 Anju Jacob1Shameer M. Habeeb1,2Leal Herlitz3Eva Simkova1,2
Anju Jacob1Shameer M. Habeeb1,2Leal Herlitz3Eva Simkova1,2 Jwan F. Shekhy1Alan Taylor1,4Walid Abuhammour1,5
Jwan F. Shekhy1Alan Taylor1,4Walid Abuhammour1,5 Ahmad Abou Tayoun1,4,6
Ahmad Abou Tayoun1,4,6 Martin Bitzan1,2*
Martin Bitzan1,2*- 1Department of Pediatrics, Al Jalila Children's Specialty Hospital, Dubai, United Arab Emirates
- 2Kidney Centre of Excellence, Al Jalila Children's Speciality Hospital, Dubai, United Arab Emirates
- 3Department of Anatomic Pathology, Cleveland Clinic, Cleveland, OH, United States
- 4Al Jalila Genomics Center, Al Jalila Children's Specialty Hospital, Dubai, United Arab Emirates
- 5Section of Infectious Diseases, Al Jalila Children's Specialty Hospital, Dubai, United Arab Emirates
- 6Department of Genetics, Mohammad Bin Rashid University of Medicine and Health Sciences, Dubai, United Arab Emirates
Background: Congenital nephrotic syndrome, historically defined by the onset of large proteinuria during the first 3 months of life, is a rare clinical disorder, generally with poor outcome. It is caused by pathogenic variants in genes associated with this syndrome or by fetal infections disrupting podocyte and/or glomerular basement membrane integrity. Here we describe an infant with congenital CMV infection and nephrotic syndrome that failed to respond to targeted antiviral therapy. Case and literature survey highlight the importance of the “tetrad” of clinical, virologic, histologic, and genetic workup to better understand the pathogenesis of CMV-associated congenital and infantile nephrotic syndromes.
Case Presentation: A male infant was referred at 9 weeks of life with progressive abdominal distention, scrotal edema, and vomiting. Pregnancy was complicated by oligohydramnios and pre-maturity (34 weeks). He was found to have nephrotic syndrome and anemia, normal platelet and white blood cell count, no splenomegaly, and no syndromic features. Diagnostic workup revealed active CMV infection (positive CMV IgM/PCR in plasma) and decreased C3 and C4. Maternal anti-CMV IgG was positive, IgM negative. Kidney biopsy demonstrated focal mesangial proliferative and sclerosing glomerulonephritis with few fibrocellular crescents, interstitial T- and B-lymphocyte infiltrates, and fibrosis/tubular atrophy. Immunofluorescence was negative. Electron microscopy showed diffuse podocyte effacement, but no cytomegalic inclusions or endothelial tubuloreticular arrays. After 4 weeks of treatment with valganciclovir, plasma and urine CMV PCR were negative, without improvement of the proteinuria. Unfortunately, the patient succumbed to fulminant pneumococcal infection at 7 months of age. Whole exome sequencing and targeted gene analysis identified a novel homozygous, pathogenic variant (2071+1G>T) in NPHS1.
Literature Review and Discussion: The role of CMV infection in isolated congenital nephrotic syndrome and the corresponding pathological changes are still debated. A search of the literature identified only three previous reports of infants with congenital nephrotic syndrome and evidence of CMV infection, who also underwent kidney biopsy and genetic studies.
Conclusion: Complete workup of congenital infections associated with nephrotic syndrome is warranted for a better understanding of their pathogenesis (“diagnostic triad” of viral, biopsy, and genetic studies). Molecular testing is essential for acute and long-term prognosis and treatment plan.
Introduction
Congenital nephrotic syndrome (CNS) is a rare disease with poor renal and overall outcome. It is defined by the occurrence of large proteinuria and hypoproteinemia, resulting in generalized edema during the first 3 months of life (1). The estimated incidence is 1–3 per 100,000 children worldwide (2–4). The etiology of the CNS is heterogeneous and may present as part of a genetic syndrome.
Congenital infections, particularly CMV and Treponema pallidum (syphilis), and occasionally Toxoplasma gondii, and other pathogens, have long been associated with rare instances of CNS (5–12). Although CMV and cytomegalic inclusions have been demonstrated (predominantly) in renal tubular epithelial cells of patients with various CMV-associated glomerulopathies (7, 13), the causative role and the pathomechanism of the virus in cases of glomerulonephritis and nephrotic syndrome are still debated (4, 10, 14).
Recent surveys revealed the presence of disease causing genetic variants in up to 80% of CNS cases (4, 10, 15, 16). Bona fide pathogenic variants commonly lead to profound structural and functional abnormalities of the podocyte and/or glomerular basement membrane that compromise the glomerular filtration barrier. The prototypic, “Finnish type” congenital nephrotic syndrome is due to biallelic pathogenic variants in NPHS1 (17, 18). Genes occasionally involved in congenital or infantile nephrotic syndrome with overlapping clinical and histological phenotypes include NPHS2 (podocin), LAMB2 (beta2-laminin), and WT1 (Wilms Tumor 1 transcription factor), among others (4).
Early subtle histological alterations, such as microcystic proximal tubular dilatation, eventually lead to nephron loss and end stage kidney disease (ESKD) by the age of 2–3 years (1, 15, 19, 20). The spectrum of histological changes encompasses diffuse mesangial sclerosis (DMS), focal segmental glomerulosclerosis (FSGS), membranous glomerulopathy, and minimal change disease that are largely determined by the type of mutation and age at biopsy (1, 4, 15, 20, 21). The long-term management of patients with CNS remains challenging and may require early nephrectomy to minimize the pervasive effects of massive proteinuria, and subsequent kidney transplantation (4, 18, 22, 23).
We hypothesized that previously postulated infectious etiologies of CNS cases are biased due to the lack of biopsies and—historically not feasible—comprehensive genetic studies.
Here, we report an infant with CNS and CMV infection and demonstrate the importance of timely and complete diagnostic workup, i.e., the tetrad of clinical findings and virological/infectious disease, histopathologic, and molecular genetic studies.
