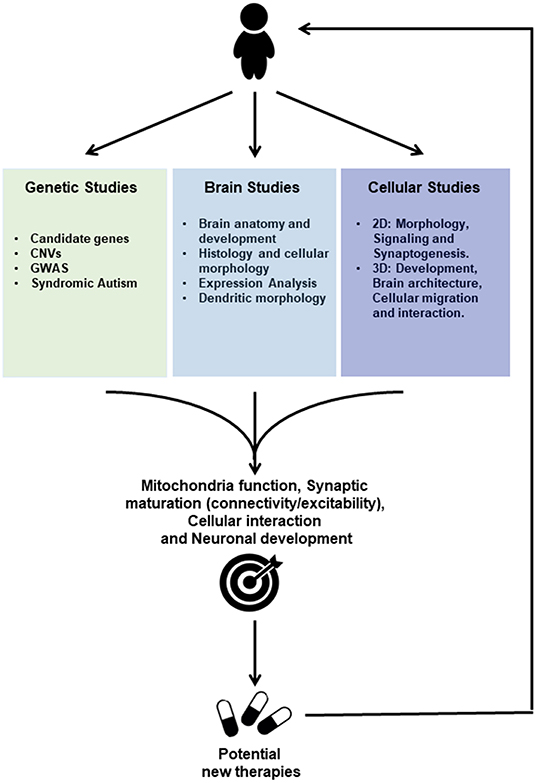
Figure 1. Scheme of ASD Drug discovery based on Genetic Studies, Post-mortem Brain Studies and Cellular Studies using induced pluripotent stem cells (IPSC) for future personalized treatment. Genetic studies reveal associations to Autism Spectrum Disorders either through candidate genes approach or Genome-wide Association Studies (GWAS), whereas genes mutated in syndromic autism have been identified. Furthermore, copy number variations (CNVs) that potentially lead to either gene disruption or duplication were more recently identified. Studies performed on post-mortem tissue provide information about brain anatomy and development, histology and cellular morphology, besides gene and protein expression profiles. Cellular Studies based on IPSC technology may be employed to generate neurons and other cell types (2D) and brain organoids (3D) using cells from patients. Those IPSC-derived models where used to investigate gene expression profiles and cellular morphology as well as cell signaling, synaptogenesis and electrophysiological properties. Moreover, 3D Studies allow to further investigate cellular migration and interaction during development. Those studies provide convergent information regarding the pathophysiology of the Autism Spectrum Disorders, toward altered mechanisms related to Mitochondria function, Synaptic maturation, Cellular interactions, and neuronal development. IPSC-derived cells are currently used to further investigate those altered functions and constitute human in vitro systems allowing drug screenings for potential targets that could lead to new therapies for ASDs.
Induced Pluripotent Stem Cells, Rescue, and Therapeutic Strategies
In the past decade, the technology has advanced to enable reprogramming of somatic cells from peripheral blood cells or skin fibroblasts into iPSCs (87) and in vitro generation of 2D or 3D neuronal culture from patients cells. While neurons grown in a dish enable researchers to investigate cell type-specific expression profiles and analyse electrophysiological properties of patient and control neurons, 3D organoids create a microenvironment that promotes interactions between cell types, leading to a more complete neuronal maturation (88) that is capable of recapitulating the organization of cortical layers observed during the neurogenesis (89).
Human iPSC-derived cells from patients with syndromic forms of autism such as Fragile X, Rett syndrome and tuberous sclerosis have been investigated and partially recapitulate phenotypes observed in post-mortem brain tissue or with pathological studies of rodent models of the diseases. Due to a lack of translational repression of mGluR5 mRNA at the synapse, iPSC-derived neurons from patients with Fragile X have an increased response to a group I mGluR agonist combined with a preferential differentiation into glutamatergic neurons (78); treatment with a mGLUR5 antagonist reduces the activation of the receptor and lead the Fragile X progenitors cells toward glial differentiation. A second study showed an increased response to Ca2+ permeable AMPA and NMDA receptors in iPSC-derived neurons from Fragile X patients (90), while treatment with a GluA2-lacking/calcium permeable-AMPARs antagonist reduces the length of the dendrites.
