 Joonhong Park
Joonhong Park Yong Gon Cho1,2†
Yong Gon Cho1,2† Jung Sun Cho
Jung Sun Cho- 1Department of Laboratory Medicine, Jeonbuk National University Medical School and Hospital, Jeonju, South Korea
- 2Research Institute of Clinical Medicine of Jeonbuk National University-Biomedical Research Institute of Jeonbuk National University Hospital, Jeonju, South Korea
- 3Department of Cardiology, College of Medicine, The Catholic University of Korea, Seoul, South Korea
Left ventricular non-compaction (LVNC) is a very rare primary cardiomyopathy with a genetic etiology, resulting from the failure of myocardial development during embryogenesis, and it carries a high risk of left ventricular dysfunction, thromboembolic phenomenon, and malignant arrhythmias. Here, we report the first case of familial LVNC in Korea, caused by a novel ACTN2 missense variant. We performed duo exome sequencing (ES) to examine the genome of the proband and his father. A 15-year-old boy was admitted for the evaluation of exertional dyspnea for 2 weeks. He was diagnosed with LVNC with a dilated cardiomyopathy phenotype [left ventricular end-diastolic dimension 60 mm, interventricular septal dimension 8.2 mm by transthoracic echocardiography (TTE)]. For the screening of familial cardiomyopathy, TTE and cardiac magnetic resonance imaging (cMRI) were performed, which revealed hypertrophic and isolated LVNC in the proband's father and sister, respectively. In particular, the cMRI revealed dense hypertrabeculation with focal aneurysmal changes in the apical septal wall in the proband's father. ES of the father–son duo identified a novel heterozygous c.668T>C variant of the ACTN2 gene (NM_001103.3:c.668T>C, p.Leu223Pro; no rsID) as the candidate cause of autosomal dominant LVNC. Sanger sequencing confirmed this novel variant in the proband, his father, and sister, but not in the proband's grandmother. Even within families harboring the same variant, a variable risk of adverse outcomes is common. Therefore, familial screening for patients with LVNC associated with ACTN2 variant should be performed for early detection of the LVNC phenotype associated with poor outcomes, such as dilated LVNC.
Introduction
Left ventricular non-compaction (LVNC) is a rare cardiomyopathy due to the failure of myocardial development during embryogenesis, and it is characterized by a high risk of left ventricular dysfunction, malignant arrhythmias, and thromboembolic phenomenon (1, 2). The prevalence of LVNC has been reported as 3 to 4% among patients with heart failure (3). During development, most of the heart muscle is a sponge-like meshwork of interwoven myocardial fibers (2, 4). As normal development progresses, these trabeculated structures undergo significant compaction that transforms them from spongy to solid fibers, which is particularly apparent in the ventricles, particularly in the left ventricle. To date, this type of hereditary cardiomyopathy is not fully understood; however, it was classified as primary cardiomyopathy of genetic origin by the American Heart Association in 2006, as well as described as a distinct entity by MOGE(S) classification (5). The European Society of Cardiology categorizes it as an unclassified cardiomyopathy (6). The disease is caused by heterozygous mutations in different genes coding for the cardiac sarcomere; calcium handling; and other cardiomyopathy related genes, including MYH7, MYBPC3, TPM1, ACTC1, TNNT2, LMNA, LDB3, TMP1, TNNI3, and ACTN2, of which, the most commonly affected are the sarcomere genes MYH7, MYBPC3, and TPM1 (7–9). Mutations in the genes encoding the Z-disc or calcium-handling proteins account for <1% of the cases (10). Among them, mutations in the ACTN2 have been implicated in mild to moderate forms of hypertrophic and dilated cardiomyopathy (#612158) (11). However, in the case of LVNC with congenital heart disease, disturbance of the NOTCH signaling pathway seems to be a part of the final common pathway for this form of the disease (12, 13).
In this report, we describe a heterogeneous phenotype in familial LVNC with a novel ACTN2 missense variant identified by exome sequencing (ES).
