 Anisha Hegde
Anisha Hegde Michelle R. Denburg2
Michelle R. Denburg2 Dorey A. Glenn
Dorey A. Glenn- 1Department of Pediatrics, University of North Carolina Hospitals, Chapel Hill, NC, United States
- 2Division of Nephrology, The Children's Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 3Division of Nephrology, University of North Carolina School of Medicine, Chapel Hill, NC, United States
Acute kidney injury (AKI) has been associated with deleterious impacts on a variety of body systems. While AKI is often accompanied by dysregulation of mineral metabolism—including alterations in calcium, phosphate, vitamin D, parathyroid hormone, fibroblast growth factor 23, and klotho—its direct effects on the skeletal system of children and adolescents remain largely unexplored. In this review, the pathophysiology of dysregulated mineral metabolism in AKI and its potential effects on skeletal health are discussed, including data associating AKI with fracture risk.
Introduction
Acute kidney injury (AKI) results from a spectrum of insults that lead to a sudden decrease in kidney function and is responsible for significant morbidity and mortality (1–3). The worldwide incidence of pediatric AKI in hospital settings ranges from 26.9 to 41.3% (2, 4). Associated mortality ranges from 8.8 to 21% and is inversely related to gross national income per capita (2, 4, 5). AKI is accompanied by systemic inflammation (6) and has been associated with distant-organ injury—including injury to the lungs (7–9), heart (10), intestines (11), liver (11, 12), and brain (13)—that contributes to short- and long-term adverse patient outcomes (14, 15). AKI is frequently accompanied by dysregulated mineral metabolism (16), supporting the hypothesis that AKI may also be accompanied by alterations in skeletal structure and function. Commonly encountered dysregulation in mineral metabolism during AKI includes hypocalcemia, hyperphosphatemia, elevated parathyroid hormone (PTH), decreased 1,25-dihydroxyvitamin D (1,25D), elevated fibroblast growth factor 23 (FGF23), and decreased klotho. These patterns are similar to those seen in individuals with chronic kidney disease–mineral bone disorder (CKD-MBD), a condition known to alter skeletal and vascular biology. Compared to CKD-MBD, much less is known about the effects of mineral dysregulation and inflammation in AKI on short- and long-term skeletal health (16–18).
Identifying independent risk factors for skeletal injury in AKI is challenging for a number of reasons. First, AKI is a heterogeneous diagnosis with a myriad of inciting and exacerbating factors. Second, short-term biomarkers of skeletal injury are lacking, and the timeframe over which skeletal outcomes occur is long. Nonetheless, in this review, we aim to summarize the existing literature in three areas: (1) current data on fracture risk following AKI; (2) potential mechanisms of dysregulation in AKI, including inflammation, which could impact skeletal health; and (3) areas for further research.
Fracture Risk and AKI
There is a striking paucity of literature exploring the association between AKI and risk of skeletal fracture. A single population-based matched cohort study conducted among 448 Taiwanese adult patients who survived dialysis-requiring AKI and 1,792 controls demonstrated a 1.25-fold increased risk of bone fracture (p = 0.049) in the AKI recovery group, even after controlling for progression to end-stage renal disease (19). Enrollees were identified through national database entries over 8 years and were prospectively followed for at least 1 year after hospital discharge. Incidence of bone fracture was 320 per 10,000 person-years in the AKI recovery group and 93 per 10,000 person-years in the control group (19). Patients with skeletal fracture also experienced increased long-term mortality (hazard ratio = 1.43, 95% confidence interval = 1.19–1.71, p < 0.001) (19). This is the only study to date that has evaluated long-term impacts of AKI on risk of skeletal fracture. Individuals with non–dialysis-dependent AKI and individuals younger than 18 years were not included in the study. These findings should be replicated in broader cohorts of adults with milder degrees of AKI and in children, as mineral dysregulation after AKI has been demonstrated in both groups (16, 20). There is also little published literature describing histologic changes in bone during or following AKI, with only one report describing findings from bone biopsy, including mild increases in bone resorption without increased osteoid (21).
Mineral Dysregulation and AKI
Hypocalcemia and Hyperphosphatemia
The majority of total body calcium and phosphate is stored as skeletal hydroxyapatite, with extracellular calcium representing ~1% and extracellular phosphate representing <1% of total body stores (22, 23). Extracellular calcium and phosphate concentrations are primarily regulated by three hormones: 1,25D, PTH, and FGF23 [Figure 1; (24)]. Dysregulation of calcium and phosphate homeostasis is a consistent finding in studies of adult patients with varying severities of AKI (25–33). In a study of 400 critically ill adults with AKI, patients had a median phosphate level of 5.2 mg/dL with an interquartile range of 4.0–6.7 mg/dL (29). Additionally, multiple studies comparing mineral metabolism in patients with AKI to hospitalized control groups without AKI demonstrated significantly higher phosphate levels in AKI groups (28, 30, 31, 33). For example, in a cohort of 51 children who underwent elective cardiac surgery, serum phosphate increased significantly over a 24-h period in those who developed AKI compared to those who did not (p = 0.01) (34).