Case Description
A 9-week-old male (ex 34 weeks gestational age, corrected age 3 weeks) had been referred to our Emergency Department with suspected surgical abdomen. He presented a 3-day history of non-bilious, non-bloody vomiting and a 2-week-history of increasing abdominal distention and scrotal swelling. The patient was the second child of his parents who are from Kerala, India, and distantly related (3rd degree cousins). The couple's firstborn son is healthy.
Pregnancy was complicated by moderate oligohydramnios and decreased fetal movements which led to urgent C-section. Mother denied fever or rash during pregnancy, and her urinalysis was normal.
Birth weight was 1970 g (20th weight percentile). The weight of the placenta is not known. APGAR was 8 and 8 after 1 and 5 min, respectively. He received CPAP and then oxygen via nasal cannula for a total of 3 days. A post-natal brain ultrasound study was reportedly normal, TSH was 12 mU/L, and he was discharged home after 6 days. Immunizations were up-to-date, including PCV13.
Clinical Exam
At presentation, the infant was alert, irritable, pale and grunting. He was normocephalic without dysmorphic features. There was no rash, no jaundice, no petechiae or ecchymoses. He was tachycardic at 175 beats per minute and tachypneic. The remainder of the cardiovascular and pulmonary findings was unremarkable. The abdomen was distended, tense and shiny with large ascites and no palpably enlarged liver or spleen. Substantial scrotal edema was noted. Weight (with edema) was 3.5 kg (0.01%, z score −3.83), length 52.5 cm (0.00%, z score −4.31), and head circumference 35.5 cm (0.04%, z score −3.35).
Investigations
At the time of admission, there was large proteinuria, hypoalbuminemia, and hypercholesterolemia, consistent with nephrotic syndrome. Nephrotic range proteinuria was defined as a spot urine protein-to-creatinine ratio of >0.23 g/mmol (corresponding to >2.0 g protein/g creatinine). For details and definitions, see Table 1). The hemoglobin (Hb) level dropped from 100 to 69 g/L over the first 10 days of admission, with inadequate reticulocyte response and normal serum ferritin and iron levels. Serum C3 and C4 concentrations, measured on day 11 of admission, were decreased. The concomitant direct agglutination (Coombs) test was negative. Secondary findings were hypogammaglobulinemia, hypothyroidism with elevated TSH and low free T4 levels, hypovitaminosis D, low-normal serum ionized calcium (1.17 mmol/L) and moderately elevated intact PTH. ALT and AST were normal. GGT and bilirubin concentrations peaked during the first few days after admission (see Table 1).
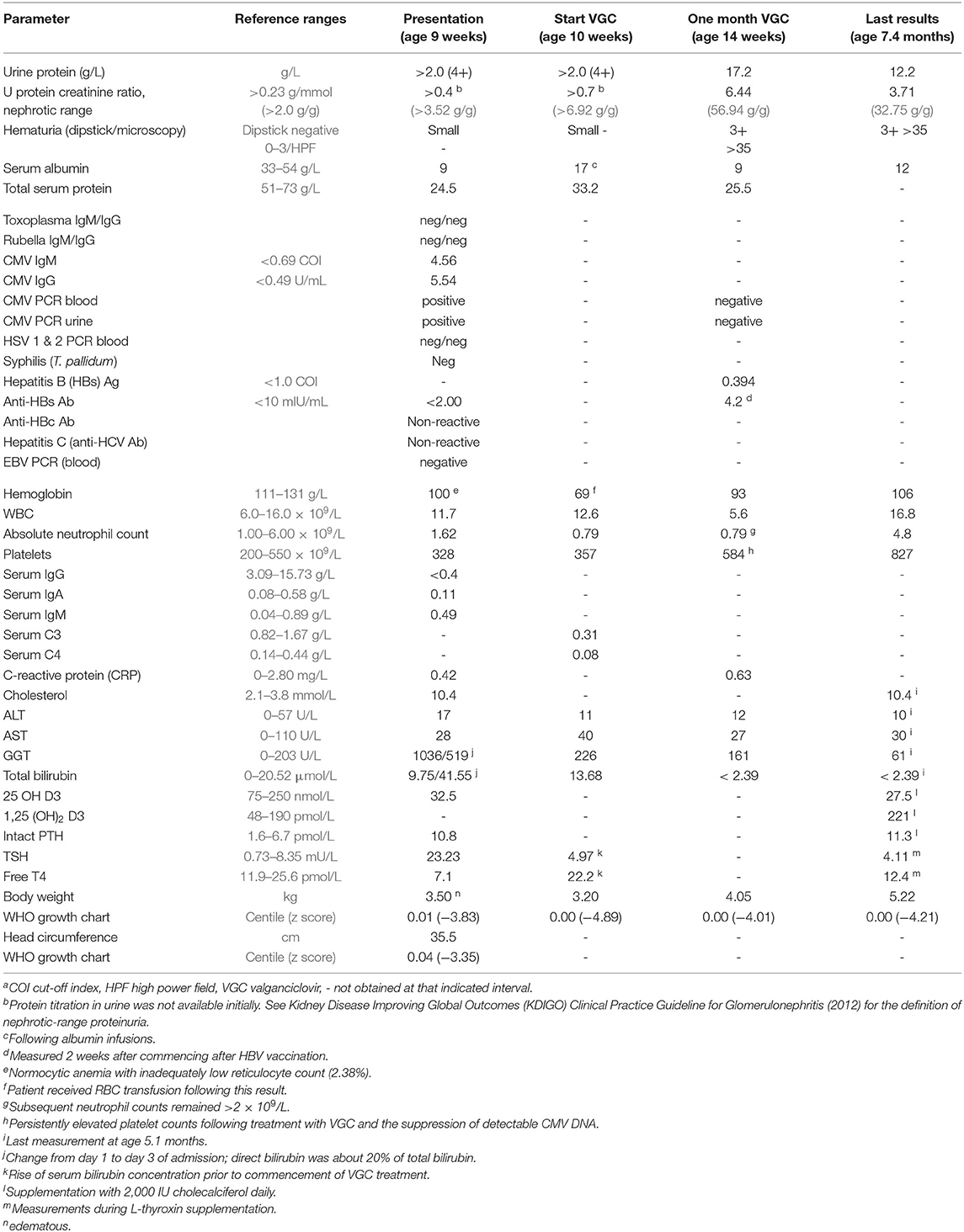
Table 1. Laboratory results during the disease coursea.