IPSC-derived Purkinje cells from patients diagnosed with tuberous sclerosis present with an increasing proportion of KI67 positive cells, decreased FMRP expression and an increased number of neurites (91). Human iPSC-derived neurons from patients with TSC2+/− mutations were defective in mitophagy with (i) an accumulation of mitochondria in the dendrites and (ii) a reduced mitochondrial potential membrane (92). These findings corroborate observations made on the temporal lobe from patients with tuberous sclerosis where an accumulation of autophagy proteins such as cargo protein p62, LC3, and ATG12 were found (93). Interestingly, phenotypes observed in iPSC-derived Purkinje cells or neurons from TSC patients were reversed with rapamycin treatment (91, 92).
IPSCs and organoids generated from patients diagnosed with Rett syndrome present with (i) a down-regulation in the expression of genes involved in neuronal development and cell signaling (94) and (ii) decreased expression in genes expressing neuronal markers that include MAP2 and DCX that should reflect deficits in neuronal differentiation (95). Interestingly, inhibition of specific microRNAs 199 and 214 restored MAP2 and DCX expression (Supplementary Table 1).
Two studies that investigated differential gene expression in iPSC-derived neurons from patients diagnosed with idiopathic forms of autism showed a significant downregulation of genes involved in neuronal development and synaptic function (96, 97). A third study focusing on idiopathic autism combined with macrocephaly demonstrated an increased proportion of proliferative cells and a decreased proportion of cells expressing markers of neuronal differentiation (98). Interestingly, differential analyses of whole transcriptomes identified a significant enrichment in ASD-related genes. All these studies demonstrated a decrease in electrophysiological activities (spontaneous activity; calcium transient; or decreased numbers of spikes and bursts) in iPSC-derived neurons from patients affected by ASDs. Application of IGF1 on cultures of iPSC-derived neurons from patients with ASDs led to an increase in the number of spikes (98). A similar treatment was applied to iPSC-derived neurons from a patient carrying a TRPC6 gene translocation. IGF1 treatment restored dendritic arborization that was decreased in TPRC6-disrupted neurons which also had decreased expression in synaptic proteins such as Homer and PSD95 (79). Several phase II clinical trial studies targeting the IGF1 pathway have been performed in Rett, Fragile X, and Phelan McDermid syndromes testing safety and primary efficacy (99).
A screening of 202 compounds was performed on iPSC-derived cortical neurons from patients with SHANK3 mutations. Two of those molecules, valproic acid (VPA)—an HDAC inhibitor and an antiepileptic drug commonly used for bipolar disease treatment—and lithium, led to an increase in SHANK3 expression and its recruitment to post-synaptic sites (100) as well as increased spontaneous calcium oscillations, improving neuronal network connectivity.
Studies using iPSC-derived cells have shown that common processes are deregulated in syndromic and idiopathic forms of autism characterized by impairment in neuronal differentiation process, an imbalance between inhibitory and excitatory neurotransmission as well as mitochondrial deficits which corroborate genetic data as well as post-mortem brain studies. Several molecules such as rapamycin and IGF1, that have been also tested in mouse models seem to be effective in human iPSC-derived cells. The literature concerning the potential effect of VPA and lithium on mouse models is inconclusive. IPSC-derived cells from patients constitute a critical in vitro model for studying the impact of ASD-associated mutations and genetic vulnerability factors on the development and progression of the disease. Thus, many reasons can justify using human iPSC-derived models to increase our understanding of ASDs.
Interestingly, differential expression of human-specific genes was shown between control and brains of patients with ASDs (101). Along these lines, specific families of genes such as transcription factors as well as genes involved in brain size and in the acquisition of language have been submitted to positive selection in the human genome (102, 103). Thus, investigating these genes may not be possible in rodents or non-human primates. Finally, while our current knowledge in epigenetic regulation and non-coding transcriptome is growing (104), it appears that some histone modifications, microRNA or long non-coding RNAs, all of which are potential therapeutic targets to the same extent as proteins, are only observed in human (105, 106).