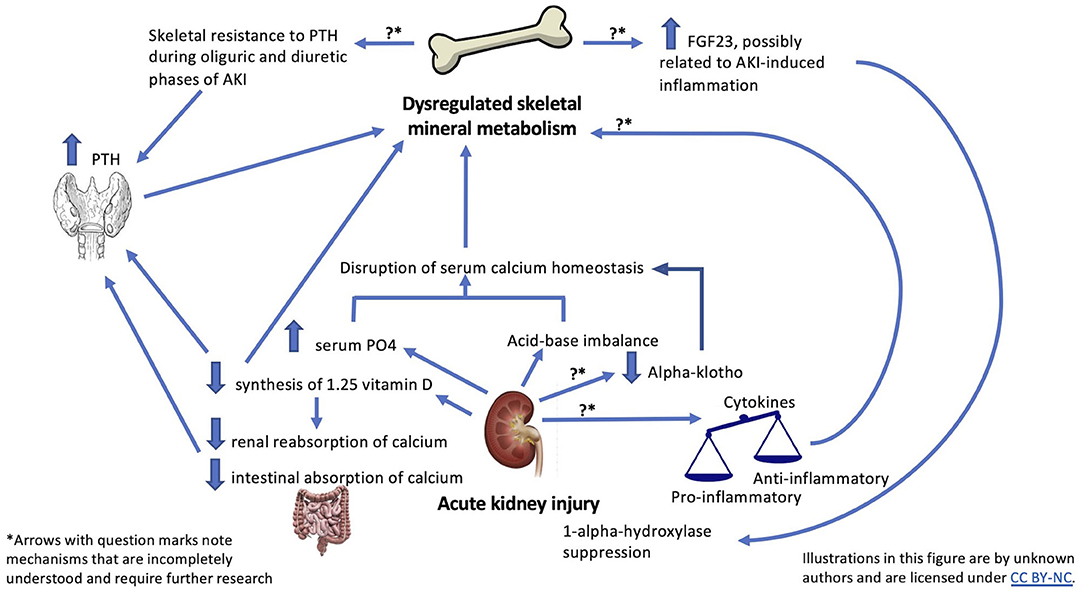
Figure 1. Summary of possible mechanisms linking AKI and skeletal outcomes.
Of the aforementioned case-control studies that reported significantly elevated phosphate levels, two measured serum calcium levels. Leaf et al. reported significantly lower calcium levels in 30 adult patients with all-cause AKI compared to 30 patients without AKI (p = 0.004) (30). Patients with AKI had a serum calcium interquartile range of 7.5–8.6 mg/dL, and patients without AKI had an interquartile range of 8.1–9.0 mg/dL (30). In contrast, Zhang et al. did not detect a statistically significant difference in ionized calcium levels between critically ill adult patients with and without AKI (p = 0.41) (28). Patients with and without AKI had mean ionized calcium levels of 1.19 ± 0.1 mmol/L and 1.15 ± 0.08, respectively. Both studies are limited by small sample size, although differing findings could be explained by heterogeneous study populations (all hospitalized patients vs. only critically ill patients) and methods of measuring calcium (total serum vs. ionized). Hypocalcemia would be expected to follow significant hyperphosphatemia as elevated serum phosphate sequesters calcium. However, patients who are critically ill are also more likely to have aberrations in albumin and acid–base balance (22), causing a potential change in ionized calcium but not in total serum calcium. In addition to affecting ionized calcium levels, metabolic acidosis in vitro has been demonstrated to directly trigger osteoblast inhibition and osteoclast stimulation in mouse models (35).
Hypocalcemia and hyperphosphatemia may have acute as well as subacute clinical relevance (22). Acute hypocalcemia has well-described effects on decreasing peripheral vascular resistance (36), decreasing myocardial contractility (37), and increasing neuromuscular reactivity (22). Further research is needed to better understand the clinical impact that mineral alterations in AKI have on skeletal health, particularly among children undergoing rapid skeletal growth.
Mechanisms of Mineral Dysregulation in AKI
1,25 Vitamin D
Alteration in regulatory hormones contributes to calcium and phosphate derangements following AKI. Decreased levels of 1,25D, and less frequently its precursor 25-hydroxyvitamin D (25D), have been described in multiple adult AKI cohorts, including in patients status post-cardiac surgery (26), those with rhabdomyolysis (27), and those with critical illness (29). There is growing evidence in adults that a history of critical illness is itself a strong risk factor for osteopenia and osteoporosis likely due to a combination of inflammation, undernutrition, immobilization, and vitamin D deficiency (38)—all of which are compounded by AKI. Similarly, vitamin D deficiency is common in critically ill children, with a pooled prevalence of 54.6% in a recent systematic review of hospitalized children with acute or critical conditions (39). Vitamin D deficiency, especially in critically ill patients, is linked to increased risk of AKI and mortality (40, 41). This link underscores the salience of investigating how vitamin D dysregulation during AKI superimposed on frequently reported baseline vitamin D deficiency may be contributing to adverse outcomes, including adverse skeletal outcomes.
Studies comparing 1,25D and 25D levels in adults with AKI to hospitalized controls without AKI have demonstrated significantly lower levels of 1,25D but not 25D (31, 42, 43). However, Tingting et al. reported lower levels of both 1,25D (p < 0.0001) and 25D (p < 0.0001) among 34 patients with critical illness and AKI compared to 12 healthy controls (44). Demographic differences in gender, age, and race between groups did not rise to statistical significance (44). This study's findings are perhaps explained by their use of healthy controls, as studies utilizing hospitalized controls have not shown differences in 25D, suggesting that low 25D may be a marker of global disease severity more so than of acute mineral dysregulation. Comparatively, Lai et al. enrolled 200 patients with AKI, 13 critically ill patients without AKI, and 17 healthy participants and reported that 1,25D levels but not 25D levels were significantly lower in the AKI group vs. the other groups (42). Taken together, these data suggest that AKI is associated with suppression of 1,25D and variable changes in 25D (16).