The ultrasound (US) scan showed enlarged kindeys with mildly increased cortical echogenicity. Liver and spleen were of normal size and echotexture. The brain US study was normal, except the presence of thalamostriate mineralizing vasculopathy.
Cardiac echography demonstrated a small perimembranous ventricular septal defect of 2–3 mm with left-to-right shunt and patent foramen ovale, none requiring specific interventions.
Infectious disease workup revealed anti-CMV IgM antibodies and CMV DNA (by PCR) in blood and urine. Maternal screening during pregnancy for toxoplasma (IgG and IgM), rubella (IgM), HIV 1 & 2 antibodies/P24 antigen, hepatitis B (HBsAg) and hepatitis C (antibodies), and syphilis was negative. Subsequent CMV testing showed high maternal serum concentrations of CMV IgG, but no anti-CMV IgM.
Treatment of the Patient
Treatment consisted of frequent albumin infusions, diuretics and ACE inhibition, with improvement of ascites and peripheral edema. He also received L-thyroxin, vitamin D and oral penicillin prophylaxis, low-dose acetyl salicylic acid, iron, and indomethacin. In addition, he was vaccinated against Streptococcus pneumoniae (PCV13, twice) and hepatitis B.
Valganciclovir treatment was initiated 9 days after presentation at a dose of 22 mg/kg/day. CMV DNA became undetectable in plasma and urine after 1 month of antiviral therapy, however proteinuria failed to improve (Table 1) strengthening the presumptive clinical diagnosis of a genetic form of congenital nephrotic syndrome.
The patient's energy and protein intake remained precariously inadequate. However, parents were reluctant to agree to g-tube insertion or (unilateral) nephrectomy. Tragically, at the age of 7.9 months, following a few days of lapsed penicillin administration due to vomiting and diarrhea, the patient succumbed to fulminant sepsis, within hours after arrival in the Emergency Department, caused by pan-sensitive S. pneumoniae.
Kidney Biopsy
A kidney biopsy was performed 15 days after hospitalization (6 days after initiation of VGC treatment) to differentiate the pathohistological changes underlying the nephrotic presentation (1, 2, 4, 12). The majority of the >100 sampled glomeruli showed mild mesangial hypercellularity with patent capillaries and normal glomerular basement membrane (GBM) contours. Ten percent of the glomeruli were globally sclerosed or segmentally scarred. Three glomeruli showed active cellular or fibrocellular crescents with proliferation in the Bowman space and focal ruptures of GBM. There was a patchy, mononuclear tubulointerstitial inflammatory infiltrate. Infiltrating interstitial lymphocytes stained positive for CD3 and CD20, respectively, indicating the presence of T- and B-cells. Trichrome staining showed fibrosis in <5% of the cortex. Arteries and arterioles were histologically normal. Immunohistochemical staining for CMV was negative. Routine immunofluorescence showed faint, likely non-specific staining for IgM and C3. Electron microscopy revealed diffuse podocyte foot process effacement with prominent microvillous transformation, but no immune-type deposits. Endothelial fenestrations were intact, and no viral or endothelial tubuloreticular inclusions were noted (Figure 1).
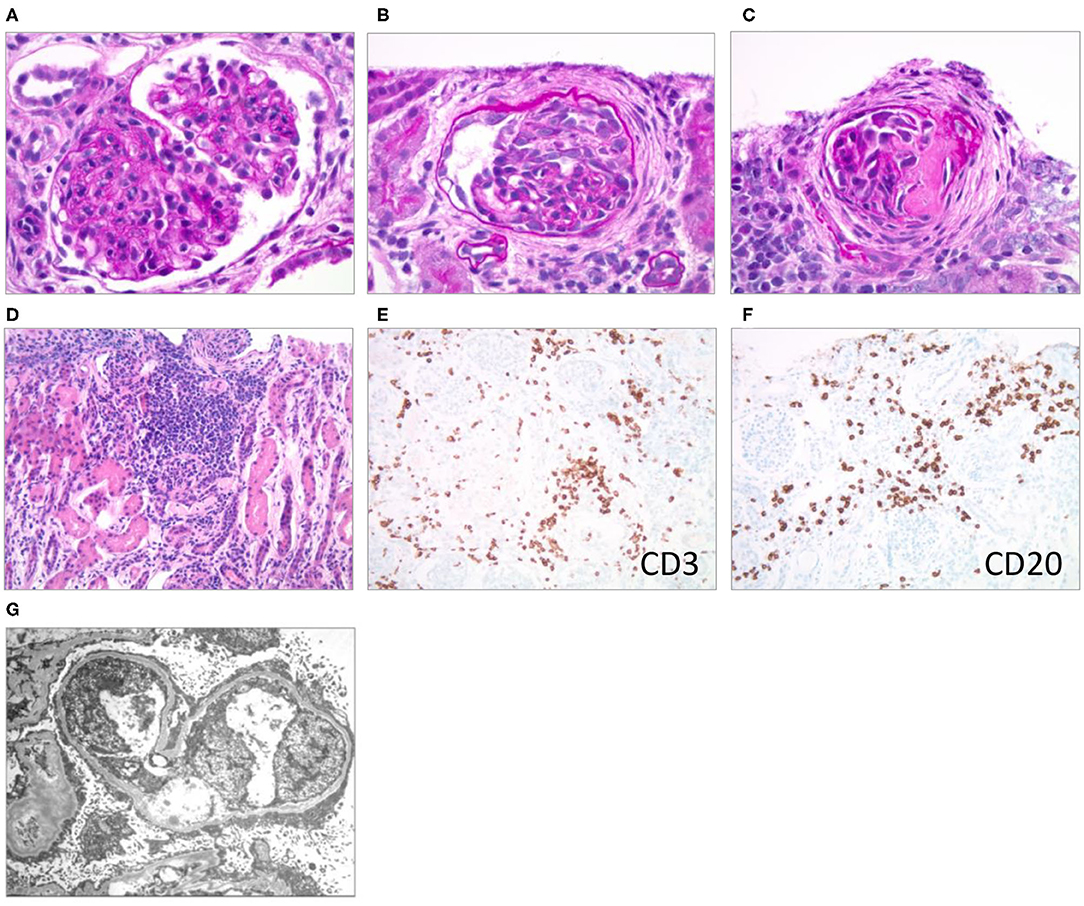
Figure 1. Renal pathological presentation (kidney biopsy of the proband 15 days after admission). (A–C) Brightfield microscopy (PAS stain x600); (D) brightfield (H & E stain x200); (E,F) immunohistochemistry (x200); (G) electron microscopy. (A) Mesangial and endocapillary hypercellularity, (B) cellular crescent, (C) segmental scar/fibrous crescent, (D–F) interstitial infiltrates, (G) diffuse podocyte foot process effacement with prominent microvillous transformation.
