 Miriam E. Imafidon1
Miriam E. Imafidon1 Birgit Sikkema-Raddatz1Kristin M. Abbott1Martine T. Meems-Veldhuis1Morris A. Swertz1,2K. Joeri van der Velde1,2Gea Beunders1
Birgit Sikkema-Raddatz1Kristin M. Abbott1Martine T. Meems-Veldhuis1Morris A. Swertz1,2K. Joeri van der Velde1,2Gea Beunders1 Dennis K. Bos1
Dennis K. Bos1 Nine V. A. M. Knoers1
Nine V. A. M. Knoers1 Wilhelmina S. Kerstjens-Frederikse1†
Wilhelmina S. Kerstjens-Frederikse1† Cleo C. van Diemen1*†
Cleo C. van Diemen1*†- 1Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, Netherlands
- 2Genomics Coordination Center, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
Background: Genetic disorders are a substantial cause of infant morbidity and mortality and are frequently suspected in neonatal intensive care units. Non-specific clinical presentation or limitations to physical examination can result in a plethora of genetic testing techniques, without clear strategies on test ordering. Here, we review our 2-years experiences of rapid genetic testing of NICU patients in order to provide such recommendations.
Methods: We retrospectively included all patients admitted to the NICU who received clinical genetic consultation and genetic testing in our University hospital. We documented reasons for referral for genetic consultation, presenting phenotypes, differential diagnoses, genetic testing requested and their outcomes, as well as the consequences of each (rapid) genetic diagnostic approach. We calculated diagnostic yield and turnaround times (TATs).
Results: Of 171 included infants that received genetic consultation 140 underwent genetic testing. As a result of testing as first tier, 13/14 patients received a genetic diagnosis from QF-PCR; 14/115 from SNP-array; 12/89 from NGS testing, of whom 4/46 were diagnosed with a small gene panel and 8/43 with a large OMIM-morbid based gene panel. Subsequent secondary or tertiary analysis and/or additional testing resulted in five more diagnoses. TATs ranged from 1 day (QF-PCR) to a median of 14 for NGS and SNP-array testing, with increasing TAT in particular when many consecutive tests were performed. Incidental findings were detected in 5/140 tested patients (3.6%).
Conclusion: We recommend implementing a broad NGS gene panel in combination with CNV calling as the first tier of genetic testing for NICU patients given the often unspecific phenotypes of ill infants and the high yield of this large panel.
Introduction
Approximately 1 in 47 children in the Netherlands is born with one or more congenital malformations, and roughly 25% of these congenital malformations are caused by genetic aberrations (1). A number of these children will require admittance to the neonatal intensive care unit (NICU), where clinical diagnostics may be complicated by a non-specific clinical presentation or limitations to physical examination, especially in the setting of critical clinical conditions. The differential diagnosis process in the NICU is thus often time-consuming, may be burdensome for the patient, and may be ineffective in reaching a genetic diagnosis. The classical genetic diagnostics workflow included molecular and cytogenetic testing to identify aneuploidies and copy number variations and, to a lesser extent, single gene analysis in case of a clear clinical diagnosis. We and others have previously shown that adding rapid next generation sequencing (rNGS) for detection of multiple monogenic diseases adds to the diagnostic yield and lowers the turnaround times (TATs) for diagnoses, thereby improving care for these children and their parents (2–4).
When implementing rNGS, a genetic diagnostic laboratory has to choose a sequencing approach, which can range from small targeted sequencing gene panels, to exome sequencing (ES) using a “virtual” gene panel filter, to whole genome sequencing (WGS) without extensive filtering. Each of these approaches has its benefits and its limitations and a different impact on the laboratory process. While targeted gene panels that investigate a relatively small number of genes can provide a fast TAT, these panels are limited in their range of diagnoses and they cannot be altered easily. On the other side of the spectrum, ES or WGS without stringent filtering may result in a higher diagnostic yield, but their detection of a far larger number of variants generally requires a longer stage of variant interpretation and will result in more incidental findings (5).
In our laboratory at the University Medical Centre Groningen (UMCG), we have now implemented rNGS for NICU patients in two approaches based on parent-child trio ES: the clinical geneticist can request either (1) a dedicated virtual gene panel based on the specific phenotype of the patient, or (2) a broad gene panel (OMIM morbid gene panel) that includes most disease-associated OMIM genes (6) when the patient's phenotype is unspecific and/or unclear. Both approaches can be requested in parallel with SNP-array for copy number variation (CNV) analysis (7), with or without prior QF-PCR for aneuploidy detection. To review and possibly revisit the strategies used in our laboratory, we retrospectively analyzed all the techniques requested for all the patients who received a consultation by a clinical geneticist in the NICU of our University hospital for a period of 2 years. Here, we describe the techniques and approaches chosen, TATs, yields and clinical implications and make recommendations for efficient implementation of rNGS. In addition, we will describe developments that have helped fine-tune our workflows protocols and improve TATs.
Materials and Methods
Study Design and Inclusion of Patients
We retrospectively included all patients admitted to the NICU who received clinical genetic consultation and genetic testing in the University Medical Center Groningen (UMCG) between December 2017 and January 2020. Patient records and clinical notes in electronic health records for each patient were accessed to document the reasons for referral for genetic consultation, presenting phenotypes, differential diagnoses, genetic testing requested by the clinical geneticist, reports with results of genetic testing, outcome of cases and post-test counseling information. The data collected were stored on a secure server within the UMCG, and the information was only accessible by those involved in this study. All data were analyzed anonymously. All parents gave informed consent for the gathering of data for research purposes, and the medical ethical board of the UMCG approved the study protocol (waiver number M20.258501).