Case Presentation
A 15-year-old boy (III-2 in Figure 1A) was admitted to the Department of Cardiology, Daejeon St. Mary's Hospital (Daejeon, Korea) for the evaluation of exertional dyspnea for 2 weeks. He used to practice fencing and complained of relatively mild exertional discomfort for several years. He did not have any other metabolic or clinical abnormality except for obesity (body mass index: 31.8 Kg/m2). The proband underwent transthoracic echocardiography (TTE), which showed a non-compacted myocardium with a dilated cardiomyopathy phenotype such as left ventricular end-diastolic dimension (LVEDD) 60 mm, interventricular septal dimension (IVS) 8.2 mm, left ventricular ejection fraction (LVEF) 25% (Figures 2A–C). Cardiac magnetic resonance imaging (cMRI) revealed LVNC with dilated cardiomyopathy (Figures 2D–F). His electrocardiogram (ECG) and Holter monitoring showed sinus arrhythmia and non-specific intraventricular conduction delay, as well as rare atrial premature contraction (Figure 2G). He did not complain of any skeletal muscle symptoms, and serum muscle enzyme levels were normal. As previously mentioned, LVNC has been classified by the American Heart Association (14) as a primary cardiomyopathy of genetic origin, presenting in patients with marked clinical heterogeneity. As such, clinical screening of the proband's family for an inherited cardiomyopathy became necessary. The proband's 48-year-old father (II-1 in Figure 1A) had a medical history of diabetes and hypertensive heart disease. He was admitted to another hospital 2 months previously because of pleural effusion and congestive heart failure. At that time, his serum NT-proBNP level was elevated (532 pg/mL). His TTE showed an echogenic mass or trabeculation and a LVEF of 35% with aneurysmal changes in the apical wall (Figures 3A–C). A coronary artery angiogram was performed to evaluate the apical aneurysm, and it demonstrated no coronary artery stenosis. In addition, cMRI revealed dense hypertrabeculation with focal aneurysmal changes in the apical septal wall. These findings could be considered as the hypertrophic phenotype of LVNC (Figures 3D–F). The proband's 18-year-old sister (III-1 in Figure 1A) did not have any symptoms; however, her TTE revealed LVNC with normal LVEF, which was consistent with isolated LVNC (Figures 3G–I). All affected individuals did not present with skeletal muscle myopathies. The clinical characteristics of affected individuals diagnosed with LVNC are described in Table 1.
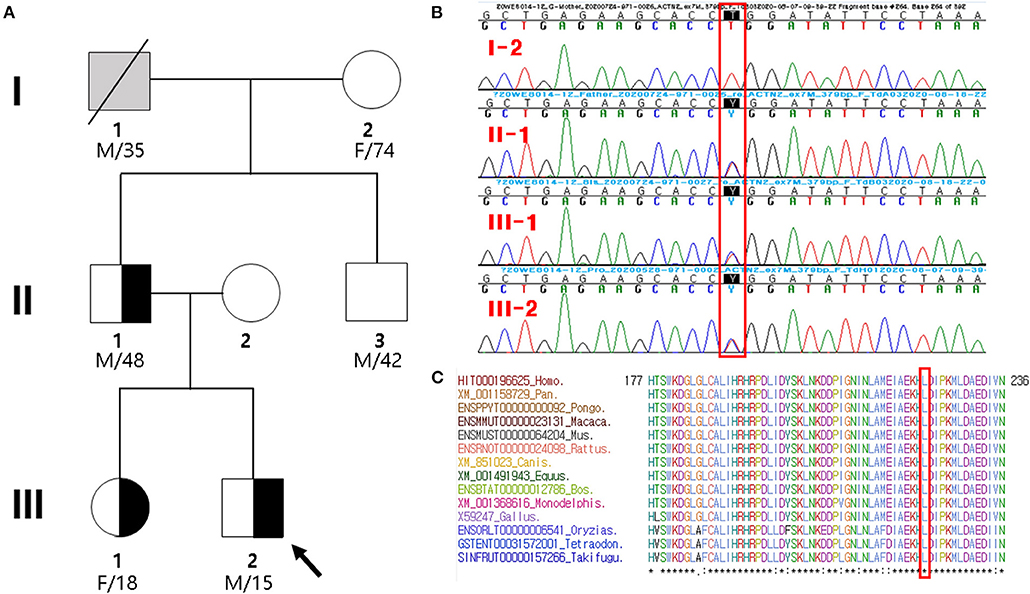
Figure 1. Pedigree analysis and sequencing results. (A) Pedigree of the proband (arrow) and his family members with heterozygous ACTN2 variant. Gray symbol indicates that the presence of ACTN2 variant was not determined. (B) Sanger sequencing confirmed a novel missense mutation (c.668T>C; p.Leu223Pro) in ACTN2, which was of paternal origin. It is highlighted in the red box. (C) Sequence alignment of the conserved cytoplasmic domain of the ACTN2 protein in multiple vertebrate species. Protein sequence of the Leu223Pro residue is highly conserved across the compared species. It is highlighted in the red box.