Conclusion
The current review aims to bring together findings in the field of ASDs provided by genetics, mouse models, post-mortem brain studies and iPSC-derived studies. All these approaches provide complementary information suggesting that ASDs are underpinned by dysregulation in the brain developmental program which include alterations of activity-dependent development, mitochondrial function as well as an imbalanced excitation-inhibition equilibrium (Figure 1; Supplementary Table 1). Those studies have also highlighted therapeutic strategies that are often primarily performed in mice but remain to be fully translated into humans. However, the human iPSC model seems to be an ideal model system for integrating all the genes and other factors that are implicated as causative factors in the development of ASDs. Furthermore, this review has highlighted the importance of human-specific gene regulation and expression involved in high cognitive and behavioral functions for ASDs. Further investigations involving functional analyses on human models are required to identify molecules and design therapeutic strategies that could be translated to patients.
Author Contributions
TD, GM, and CR defined the scope of the review. GM and CR wrote the manuscript and prepared the tables. GB, TD, and LB corrected, edited, and formatted the manuscript.
Funding
This work was supported through funding by the Alain and Sandra Bouchard Intellectual Disabilities platform and the Djavid Mowfaghian foundation. GB has received the New Investigator Salary Award from the Canadian Institutes of Health Research (2017–2022).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2019.00225/full#supplementary-material
References
2. Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. (2007) 17:103–11. doi: 10.1016/j.conb.2007.01.009
3. Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. (1994) 24:659–85. doi: 10.1007/BF02172145
4. Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC, et al. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. (2000) 30:205–23. doi: 10.1023/A:1005592401947
5. Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. (2015) 16:551–63. doi: 10.1038/nrn3992
6. O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. (2012) 338:1619–22. doi: 10.1126/science.1227764
7. Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. (2012) 149:525–37. doi: 10.1016/j.cell.2012.03.028
8. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. (2007) 316:445–9. doi: 10.1126/science.1138659
9. Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. (1991) 66:817–22. doi: 10.1016/0092-8674(91)90125-I
10. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. (1999) 23:185–8. doi: 10.1038/13810
11. Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. (2007) 9:88–100. doi: 10.1097/GIM.0b013e31803068c7
12. Lin C, Franco B, Rosner MR. CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum Mol Genet. (2005) 14:3775–86. doi: 10.1093/hmg/ddi391
13. Lin L, Lesnick TG, Maraganore DM, Isacson O. Axon guidance and synaptic maintenance: preclinical markers for neurodegenerative disease and therapeutics. Trends Neurosci. (2009) 32:142–9. doi: 10.1016/j.tins.2008.11.006
14. Lambert L, Bienvenu T, Allou L, Valduga M, Echenne B, Diebold B, et al. MEF2C mutations are a rare cause of Rett or severe Rett-like encephalopathies. Clin Genet. (2012) 82:499–501. doi: 10.1111/j.1399-0004.2012.01861.x
15. Thiele EA. Managing epilepsy in tuberous sclerosis complex. J Child Neurol. (2004) 19:680–6. doi: 10.1177/08830738040190090801
16. Phelan MC, Rogers RC, Saul RA, Stapleton GA, Sweet K, McDermid H, et al. 22q13 deletion syndrome. Am J Med Genet. (2001) 101:91–9. doi: 10.1002/1096-8628(20010615)101:2<91::AID-AJMG1340>3.0.CO;2-C
17. Jacob FD, Ramaswamy V, Andersen J, Bolduc FV. Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet. (2009) 17:1577–81. doi: 10.1038/ejhg.2009.95
18. Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. (2008) 9:341–55. doi: 10.1038/nrg2346