Genetic Counseling
Genetic consultations were requested by NICU physicians when a genetic cause was suspected for the congenital malformations and/or abnormalities seen in the patient during admittance. After in-depth physical examination of the patient, parents were counseled about genetic testing in general, risk of incidental findings, risk of uncertain or unclear findings (VUSs) and about the recommended tests for each case. Actionable incidental findings, as defined by the ACMG (8), were reported to the parents. However, parents could opt out of genetic diagnostics at any time and were given the option to not be notified in case of incidental findings. If genetic testing was not deemed to be informative for the observed congenital abnormality, no testing took place and the patient was not included in this study.
Genetic Diagnostics Workflow
DNA was extracted from peripheral blood of the child and parents using standard diagnostic procedures. QF-PCR (Devyser Complete v2; Devyser) was performed according to standard protocol for trisomies 13, 18, 21, and the X and Y chromosomes (maximum 4 days TAT). Depending on the diagnostic request, routine-care SNP-array (using Global Screening Array (GSA) on an Illumina iSCAN) and ES were either done sequentially, or in parallel, in a patient-parent trio design (maximum 28 days TAT). ES was performed using SureSelect Human All Exon V6 (Agilent, USA) target enrichment, according to standard procedures, on Bravo automated liquid handling robots (Agilent), and then sequenced on an Illumina NextSeq500 sequencer. For reliable results the aim is 20× coverage for 95% of the target genes.
Requested virtual NGS gene panels ranged from phenotype based (PB, ranging from 5 to 329 genes) to a large panel based on all OMIM morbid genes (~4,200 genes); all gene panels used are listed in Supplementary Table 1. In addition to our rNGS diagnostic workflow, a trio-based open-exome analysis of all protein coding genes (~23,000 genes), similar to whole exome sequencing, could also be requested for cases without a diagnosis after rNGS. This test is not offered as rNGS as the interpretation of variants prolongs the TAT.
Data Analysis
SNP array data was processed in NxClinical/Nexus software (BioDiscovery, Inc., USA). In general, copy number deletions >50 kb containing an OMIM morbid gene, deletions >150 kb, duplications >200 kb and homozygous regions of at least 10 Mb were reported. CNVs seen in 10% or more of our in-house database of healthy controls were considered common (9).
Raw ES data (VCF file) was processed according to standardized protocols, as described previously (10). Sequence variants (single nucleotide variants (SNVs) and small indels) were automatically filtered using Alissa Interpret software (Agilent Technologies, Inc., USA) with the requested virtual gene panel. In general, the filter steps in Alissa included in-house, national and international databases of previously classified variants, the GAVIN (Gene-Aware Variant Interpretation) prioritization tool (11), minor allele frequencies from GnomAD, the disease mode of inheritance reported in OMIM, the Human Gene Mutation Database (HGMD) and OMIM-reported modes of inheritance using MOLGENIS (version 1.4.0) (11–15). Variants that remained after these filtering steps were evaluated by the operating technician, a genetic laboratory specialist and a clinical geneticist in matching the phenotype. A detailed description of the variant-filtering process was published previously (10).
The optional trio-based open-exome analysis examines all protein coding genes in the exome and is performed with an additional filtering step based on a variant's mode of inheritance in the patient (i.e., compound heterozygous variants inherited one from each of the parents). Once filtered, all candidate genes are examined for human disease associations in OMIM and HGMD, and in the citation database of PubMed (NCBI, USA). The literature was also checked for information on relevant animal models.
Reporting of Variants
All variants that might explain the patient phenotype, and all incidental findings were classified using Alamut software (Interactive BioSoftware/SophiaGenetics) according to standardized guidelines based on Richards et al., while taking into account the HGMD, CADD score and population frequency (9, 16, 17). Variants classified as pathogenic and likely pathogenic were always communicated to parents (8). Variants of unknown significance (VUSs) were communicated after a multidisciplinary consultation where the clinical geneticist deemed the gene with the VUS matched (part of) the phenotype and the laboratory specialist found sufficient evidence for possible pathogenicity of the variant (i.e., allele frequency, conservation, de novo).
The incidental findings we reported fell into two categories: (1) (likely) pathogenic variants/CNVs predicted to cause an actionable disease, knowledge of which could lead to health benefits for the child and/or parents (8); (2) (likely) pathogenic variants in genes/CNVs associated with diseases unrelated to the phenotype, for which currently no therapy is available (i.e., developmental delay and/or intellectual disability).
Study Outcomes
We measured diagnostic yield, TATs and the clinical consequences of the (rapid) genetic diagnostic approach. We distinguished diagnostic yield into three categories:
- Genetic diagnosis: genetic test result explains (most of) the phenotype
- Potential genetic diagnosis: reported VUS
- No diagnosis: no genetic cause was identified by genetic testing.
TAT was measured as the number of days between the first genetic consultation by the physician and the definitive report of genetic diagnostic results sent to the parents by the physician.
Results
Inclusion and Characteristics of Patients
Between December 2017 and January 2020, 171 infants received genetic consultation and 140 of these underwent genetic testing. Patients were retrospectively included in this study (see Table 1 for baseline characteristics). Most patients (73%) were admitted on first day of life (range 0–54 days) and most of the patients (98%) who were not admitted on the day of birth, had been admitted to a regional hospital before being transferred to the NICU of the UMCG. Of the 171 included patients, 26 have died during the 2-years study period: 21 patients died in the NICU and five patients died after discharge. The age at death ranged from 1 to 335 days, with a median of 22 days.
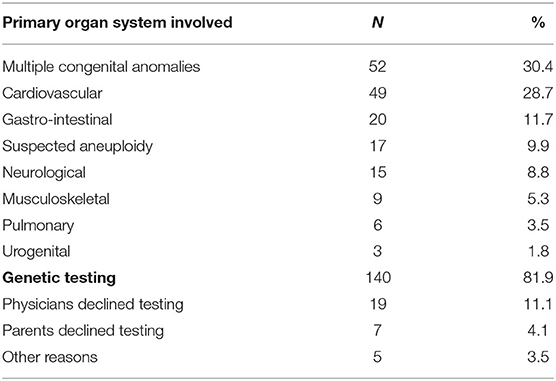
Table 1. Characteristics of 171 included patients.