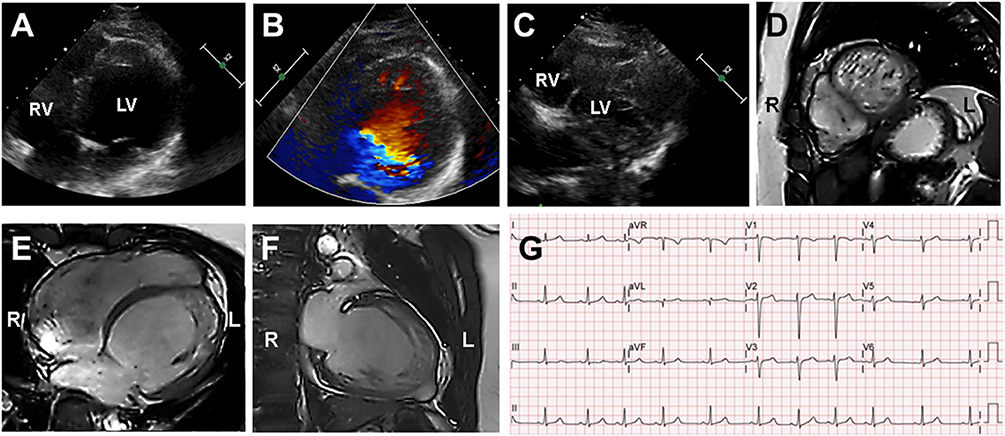
Figure 2. Echocardiography and cardiac magnetic resonance imaging (cMRI) in the proband (III-2). (A–C) Echocardiography and Color Doppler showed dilated left ventricle chamber and prominent trabeculation and flow within the deep intertrabecular recesses. (D–F) cMRI revealed that the non-compacted to compacted ratio ≥2 and severely dilated biventricular myocardium that was compatible with dilated LVNC. (G) Proband's electrocardiogram demonstrated sinus arrhythmia and non-specific intraventricular conduction delay.
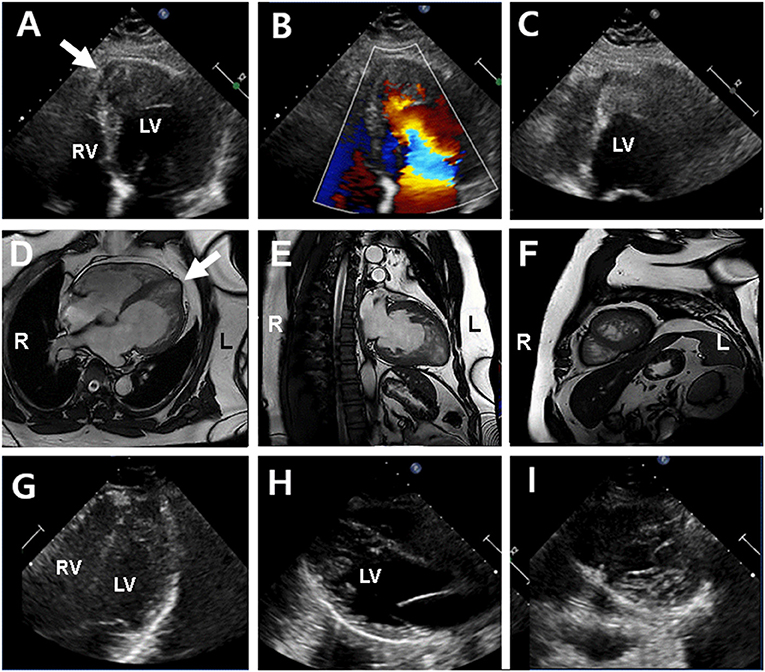
Figure 3. Echocardiography and cardiac magnetic resonance imaging (cMRI) in the proband's father (II-1) (upper and middle panel) and sister (III-1) (lower panel). (A–C) Echocardiography showed left ventricular hypertrophy and apical echogenic mass or dense trabeculation and apical septal aneurysm (arrow) in the proband's father. (D–F) cMRI determined dense trabeculation and focal apical septal aneurysm that considered as hypertrophic LVNC in the proband's father. (G–I) Echocardiography in proband's sister showed normal left ventricular function and geometry except for prominent trabeculation of left ventricle that diagnosed with isolated LVNC in the proband's sister.
