 Nathan Grant1
Nathan Grant1 J. Michael Taylor2Zach Plummer1Kasiani Myers3Thomas Burrow4Lori Luchtman-Jones3Anna Byars2Adrienne Hammill3Katie Wusick3
J. Michael Taylor2Zach Plummer1Kasiani Myers3Thomas Burrow4Lori Luchtman-Jones3Anna Byars2Adrienne Hammill3Katie Wusick3 Edward Smith5James Leach6
Edward Smith5James Leach6 Sudhakar Vadivelu1,6*
Sudhakar Vadivelu1,6*- 1Division of Pediatric Neurosurgery, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 2Division of Neurology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 3Division of Hematology – Oncology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 4Division of Human Genetics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 5Department of Neurosurgery, Boston Children's Hospital, Boston, MA, United States
- 6Department of Radiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
Mucopolysaccharidosis (MPS) type I is a rare lysosomal storage disorder caused by an accumulation of glycosaminoglycans (GAGs) resulting in multisystem disease. Neurological morbidity includes hydrocephalus, spinal cord compression, and cognitive decline. While many neurological symptoms have been described, stroke is not a widely-recognized manifestation of MPS I. Accordingly, patients with MPS I are not routinely evaluated for stroke, and there are no guidelines for managing stroke in patients with this disease. We report the case of a child diagnosed with MPS I who presented with overt stroke and repeated neurological symptoms with imaging findings for severe ventriculomegaly, infarction, and bilateral terminal carotid artery stenosis. Direct intracranial pressure evaluation proved negative for hydrocephalus. The patient was subsequently treated with cerebral revascularization and at a 3-year follow-up, the patient reported no further neurological events or new ischemia on cerebral imaging. Cerebral arteriopathy in patients with MPS I may be associated with GAG accumulation within the cerebrovascular system and may predispose patients to recurrent strokes. However, further studies are required to elucidate the etiology of cerebrovascular arteriopathy in the setting of MPS I. Although the natural history of steno-occlusive arteriopathy in patients with MPS I remains unclear, our findings suggest that cerebral revascularization is a safe treatment option that may mitigate the risk of future strokes and should be strongly considered within the overall management guidelines for patients with MPS I.
Introduction
Mucopolysaccharidosis (MPS) type I is a rare, autosomal recessive, lysosomal storage disorder caused by a deficiency of the enzyme alpha-L-iduronidase with resultant accumulation of glycosaminoglycans (GAGs) within the lysosomes (1, 2). MPS I is classified into two clinical entities based on disease severity: severe [Hurler syndrome (OMIM #607014)] and attenuated [Hurler-Scheie (OMIM #607015) and Scheie syndromes (OMIM #607016)]. Clinical features include coarse facial appearance, corneal clouding, hepatosplenomegaly, hearing loss, hydrocephalus, cardiac valvular disease, airway obstruction, spinal cord compression, dysostosis multiplex, and cognitive decline (2). Without treatment, individuals with severe MPS I may die within the first decade of life, usually from cardiorespiratory failure or progressive neurological disease (2, 3). Currently, short-term administration of enzyme replacement therapy (ERT) in combination with hematopoietic stem cell transplantation (HSCT) is recommended for patients with severe MPS I and is associated with improved mortality and engraftment rates, reduced urinary GAG excretions, regression of cerebral ventriculomegaly, and stabilization of cardiac ventricular dysfunction (4–7). ERT, however, does not cross the blood-brain barrier in sufficient quantities to address neurological disease (2, 8, 9). Accordingly, the natural history of MPS I may be characterized by progressive neurological morbidity (2, 8–11).
Stroke is not a widely recognized manifestation of MPS I (2, 12, 13). Accordingly, patients with MPS I are not routinely screened for stroke and there are no established guidelines for managing stroke in this population. We report the first case of a 17-month-old male with MPS I who developed an acute ischemic stroke secondary to progressive multifocal cerebral arteriopathy. Cerebral revascularization was performed and long-term follow-up indicated successful engraftment and protection from further stroke. Our findings indicate that cerebral revascularization can be considered an appropriate treatment option for patients with MPS I at risk of stroke.