19. Gilbert J, Man HY. Fundamental elements in autism: from neurogenesis and neurite growth to synaptic plasticity. Front Cell Neurosci. (2017) 11:359. doi: 10.3389/fncel.2017.00359
20. Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. (2008) 320:1224–9. doi: 10.1126/science.1153252
21. Kelleher RJ III, Bear MF. The autistic neuron: troubled translation? Cell. (2008) 135:401–6. doi: 10.1016/j.cell.2008.10.017
22. Mullins C, Fishell G, Tsien RW. Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron. (2016) 89:1131–56. doi: 10.1016/j.neuron.2016.02.017
23. Badcock C, Crespi B. Imbalanced genomic imprinting in brain development: an evolutionary basis for the aetiology of autism. J Evol Biol. (2006) 19:1007–32. doi: 10.1111/j.1420-9101.2006.01091.x
24. Maussion G, Carayol J, Lepagnol-Bestel AM, Tores F, Loe-Mie Y, Milbreta U, et al. Convergent evidence identifying MAP/microtubule affinity-regulating kinase 1 (MARK1) as a susceptibility gene for autism. Hum Mol Genet. (2008) 17:2541–51. doi: 10.1093/hmg/ddn154
25. Chapleau CA, Calfa GD, Lane MC, Albertson AJ, Larimore JL, Kudo S, et al. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis. (2009) 35:219–33. doi: 10.1016/j.nbd.2009.05.001
26. Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry. (2012) 17:71–84. doi: 10.1038/mp.2011.57
27. Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. (2013) 493:327–37. doi: 10.1038/nature11860
28. Oddi D, Subashi E, Middei S, Bellocchio L, Lemaire-Mayo V, Guzman M, et al. Early social enrichment rescues adult behavioral and brain abnormalities in a mouse model of fragile X syndrome. Neuropsychopharmacology. (2015) 40:1113–22. doi: 10.1038/npp.2014.291
29. Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. (2012) 76:325–37. doi: 10.1016/j.neuron.2012.07.022
30. Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. (2012) 15:431–40, S1. doi: 10.1038/nn.3033
31. D'Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. (2007) 30:425–31. doi: 10.1016/j.tins.2007.06.003
32. Olmos-Serrano JL, Corbin JG, Burns MP. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev Neurosci. (2011) 33:395–403. doi: 10.1159/000332884
33. Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anilkumar S, et al. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med. (2013) 19:1473–7. doi: 10.1038/nm.3353
34. Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. (2010) 468:263–9. doi: 10.1038/nature09582
35. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. (2007) 315:1143–7. doi: 10.1126/science.1138389
36. Samaco RC, McGraw CM, Ward CS, Sun Y, Neul JL, Zoghbi HY. Female Mecp2(+/-) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum Mol Genet. (2013) 22:96–109. doi: 10.1093/hmg/dds406
37. Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. (2008) 28:4104–15. doi: 10.1128/MCB.00289-08
38. Bartolome A, Garcia-Aguilar A, Asahara SI, Kido Y, Guillen C, Pajvani UB, et al. MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol Cell Biol. (2017) 37: e00441–17. doi: 10.1128/MCB.00441-17
39. Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, et al. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. (2012) 3:1292. doi: 10.1038/ncomms2295
40. Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis. (2013) 51:93–103. doi: 10.1016/j.nbd.2012.10.014
41. Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. (2007) 39:25–7. doi: 10.1038/ng1933
42. Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L, et al. Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. (2015) 11:1400–13. doi: 10.1016/j.celrep.2015.04.064
43. Speed HE, Kouser M, Xuan Z, Reimers JM, Ochoa CF, Gupta N, et al. Autism-Associated Insertion Mutation (InsG) of Shank3 Exon 21 causes impaired synaptic transmission and behavioral deficits. J Neurosci. (2015) 35:9648–65. doi: 10.1523/JNEUROSCI.3125-14.2015
44. Qin L, Ma K, Wang ZJ, Hu Z, Matas E, Wei J, et al. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat Neurosci. (2018) 21:564–75. doi: 10.1038/s41593-018-0110-8