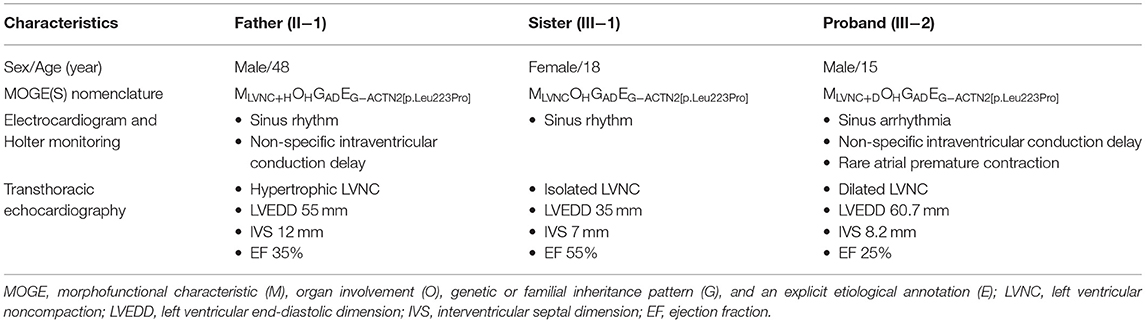
Table 1. Clinical characteristics of individuals harboring novel ACTN2 variant diagnosed as left ventricular non-compaction.
The proband's grandfather (I-1 in Figure 1A) died at the age of 35 years due to gastric cancer, and his cardiac history was unknown. The proband's 74-year-old grandmother (I-2 in Figure 1A) did not have any cardiac disease except for hypertension. She did not present with any cardiac symptoms, and her TTE showed normal cardiac function and geometry. Thus, the inheritance of the proband's father was not determined presumptively, whether inherited or sporadic. The father's brother (II-3 in Figure 1A) had no clinical symptoms, lived in another area, and did not agree to be tested. The proband's mother was completely normal through birth to adolescent and showed no skeletal muscle myopathies or cardiac symptoms and was not investigated.
Exome Sequencing of the Father–Son Duo
The study protocol was approved by the Institutional Review Board of the Catholic University of Korea. Written informed consent was obtained from the legal guardian/next of kin of the individual(s) and minor(s) involved in the study for the publication of any potentially identifiable images or data included in this article. The exomic DNA of the proband and his father was enriched using Agilent's SureSelect XT Human All Exon v5 (Agilent Technologies, Santa Clara, CA, USA). Paired-end sequencing was conducted on the Illumina HiSeq2500 (Illumina, San Diego, CA, USA) for detection of the variant, given the suspicion of familial disease at the Green Cross Genome (Yongin, Korea). Base calling, alignment, variant calling, annotation, and quality control reporting were performed using a GATK Best Practices workflow for germline short variant discovery and were manually reviewed by medical laboratory geneticists. Variants that pass the filtering criteria are as follows: Phred quality score >20, no Fisher strand bias, read depth >30 ×, allele frequency <0.1%, non-synonymous substitution or indel occurred in coding region and exon–intron boundaries, heterozygous variant in both father and son in an autosomal dominant manner, and reported to be associated with cardiac disease in the OMIM database. In particular, annotation of the identified variants with respect to the results of their variant on the reported genes and the variant effect predicted by in silico computational tools, such as MutationAssessor (http://mutationassessor.org/r3/) (15), Sorting Intolerant from Tolerant (https://sift.bii.a-star.edu.sg/) (16), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) (17), Likelihood Ratio Test (http://www.genetics.wustl.edu/jflab/lrt_query.html) (18), and MutationTaster (http://www.mutationtaster.org/) (19), and public genome databases from gnomAD (https://gnomad.broadinstitute.org/) and KRGDB (http://coda.nih.go.kr/coda/KRGDB/index.jsp), were estimated using the dbNSFP 2.4 (20) and Ensembl Variant Effect Predictor (21).