The most frequent reasons for referral for genetic consultation were the presence of multiple congenital anomalies involving at least two organ systems (30%, 52/171), congenital cardiovascular anomalies (29%, 49/171), followed by gastro-intestinal anomalies (12%, 20/171). The request for genetic consultation was made at a median age of 1 day of life (range 0–70 days), most often the day after admittance. For 19 patients, selected genetic testing had taken place prenatally. After birth, a second genetic consultation was requested if this was warranted by the postnatal phenotype of the patient. A total of 31 cases were excluded, because patients did not have genetic testing after consultation for the following reasons: (i) the parents declined testing, most commonly stating that genetic testing would not have an added value to them (n = 7); (ii) the clinical geneticists deemed genetic testing to be unnecessary as the condition was not likely to be caused by a genetic aberration (n = 19); (iii) the patient was transferred to a different University Medical Center (n = 3); (iv) a confirmed antenatal diagnosis (n = 1); and (v) insurance problems (n = 1) (see Table 1).
Diagnostic Workflow: Phenotype-Based Sequencing vs. OMIM Morbid Gene Panel
A total of 34 patients were offered SNP-array and the OMIM morbid gene panel as first tier diagnostic tests, because of the patient's complex phenotype. Nine out of these 29 patients were diagnosed: seven were diagnosed via the OMIM morbid gene panel and two showed a chromosomal abnormality with SNP-array. One patient was diagnosed after further testing, i.e., by analysis of the DMPK gene, associated with myotonic dystrophy, performed outside of the UMCG (a trinucleotide repeat as causal variant are not detected by ES). The median turnaround time till genetic diagnosis was 28 days (range 12–63 days) (Table 2). After OMIM morbid/SNP-array as first tier, 22 patients remained undiagnosed. Ten patients that could not be diagnosed by the OMIM morbid gene panel were offered open-exome analysis, but none were diagnosed with this technique (Figure 1). The 22 genetically undiagnosed patients had a median of three requested genetic tests (range 2–5). It took a median of 82 days (range 16–582 days) to complete all requested genetic tests.
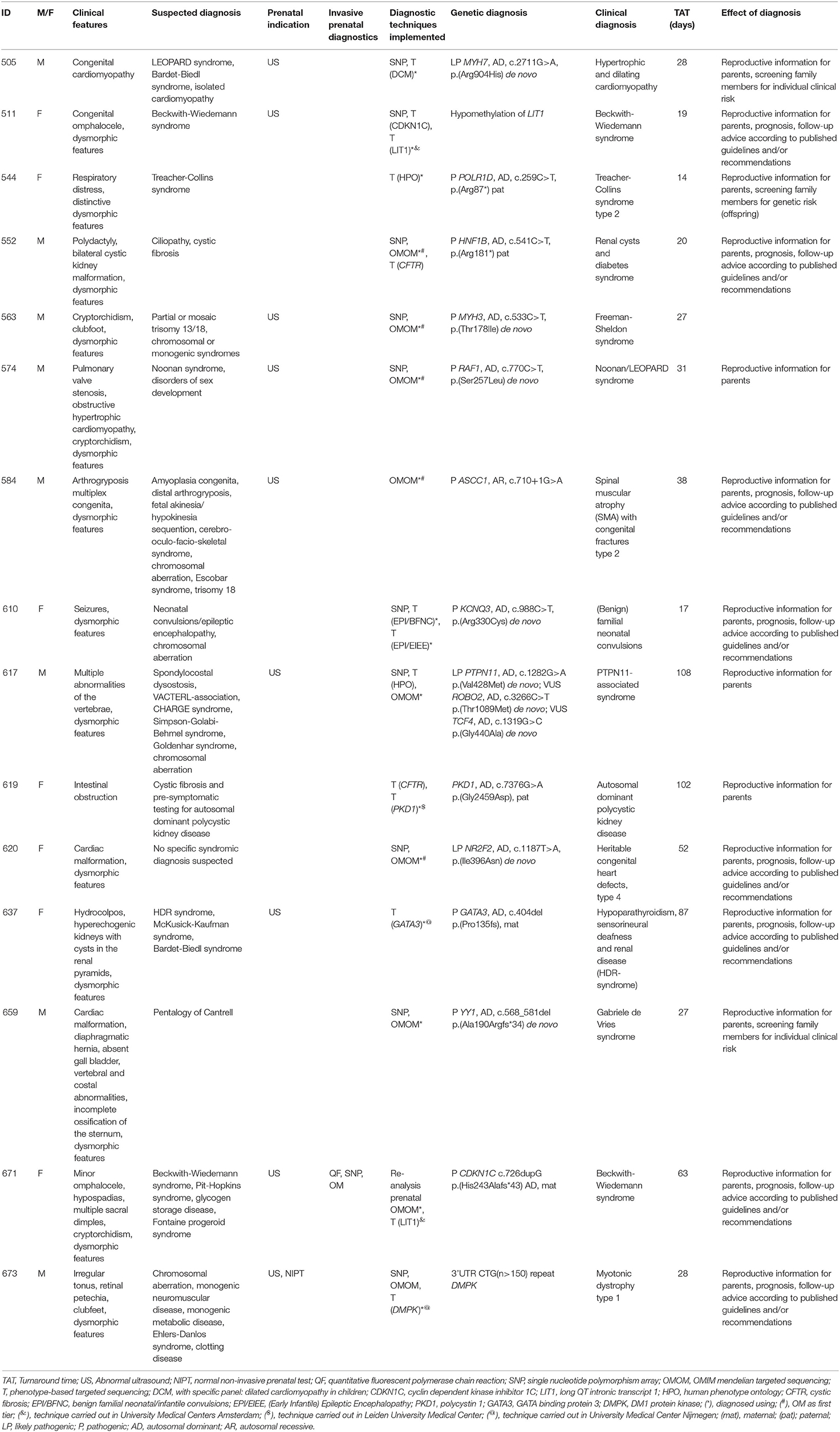
Table 2. Diagnoses made with Next Generation Sequencing based tests.