45. Koziol LF, Budding D, Andreasen N, D'Arrigo S, Bulgheroni S, Imamizu H, et al. Consensus paper: the cerebellum's role in movement and cognition. Cerebellum. (2014) 13:151–77. doi: 10.1007/s12311-013-0511-x
46. Pierce K, Courchesne E. Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biol Psychiatry. (2001) 49:655–64. doi: 10.1016/S0006-3223(00)01008-8
47. Alvarez JA, Emory E. Executive function and the frontal lobes: a meta-analytic review. Neuropsychol Rev. (2006) 16:17–42. doi: 10.1007/s11065-006-9002-x
48. Rushworth MF, Noonan MP, Boorman ED, Walton ME, Behrens TE. Frontal cortex and reward-guided learning and decision-making. Neuron. (2011) 70:1054–69. doi: 10.1016/j.neuron.2011.05.014
49. Goldman-Rakic PS. Development of cortical circuitry and cognitive function. Child Dev. (1987) 58:601–22. doi: 10.2307/1130201
50. Pinkham AE, Hopfinger JB, Pelphrey KA, Piven J, Penn DL. Neural bases for impaired social cognition in schizophrenia and autism spectrum disorders. Schizophr Res. (2008) 99:164–75. doi: 10.1016/j.schres.2007.10.024
51. Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, et al. Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC Autopsy Research Report. Am J Psychiatry. (1986) 143:862–6. doi: 10.1176/ajp.143.7.862
52. Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain. (1998) 121:889–905. doi: 10.1093/brain/121.5.889
53. Fatemi SH, Halt AR, Realmuto G, Earle J, Kist DA, Thuras P, et al. Purkinje cell size is reduced in cerebellum of patients with autism. Cell Mol Neurobiol. (2002) 22:171–5. doi: 10.1023/A:1019861721160
54. Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology. (2002) 58:428–32. doi: 10.1212/WNL.58.3.428
55. Buxhoeveden DP, Semendeferi K, Buckwalter J, Schenker N, Switzer R, Courchesne E. Reduced minicolumns in the frontal cortex of patients with autism. Neuropathol Appl Neurobiol. (2006) 32:483–91. doi: 10.1111/j.1365-2990.2006.00745.x
56. Hardan AY, Muddasani S, Vemulapalli M, Keshavan MS, Minshew NJ. An MRI study of increased cortical thickness in autism. Am J Psychiatry. (2006) 163:1290–2. doi: 10.1176/ajp.2006.163.7.1290
57. Lainhart JE, Bigler ED, Bocian M, Coon H, Dinh E, Dawson G, et al. Head circumference and height in autism: a study by the Collaborative Program of Excellence in Autism. Am J Med Genet A. (2006) 140:2257–74. doi: 10.1002/ajmg.a.31465
58. Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, et al. Neuron number and size in prefrontal cortex of children with autism. JAMA. (2011) 306:2001–10. doi: 10.1001/jama.2011.1638
59. Minshew NJ, Williams DL. The new neurobiology of autism: cortex, connectivity, and neuronal organization. Arch Neurol. (2007) 64:945–50. doi: 10.1001/archneur.64.7.945
60. Lewis JD, Theilmann RJ, Townsend J, Evans AC. Network efficiency in autism spectrum disorder and its relation to brain overgrowth. Front Hum Neurosci. (2013) 7:845. doi: 10.3389/fnhum.2013.00845
61. Belichenko PV, Oldfors A, Hagberg B, Dahlstrom A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. (1994) 5:1509–13. doi: 10.1097/00001756-199407000-00025
62. Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. (2000) 10:1038–44. doi: 10.1093/cercor/10.10.1038
63. Skaar DA, Shao Y, Haines JL, Stenger JE, Jaworski J, Martin ER, et al. Analysis of the RELN gene as a genetic risk factor for autism. Mol Psychiatry. (2005) 10:563–71. doi: 10.1038/sj.mp.4001614
64. Fatemi SH. Reelin glycoprotein in autism and schizophrenia. Int Rev Neurobiol. (2005) 71:179–87. doi: 10.1016/S0074-7742(05)71008-4
65. Zhubi A, Chen Y, Guidotti A, Grayson DR. Epigenetic regulation of RELN and GAD1 in the frontal cortex (FC) of autism spectrum disorder (ASD) subjects. Int J Dev Neurosci. (2017) 62:63–72. doi: 10.1016/j.ijdevneu.2017.02.003
66. Soghomonian JJ, Zhang K, Reprakash S, Blatt GJ. Decreased parvalbumin mRNA levels in cerebellar Purkinje cells in autism. Autism Res. (2017) 10:1787–96. doi: 10.1002/aur.1835