1,25D deficiency contributes to decreased intestinal absorption of calcium and decreased renal reabsorption of calcium (24, 45). These changes in AKI are thought to be mediated by decreased renal synthesis of 1,25D secondary to proximal tubule injury or from FGF23-induced suppression of 1-α-hydroxylase (46). Substrate delivery of 25D to the proximal tubule in AKI may also decrease because of lower levels of circulating vitamin D–binding protein (DBP), decreased filtration of 25D-DBP complexes due to reduced glomerular filtration rate (GFR), and decreased uptake of these complexes for processing due to proximal tubule injury (16).
Altered vitamin D metabolism in AKI, in addition to vitamin D deficiency potentially present on admission or attributed to critical illness, may be clinically relevant in multiple ways. Vitamin D plays a key role in skeletal health, mitigating fracture risk, and optimizing bone density and content through direct mineral regulation (47). 1,25D, through binding to its nuclear receptor, may also play a role in immune and inflammatory regulation (48). Even though vitamin D deficiency in AKI is likely transient, resolving with renal recovery (26, 27, 49), it has been associated with endothelial dysfunction (50), oxidative stress, and inflammation—both in the renal microvasculature and in distant organs (14, 51, 52). Arfian et al. demonstrated renoprotective effects of vitamin D—including reducing myofibroblasts and renal inflammation—in mice with kidney ischemia–reperfusion injuries (IRIs), although protective effects of vitamin D on distant organs such as bone following AKI have not yet been elucidated (52).
Parathyroid Hormone
PTH is the primary regulator of serum calcium, and elevated PTH levels have been frequently reported in patients with AKI (16, 17, 26, 27, 29). Studies have consistently demonstrated significantly elevated PTH in individuals with AKI compared to healthy controls (44, 53, 54). These elevations are thought to be secondary to hypocalcemia and low serum 1,25D levels as discussed above (24). Upregulation of the parathyroid calcium-sensing receptor due to acute inflammation may additionally affect the calcium-PTH set-point (16, 24).
Skeletal and renal resistance to PTH has been described in humans following AKI (25, 29, 30), potentially contributing to dysregulated hormonal control of calcium and phosphate. In patients with CKD, skeletal resistance to PTH is thought to be secondary to downregulated PTH receptors (16); however, specific mechanisms of PTH resistance in AKI have not been studied. Massry et al. demonstrated that patients with AKI failed to respond appropriately to exogenous PTH infusion during their oliguric and diuretic phases of AKI but produced appropriate increases in serum calcium once renal function recovered (25).
Thus, skeletal resistance to PTH in AKI is likely a transient phenomenon, as is elevated PTH (26, 30). For example, in a study comparing PTH levels in hospitalized adults with and without all-cause AKI, PTH levels were initially higher in the AKI group (p = 0.004) compared to the non-AKI group but were no longer different by day 5 of enrollment (p = 0.56) (30). Additional studies of hospitalized adults with and without AKI following cardiac surgery and critical illness (28, 31) have reported that PTH was comparatively elevated (and occasionally severely, with PTH >250 mg/dL) in AKI groups but did not rise to statistical significance. Small sample sizes and heterogeneous patient populations may explain these inconsistent findings. Likewise, findings may be confounded by elevations of PTH due to critical illness (55, 56).
Fibroblast Growth Factor 23 and Klotho
FGF23 is a bone-derived protein, produced by osteoblasts and osteocytes and known to regulate vitamin D metabolism and phosphate homeostasis (57–59). FGF23 was first identified in patients with tumor-induced osteomalacia and mineralization defects and has since become an important link connecting renal and skeletal physiology (60). Studies have consistently demonstrated that FGF23 levels increase rapidly after onset of AKI (28–31, 61–66). In mouse models of folic acid–induced AKI, FGF23 levels have been shown to rise within 1 h following AKI (62). In adults with AKI after cardiac surgery, FGF23 levels demonstrated more than a 15-fold increase at 24 h post-surgery (62).
While there is strong evidence of a role for FGF23 in mediating phosphate and vitamin D dysregulation in early-stage CKD (67), the effects of FGF23 on skeletal health following AKI have not been studied (61, 68). Rise in FGF23 following AKI appears to be independent of PTH and 1,25D levels (20, 58, 62, 69), but resultant effects of FGF23 on PTH and 1,25D levels in AKI have yet to be conclusively demonstrated. FGF23 is also thought to play a role in the inflammatory response following AKI (58). In mice with folic acid–induced AKI, very high FGF23 mRNA expression was detected in thymus and spleen, suggesting that FGF23 may be associated with increased TNF expression and elevated inflammatory cytokine levels through effects on lymphoid organs (58).
FGF23 elevation is likely altered in a timeframe consistent with reduced renal function. For example, in 32 pediatric patients who underwent cardiac surgery requiring cardiopulmonary bypass, those children who developed post-operative AKI demonstrated a significant increase in intact FGF23 at 2 h post-reperfusion compared to those who did not develop AKI (p = 0.04). At 48 h, intact FGF23 levels between these groups were not significantly different (p = 0.19), although C-terminal FGF23 levels were significantly elevated (p = 0.006) in the AKI group (20). Animal models of AKI due to folic acid nephropathy have demonstrated increased FGF23 expression in femur lysates, but the factors responsible for this elevation in bone during a period of acutely reduced renal function have not been identified (58, 62, 64).
α-Klotho, a transmembrane protein mainly produced by renal tubular epithelial cells, plays a critical role in FGF23 receptor binding (16) and, through its actions on the kidney and parathyroid glands, plays an important role in mineral metabolism (64, 70, 71). Its extracellular domain can modulate renal calcium and potassium absorption independently of FGF23 (16, 72) in the distal tubule and can be cleaved into the circulation, facilitating distant organ effects of FGF23 (64). In CKD mouse models, klotho has been shown to decrease ectopic calcification (including soft tissue calcification), likely through tight coregulation of phosphorous with FGF23 (70). Klotho deficiency has been demonstrated in animal models of AKI (73, 74), although studies in humans are lacking (64, 71, 73, 75–78). Cytokines produced during AKI, including tumor necrosis factor α (TNF-α), may downregulate renal expression of klotho (79). No studies have yet examined whether klotho deficiency may contribute to aberrant calcification in AKI.