Case Description
History and Presentation
The patient was born at 40 weeks to non-consanguineous parents who are carriers of MPS I. At 9 months, the patient developed dysostosis multiplex, kyphosis, macrocephaly, and coarse facial features. Enzyme analysis revealing abnormally low alpha-L-iduronidase activity and elevated GAG levels confirmed the diagnosis of MPS I (Hurler syndrome) (14). The patient commenced the recommended treatment for MPS I with 8 weeks of ERT followed by HSCT (5, 6). He experienced graft failure following his first HSCT; however, a second round of this combinatorial therapy resulted in successful donor engraftment.
Subsequent urinary analysis demonstrated GAG-level reduction consistent with the treatment protocol (4). There was no evidence of graft-versus-host disease, thrombotic microangiopathy, or adverse events related to treatment with ERT and HSCT. Prior to the patient's second HSCT, neuroimaging was obtained for macrocephaly and findings were consistent (15) with MPS I (Figure 1). However, the patient showed no clinical evidence of neurological symptoms at this time.
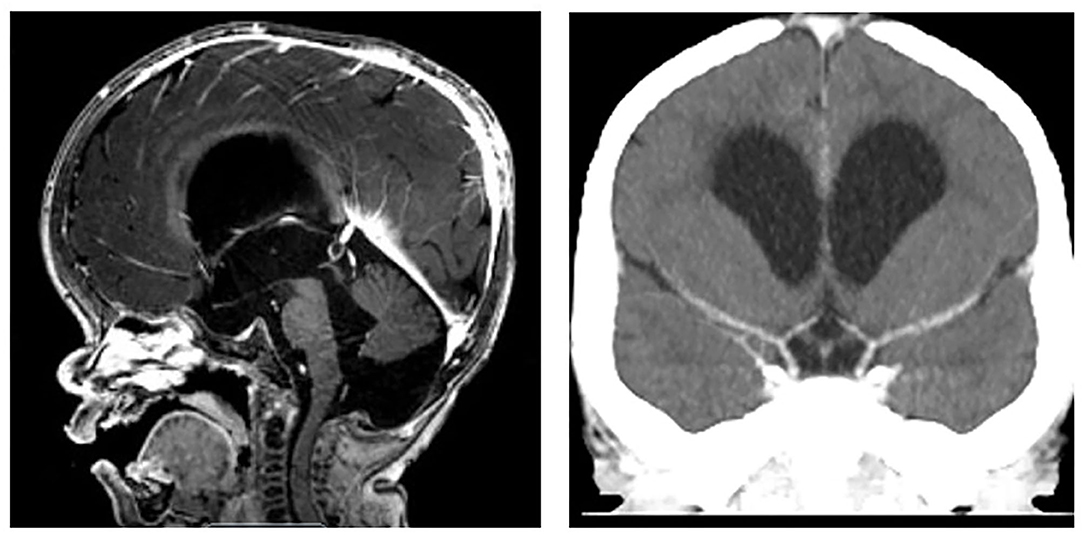
Figure 1. MPS I prior to stroke. (Left) MRI Head demonstrating imaging findings of macrocephaly, ventriculomegaly, small foramen magnum, and mild narrowing of cervical spine without cord compression. (Right) CT Head with contrast demonstrating bilateral internal carotid termini without stenosis.
One month after his second HSCT, at 17 months of age, the patient reported new right-sided weakness, emesis, and a decrease in appetite that were concerning for stroke. Brain MRI revealed an acute left middle cerebral artery (MCA) territory infarct and right cerebral ischemia. MR and CT angiography demonstrated complete absence of blood flow into the distal left MCA segments and bilateral carotid terminus narrowing (Figure 2). The patient was given 81 mg aspirin daily.