Genetic Studies
Genomic DNA, extracted from peripheral blood cells, underwent a series of ultra-sonication, chemical, and enzymatic steps to generate a sequencing-ready library of short fragments (300–400 bp) using the SureSelectXT kit (Agilent, USA). RNA capture probes targeting all coding regions were used to enrich for whole exome regions using the SureSelect Clinical Research Exome V2 kit (Agilent, USA). The enriched library was then subjected to next generation sequencing (2 × 150 bp) using the SP flow cell and the NovaSeq platform (Illumina, USA). Sequencing data were then processed using an in-house custom made bioinformatics pipeline to retain high quality sequencing reads with at least 100X coverage across all coding regions. Rare variants in 99 genes associated with nephrotic syndrome (Supplementary Table 1) were filtered for analysis and interpretation using the American College of Medical Genetics and Genomics (ACMGG) Sequence variant interpretation guideline (24). Two variants in the NPHS1 and the ITSN2 genes met the ACMGG criteria for reporting. NPHS1 encodes nephrin which is central for the integrity of the podocyte slit diaphragm. Pathogenic variants in NPHS1 cause classical Finnish-type congenital nephrotic syndrome (17). The apparently homozygous 2071+1G>T variant in NPHS1 (NM_004646.3) has not been previously reported in individuals with disease and is absent from large population studies such as the Genome Aggregation Database (genomAD) and the Greater Middle East (GME) Variome database. This variant occurs in the conserved region (±1,2) of the splice consensus sequence of the only known NPHS1 transcript, and is predicted to cause altered splicing leading to an abnormal or absent protein (25). The mutation is apparently homozygous, although a large deletion of the second allele could not be ruled out since parental testing was not conducted.
A heterozygous c.2713G>A p. (Ala905Thr) missense variant was also reported in ITSN2 (NM_006277.2), the gene encoding intersectin 2, a member of the guanine exchange factor (GEF) family of proteins that activate Cdc42. This variant was classified as uncertain due to lack of sufficient evidence supporting its clinical significance. Bi-allelic pathogenic variants in ITSN2 have been reported as a novel cause of nephrotic syndrome (26). The identified variant is absent from large population studies such as the Genome Aggregation Database (gnomAD) and the Greater Middle East (GME) Variome database. Computational prediction tools did not provide evidence for or against pathogenicity. No variants were discovered in complement-related genes or genes associated with (other) immunodeficiencies.
Survey of the Literature
CMV-Associated Congenital and Infantile Nephrotic Syndrome
Following the identification of a pathogenic NPHS1 variant as the genetic cause of the patient's nephrotic syndrome, we wondered whether recognizable histopathological features would allow differentiating CMV-associated lesions (and CMV CNS) from CNS due to defined genetic variants. We therefore searched the available literature (PubMed and Google Scholar, without language restriction) for CMV-associated/infantile nephrotic syndrome with histological and genetic findings (“tetrad” of documented CMV infection and clinical features, kidney biopsy, and mutation screen).
Our survey identified only three cases with a complete tetrad: one patient with a homozygous NPHS2 pathogenic variant, one patient with reported pathogenic variants in both NPHS1 and COL4A5 (however the number of variants and the phase of the variants in each gene was not detailed), and one with no detectable variants in NPH1, NPH2, and WT1 (Table 2; patients #3, 7, and 6, respectively) (12, 14, 31). All three patients were treated with ganciclovir (GCV). Only patient #6 achieved sustained remission of proteinuria (31).