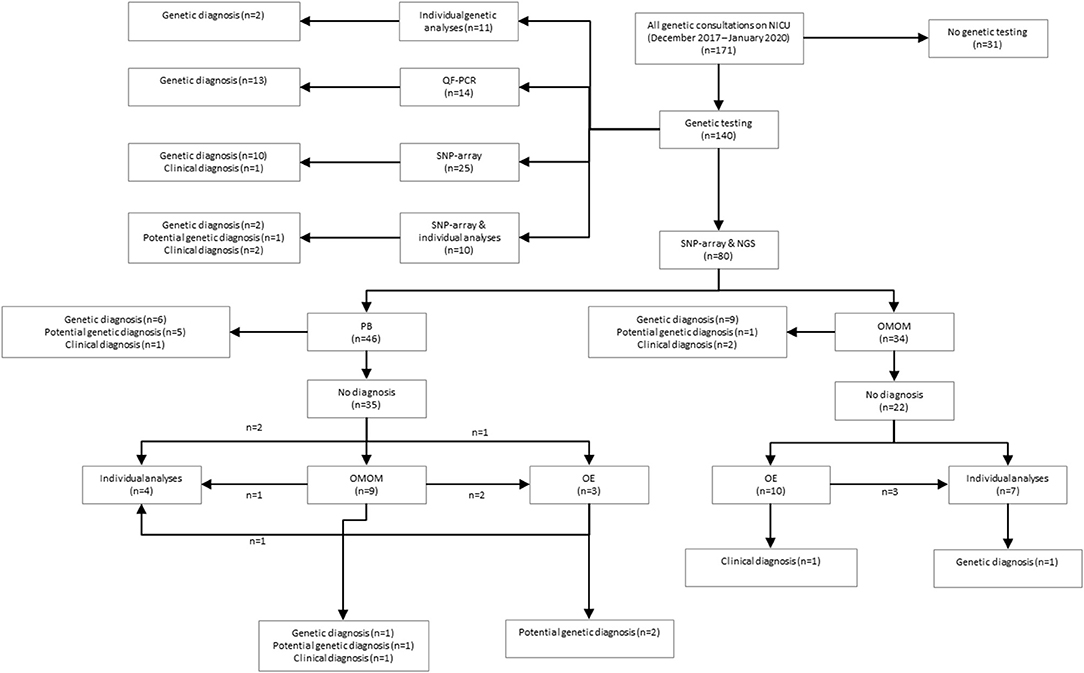
Figure 1. Schematic overview of study results. QF, quantitative fluorescent polymerase chain reaction; SNP, single nucleotide polymorphism array; NGS, next-generation sequencing; PB, phenotype-based targeted sequencing, OMOM, OMIM mendelian targeted sequencing; OE, open-exome analysis.
A total of 46 patients were offered SNP-array and PB sequencing as first tier diagnostic techniques. Of these 46 patients, six were diagnosed: five were diagnosed using PB sequencing and one showed a chromosomal abnormality with SNP-array. For these six patients with a genetic diagnosis, a median of three (range two to four) genetic tests were requested. The median total turnaround time for these patients was 21 days (range 1–32 days) (Table 2). After PB sequencing/SNP-array as first tier, 35 patients remained undiagnosed. Of these, 26 patients had no further genetic testing, in 21 because the probable clinical diagnosis was a non-syndromic congenital heart defect and in five because features were concluded to be non-genetic (for instance post-ischemic or infectious). Nine patients who could not be diagnosed with this PB sequencing were offered the OMIM morbid gene panel, and of those nine, two patients were subsequently offered open-exome analysis. One patient was offered open-exome analysis immediately after PB sequencing. However, only one patient was diagnosed with one of these additional techniques (Figure 1). The 35 genetically undiagnosed patients had a median of two requested genetic tests (range 1–6). It took a median of 114 days (range 19–320 days) to complete all requested genetic testing.
To illustrate how TATs can become long, we describe one case in which subsequent phenotype-based analyses were requested. Patient 566 was born in a general hospital after an otherwise uncomplicated pregnancy. After birth, the patient was transferred to the UMCG, because of multiple congenital abnormalities. Genetic consultation was requested on the day of admittance to the NICU. Several clinical findings were reported, such as radial aplasia, multiple anomalies of the vertebra, esophagus atresia with fistula and dysmorphic features. Based on the clinical features, several syndromic causes were considered, including CHARGE syndrome, TAR syndrome, VACTERL-association, Nager syndrome and Treacher-Collins syndrome. SNP-array and the OMIM morbid gene panel tests were negative after 9 and 29 days, respectively. Open-exome analysis was requested for this patient and, after 126 days, no genetic cause could be detected. During this time it was also revealed that the patient suffered from an ASD/VSD. After this additional information it was concluded that the most probable diagnosis was VACTERL-association. The case was closed after 167 days.
Diagnostic Yield
The overall yield of genetic testing in our cohort was 31% (44/140). QF-PCR and SNP array provided a diagnosis in 13/14 (93%) and 14/115 (12%) patients, respectively (Table 3). Phenotype-based sequencing provided a diagnosis in 4/46 (9%) patients, and the broader OMIM morbid gene panel provided a diagnosis in 9/43 (21%). Five patients (16%) were diagnosed after analysis of an individual gene such as LIT1 and GATA3 (Figure 1). After QF-PCR 10 patients were diagnosed with Down syndrome, two patients with Edwards syndrome and one with Patau syndrome.