Results
By estimating sequence quality along all sequences, 6,119 and 5,241 million reads were generated from the proband and his father's samples, respectively. The % bases above average 30 × were achieved for 93.6 and 91% of the target region, and the mean read depth (×) was 123 and 104, respectively. ES of the father–son duo identified eight heterozygous variants as candidate causes of autosomal dominant inherited diseases (Supplementary Table 1). Because there was no phenotypic description related to LVNC in the OMIM database, except for the ACTN2 variant (chr1:236894585T/C), the ACTN2 variant seems to be pathogenic. In the proband and his father presenting with a similar phenotype, the missense ACTN2 variant causing a codon change of leucine to proline at position 223 (NM_001103.3:c.668T>C, p.Leu223Pro; no rsID) was newly identified. To confirm the presence of the ACTN2 c.668T>C variation in the affected members, Sanger sequencing was performed. It confirmed the heterozygous c.668T>C variant in the proband, his father, and sister, while it was absent from the blood DNA of the proband's grandmother (Figure 1B). Thus, the variant identified from the affected individuals demonstrated to cause an autosomal dominant cardiomyopathy. In addition, this missense variant of the ACTN2 was predicted to be “deleterious,” “damaging,” or “disease causing” by computational in silico prediction. Cross-species sequence comparisons (phastCons, SiPhy, and GERP) of amino acid sequences of ACTN2 protein revealed that this mutated site was highly conserved in vertebrates (phastCons 1 > cutoff of 0.8, SiPhy 14.464 > 12.17, and GERP 4.68 > 4.4) (Figure 1C). The variant is not listed in gnomAD or KRGDB. Paternity and kinship analysis was conducted using short tandem repeat (STR) multiplex assay (AmpFLSTR® Identifiler; Applied Biosystems, Foster City, CA, USA), and STR analysis confirmed the biological association of the parents and sibling with the proband.
Discussion
LVNC is classified into the following subtypes: hypertrophic, hypertrophic dilated, dilated, restrictive, isolated, and biventricular (22). Our cases were consistent with hypertrophic LVNC (II-1), dilated LVNC (III-2), and isolated LVNC (III-1) in a single family, which are heterogeneous phenotypes of LVNC. It is essential to accurately assess the phenotype of LVNC because different phenotypes are associated with variable outcomes and necessitate different surveillance strategies (23). Routinely, the diagnosis of LVNC relies on non-invasive imaging studies, such as TTE and cMRI. The diagnostic criteria for LVNC are based on the ratio of thickness of the non-compacted layer to that of the compacted layer, with a ratio >2–3:1 at the end of the typical diastole (22, 24). TTE is the most common diagnostic modality because of its availability. However, cMRI can delineate the extent of LVNC more precisely and also provide additional morphological characterization of the myocardium (23, 25).
Diverse clinical phenotypes were observed in this family, including hypertrophic, dilated, and isolated LVNC. In particular, it was difficult to distinguish the dense myocardial trabeculation from LV thrombus in the proband's father because he had focal aneurysmal changes at the LV apex. In this case, cMRI could provide a high degree of spatial resolution of the LV apex. In addition to imaging studies, genetic diagnosis can be useful in elucidating the underlying genetic cause of the disease, and it allows for predictive testing of other asymptomatic at-risk members in families with such variable clinical presentations. Our cases were the first Korean cases of familial LVNC caused by a novel ACTN2 missense variant and were identified by a duo ES approach to examine the genome of the proband and his father. As with the universal TNM staging for tumors, MOGE(S) nomenclature is a descriptive nosology that combines morphofunctional traits and organ/system involvement with familial inheritance patterns, identified genetic defects, or other etiologies (26). The MOGE(S) nomenclature for this family is shown in Table 1. The MOGE(S) system distinguishes LVNC from LV dilation and dysfunction (MLVNC+D) or LV hypertrophy (MLVNC+H) from isolated LVNC (MLVNC).