Inflammation in AKI
Acute intrarenal and systemic inflammation, including the release of proinflammatory cytokines, has been well-described following AKI (6, 7, 11–14, 80–84). Inflammation is a crucial biologic response for eliminating pathogens and repairing injured tissue. However, the balance between proinflammatory and anti-inflammatory responses is often abnormal in AKI (83). In renal IRI, cellular damage triggers an inflammatory response that includes oxidative stress in renal tubular epithelial cells, necrotic cells that release a variety of molecules to signal damage, and cytokine release from activated renal parenchymal cells and dendritic cells that recruit innate and adaptive immune mediators (83).
Increased serum levels of proinflammatory cytokines, including TNF-α, interleukin 6 (IL-6), and IL-8, have been described during AKI in animal models of renal IRI (80). These animal models have demonstrated dramatic increases in plasma cytokine levels compared to non-IRI animal models, including sham surgery and bilateral nephrectomy (80). Similar increases in cytokine levels have been noted in trauma patients with AKI compared to those without AKI early on in their hospital courses (80). Circulating cytokines during AKI have been shown to impact distant organs, including the brain (13), liver (11), and lungs (8). In addition to increased cytokine production, experimental models indicate that cytokine clearance is also decreased in AKI (80).
Although direct effects of proinflammatory cytokines specifically on bone during AKI have not been studied, proinflammatory cytokines have been associated with adverse effects on bone formation and resorption [Figure 1; (85–87)] For example, TNF-α and IL-6 have been shown to activate the parathyroid calcium-sensing receptor (88) and to inhibit renal expression of 1-α-hydroxylase (89), which could contribute to hormonal dysregulation of mineral metabolism in AKI. TNF-α has been reported to inhibit PHEX gene expression. PHEX gene is expressed mostly in osteoblasts, and loss of PHEX function has been linked to defective mineralization (90). Additionally, data from the Women's Health Initiative Study (91) suggest that TNF-α plays a role in mediating fracture risk, as association between estimated GFR and fracture risk was eliminated after adjustment for TNF-α receptor levels.
Anti-inflammatory cytokines are also increased in AKI (83). IL-10 is an anti-inflammatory cytokine that may have osteoprotective effects and has emerged along with IL-6 as a key player in signaling distant effects of acute renal inflammation (81). IL-10 is produced by T-regulatory cells (60) that in vitro directly inhibit osteoclast activity, including their differentiation and function, and in vivo have been shown to protect against TNF-α-induced bone loss in mice (92). The net outcome of this interplay between osteotoxic and osteoprotective factors on bone in the setting of AKI has yet to be determined (83).
Transcriptomics research in a murine model of ischemic AKI identified increased levels of IL-10 and IL-6 in lung tissue after AKI, in addition to global transcriptomic changes and histologic injury (81). Similar to the lung, the skeletal system has an extensive capillary network, including in the metabolically active skeletal system of children and adolescents undergoing periods of rapid growth (93). Thus, the relationship of inflammation in AKI and its potential direct consequences on bone health is an area ripe for additional investigation (80, 84).
Areas for Future Research
1. Epidemiological studies are needed to further characterize the burden of and risk factors for skeletal complications, including fractures, in patients with AKI. These studies should include pediatric patients, who are experiencing AKI during times of bone mineral accrual and linear growth (24).
2. Larger studies are needed to better characterize which aspects of dysregulated mineral metabolism, if any, persist after renal recovery following AKI.
3. The use of bone imaging (dual energy x-ray absorptiometry / high resolution peripheral quantitative computed tomography) lends itself to investigations into whether changes in bone structure and microarchitecture are seen in the acute or subacute phases of AKI.
4. Further investigation is needed regarding the extent to which systemic vascular and inflammatory changes detected following AKI, including vitamin D–associated changes, alter bone epigenetics and transcriptomics and might contribute to effects on long-term skeletal health. Metabolomics studies in AKI may identify downstream targets relevant for additional investigation (10, 94).
5. Although the pathophysiology of AKI generally involves a common cascade of inflammation secondary to ischemia, reperfusion, cell injury, and cell death, AKI remains a clinically heterogeneous diagnosis with varying therapies. The potential compounding effect of frequently used therapies in AKI, such as diuretics and other medications (87), on mineral dysregulation and bone health has not been evaluated.
Author Contributions
AH and DG contributed to the literature review and layout of this article. AH wrote the first draft of the manuscript, which was revised by DG. AH, DG, and MD contributed to final manuscript revision and have approved the submitted version. All authors contributed to the article and approved the submitted version.
Conflict of Interest
MD reports funding from Mallinckrodt Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Goldstein SL, Adrogue HE. Chapter 365: Acute kidney injury. In: McInerny T, Adam H, Campbell D, DeWitt T, Foy J, Kamat D, editors. American Academy of Pediatrics Textbook of Pediatric Care. 2nd ed. (2017). Available online at: https://pediatriccare.solutions.aap.org/chapter.aspx?sectionid=138300635&bookid=1626 (accessed December 01, 2020).