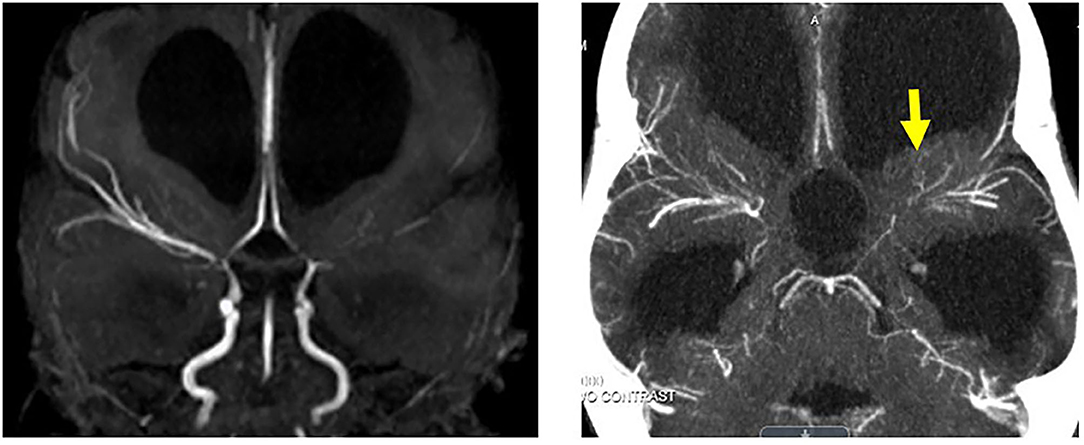
Figure 2. MPS I arteriopathy after left cerebral stroke. (Left) MR angiogram 8 weeks after stroke with left MCA occlusion and bilateral ICA termini steno-occlusive disease. (Right) New left arm weakness episodes 3 months after overt stroke with persisting bilateral steno-occlusive disease and adjacent irregular collaterals identified on CT angiogram (yellow arrow).
Serial follow-up MR imaging >6 months post stroke demonstrated continued stroke evolution and bilateral internal carotid artery stenoses (Figure 3, upper panel). Digital subtraction angiography was not performed because of the patient's abdominal aortic aneurysm and renal artery stenosis. Cardiac echocardiograms demonstrated mild mitral regurgitation without evidence of vegetation and EKG, thrombophilia, and platelet function testing were unremarkable. The patient subsequently underwent direct intracranial pressure measurement and therapeutic drainage trials, which revealed normal intracranial pressures without clinical or ventricular improvement. Hydration and hemoglobin status remained stable. Therefore, a diagnosis of bilateral intracranial steno-occlusive arteriopathy was established in the setting of MPS I.
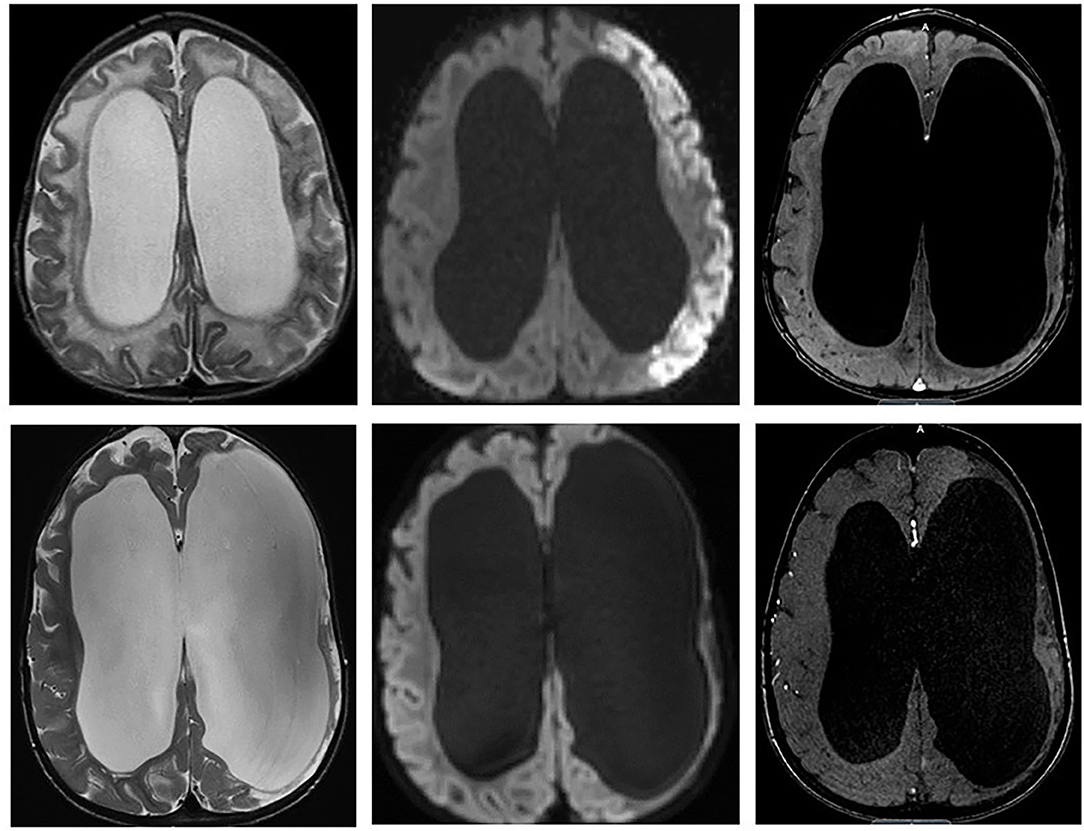
Figure 3. (Upper) Initial MRI of left MCA overt stroke identified on T2, diffusion, and MRA. (Lower) Two years after right cerebral revascularization demonstrating no new ischemic or infarcted territories and increased right cerebral cortical vascularity.
Surgical Treatment
To our knowledge, there is no literature describing surgical outcomes for steno-occlusive cerebrovascular disease in patients with MPS I. Accordingly, a multidisciplinary review in our Cerebrovascular Center at Cincinnati Children's Hospital and a secondary review with Boston Children's Hospital were convened to discuss treatment options. Given the patient's repetitive presentations of arm weakness and irritability, right steno-occlusive disease, and absence of elevated ICP, we collectively recommended right cerebral revascularization. Left cerebral revascularization was not recommended due to the patient's large-territory, completed stroke.