The ACTN2 mutation, as a sarcomere mutation, has rarely been reported among families with LVNC (12, 27–29). The ACTN2 encodes alpha-actinin2, which is the only muscle isoform of α-actinin expressed in the cardiac muscle. ACTN2 plays important roles, such as a structure anchor and stretch sensor, and is involved in ion channel organization in the Z-disc of cardiomyocytes (12). Moreover, ACTN2 interacts directly with the cardiac, potassium, and sodium ion channels. Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2 has been reported (30). Previous studies reported that the ACTN2 mutation may lead to diverse cardiomyopathy, including dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, and LVNC, as well as arrhythmia, such as idiopathic ventricular fibrillation, juvenile atrial arrhythmias, and sudden unexplained death (28, 31–34). Notably, the variant ACTN2 c.683T>C (p.Met228Thr) was described by Girolami et al. (28) in a family with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias. In this study, the identified variant ACTN2 c.668T>C (p.Leu223Pro) is next to this variant (p.Met228Thr), and ECG and Holter monitoring of the juvenile proband revealed non-specific intraventricular conduction delay, sinus arrhythmia, and premature atrial contraction. On the other hand, Alpha-actinin-2 encoded by the ACTN2 is highly abundant in cardiac and skeletal muscle, where it plays several functional and structural roles in the sarcomeres (34). Thereby underlies the diverging pathomechanisms resulting in not only cardiomyopathy but also skeletal myopathy (35). Recently, Savarese et al. (36) have described mutations in the ACTN2 gene causing a late onset distal myopathy. Thus, we cannot exclude the occurrence of a late onset skeletal myopathy someday in our affected individuals with an ACTN2-related cardiomyopathy.
Even within families with LVNC harboring the same mutation, adverse outcomes are variable (12, 28). Despite the low genotype–phenotype correlation in cardiomyopathies and cardiac channelopathies, recent guidelines and experts recommend family screening. Therefore, family screening for patients with LVNC associated with ACTN2 mutation should be performed for early detection of the LVNC phenotype with poor outcomes, such as dilated LVNC (22, 37). While diverse types of LV morphology have also been identified in patients with mutations in ACTN2 (28, 31, 32), this is exemplified by our study on the structural pathologies caused by Leu223Pro in the ACTN2 actin binding domain (ABD). This comprises two calponin homology domains connected via a flexible linker, which allows for conformational flexibility in the ABDs, to a rod domain on ACTN2 (11). For example, Gly111Val and Ala119Thr on the CH1 domain have small effects on the structure, function, and behavior and might contribute to a mild phenotype for this disease. In addition, digenic inheritance and genetic modifiers exert a major effect and can explain the pleiotropic phenotype (38): two genes in ACTN2 and CMYA5 contribute to the development of cardiomyopathy (39).
In conclusion, we described a Korean family with a novel likely pathogenic ACTN2 variant, with LVNC manifesting as distinct heterogeneous phenotypes, such as hypertrophic, dilated, and isolated phenotypes, within the same family. Genetic screening of the family for congenital cardiomyopathy is necessary for patients with LVNC associated with ACTN2 mutation, even if the patient has isolated LVNC that is expected to have a clinically good prognosis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The study protocol was approved by the Institutional Review Board of the Catholic University of Korea. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JP and YC made substantial contributions to the analysis and interpretation of the NGS data and were involved in drafting the manuscript. HP made substantial contributions to the interpretations of clinical medical records. JC was involved in drafting the manuscript and in critically revising important intellectual content. All authors have read and approved the manuscript for submission, contributed to the article, and approved the submitted version.