2. Lameire N, Van Biesen W, Vanholder R. Epidemiology of acute kidney injury in children worldwide, including developing countries. Pediatr Nephrol. (2017) 32:1301–14. doi: 10.1007/s00467-016-3433-2
3. Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. (2012) 120:c179–84. doi: 10.1159/000339789
4. Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, et al. World incidence of AKI: A meta-analysis. Clin J Am Soc Nephrol. (2013) 8:1482–93. doi: 10.2215/CJN.00710113
5. Fortrie G, De Geus HRH, Betjes MGH. The aftermath of acute kidney injury: a narrative review of long-term mortality and renal function. Crit Care. (2019) 23:24. doi: 10.1186/s13054-019-2314-z
6. Gyurászová M, Kovalčíková AG, Renczés E, Kmetová K, Celec P, Bábíčková J, et al. Oxidative stress in animal models of acute and chronic renal failure. Dis Markers. (2019) 2019:8690805. doi: 10.1155/2019/8690805
7. Andres-Hernando A, Altmann C, Bhargava R, Okamura K, Bacalja J, Hunter B, et al. Prolonged acute kidney injury exacerbates lung inflammation at 7 days post-acute kidney injury. Physiol Rep. (2014) 2:e12084. doi: 10.14814/phy2.12084
8. Faubel S, Edelstein CL. Mechanisms and mediators of lung injury after acute kidney injury. Nat Rev Nephrol. (2016) 12:48–60. doi: 10.1038/nrneph.2015.158
9. Teixeira JP, Ambruso S, Griffin BR, Faubel S. Pulmonary consequences of acute kidney injury. Semin Nephrol. (2019) 39:3–16. doi: 10.1016/j.semnephrol.2018.10.001
10. Fox BM, Gil HW, Kirkbride-Romeo L, Bagchi RA, Wennersten SA, Haefner KR, et al. Metabolomics assessment reveals oxidative stress and altered energy production in the heart after ischemic acute kidney injury in mice. Kidney Int. (2019) 95:590–610. doi: 10.1016/j.kint.2018.10.020
11. Park SW, Chen SW, Kim M, Brown KM, Kolls JK, D'Agati VD, et al. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab Investig. (2011) 91:63–84. doi: 10.1038/labinvest.2010.151
12. Shang Y, Madduma Hewage S, Wijerathne CUB, Siow YL, Isaak CK, Karmin O. Kidney ischemia-reperfusion elicits acute liver injury and inflammatory response. Front Med. (2020) 7:201. doi: 10.3389/fmed.2020.00201
13. Liu M, Liang Y, Chigurupati S, Lathia JD, Pletnikov M, Sun Z, et al. Acute kidney injury leads to inflammation and functional changes in the brain. J Am Soc Nephrol. (2008) 19:1360–70. doi: 10.1681/ASN.2007080901
14. Druml W. Systemic consequences of acute kidney injury. Curr Opin Crit Care. (2014) 20:613–9. doi: 10.1097/MCC.0000000000000150
15. Uber AM, Sutherland SM. Acute kidney injury in hospitalized children: consequences and outcomes. Pediatr Nephrol. (2020) 35:213–20. doi: 10.1007/s00467-018-4128-7
16. Leaf DE, Christov M. Dysregulated mineral metabolism in AKI. Semin Nephrol. (2019) 39:41–56. doi: 10.1016/j.semnephrol.2018.10.004
17. Wesseling-Perry K. Bone disease in pediatric chronic kidney disease. Pediatr Nephrol. (2013) 28:569–76. doi: 10.1007/s00467-012-2324-4
18. Yang T, Wang W, Tang X, Shi P, Zhang L, Yu W, et al. Association between mineral and bone disorder in patients with acute kidney injury following cardiac surgery and adverse outcomes. BMC Nephrol. (2019) 20:369. doi: 10.1186/s12882-019-1572-y
19. Wang WJ, Chao C Ter, Huang YC, Wang CY, Chang CH, Huang TM, et al. The impact of acute kidney injury with temporary dialysis on the risk of fracture. J Bone Miner Res. (2014) 29:676–84. doi: 10.1002/jbmr.2061
20. Hanudel MR, Wesseling-Perry K, Gales B, Ramos G, Campbell V, Ethridge K, et al. Effects of acute kidney injury and chronic hypoxemia on fibroblast growth factor 23 levels in pediatric cardiac surgery patients. Pediatr Nephrol. (2016) 31:661–9. doi: 10.1007/s00467-015-3257-5
21. Zech P. Bone biopsies in patients with acute renal failure. In: Proceedings of the International Symposium “The Kidney and Calcium” (Les Trois Epis) (1972).
22. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. (2008) 35:215. doi: 10.1016/j.pop.2008.01.007
23. Fukumoto S. Phosphate metabolism and vitamin D. Bonekey Rep. (2014) 3:497. doi: 10.1038/bonekey.2013.231
24. Underland L, Markowitz M, Gensure R. Calcium and phosphate hormones: vitamin D, parathyroid hormone, and fibroblast growth factor 23 education gap. Pediatrics Rev. (2020) 41:3–11. doi: 10.1542/pir.2018-0065
25. Massry SG, Arieff AI, Coburn JW, Palmieri G, Kleeman CR. Divalent ion metabolism in patients with acute renal failure: studies on the mechanism of hypocalcemia. Kidney Int. (1974) 5:437–445. doi: 10.1038/ki.1974.62
26. Pietrek J, Kokot F, Kuska J. Serum 25-hydroxyvitamin D and parathyroid hormone in patients with acute renal failure. Kidney Int. (1978) 13:178–85. doi: 10.1038/ki.1978.25
27. Llach F, Felsenfeld AJ HM. The pathophysiology of altered calcium metabolism in rhabdomyolysis-induced acute renal failure. N Engl J Med. (1981) 305:117–23.