The patient underwent right-sided indirect revascularization with pial synangiosis and dural inversion, which was well-tolerated without post-operative complication (16, 17). A 2-year postoperative MRI demonstrated increased cortical vascularity without progressive ischemic changes (Figure 3, Lower panel). At a 3-year follow-up, the patient was clinically well with resolution of paresis and without recurrent symptoms.
Discussion
For the first time, we describe a child with MPS I who presented with an acute ischemic stroke and underlying cerebral arteriopathy. Although strokes have been reported in patients with MPS I (12, 13, 18–20), stroke is not a widely-recognized clinical manifestation of MPS I and there are no reviews or guidelines for managing stroke in patients with this disease (2, 12, 13). This report, to our knowledge, is the first clinical description of the development of progressive cerebral arteriopathy treated via revascularization surgery in a patient with MPS I. Our findings suggest that cerebral revascularization is a safe treatment option that may mitigate future stroke risk and should be considered within the overall management guidelines for patients with MPS I.
Stroke in MPS I
Neurological manifestations of MPS I include hydrocephalus, spinal cord compression, and cognitive decline (2, 10, 11, 21). Cardiac manifestations include hypertension, arrhythmia, valvular disease, and coronary artery disease (2, 7, 22). Stroke is not a widely recognized manifestation of MPS I, and patients with MPS I are not routinely evaluated for stroke (2, 12, 13). However, a few cases of stroke have been reported in patients with this disease (Table 1) (12, 13, 18–20). A cardioembolic infarction disrupting the left internal carotid artery was the reported cause of stroke in a 41-year-old female with MPS I. After treatment with a recombinant tissue plasminogen activator (rtPA), this patient demonstrated signs of clinical improvement with recanalization of the left internal carotid artery (12). Another child with MPS I presented with an MCA territory occlusion and was treated with 7 days of heparinization, though no neurological changes were observed (13). Along with our current report, these studies indicate that stroke occurs in MPS I; however, it is uncertain whether rtPA therapy and anticoagulants will mitigate the risk of future strokes in patients with this disease. More research into the pathophysiological cause of stroke in MPS I and long-term treatment outcomes can help develop guidelines for managing stroke in this population.