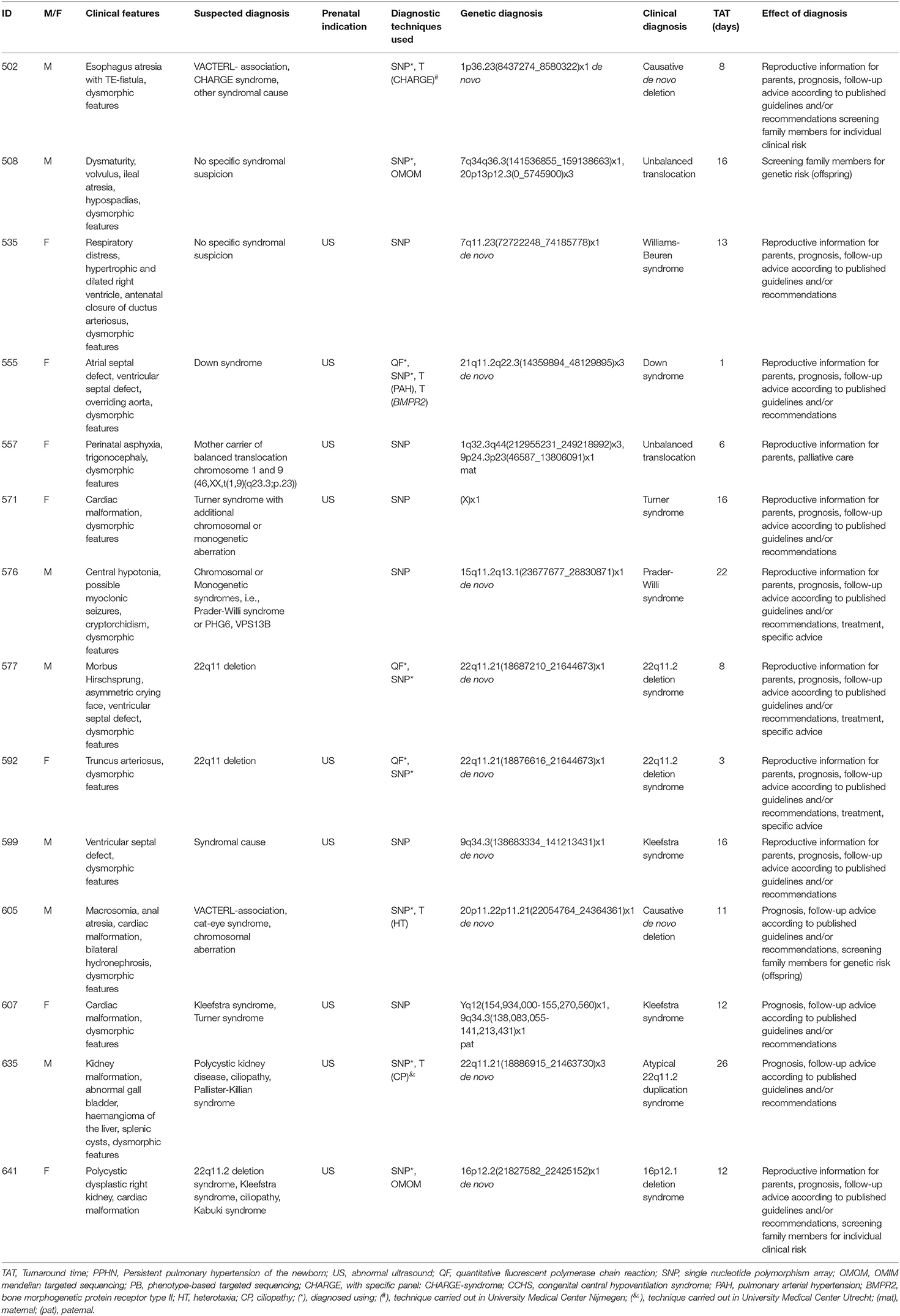
Table 3. Diagnoses made with SNP-array.
In addition to (likely) pathogenic variants, to ten patients, one or multiple VUS(s) were reported as the potential genetic diagnosis. Including these potential genetic diagnoses, the diagnostic yield was 54/140 (39%). VUSs were reported after PB sequencing to five patients, two after the OMIM morbid gene panel, two after open-exome analysis and one after exome sequencing in another academic center (Table 4).