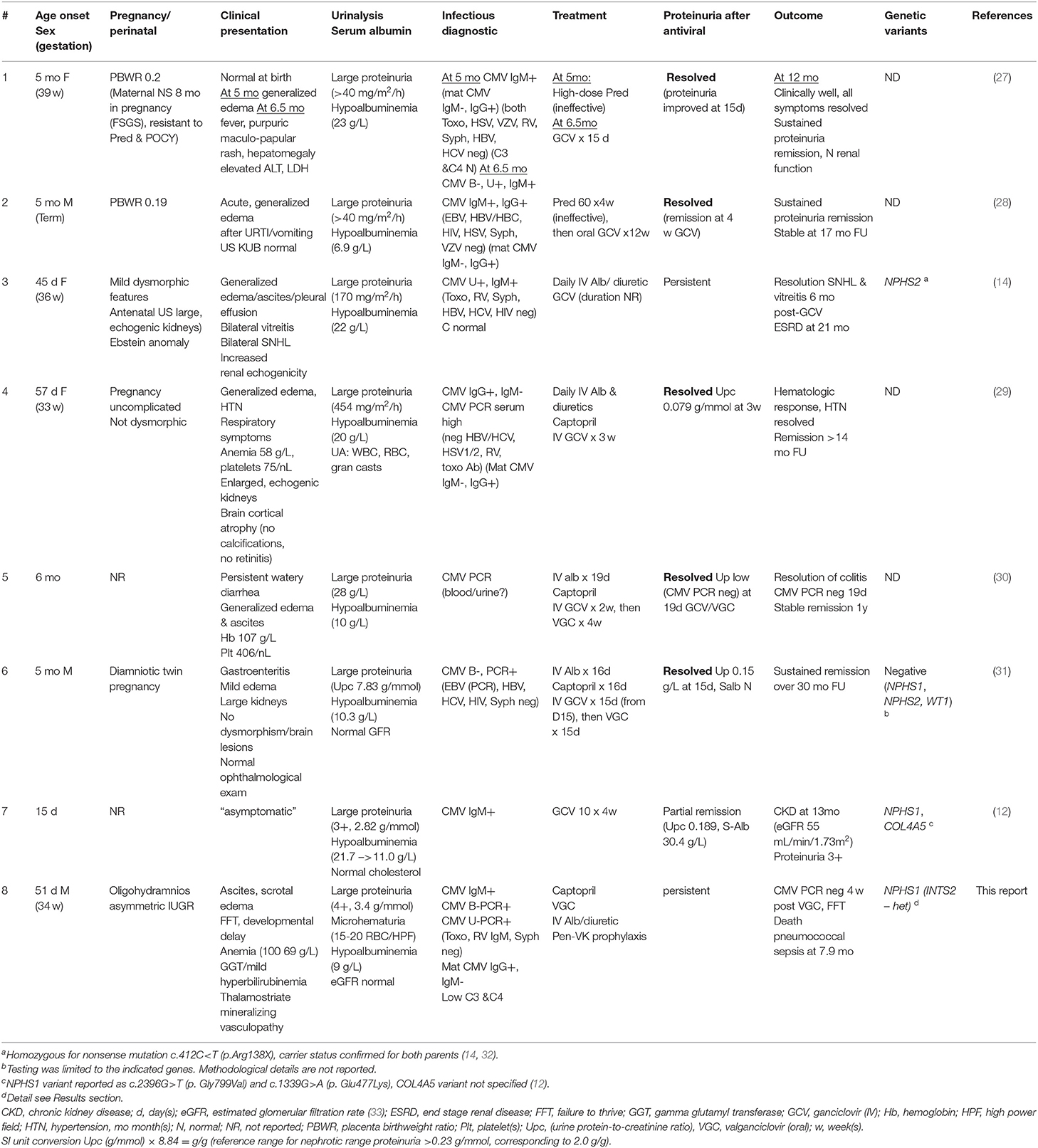
Table 2. CMV-associated congenital and infantile nephrotic syndrome (Literature review and proband).
We then searched for reports of patients with CMV-associated congenital or infantile nephrotic syndrome, who also had a kidney biopsy. All identified cases (#1, 2, 4, 5) were treated with GCV (or GCV, followed by VGC), and all entered stable, proteinuria-free remission (Table 2). Mean antiviral treatment duration was 38 ± 27 (median 30) days; proteinuria improved substantially or disappeared after a mean of 20 ± 5 (median 19) days. The nephrotic syndrome was GCV-resistant in three cases (#3, 7, and 8). These patients were significantly younger at nephrotic syndrome onset (median 1.5, range 0.5–1.7 months) than GCV “sensitive” patients (median 5, range 1.9–6 months) (P < 0.05, unpaired t-test). There was no apparent difference in the presenting nephrotic features (urine protein excretion and serum albumin concentrations) between GCV/VGC responsive and refractory patients.
Kidney Biopsy Results in the Literature
Results of the biopsy survey are detailed in Table 3. In this analysis we included also patients without genetic results under the assumption that stable remission from CMV-associated nephrotic syndrome indicates the absence of podocyte gene mutations. We identified and evaluated eight patients (including our case) with a total of 9 biopsies. The biopsies of four of the five GCV “sensitive” (mutation “negative”) patients (#1, 2, 4, and 6) (27–29, 31) revealed mild-moderate mesangial cell proliferation with or without (mesangial) matrix increase. One of these biopsies also demonstrated mesangial sclerosis (#4). This contrasts with the findings in patients with GCV “resistant” (mutation “positive”) CNS (#3, 7, and 8) (12, 14), where light microscopic findings demonstrate a spectrum of normal-appearing glomeruli (#3a), glomerular sclerosis (#3b and 8) and occasional cellular or fibrocellular crescents, as well as focal mesangial proliferation (#7 and 8).