28. Zhang M, Hsu R, Hsu C, Kordesch K, Nicasio E, Cortez A, et al. FGF-23 and PTH levels in patients with acute kidney injury: a cross-sectional case series study. Ann Intensive Care. (2011) 1:21. doi: 10.1186/2110-5820-1-21
29. Leaf DE, Siew ED, Eisenga MF, Singh K, Mc Causland FR, Srivastava A, et al. Fibroblast growth factor 23 associates with death in critically ill patients. Clin J Am Soc Nephrol. (2018) 13:531–541. doi: 10.2215/CJN.10810917
30. Leaf DE, Wolf M, Waikar SS, Chase H, Christov M, Cremers S, et al. FGF-23 levels in patients with AKI and risk of adverse outcomes. Clin J Am Soc Nephrol. (2012) 7:1217–23. doi: 10.2215/CJN.00550112
31. Leaf DE, Christov M, Jüppner H, Siew E, Ikizler TA, Bian A, et al., Fibroblast growth factor 23 levels are elevated and associated with severe acute kidney injury and death following cardiac surgery. Kidney Int. (2016) 89:939–48. doi: 10.1016/j.kint.2015.12.035
32. Jung SY, Kim H, Park S, Jhee JH, Yun HR, Kim H, et al. Electrolyte and mineral disturbances in septic acute kidney injury patients undergoing continuous renal replacement therapy. Medicine. (2016) 95:e4542. doi: 10.1097/MD.0000000000004542
33. Saour M, Zeroual N, Ridolfo J, Nogue E, Picot M-C, Gaudard P, et al. Serum phosphate kinetics in acute kidney injury after cardiac surgery: an observational study. J Cardiothorac Vasc Anesth. (2020) 34: 2964–72. doi: 10.1053/j.jvca.2020.05.023
34. Burra V, Nagaraja PS, Singh NG, Prabhakar V, Manjunatha N. Early prediction of acute kidney injury using serum phosphorus as a biomarker in pediatric cardiac surgical patients. Ann Card Anaesth. (2018) 21:455–9. doi: 10.4103/aca.ACA_14_18
35. Krieger NS, Sessler NE, Bushinsky DA. Acidosis inhibits osteoblastic and stimulates osteoclastic activity in vitro. Am J Physiol Physiol. (1992) 262(3 Pt 2):F442–8. doi: 10.1152/ajprenal.1992.262.3.F442
36. Scheidegger D, Drop LJ, Schellenberg J-C. Role of the systemic vasculature in the hemodynamic response to changes in plasma ionized calcium. Arch Surg. (1980). 115:206–11. doi: 10.1001/archsurg.1980.01380020072017
37. Szent-Györgyi AG. Calcium regulation of muscle contraction. Biophys J. (1975) 15:707–23. doi: 10.1016/S0006-3495(75)85849-8
38. Rousseau AF, Kerschan-Schindl K, Scherkl M, Amrein K. Bone metabolism and fracture risk during and after critical illness. Curr Opin Crit Care. (2020) 26:379–85. doi: 10.1097/MCC.0000000000000734
39. Cariolou M, Cupp MA, Evangelou E, Tzoulaki I, Berlanga-Taylor AJ. Importance of Vitamin D in acute and critically ill children with subgroup analyses of sepsis and respiratory tract infections: a systematic review and meta-analysis. BMJ Open. (2019) 9:e027666. doi: 10.1136/bmjopen-2018-027666
40. Braun A, Christopher K. Vitamin D in acute kidney injury. Inflamm Allergy-Drug Targets. (2013) 12:262–72. doi: 10.2174/18715281113129990044
41. Braun AB, Litonjua AA, Moromizato T, Gibbons FK, Giovannucci E, Christopher KB. Association of low serum 25-hydroxyvitamin D levels and acute kidney injury in the critically ill. Crit Care Med. (2012) 40:3170–9. doi: 10.1097/CCM.0b013e318260c928
42. Lai L, Qian J, Yang Y, Xie Q, You H, Zhou Y, et al. Is the serum vitamin D level at the time of hospital-acquired acute kidney injury diagnosis associated with prognosis? PLoS ONE. (2013) 8:e64964. doi: 10.1371/journal.pone.0064964
43. Leaf DE, Waikar SS, Wolf M, Cremers S, Bhan I, Stern L. Dysregulated mineral metabolism in patients with acute kidney injury and risk of adverse outcomes. Clin Endocrinol. (2013) 79:491–8. doi: 10.1111/cen.12172
44. Tingting L, Vijayan A, Dusso A, Jain S, Coyne DW. Relationship of 1,25 dihydroxy vitamin D levels to clinical outcomes in critically ill patients with acute kidney injury. J Nephrol Ther. (2014) 5:190. doi: 10.4172/2161-0959.1000190
45. Slatopolsky E, Weerts C, Thielan J, Horst R, Harter H, Martin KJ. Marked suppression of secondary hyperparathyroidism by intravenous administration of 1,25-dihydroxycholecalciferol in uremic patients. J Clin Invest. (1984) 74:2136–43. doi: 10.1172/JCI111639
46. Hsu CH, Patel S, Young EW, Simpson RU. Production and metabolic clearance of calcitriol in acute renal failure. Kidney Int. (1988) 33:530–5. doi: 10.1038/ki.1988.30
47. Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. (2011) 96:53–8. doi: 10.1210/jc.2010-2704
48. Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. (2010) 20:1352–60. doi: 10.1101/gr.107920.110
49. Cameron LK, Lei K, Smith S, Doyle NL, Doyle JF, Flynn K, et al. Vitamin D levels in critically ill patients with acute kidney injury: a protocol for a prospective cohort study (VID-AKI). BMJ Open. (2017) 7:e016486. doi: 10.1136/bmjopen-2017-016486
50. de Bragança AC, Volpini RA, Mehrotra P, Andrade L, Basile DP. Vitamin D deficiency contributes to vascular damage in sustained ischemic acute kidney injury. Physiol Rep. (2016) 4:e12829. doi: 10.14814/phy2.12829
51. Reis NG, Francescato HDC, de Almeida LF, da Silva CGA, Costa RS, Coimbra TM. Protective effect of calcitriol on rhabdomyolysis-induced acute kidney injury in rats. Sci Rep. (2019) 9:7090. doi: 10.1038/s41598-019-43564-1
52. Arfian N, Budiharjo S, Prasetyo Wibisono D, Ananda Wahyu Setyaningsih W, Mansyur Romi M, Laila Afifa An-nur Willya Saputri R, et al. Vitamin D ameliorates kidney ischemia reperfusion injury via reduction of inflammation and myofibroblast expansion. Kobe J Med Sci. (2019) 65:E138–43. doi: 10.24546/81012029
53. Druml W, Schwarzenhofer M, Apsner RHW. Fat-soluble vitamins in patients with acute renal failure. Min Electrolyte Metab. (1998) 24:220–6.