67. Ramoz N, Reichert JG, Smith CJ, Silverman JM, Bespalova IN, Davis KL, et al. Linkage and association of the mitochondrial aspartate/glutamate carrier SLC25A12 gene with autism. Am J Psychiatry. (2004) 161:662–9. doi: 10.1176/appi.ajp.161.4.662
68. Correia CT, Coutinho AM, Sequeira AF, Sousa IG, Lourenco Venda L, Almeida JP, et al. Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TrkB signaling in autism. Genes Brain Behav. (2010) 9:841–8. doi: 10.1111/j.1601-183X.2010.00627.x
69. Wang M, Chen H, Yu T, Cui G, Jiao A, Liang H. Increased serum levels of brain-derived neurotrophic factor in autism spectrum disorder. Neuroreport. (2015) 26:638–41. doi: 10.1097/WNR.0000000000000404
70. Lepagnol-Bestel AM, Maussion G, Boda B, Cardona A, Iwayama Y, Delezoide AL, et al. SLC25A12 expression is associated with neurite outgrowth and is upregulated in the prefrontal cortex of autistic subjects. Mol Psychiatry. (2008) 13:385–97. doi: 10.1038/sj.mp.4002120
71. Maussion G, Moalic JM, Simonneau M, Gorwood P, Ramoz N. Increased expression of BDNF mRNA in the frontal cortex of autistic patients. Behav Brain Res. (2019) 359:903–9. doi: 10.1016/j.bbr.2018.06.023
72. Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. (2008) 30:303–11. doi: 10.1016/j.nbd.2008.01.012
73. Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. (2011) 474:380–4. doi: 10.1038/nature10110
74. Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L, et al. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. (2012) 8:e1002592. doi: 10.1371/journal.pgen.1002592
75. Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. (2016) 540:423–7. doi: 10.1038/nature20612
76. Gharani N, Benayed R, Mancuso V, Brzustowicz LM, Millonig JH. Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol Psychiatry. (2004) 9:474–84. doi: 10.1038/sj.mp.4001498
77. Fatemi SH, Folsom TD, Kneeland RE, Liesch SB. Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both Fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec. (2011) 294:1635–45. doi: 10.1002/ar.21299
78. Achuta VS, Grym H, Putkonen N, Louhivuori V, Karkkainen V, Koistinaho J, et al. Metabotropic glutamate receptor 5 responses dictate differentiation of neural progenitors to NMDA-responsive cells in fragile X syndrome. Dev Neurobiol. (2017) 77:438–53. doi: 10.1002/dneu.22419
79. Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry. (2015) 20:1350–65. doi: 10.1038/mp.2014.141
80. Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. (2011) 89:551–63. doi: 10.1016/j.ajhg.2011.09.011
81. Gigek CO, Chen ES, Ota VK, Maussion G, Peng H, Vaillancourt K, et al. A molecular model for neurodevelopmental disorders. Transl Psychiatry. (2015) 5:e565. doi: 10.1038/tp.2015.56
82. Chen ES, Gigek CO, Rosenfeld JA, Diallo AB, Maussion G, Chen GG, et al. Molecular convergence of neurodevelopmental disorders. Am J Hum Genet. (2014) 95:490–508. doi: 10.1016/j.ajhg.2014.09.013
83. O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. (2012) 485:246–50. doi: 10.1038/nature10989
84. Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci USA. (2014) 111:E4468–4477. doi: 10.1073/pnas.1405266111
85. Bell S, Maussion G, Jefri M, Peng H, Theroux JF, Silveira H, et al. Disruption of GRIN2B impairs differentiation in human neurons. Stem Cell Reports. (2018) 11:183–96. doi: 10.1016/j.stemcr.2018.05.018
86. Schwede M, Nagpal S, Gandal MJ, Parikshak NN, Mirnics K, Geschwind DH, et al. Strong correlation of downregulated genes related to synaptic transmission and mitochondria in post-mortem autism cerebral cortex. J Neurodev Disord. (2018) 10:18. doi: 10.1186/s11689-018-9237-x
87. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. (2007) 131:861–72. doi: 10.1016/j.cell.2007.11.019
88. McMurtrey RJ. Roles of diffusion dynamics in stem cell signaling and three-dimensional tissue development. Stem Cells Dev. (2017) 26:1293–303. doi: 10.1089/scd.2017.0066
89. Qian X, Jacob F, Song MM, Nguyen HN, Song H, Ming GL. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat Protoc. (2018) 13:565–80. doi: 10.1038/nprot.2017.152