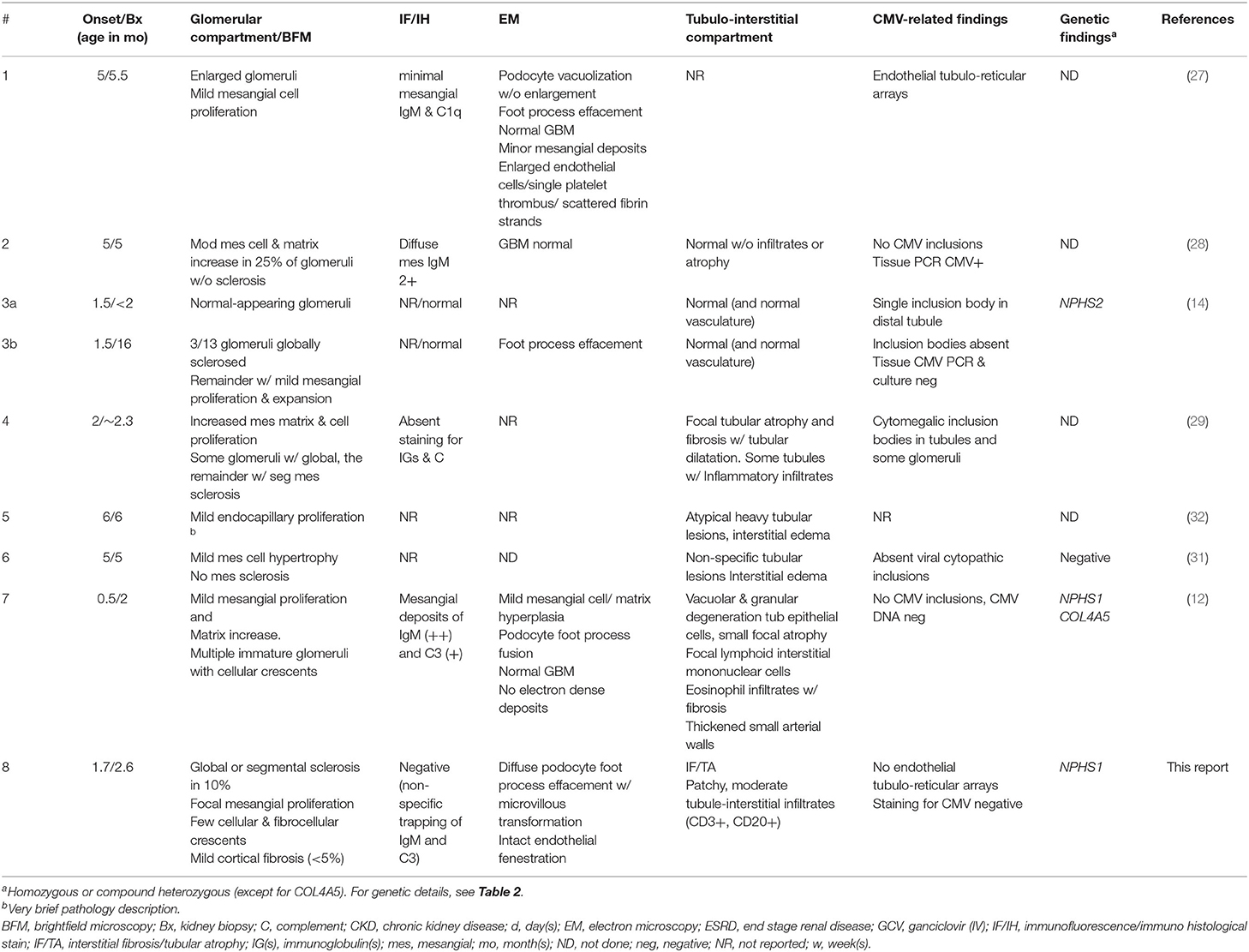
Table 3. Renal biopsy findings (Literature review and proband).
Discussion
The presented case demonstrates features of classical (Finnish-type) congenital nephrotic syndrome (CNS) due to a homozygous NPHS1 pathogenic variant, complicated by congenital CMV infection. There was no retinopathy or microcephaly, and apart from (non-specific) thalamostriate mineralizing vasculopathy (34, 35), the infant had no detectable cerebral abnormalities by ultrasound. However, he had substantial anemia (resulting in RBC transfusion), neutropenia (without thrombocytopenia) and temporary GGT and bilirubin elevation (without hepato- or splenomegaly). The C3 and C4 hypocomplementemia was surprising and deserves further inquiry in comparable cases of congenital/Finnish type nephrotic syndrome and of congenital CMV infections, whereas the extreme hypogammaglobulinemia resulted in all likelihood from massive protein loss via the disrupted glomerular filtration barrier.
Intrauterine or perinatal CMV exposure may have been responsible, at least in part, for the presenting signs and symptoms in our patient. However, there are no criteria to predict the role of CMV in this or similar patients' nephrotic syndromes. Due to health insurance reasons, we were unable to pursue early genetic testing. However, we proceeded with a kidney biopsy for diagnostic and therapeutic guidance (1, 4).
The histological findings were consistent with what has been described in CMV-related congenital nephrotic syndrome, including proliferative lesions and diffuse podocyte effacement. Lack of CMV detection (or inclusion bodies) in renal tissue makes it less likely that the virus was the direct cause of the observed morphological changes. Some of the demonstrated lesions may have arisen as part of the inflammatory and cytokine response of the host, albeit without virus or interferon-gamma-induced (endothelial) tubuloreticular arrays (36, 37). Interestingly, serum C3 and C4 protein levels were decreased during the early course, yet we failed to observe unequivocal immune deposits in the kidney biopsy.
Antiviral treatment was combined with supportive measures, mainly diuretic-assisted albumin infusions, thyroid hormone and vitamin D which controlled the profound edema and hormonal deficiencies. Persistence of large proteinuria and dependency on albumin infusions after the CMV PCR had become negative, favored a genetic cause of the nephrotic syndrome. Attempts to convince the parents to optimize nutrition (g-tube feeding) and (unilateral) nephrectomy to reduce protein losses (2, 4, 22) remained unsuccessful. Unfortunately, he succumbed to foudroyant pneumococcal sepsis at almost 8 months of age, despite repeat vaccination against S. pneumoniae and the prescription of antibiotic prophylaxis, which highlights the highly immunocompromised status of infants with severe nephrotic syndrome. We were eventually able to perform whole exome sequencing and can at least offer genetic counseling for the family.
The occurrence of NS in newborns with intrauterine exposure to CMV has been known since more than 50 years (7, 13). While infants with overwhelming congenital CMV disease demonstrate CMV invasion and proliferation in the kidney along with many other tissues (7), the mechanism of CMV-induced glomerular injury and podocytopathy is not well-defined. Previous studies have addressed direct, virus-mediated tissue injury and injury induced by the host immune response, such as T cell infiltration or immune complex formation (38, 39). However, the natural history and outcome of theses lesions has not been adequately documented, and there is a paucity of cases correlating virological and serological results with renal tissue and genetic studies.
The historical attribution of (severe) CNS to concurrent (congenital) CMV infection, including some of the histological changes, needs be revisited (4, 14). This notion does not negate the possible occurrence of (large) proteinuria and edema in newborns with severe, CMV-induced injuries, such as microcephaly or sensorineural hearing loss, with or without hepato- and splenomegaly and cytopenia.
Although the number of reported cases of CMV-associated CNS with a complete diagnostic “tetrad” is surprisingly small (Table 2), our analysis seems to confirm that an onset of nephrotic syndrome within 3 months of life predicts an underlying variant within podocyte or GBM-related genes, regardless of a coincidental CMV infection. In contrast, onset of nephrotic syndrome between 4 and 12 months and active CMV proliferation in previously asymptomatic infants appears to increase the likelihood of CMV as the remediable cause of the nephrotic syndrome. Neither the severity of proteinuria or hypoalbuminemia at presentation, nor (estimated) GFR or hematological parameters (anemia or thrombocytopenia)–where reported–appear to have discriminatory power in this small cohort.