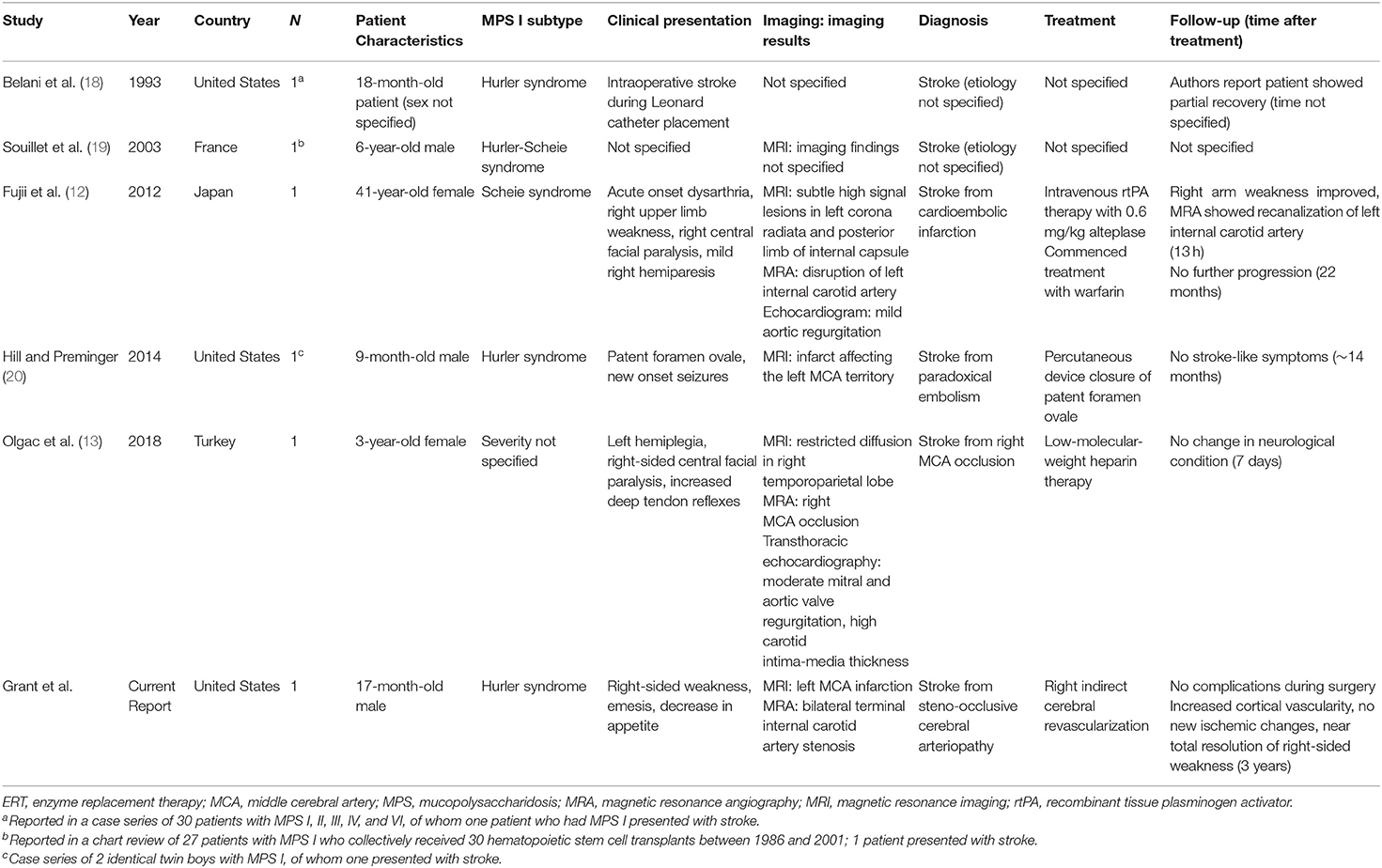
Table 1. Stroke reported in patients with mucopolysaccharidosis (MPS) type I.