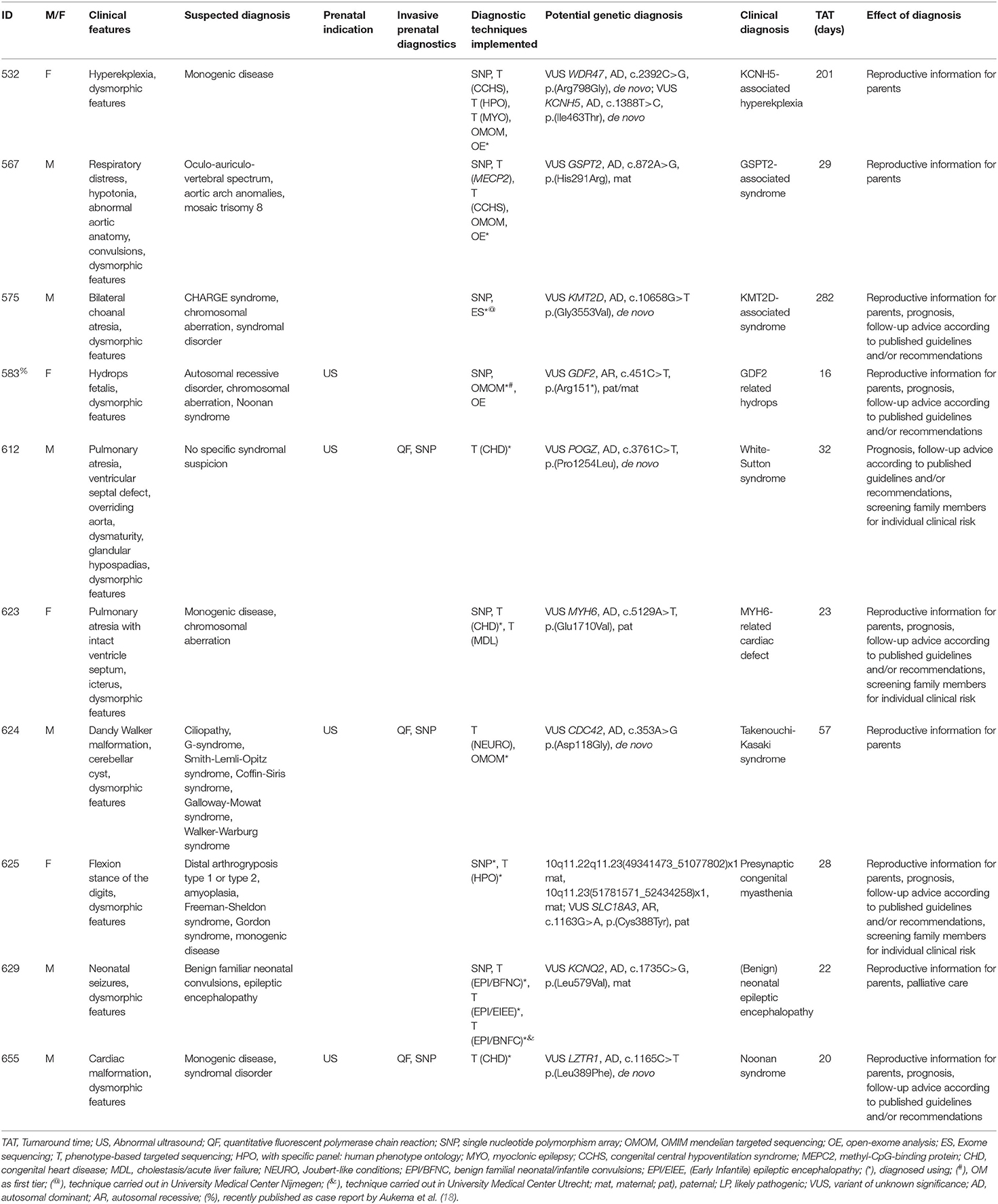
Table 4. Reported variants of unknown significance.
Incidental Findings
Incidental findings (IFs) were found in six out of 140 tested patients (4%). Of these, three were identified in 43 OMIM morbid panel analyses (7%) and three in 115 SNP-arrays (3%). Two of the IF's in OMIM morbid gene panels were actionable IFs (MYH7, PSKH9) in accordance with the ACMG (8). No IF's were found with QF, PB sequencing, open-exome analysis or individual gene analysis (Table 5).

Table 5. Reported incidental findings.
Discussion
The results of our retrospective study demonstrate that a broad gene panel including all OMIM Mendelian genes is the favorable exome sequencing approach in rapid genetic diagnostics in neonatal intensive care. This broad OMIM morbid gene panel resulted in a higher diagnostic yield than a dedicated phenotype-based gene panel, namely a 21 vs. 9% yield. The TATs for both as primary test were comparable. The most probable explanation for the low diagnostic yield using a targeted panel is that we are not as good as we like to believe in making clinical diagnoses in this group of patients: most patients in the NICU have complex phenotypes, which are not easily recognized early after birth, a problem which is aggravated by the limitations of physical examination in a critical care setting.
The overall diagnostic yield of genetic testing in our cohort was 31%, but this differed per test. CNV detection using SNP-array and chromosome number anomaly detection using QF-PCR resulted in the most diagnoses, 14 and 13, respectively (32 and 30% of the diagnostic yield, respectively), followed by the broad OMIM morbid gene panel, with nine diagnoses (20% of the diagnostic yield). Comparison with other studies is difficult because the inclusion criteria, numbers of patients and data analyses are different in each study. A large cohort of 129 patients showed 20% diagnostic yield, regardless of rapid ES or rapid WGS, and found 25 SNV/indel (13%), 10 CNV (6%), and two aneuploidy (1%) diagnoses (4). In the RAPIDOMICS approach (19), which included 25 patients, the diagnostic yield was 72% using trio analyses and an OMIM based gene panel. In a comparable study, the diagnostic yield was 50% in 20 patients (20). A multi-center based Australian study included 108 neonatal and pediatric ICU patients and achieved a diagnostic yield of 51% (21). Another Dutch study reported an overall genetic diagnostic yield of 24.2% in 132 patients from the NICU/PICU/medium neonatal care units; of these 132 patients, 31 patients had an exome sequencing test with a yield of 17% (22). In general, the diagnostic yield of rNGS in NICU care is strongly dependent on inclusion criteria and genetic tests performed before.
In addition to (likely) pathogenic variants, VUSs were reported to patients in 10 cases. These are variants in genes that match the phenotypic features of the patient, but for which there is not enough current evidence to classify them as likely pathogenic. Reporting these strongly suggestive variants allows for follow-up studies. Looking for other patients in Genematcher (23), segregation studies, or functional studies might help to re-classify such VUSs.