90. Achuta VS, Moykkynen T, Peteri UK, Turconi G, Rivera C, Keinanen K, et al. Functional changes of AMPA responses in human induced pluripotent stem cell-derived neural progenitors in fragile X syndrome. Sci Signal. (2018) 11:eaan8784. doi: 10.1126/scisignal.aan8784
91. Sundberg M, Tochitsky I, Buchholz DE, Winden K, Kujala V, Kapur K, et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol Psychiatry. (2018) 23:2167–83. doi: 10.1038/s41380-018-0018-4
92. Ebrahimi-Fakhari D, Saffari A, Wahlster L, Di Nardo A, Turner D, Lewis TL Jr, et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. (2016) 17:1053–70. doi: 10.1016/j.celrep.2016.09.054
93. Yasin SA, Ali AM, Tata M, Picker SR, Anderson GW, Latimer-Bowman E, et al. mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol. (2013) 126:207–18. doi: 10.1007/s00401-013-1135-4
94. Tanaka Y, Kim KY, Zhong M, Pan X, Weissman SM, Park IH. Transcriptional regulation in pluripotent stem cells by methyl CpG-binding protein 2. (MeCP2). Hum Mol Genet. (2014) 23:1045–55. doi: 10.1093/hmg/ddt500
95. Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. (2018) 23:1051–65. doi: 10.1038/mp.2017.86
96. Liu X, Campanac E, Cheung HH, Ziats MN, Canterel-Thouennon L, Raygada M, et al. Idiopathic autism: cellular and molecular phenotypes in pluripotent stem cell-derived neurons. Mol Neurobiol. (2017) 54:4507–23. doi: 10.1007/s12035-016-9961-8
97. DeRosa BA, El Hokayem J, Artimovich E, Garcia-Serje C, Phillips AW, Van Booven D, et al. Convergent pathways in idiopathic autism revealed by time course transcriptomic analysis of patient-derived neurons. Sci Rep. (2018) 8:8423. doi: 10.1038/s41598-018-26495-1
98. Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry. (2017) 22:820–35. doi: 10.1038/mp.2016.95
99. Vahdatpour C, Dyer AH, Tropea D. Insulin-like growth factor 1 and related compounds in the treatment of childhood-onset neurodevelopmental disorders. Front Neurosci. (2016) 10:450. doi: 10.3389/fnins.2016.00450
100. Darville H, Poulet A, Rodet-Amsellem F, Chatrousse L, Pernelle J, Boissart C, et al. Human pluripotent stem cell-derived cortical neurons for high throughput medication screening in autism: a proof of concept study in SHANK3 haploinsufficiency syndrome. Ebio Med. (2016) 9:293–305. doi: 10.1016/j.ebiom.2016.05.032
101. Liu X, Han D, Somel M, Jiang X, Hu H, Guijarro P, et al. Disruption of an evolutionarily novel synaptic expression pattern in autism. PLoS Biol. (2016) 14:e1002558. doi: 10.1371/journal.pbio.1002558
102. Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, Glanowski S, et al. Natural selection on protein-coding genes in the human genome. Nature. (2005) 437:1153–7. doi: 10.1038/nature04240
103. Fisher SE, Marcus GF. The eloquent ape: genes, brains and the evolution of language. Nat Rev Genet. (2006) 7:9–20. doi: 10.1038/nrg1747
104. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. (2009) 136:629–41. doi: 10.1016/j.cell.2009.02.006
105. Shulha HP, Crisci JL, Reshetov D, Tushir JS, Cheung I, Bharadwaj R, et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. (2012) 10:e1001427. doi: 10.1371/journal.pbio.1001427
Keywords: autism spectrum disorder, genetics, post-mortem brain studies, human induced pluripotent stem cells, therapeutic strategy
Citation: Maussion G, Rocha C, Bernard G, Beitel LK and Durcan TM (2019) Patient-Derived Stem Cells, Another in vitro Model, or the Missing Link Toward Novel Therapies for Autism Spectrum Disorders? Front. Pediatr. 7:225. doi: 10.3389/fped.2019.00225
Received: 24 December 2018; Accepted: 20 May 2019;
Published: 06 June 2019.
Edited by:
Jaime Imitola, Department of Neuroscience, The Ohio State University, United StatesReviewed by:
Renata Rizzo, University of Catania, ItalyMaria Marchetto, Salk Institute for Biological Studies, United States
Copyright © 2019 Maussion, Rocha, Bernard, Beitel and Durcan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas M. Durcan, dGhvbWFzLmR1cmNhbkBtY2dpbGwuY2E=