54. Shieh SD, Lin YF, Lin SH LK. A prospective study of calcium metabolism in exertional heat stroke with rhabdomyolysis and acute renal failure. Nephron. (1995) 71:428–32.
55. Terzioglu-Usak S, Elibol B, Dalli T, Guler C, Aysan E. Effect of restraint stress on plasma PTH concentration and its molecular targets expressions in Wistar rats. Int J Endocrinol Metab. (2018) 16:e66979. doi: 10.5812/ijem.66979
56. Zaloga GP, Teres D. Critical illness is associated with elevated parathyroid hormone. Crit Care. (2001) 5:P208. doi: 10.1186/cc1275
57. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. (2013) 75:503–33. doi: 10.1146/annurev-physiol-030212-183727
58. Egli-Spichtig D, Zhang MYH, Perwad F. Fibroblast growth factor 23 expression is increased in multiple organs in mice with folic acid-induced acute kidney injury. Front Physiol. (2018) 9:1494. doi: 10.3389/fphys.2018.01494
59. Neyra JA, Moe OW, Hu MC. Fibroblast growth factor 23 and acute kidney injury. Pediatr Nephrol. (2015) 30:1909–18. doi: 10.1007/s00467-014-3006-1
60. Zaidi M, Yuen T, Sun L, Rosen CJ. Regulation of skeletal homeostasis. Endocr Rev. (2018) 39:701–18. doi: 10.1210/er.2018-00050
61. Christov M. Fibroblast growth factor 23 in acute kidney injury. Curr Opin Nephrol Hypertens. (2014) 23:340–5. doi: 10.1097/01.mnh.0000447021.51722.2f
62. Christov M, Waikar SS, Pereira RC, Havasi A, Leaf DE, Goltzman D, et al. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int. (2013) 84:776–85. doi: 10.1038/ki.2013.150
63. Leaf DE, Jacob KA, Srivastava A, Chen ME, Christov M, Jüppner H, et al. Fibroblast growth factor 23 levels associate with AKI and death in critical illness. J Am Soc Nephrol. (2017) 28:1877–85. doi: 10.1681/ASN.2016080836
64. Christov M, Neyra JA, Gupta S, Leaf DE. Fibroblast growth factor 23 and klotho in AKI. Semin Nephrol. (2019) 39:57–75. doi: 10.1016/j.semnephrol.2018.10.005
65. Fayed A, Radwan WA, Amin M, Gamal A. Prediction of mortality and need for renal replacement therapy in patients of acute kidney injury using fibroblast growth factor 23. Saudi J Kidney Dis Transpl. (2019) 30:1044–51. doi: 10.4103/1319-2442.270259
66. Meurer M, Höcherl K. Renal ischemia-reperfusion injury impairs renal calcium, magnesium, and phosphate handling in mice. Pflugers Arch Eur J Physiol. (2019) 471:901–14. doi: 10.1007/s00424-019-02255-6
67. Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. (2010) 78:975–80. doi: 10.1038/ki.2010.313
68. Kronenberg H. Npt2a–the key to phosphate homeostasis. N Engl J Med. (2002) 347:1022–24. doi: 10.1056/NEJMe020098
69. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. (2004) 19:429–35. doi: 10.1359/JBMR.0301264
70. Hu M-C, Kuro-O M, Moe OW. Klotho and kidney disease. J Nephrol. (2010) 23:(Suppl. 16):S136–44. doi: 10.1159/000346778
71. Castellano G, Intini A, Stasi A, Divella C, Gigante M, Pontrelli P, et al. Complement modulation of anti-aging factor klotho in ischemia/reperfusion injury and delayed graft function. Am J Transplant. (2016) 16:325–33. doi: 10.1111/ajt.13415
72. Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, et al. Alpha-klotho as a regulator of calcium homeostasis. Science. (2007) 316: 1615–8. doi: 10.1126/science.1135901
73. Hu MC, Shi M, Zhang J, Quiones H, Kuro-O M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. (2010) 78:1240–51. doi: 10.1038/ki.2010.328
74. Sugiura H, Yoshida T, Tsuchiya K, Mitobe M, Nishimura S, Shirota S, et al. Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrol Dial Transplant. (2005) 20:2636–45. doi: 10.1093/ndt/gfi165
75. Kim AJ, Ro H, Kim H, Chang JH, Lee HH, Chung W, et al. Klotho and S100A8/A9 as discriminative markers between pre-renal and intrinsic acute kidney injury. PLoS ONE. (2016) 11:e0147255. doi: 10.1371/journal.pone.0147255
76. Seo MY, Yang J, Lee JY, Kim K, Kim SC, Chang H, et al. Renal klotho expression in patients with acute kidney injury is associated with the severity of the injury. Korean J Intern Med. (2015) 30:489–95. doi: 10.3904/kjim.2015.30.4.489
77. Neyra JA, Li X, Mescia F, Ortiz-Soriano V, Adams-Huet B, Pastor J, et al. Urine klotho is lower in critically ill patients with versus without acute kidney injury and associates with major adverse kidney events. Crit Care Explor. (2019) 1:e0016. doi: 10.1097/cce.0000000000000016