Diagnostic Evaluation and Stroke Management
The patient described in this report was diagnosed with MPS I at 9 months and presented with a stroke at 17 months of age. The patient's symptoms and imaging are indicative of cerebral arteriopathy with bilateral internal carotid artery stenosis and left MCA occlusion. MRI examinations also demonstrated significant cerebral ventriculomegaly. The possibility of hydrocephalus-related vascular compression was evaluated through repeat lumbar punctures and intracranial pressure monitoring; however, all procedures confirmed normal intracranial pressure. The patient's cerebral ventriculomegaly was therefore consistent with ex vacuo changes of ischemic stroke rather than hydrocephalus.
Although the patient's stroke in association with bilateral internal carotid artery stenosis resembled moyamoya, we could not perform a conventional digital subtraction cerebral arteriogram in this patient to diagnose moyamoya (23). Nevertheless, the patient exhibited critical stenoses and distal ischemia on MRI with waxing and waning clinical presentation. It is unlikely the serial MRAs overestimated the high degree of steno-occlusive arteriopathy given the patient's stable hydration and hemoglobin status. Furthermore, there was no evidence for cerebral vasculitis or cardiac abnormalities indicative of cardioembolic stroke. Therefore, a multidisciplinary team was convened to discuss treatment options for this patient. Continued clinical surveillance amid repetitive stroke-like presentations was considered of great risk; thus, cerebral revascularization was collectively recommended to mitigate the risk for recurrent stroke.
Potential Etiology
Our findings are consistent with a steno-occlusive disease, although the exact etiology of cerebral arteriopathy in patients with MPS I remains unclear. GAG accumulation is common in patients with MPS disorders and has been associated with many neurological and cardiac manifestations of MPS I (12, 21, 24). It is possible that an accumulation of GAGs contributed to this patient's cerebral arteriopathy, ultimately resulting in stroke. Proteoglycan imbalance may affect the arterial smooth muscle cell proliferation rate during MPS I elastogenesis (25), which may be like other intracranial steno-occlusive diseases (26). As we cannot demonstrate a molecular pathway for causation here, further studies will be necessary to determine the exact etiology of cerebral arteriopathy and stroke in patients with MPS I.
Treatment Recommendations and Outcomes
As cerebral arteriopathy predisposes patients to stroke, treatment is needed to mitigate the risk of recurrent strokes in MPS I. Surgical revascularization prevents further ischemic injury by increasing collateral blood flow to areas of insufficient perfusion due to arteriopathy using external circulation as a donor supply (17, 23, 27). Indirect cerebral revascularization is considered safe and effective for revascularizing the pediatric brain and preventing future stroke (17, 27, 28). While revascularization surgery has been documented in one patient with MPS I, this finding is listed in a large cohort study without clinical presentation, imaging results, and postoperative outcomes (29). The lack of MPS I revascularization evidence made management challenging, necessitating multidisciplinary review. Our findings demonstrate that cerebral revascularization is a safe and effective treatment option to mitigate the risk of recurrent stroke in children with MPS I. Cerebral revascularization should be considered within the overall management of this disease.
Conclusion
This report provides two important new findings. First, we demonstrate that patients with MPS I are at risk of significant overt stroke secondary to cerebral arteriopathy. As symptoms of MPS I typically emerge in early childhood (2, 11), pediatric providers should consider screening for stroke in children with this disease. Second, this is the first clinical description of cerebral revascularization in a patient with MPS I, supported by long-term follow-up that confirmed favorable tolerance of surgery without any transient or neurological stroke events. Further investigation will help establish guidelines for identifying and treating stroke in patients with MPS I.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article. Informed consent was obtained from the patient for being included in this case report.
Author Contributions
NG collected data, analyzed and interpreted the data, drafted the initial manuscript, reviewed, and revised the manuscript. JT conceptualized and organized the case report, analyzed and interpreted the data, and critically revised the manuscript for important intellectual content. ZP, KM, TB, LL-J, AB, AH, KW, and ES analyzed and interpreted the data and critically revised the manuscript for important intellectual content. JL and SV conceptualized and organized the case report, analyzed and interpreted the data, drafted the initial manuscript, and critically revised the manuscript for important intellectual content. All authors have approved the final manuscript and agree to be accountable for all aspects of the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.606905/full#supplementary-material
Abbreviations
ERT, enzyme replacement therapy; GAGs, glycosaminoglycans; HSCT, hematopoietic stem cell transplantation; ICP, intracranial pressure; IONM, intraoperative neurophysiologic monitoring; MCA, middle cerebral artery; MMA, middle meningeal artery; MPS, mucopolysaccharidosis; rtPA, recombinant tissue plasminogen activator; STA, superficial temporal artery.