Table 3 juxtaposes the histological findings of CMV-associated congenital and infantile NS. All but one patient demonstrating sustained remission of proteinuria following antiviral therapy, presented after 3 months of age. One of the common features of all patients with CMV-associated congenital or infantile nephrotic syndrome appears to be (usually mild) mesangial proliferation with or without matrix accumulation. Segmental and global glomerulosclerosis (40) and crescent formation is only noted in the two NPHS1 patients. Given the paucity of completely defined cases and the heterogeneity of renal pathology among patients with autosomal recessive forms of CNS, the histological differentiation between CMV-induced congenital or infantile NS and patients with an underlying genetic variant remains challenging.
The NPHS1 mutation identified in our patient is apparently homozygous, although a large deletion of the second allele could not be ruled out as parental testing was not possible. The recurrence risk would remain the same [25%] if the variant was homozygous, or compound heterozygous with a pathogenic deletion. Identifying the exact variant(s) allows for targeted variant analysis either prenatally or via in vitro fertilization and pre-implantation genetic diagnosis) for any future pregnancies.
The strength of this report is the complete characterization of the case and others identified in a comprehensive literature survey. Its limitation—and reason for publication—is the small number of fully described cases.
Conclusions
Genetic testing is warranted in all patients presenting with isolated or syndromic nephrotic syndrome immediately after birth or within the first 2–3 months of life, whether CMV is present or not. To further clarify the specific contributions of CMV and CMV disease to the histological changes in the kidney and severe, persistent nephrotic syndrome, a correlation (tetrad) of clinical and laboratory findings, kidney biopsy with all staining modalities and/or molecular tools, and genetic work-up are needed.
Ethics Statement
Written informed consent was obtained from the minors' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
AJ collected the data, reviewed the literature, and wrote the first draft of the manuscript. SH, ES, JFS, and AJ participated in the care of the patient. LH read and interpreted the kidney biopsy and provided the photographs. AT and AA were responsible for genetic counseling, performed whole exome sequencing, and analyzed and interpreted the molecular data. WA provided expert infectious disease consultation. MB supervised the care of the patient, reviewed all clinical and laboratory data, performed an independent comprehensive literature review, and wrote the final version of the manuscript. All authors critically reviewed and edited the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thanked Dr. Muhammad Anwar for his help with the kidney biopsy and all physicians, nurses, and allied health professionals who contributed to the care and well-being of the described patient and his family.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.580178/full#supplementary-material
References
1. Habib R. Nephrotic syndrome in the 1st year of life. Pediatr Nephrol. (1993) 7:347–53. doi: 10.1007/BF00857534
2. Holmberg C, Laine J, Ronnholm K, Ala-Houhala M, Jalanko H. Congenital nephrotic syndrome. Kidney Int Suppl. (1996) 53:S51–6.
3. Holmberg C, Antikainen M, Ronnholm K, Ala Houhala M, Jalanko H. Management of congenital nephrotic syndrome of the Finnish type. Pediatr Nephrol. (1995) 9:87–93. doi: 10.1007/BF00858984
4. Reynolds BC, Oswald RJA. Diagnostic and management challenges in congenital nephrotic syndrome. Pediatric Health Med Ther. (2019) 10:157–67. doi: 10.2147/PHMT.S193684
5. Suskind R, Winkelstein JA, Spear GA. Nephrotic syndrome in congenital syphilis. Arch Dis Child. (1973) 48:237–9. doi: 10.1136/adc.48.3.237
6. Shahin B, Papadopoulou ZL, Jenis EH. Congenital nephrotic syndrome associated with congenital toxoplasmosis. J Pediatr. (1974) 85:366–70. doi: 10.1016/S0022-3476(74)80117-4
7. Beneck D, Greco MA, Feiner HD. Glomerulonephritis in congenital cytomegalic inclusion disease. Hum Pathol. (1986) 17:1054–9. doi: 10.1016/S0046-8177(86)80090-9
8. Roussel B, Pinon JM, Birembaut P, Rullier J, Pennaforte F. Congenital nephrotic syndrome associated with congenital toxoplasmosis. Arch Fr Pediatr. (1987) 44:795–7.