78. Seibert E, Radler D, Ulrich C, Hanika S, Fiedler R, Girndt M. Serum klotho levels in acute kidney injury. Clin Nephrol. (2017) 87:173–9. doi: 10.5414/CN108970
79. Ruiz-Andres O, Dolores Sanchez-Niño M, Antonio Moreno J, Ruiz-Ortega M, Mario Ramos A, Belen Sanz A, et al. Downregulation of kidney protective factors by inflammation: role of transcription factors and epigenetic mechanisms. Am J Physiol Ren Physiol. (2016) 311:1329–40. doi: 10.1152/ajprenal.00487.2016
80. Singbartl K, Formeck CL, Kellum JA. Kidney-immune system crosstalk in AKI. Semin Nephrol. (2019) 39:96–106. doi: 10.1016/j.semnephrol.2018.10.007
81. Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol. (2008) 19:547–58. doi: 10.1681/ASN.2007040469
82. Ratliff BB, Rabadi MM, Vasko R, Yasuda K, Goligorsky MS. Messengers without borders: mediators of systemic inflammatory response in AKI. J Am Soc Nephrol. (2013) 24:529–36. doi: 10.1681/ASN.2012060633
83. Rabb H, Griffin MD, McKay DiB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. (2016) 27:371–9. doi: 10.1681/ASN.2015030261
84. Darisipudi MN, Knauf F. An update on the role of the inflammasomes in the pathogenesis of kidney diseases. Pediatr Nephrol. (2016) 31:535–44. doi: 10.1007/s00467-015-3153-z
85. Gilbert L, He X, Farmer P, Rubin J, Drissi H, Van Wijnen AJ, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2αA) is inhibited by tumor necrosis factor-α. J Biol Chem. (2002) 277:2695–701. doi: 10.1074/jbc.M106339200
86. Ahuja SS, Zhao S, Bellido T, Plotkin LI, Jimenez F, Bonewald LF. CD40 ligand blocks apoptosis induced by tumor necrosis factor α, glucocorticoids, and etoposide in osteoblasts and the osteocyte-like cell line murine long bone osteocyte-Y4. Endocrinology. (2003) 144:1761–9. doi: 10.1210/en.2002-221136
87. Glenn DA, Denburg MR. Bone health in glomerular kidney disease. Curr Osteoporos Rep. (2019) 17:570–9. doi: 10.1007/s11914-019-00531-z
88. Hendy GN, Canaff L. Calcium-sensing receptor, proinflammatory cytokines and calcium homeostasis. Semin Cell Dev Biol. (2016) 49:37–43. doi: 10.1016/j.semcdb.2015.11.006
89. Liu N, Nguyen L, Chun RF, Lagishetty V, Ren S, Wu S, et al. Altered endocrine and autocrine metabolism of vitamin D in a mouse model of gastrointestinal inflammation. Endocrinology. (2008) 149:4799–808. doi: 10.1210/en.2008-0060
90. Uno JK, Kolek OI, Hines ER, Xu H, Timmermann BN, Kiela PR, et al. The role of tumor necrosis factor α in down-regulation of osteoblast phex gene expression in experimental murine colitis. Gastroenterology. (2006) 131:497–509. doi: 10.1053/j.gastro.2006.05.020
91. Ensrud KE, Barbour K, Canales MT, Danielson ME, Boudreau RM, Bauer DC, et al. Renal function and nonvertebral fracture risk in multiethnic women: the Women's Health Initiative (WHI). Osteoporos Int. (2012) 23:887–99. doi: 10.1007/s00198-011-1667-1
92. Yuan FL, Li X, Lu WG, Xu RS, Zhao YQ, Li CW, et al. Regulatory T cells as a potent target for controlling bone loss. Biochem Biophys Res Commun. (2010) 402:173–6. doi: 10.1016/j.bbrc.2010.09.120
93. Stagi S, Cavalli L, Iurato C, Seminara S, Brandi ML, De Martino M. Bone metabolism in children and adolescents: main characteristics of the determinants of peak bone mass. Clin Cases Miner Bone Metab. (2013) 10:172–9.
Keywords: AKI, skeleton, parathyroid hormone, fibroblast growth factor 23, klotho, vitamin D, mineral, inflammation
Citation: Hegde A, Denburg MR and Glenn DA (2021) Acute Kidney Injury and Pediatric Bone Health. Front. Pediatr. 8:635628. doi: 10.3389/fped.2020.635628
Received: 30 November 2020; Accepted: 30 December 2020;
Published: 09 February 2021.
Edited by:
Katja Michelle Gist, Children's Hospital Colorado, United StatesReviewed by:
David Selewski, University of South Carolina, United StatesMarissa DeFreitas, University of Miami, United States
Copyright © 2021 Hegde, Denburg and Glenn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anisha Hegde, YW5pc2hhLmhlZ2RlQHVuY2hlYWx0aC51bmMuZWR1; Dorey A. Glenn, ZG9yZXlfZ2xlbm5AbWVkLnVuYy5lZHU=