References
1. Bach G, Friedman R, Weissmann B, Neufeld EF. The defect in the Hurler and Scheie syndromes: deficiency of α-L-Iduronidase. Proc Natl Acad Sci USA. (1972) 69:2048–51. doi: 10.1073/pnas.69.8.2048
2. Muenzer J, Wraith JE, Clarke LA, the International Consensus Panel on the Management Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. (2009) 123:19–29. doi: 10.1542/peds.2008-0416
3. Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, Valle D, Childs R, Kinzler KW, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw Hill (2001) p. 3421–52.
4. Grewal SS, Wynn R, Abdenur JE, Burton BK, Gharib M, Haase C, et al. Safety and efficacy of enzyme replacement therapy in combination with hematopoietic stem cell transplantation in Hurler syndrome. Genet Med. (2005) 7:143–6. doi: 10.1097/01.GIM.0000154299.22120.6A
5. Tolar J, Grewal SS, Bjoraker KJ, Whitley CB, Shapiro EG, Charnas L, et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. (2008) 41:531–5. doi: 10.1038/sj.bmt.1705934
6. Ghosh A, Miller W, Orchard PJ, Jones SA, Mercer J, Church HJ, et al. Enzyme replacement therapy prior to haematopoietic stem cell transplantation in mucopolysaccharidosis type I: 10 year combined experience of 2 centres. Mol Genet Metab. (2016) 117:373–7. doi: 10.1016/j.ymgme.2016.01.011
7. Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis, and management. J Inherit Metab Dis. (2011) 34:1183–97. doi: 10.1007/s10545-011-9359-8
8. Tokic V, Barisic I, Huzjak N, Petkovic G, Fumic K, Paschke E. Enzyme replacement therapy in two patients with an advanced severe (Hurler) phenotype of mucopolysaccharidosis I. Eur J Pediatri. (2007) 166:727–32. doi: 10.1007/s00431-006-0316-8
9. Concolino D, Deodato F, Parini R. Enzyme replacement therapy: efficacy and limitations. Ital J Pediatr. (2018) 44:120. doi: 10.1186/s13052-018-0562-1
10. Shapiro EG, Nestrasil I, Rudser K, Delaney K, Kovac V, Ahmed A, et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: age severity, and treatment. Mol Genet Metab. (2015) 116:61–8. doi: 10.1016/j.ymgme.2015.06.002
11. Grosse SD, Lam WKK, Wiggins LD, Kemper AR. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type I. Genet Med. (2017) 19:975–82. doi: 10.1038/gim.2016.223
12. Fujii D, Manabe Y, Tanaka T, Kono S, Sakai Y, Narai H, et al. Scheie syndrome diagnosed after cerebral infarction. J Stroke Cerebrovasc Dis. (2012) 21:330–2. doi: 10.1016/j.jstrokecerebrovasdis.2010.09.006
13. Olgac A, Tumer L, Damar Ç, Hasanoglu A, Ezgü F. Acute stroke in a patient with mucopolysaccharidosis type I with increased carotid intima-media thickness. EJMO. (2018) 2:114–6. doi: 10.14744/ejmo.2018.51523
14. Johnson BA, Dajnoki A, Bodamer OA. Diagnosing lysosomal storage disorders: mucopolysaccharidosis type I. Curr Protoc Hum Genet. (2015) 84:17.17.1–8. doi: 10.1002/0471142905.hg1717s84
15. Matheus MG, Castillo M, Smith JK, Armao D, Towle D, Muenzer J. Brain MRI findings in patients with mucopolysaccharidosis types I and II and mild clinical presentation. Neuroradiology. (2004) 46:666–72. doi: 10.1007/s00234-004-1215-1
16. Adelson PD, Scott RM. Pial synangiosis for moyamoya syndrome in children. Pediatr Neurosurg. (1995) 23:26–33. doi: 10.1159/000120932
17. Smith ER, Scott RM. Surgical management of moyamoya syndrome. Skull Base. (2005) 15:15–26. doi: 10.1055/s-2005-868160