Based on our results, we suggest QF-PCR should be performed as first tier only when phenotypic features are strongly suggestive of trisomy 13, 18, or 21, and no NIPT has been performed during pregnancy, as QF-PCR is very fast and cheap. Exome sequencing should be performed with a broad gene panel for SNV and CNV detection or a separate array as CNV test. Because of the limited yield with single gene testing or a dedicated PB virtual panel, these tests are less preferable with the patient population present on the NICU. This is supported by the large number of patients (n = 9; 26%) where a targeted panel had to be followed up by the OMIM morbid gene panel. The ones that did not proceed to further testing were mainly patients with single organ anomalies (for instance congenital heart malformations). Dillon et al. even reported that using a panel may lead to missing diagnoses: in their study 23% of exome sequencing-diagnosed children would not have been diagnosed had a panel been selected (24). Xue et al. published a genetic testing algorithm for Mendelian disorders in which they distinguish between distinctive clinical features followed by single gene/panel testing vs. multiple non-specific concerns followed by array and exome sequencing trio testing (25), and our diagnostic yield per test supports this suggested workflow. However, Xue et al. do not discuss a large OMIM morbid gene panel as an intermediate between PB and exome sequencing (in which all ~23,000 genes are assessed). Moreover, it is important to keep in mind that it might not be as easy to define distinct clinical features in the special group of NICU patients.
A straightforward approach of starting with a broad gene panel, not biased by the phenotype interpretation, can reduce the total TAT because it makes sequential testing obsolete. Although the TAT for the targeted small PB panel approach and the broad OMIM morbid gene panel approach as primary test was comparable (14 days on average), the TAT for a diagnosis was much longer for the group of patients with a PB panel as initial test, due to required further testing. Because a genetic diagnosis was made in only four cases in this scenario, and other PB panels and/or an OMIM morbid gene panel were subsequently requested. This meant that total TAT until diagnosis for these cases was a median of 114 days, on average, and up to 320 days. Focusing on one OMIM morbid gene panel as primary test allows for further automation in sample handling and data interpretation, which can further reduce TATs. In our laboratory, we incorporated additional filtering based on a variant's mode of inheritance in the patient into our OMIM morbid gene panel filtering strategy, which reduced interpretation time, thereby reducing the number of variants for interpretation.
A drawback of testing a large set of genes might be the increased chance of incidental findings (IFs). In our study we found eight IFs in total, of which five were found with the OMIM morbid gene panel and three were found with SNP-array. In accordance with current Dutch guidelines for IFs, carriage of a (likely) pathogenic variant in an autosomal recessive gene are not reported. In the pretest counseling, parents are informed about IFs. Opting out for IFs associated with diseases with therapeutic options during childhood is not offered to parents. Given the higher number of genetic diagnoses using the OMIM morbid gene panel, and the only slightly increased number of actionable IFs, we think testing using a broad gene panel in NICU patients is justified. However, strategies for data filtering should be focused on not detecting IFs, for instance by filtering late onset diseases in the setting of diagnostics for infants.
The benefit of an extra open-exome approach is that it allows for the detection of novel genes and for associating known genes with complex phenotypes. However, this approach depends on the diligence of all clinical staff in keeping up with publications and contacting colleagues with patients with similar phenotypes or with variants in the same genes (data-sharing), and this is an investment that many diagnostic laboratories are not able to make. To alleviate this problem, the latest generation of bioinformatics tools may offer complementary approaches to identify potentially causal variants located outside of currently known disease genes. For instance, a prediction of clinical relevance may be assigned to unknown genes using algorithms that perform gene prioritization based on patient symptoms, provided that the tool of choice covers at least a majority of all expressed genes in the genome (26). To discover the most promising variants within these prioritized unknown genes, the latest genome-wide variant pathogenicity estimates (27) could be utilized in combination with gene-specific probabilities of intolerance to loss-of-function and missense mutations (12). Labor-intensive functional follow ups and contacting of colleagues can then be focused on only the most promising candidates, directing precious resources to those variants of which the chances of reaching a final molecular diagnosis are the highest.
In conclusion, analyzing a broad gene panel in parallel with CNV testing, ideally within one test, as the first tier of genetic testing for NICU patients is the best choice to identify the genetic cause of their condition quickly. However, data filtering strategies are necessary to minimize the chance of detecting IFs as much as possible, and informed consent of the parents and a clear policy/protocol how to handle IFs are obligatory. To shorten TATs, effort should be put into further automation of laboratory procedures and data interpretation. Careful description of phenotypic features and monitoring and reporting of VUSs that likely explain the phenotype are essential to pinpoint to novel disease-causing genes, while data sharing, segregation analyses and functional testing now need priority to re-classify these VUSs.
Data Availability Statement
The data analyzed in this study is subject to the following licenses/restrictions: The dataset is generated in a hospital setting as diagnostics. The patient data are not allowed to leave the hospital's secured server environment. Requests to access these datasets should be directed to Cleo C. van Diemen, Yy5jLnZhbi5kaWVtZW5AdW1jZy5ubA==.
Ethics Statement
The studies involving human participants were reviewed and approved by Medisch Ethische Toetsingscommissie University Medical Center Groningen. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
MI retrieved the clinical and genetic data, performed the data analysis, and drafted the manuscript. BS-R, WK-F, and CD contributed to the conception and design of the research, contributed to interpretation of the data, and co-wrote the manuscript. KA, MM-V, MS, KV, GB, DB, and NK contributed to clinical and genetic testing data collection, analysis, and interpretation of the data. All authors critically revised the manuscript and read and approved the final manuscript.
Funding
This project has received funding from the Netherlands Organization for Scientific Research NWO under VIDI grant number 917.164.455. In addition, we acknowledge support from the European Union's Horizon 2020 research and innovation program under grant agreement No. 779257 (Solve-RD) and 825575 (European Joint Program on Rare Disease).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the departments of Pediatrics and Neonatology of the UMCG and the Genomics Coordination Center, department of Genetics, UMCG for their collaboration and expertise. We would like to thank Kate McIntyre for editorial assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.600556/full#supplementary-material
References
1. EUROCAT. Prevalence Charts and Tables. Available online at: https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence_en (accessed December 10, 2019).