9. Niemsiri S. Congenital syphilitic nephrosis. Southeast Asian J Trop Med Public Health. (1993) 24:595–600.
10. Wang JJ, Mao JH. The etiology of congenital nephrotic syndrome: current status and challenges. World J Pediatr. (2016) 12:149–58. doi: 10.1007/s12519-016-0009-y
11. Kim YH, Song JH, Kim CJ, Yang EM. Congenital syphilis presenting with only nephrotic syndrome: reemergence of a forgotten disease. J Korean Med Sci. (2017) 32:1374–6. doi: 10.3346/jkms.2017.32.8.1374
12. Chen Y, Zhang Y, Wang F, Zhang H, Zhong X, Xiao H, et al. Analysis of 14 patients with congenital nephrotic syndrome. Front Pediatr. (2019) 7:341. doi: 10.3389/fped.2019.00341
13. Cozzutto C, Felici N. Unusual glomerular change in cytomegalic inclusion disease. Virchows Arch A Pathol Anat Histol. (1974) 364:365–9. doi: 10.1007/BF00432734
14. Frishberg Y, Rinat C, Feinstein S, Becker-Cohen R, Megged O, Schlesinger Y. Mutated podocin manifesting as CMV-associated congenital nephrotic syndrome. Pediatr Nephrol. (2003) 18:273–5. doi: 10.1007/s00467-003-1079-3
15. Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2015) 26:1279–89. doi: 10.1681/ASN.2014050489
16. Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. (2017) 91:937–47. doi: 10.1016/j.kint.2016.10.013
17. Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. (1998) 1:575–82. doi: 10.1016/S1097-2765(00)80057-X
18. Jalanko H. Congenital nephrotic syndrome. Pediatr Nephrol. (2009) 24:2121–8. doi: 10.1007/s00467-007-0633-9
19. Machuca E, Benoit G, Nevo F, Tete MJ, Gribouval O, Pawtowski A, et al. Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. (2010) 21:1209–17. doi: 10.1681/ASN.2009121309
20. Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. (2015) 10:592–600. doi: 10.2215/CJN.06260614
21. Kari JA, Montini G, Bockenhauer D, Brennan E, Rees L, Trompeter RS, et al. Clinico-pathological correlations of congenital and infantile nephrotic syndrome over twenty years. Pediatr Nephrol. (2014) 29:2173–80. doi: 10.1007/s00467-014-2856-x
22. Holtta T, Jalanko H. Congenital nephrotic syndrome: is early aggressive treatment needed? Yes. Pediatr Nephrol. (2020) 35:1985–90. doi: 10.1007/s00467-020-04578-4
23. Boyer O, Berody S. Congenital nephrotic syndrome: is early aggressive treatment needed?-No. Pediatr Nephrol. (2020) 35:1991–96. doi: 10.1007/s00467-020-04556-w
24. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. (2017) 19:4–23. doi: 10.1016/j.jmoldx.2016.10.002
25. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. (2018) 39:1517–24. doi: 10.1002/humu.23626
26. Ashraf S, Kudo H, Rao J, Kikuchi A, Widmeier E, Lawson JA, et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat Commun. (2018) 9:1960. doi: 10.1038/s41467-018-04193-w
27. Giani M, Edefonti A, Damiani B, Marra G, Colombo D, Banfi G, et al. Nephrotic syndrome in a mother and her infant: relationship with cytomegalovirus infection. Pediatr Nephrol. (1996) 10:73–5. doi: 10.1007/BF00863452
28. Berbel O, Vera-Sempere F, Cordoba J, Zamora I, Simon J. Cytomegalovirus nephrotic syndrome. Nefrologia. (2003) 23:451–3.
29. Besbas N, Bayrakci US, Kale G, Cengiz AB, Akcoren Z, Akinci D, et al. Cytomegalovirus-related congenital nephrotic syndrome with diffuse mesangial sclerosis. Pediatr Nephrol. (2006) 21:740–2. doi: 10.1007/s00467-006-0051-4
30. Zhang B, Fila M, Fakhoury M, Baudouin V, Deschenes G, Jacqz-Aigrain E, et al. Pharmacokinetics and dosage individualization of ganciclovir and valganciclovir in an infant with nephrotic syndrome associated with cytomegalovirus infection. J Antimicrob Chemother. (2014) 69:1150–1. doi: 10.1093/jac/dkt472
31. Hogan J, Fila M, Baudouin V, Peuchmaur M, Deschenes G, Niel O. Cytomegalovirus infection can mimic genetic nephrotic syndrome: a case report. BMC Nephrol. (2015) 16:156. doi: 10.1186/s12882-015-0152-z
32. Frishberg Y, Rinat C, Megged O, Shapira E, Feinstein S, Raas-Rothschild A. Mutations in NPHS2 encoding podocin are a prevalent cause of steroid-resistant nephrotic syndrome among Israeli-Arab children. J Am Soc Nephrol. (2002) 13:400–5.
33. Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. (2009) 20:629–37. doi: 10.1681/ASN.2008030287
34. Amir J, Schwarz M, Levy I, Haimi-Cohen Y, Pardo J. Is lenticulostriated vasculopathy a sign of central nervous system insult in infants with congenital CMV infection? Arch Dis Child. (2011) 96:846–50. doi: 10.1136/adc.2010.208405
35. Sisman J, Chalak L, Heyne R, Pritchard M, Weakley D, Brown LS, et al. Lenticulostriate vasculopathy in preterm infants: a new classification, clinical associations and neurodevelopmental outcome. J Perinatol. (2018) 38:1370–8. doi: 10.1038/s41372-018-0206-8
36. Hammar SP, Luu JY, Bockus DE, Remington FL, Luu JW, Friedman S, et al. Induction of tubuloreticular structures in cultured human endothelial cells by recombinant interferon alfa and beta. Ultrastruct Pathol. (1992) 16:211–8. doi: 10.3109/01913129209074562
37. Lee CJ, Suh KS, Kim KH, Chang YK, Na KR, Lee KW. The clinicopathologic significance of endothelial tubuloreticular inclusions in glomerular diseases. Ultrastruct Pathol. (2013) 37:386–94. doi: 10.3109/01913123.2013.814738
38. Stagno S, Volanakis JE, Reynolds DW, Stroud R, Alford CA. Immune complexes in congenital and natal cytomegalovirus infections of man. J Clin Invest. (1977) 60:838–45. doi: 10.1172/JCI108838
39. Platt JL, Sibley RK, Michael AF. Interstitial nephritis associated with cytomegalovirus infection. Kidney Int. (1985) 28:550–2. doi: 10.1038/ki.1985.163
Keywords: Finnish-type nephrotic syndrome, NPHS1, cytomegalovirus, Streptococcus pneumoniae, case report, glomerulonephritis, infantile nephrotic syndrome
Citation: Jacob A, Habeeb SM, Herlitz L, Simkova E, Shekhy JF, Taylor A, Abuhammour W, Abou Tayoun A and Bitzan M (2020) Case Report: CMV-Associated Congenital Nephrotic Syndrome. Front. Pediatr. 8:580178. doi: 10.3389/fped.2020.580178
Received: 04 July 2020; Accepted: 02 November 2020;
Published: 27 November 2020.
Edited by:
Sami Sanjad, American University of Beirut Medical Center, LebanonReviewed by:
Mario Giordano, Azienda Ospedaliero Universitaria Consorziale Policlinico di Bari, ItalyVera Maria Santoro Belangero, Campinas State University, Brazil
Copyright © 2020 Jacob, Habeeb, Herlitz, Simkova, Shekhy, Taylor, Abuhammour, Abou Tayoun and Bitzan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Bitzan, bWFydGluLmJpdHphbkBhamNoLmFl