18. Belani KG, Krivit W, Carpenter BL, Braunlin E, Buckley JJ, Liao JC, et al. Children with mucopolysaccharidosis:perioperative care, morbidity, mortality, and new findings. J Pediatr Surg. (1993) 28:403–10. doi: 10.1016/0022-3468(93)90240-L
19. Souillet G, Guffon N, Maire I, Pujol M, Taylor P, Sevin F, et al. Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. (2003) 31:1105–17. doi: 10.1038/sj.bmt.1704105
20. Hill J, Preminger T. Percutaneous PFO closure for paradoxical stroke in 8-kg twins. Catheter Cardiovasc Interv. (2014) 84:110–3. doi: 10.1002/ccd.25183
21. Dalla Corte A, de Souza CFM, Anés M, Giugliani R. Hydrocephalus and mucopolysaccharidoses: what do we know and what do we not know? Childs Nerv Syst. (2017) 33:1073–80. doi: 10.1007/s00381-017-3476-0
22. Mohan UR, Hay AA, Cleary MA, Wraith JE, Patel RG. Cardiovascular changes in children with mucopolysaccharide disorders. Acta Paediatr. (2002) 91:799–804. doi: 10.1111/j.1651-2227.2002.tb03330.x
23. Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. (2009) 360:1226–37. doi: 10.1056/NEJMra0804622
24. Cabrera GH, Fernández I, Dominguez M, Clarke LA. Left ventricular aneurysm in an adult patient with mucopolysaccharidosis type I: comment on pathogenesis of a novel complication. Mol Genet Metab. (2012) 106:470–3. doi: 10.1016/j.ymgme.2012.06.001
25. Hinek A, Wilson SE. Impaired elastogenesis in Hurler disease: dermatan sulfate accumulation linked to deficiency in elastin-binding protein and elastic fiber assembly. Am J Pathol. (2000) 156:925–38. doi: 10.1016/S0002-9440(10)64961-9
26. Milewicz DM, Kwartler CS, Papke CL, Regalado ES, Cao J, Reid AJ. Genetic variants promoting smooth muscle cell proliferation can result in diffuse and diverse vascular diseases: evidence for a hyperplastic vasculomyopathy. Genet Med. (2010) 12:196–203. doi: 10.1097/GIM.0b013e3181cdd687
27. Ellis MJ, Armstrong D, Dirks PB. Large vascular malformation in a child presenting with vascular steal phenomenon managed with pial synangiosis. J Neurosurg Pediatr. (2011) 7:15–21. doi: 10.3171/2010.10.PEDS10388
28. Scott RM, Smith JL, Robertson RL, Madsen JR, Soriano SG, Rockoff MA. Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg. (2004) 100:142–9. doi: 10.3171/ped.2004.100.2.0142
Keywords: mucopolysaccharidosis I, stroke, cerebral arteriopathy, ventriculomegaly, pial synangiosis, cerebral revascularization
Citation: Grant N, Taylor JM, Plummer Z, Myers K, Burrow T, Luchtman-Jones L, Byars A, Hammill A, Wusick K, Smith E, Leach J and Vadivelu S (2021) Case Report: Cerebral Revascularization in a Child With Mucopolysaccharidosis Type I. Front. Pediatr. 9:606905. doi: 10.3389/fped.2021.606905
Received: 16 September 2020; Accepted: 16 April 2021;
Published: 10 June 2021.
Edited by:
Andrea Gropman, Children's National Hospital, United StatesReviewed by:
Emanuele Micaglio, IRCCS Policlinico San Donato, ItalyNicolina Cristina Sorrentino, Telethon Institute of Genetics and Medicine (TIGEM), Italy
Copyright © 2021 Grant, Taylor, Plummer, Myers, Burrow, Luchtman-Jones, Byars, Hammill, Wusick, Smith, Leach and Vadivelu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sudhakar Vadivelu, c3VkaGFrYXIudmFkaXZlbHVAY2NobWMub3Jn