2. van Diemen CC, Kerstjens-Frederikse WS, Bergman KA, de Koning TJ, Sikkema-Raddatz B, van der Velde JK, et al. Rapid targeted genomics in critically ill newborns. Pediatrics. (2017) 140:e20162854. doi: 10.1542/peds.2016-2854
3. Ceyhan-Birsoy O, Murry JB, Machini K, Lebo MS, Yu TW, Fayer S, et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the babyseq project. Am J Hum Genet. (2019) 104:76–93. doi: 10.1016/j.ajhg.2018.11.016
4. Kingsmore SF, Cakici JA, Clark MM, Gaughran M, Feddock M, Batalov S, et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am J Hum Genet. (2019) 105:719–33. doi: 10.1016/j.ajhg.2019.08.009
5. Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet. (2016) 135:359–62. doi: 10.1007/s00439-015-1631-9
6. Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. (2019) 47:D1038–d43. doi: 10.1093/nar/gky1151
7. Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci USA. (2015) 112:5473–8. doi: 10.1073/pnas.1418631112
8. Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. (2017) 19:249–55. doi: 10.1038/gim.2016.190
9. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. (2017) 136:665–77. doi: 10.1007/s00439-017-1779-6
10. Corsten-Janssen N, Bouman K, Diphoorn JCD, Scheper AJ, Kinds R, El Mecky J, et al. A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound. Prenat Diagn. (2020) 40:1300–9. doi: 10.1002/pd.5781
11. van der Velde KJ, de Boer EN, van Diemen CC, Sikkema-Raddatz B, Abbott KM, Knopperts A, et al. GAVIN: gene-Aware Variant INterpretation for medical sequencing. Genome Biol. (2017) 18:6. doi: 10.1186/s13059-016-1141-7
12. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1530/ey.17.14.3
13. Swertz MA, Dijkstra M, Adamusiak T, van der Velde JK, Kanterakis A, Roos ET, et al. The MOLGENIS toolkit: rapid prototyping of biosoftware at the push of a button. BMC Bioinformatics. (2010) 11 (Suppl. 12):S12. doi: 10.1186/1471-2105-11-S12-S12
14. University of Washington H-AIfBaBIoH. CADD Scores (2013). Available online at: http://cadd.gs.washington.edu/download
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–d94. doi: 10.1093/nar/gky1016
18. Aukema SM, Ten Brinke GA, Timens W, Vos YJ, Accord RE, Kraft KE, et al. A homozygous variant in growth and differentiation factor 2 (GDF2) may cause lymphatic dysplasia with hydrothorax and nonimmune hydrops fetalis. Am J Med Genet A. (2020) 182:2152–60. doi: 10.1002/ajmg.a.61743
19. Elliott AM, du Souich C, Lehman A, Guella I, Evans DM, Candido T, et al. RAPIDOMICS: rapid genome-wide sequencing in a neonatal intensive care unit-successes and challenges. Eur J Pediatr. (2019) 178:1207–18. doi: 10.1007/s00431-019-03399-4
20. Brunelli L, Jenkins SM, Gudgeon JM, Bleyl SB, Miller CE, Tvrdik T, et al. Targeted gene panel sequencing for the rapid diagnosis of acutely ill infants. Mol Genet Genomic Med. (2019) 7:e00796. doi: 10.1002/mgg3.796
21. Lunke S, Eggers S, Wilson M, Patel C, Barnett CP, Pinner J, et al. Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the australian public health care system. Jama. (2020) 323:2503−11. doi: 10.1001/jama.2020.7671
22. van der Sluijs PJ, Aten E, Barge-Schaapveld DQCM, Bijlsma EK, Bökenkamp-Gramann R, Donker Kaat L, et al. Putting genome-wide sequencing in neonates into perspective. Genet Med. (2019) 21:1074–82. doi: 10.1038/s41436-018-0293-0
23. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. (2015) 36:928–30. doi: 10.1002/humu.22844
24. Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Melbourne Genomics Health Alliance, et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. (2018) 26:644–51. doi: 10.1038/s41431-018-0099-1
25. Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med. (2015) 17:444–51. doi: 10.1038/gim.2014.122
26. van der Velde KJ, van den Hoek S, van Dijk F, Hendriksen D, van Diemen CC, Johansson LF, et al. A pipeline-friendly software tool for genome diagnostics to prioritize genes by matching patient symptoms to literature. Adv Genet. (2020) 1:e10023. doi: 10.1002/ggn2.10023
Keywords: neonatal intensive care (unit), next generation sequencing, genetic diagnostics, copy number variation, genetic disease
Citation: Imafidon ME, Sikkema-Raddatz B, Abbott KM, Meems-Veldhuis MT, Swertz MA, van der Velde KJ, Beunders G, Bos DK, Knoers NVAM, Kerstjens-Frederikse WS and van Diemen CC (2021) Strategies in Rapid Genetic Diagnostics of Critically Ill Children: Experiences From a Dutch University Hospital. Front. Pediatr. 9:600556. doi: 10.3389/fped.2021.600556
Received: 30 August 2020; Accepted: 29 April 2021;
Published: 31 May 2021.
Edited by:
Pankaj Agrawal, Harvard University, United StatesReviewed by:
Linda De Vries, Leiden University Medical Center, NetherlandsZornitza Stark, Royal Children's Hospital, Australia
Copyright © 2021 Imafidon, Sikkema-Raddatz, Abbott, Meems-Veldhuis, Swertz, van der Velde, Beunders, Bos, Knoers, Kerstjens-Frederikse and van Diemen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cleo C. van Diemen, Yy5jLnZhbi5kaWVtZW5AdW1jZy5ubA==
†These authors have contributed equally to this work