Funding
This work was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (2020R1F1A1077316). The authors wish to acknowledge the financial support of the Catholic Medical Center Research Foundation made in the program year of 2021.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Drs. Chang-Seok Ki and Ju Sun Song (Green Cross Genome, Yongin, Korea) for their contribution in providing technical support for data analysis of next-generation sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.609389/full#supplementary-material
References
1. Pignatelli RH, McMahon CJ, Dreyer WJ, Denfield SW, Price J, Belmont JW, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. (2003) 108:2672–8. doi: 10.1161/01.CIR.0000100664.10777.B8
2. Ichida F. Left ventricular noncompaction - risk stratification and genetic consideration. J Cardiol. (2020) 75:1–9. doi: 10.1016/j.jjcc.2019.09.011
3. Kovacevic-Preradovic T, Jenni R, Oechslin EN, Noll G, Seifert B, Attenhofer Jost CH. Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience. Cardiology. (2009) 112:158–64. doi: 10.1159/000147899
4. Arbustini E, Favalli V, Narula N, Serio A, Grasso M. Left ventricular noncompaction: a distinct genetic cardiomyopathy? J Am Coll Cardiol. (2016) 68:949–66. doi: 10.1016/j.jacc.2016.05.096
5. Klenda J, Boppana LKT, Vindhyal MR. Heart failure secondary to left ventricular non-compaction cardiomyopathy in a 26-year-old male. Cureus. (2018) 10:e3011. doi: 10.7759/cureus.3011
6. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. (2008) 29:270–6. doi: 10.1093/eurheartj/ehm342
7. Hoedemaekers YM, Caliskan K, Majoor-Krakauer D, van de Laar I, Michels M, Witsenburg M, et al. Cardiac beta-myosin heavy chain defects in two families with non-compaction cardiomyopathy: linking non-compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur Heart J. (2007) 28:2732–7. doi: 10.1093/eurheartj/ehm429
8. Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. (2008) 117:2893–901. doi: 10.1161/CIRCULATIONAHA.107.746164
9. Probst S, Oechslin E, Schuler P, Greutmann M, Boyé P, Knirsch W, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. (2011) 4:367–74. doi: 10.1161/CIRCGENETICS.110.959270
10. Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. (2013) 50:228–39. doi: 10.1136/jmedgenet-2012-101270
11. Haywood NJ, Wolny M, Rogers B, Trinh CH, Shuping Y, Edwards TA, et al. Hypertrophic cardiomyopathy mutations in the calponin-homology domain of ACTN2 affect actin binding and cardiomyocyte Z-disc incorporation. Biochem J. (2016) 473:2485–93. doi: 10.1042/BCJ20160421
12. Bagnall RD, Molloy LK, Kalman JM, Semsarian C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med Genet. (2014) 15:99. doi: 10.1186/s12881-014-0099-0
13. Frustaci A, De Luca A, Guida V, Biagini T, Mazza T, Gaudio C, et al. Novel α-actin gene mutation p.(Ala21Val) causing familial hypertrophic cardiomyopathy, myocardial noncompaction, and transmural crypts. Clinical-Pathologic correlation. J Am Heart Assoc. (2018) 7:e008068. doi: 10.1161/JAHA.117.008068
14. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. (2006) 113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287
15. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. (2011) 39:e118. doi: 10.1093/nar/gkr407
16. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509
17. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013) Chapter 7:Unit7.20. doi: 10.1002/0471142905.hg0720s76
18. Yang Z. A likelihood ratio test of speciation with gene flow using genomic sequence data. Genome Biol Evol. (2010) 2:200–11. doi: 10.1093/gbe/evq011
19. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
20. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. (2016) 37:235–41. doi: 10.1002/humu.22932
21. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The ensembl variant effect predictor. Genome Biology. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
22. Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. (2015) 386:813–25. doi: 10.1016/S0140-6736(14)61282-4
23. Petersen SE. CMR and LV noncompaction: does it matter how we measure trabeculations? JACC Cardiovasc Imaging. (2013) 6:941–3. doi: 10.1016/j.jcmg.2013.03.007
24. Ikeda U, Minamisawa M, Koyama J. Isolated left ventricular non-compaction cardiomyopathy in adults. J Cardiol. (2015) 65:91–7. doi: 10.1016/j.jjcc.2014.10.005
25. Stacey RB, Caine AJ Jr, Hundley WG. Evaluation and management of left ventricular noncompaction cardiomyopathy. Curr Heart Fail Rep. (2015) 12:61–7. doi: 10.1007/s11897-014-0237-1
26. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the world heart federation. J Am Coll Cardiol. (2013) 62:2046–72. doi: 10.1016/j.jacc.2013.08.1644
27. van Waning JI, Moesker J, Heijsman D, Boersma E, Majoor-Krakauer D. Systematic review of genotype-phenotype correlations in noncompaction cardiomyopathy. J Am Heart Assoc. (2019) 8:e012993. doi: 10.1161/JAHA.119.012993
28. Girolami F, Iascone M, Tomberli B, Bardi S, Benelli M, Marseglia G, et al. Novel α-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: a massively parallel sequencing study. Circ Cardiovasc Genet. (2014) 7:741–50. doi: 10.1161/CIRCGENETICS.113.000486
29. van Waning JI, Caliskan K, Hoedemaekers YM, van Spaendonck-Zwarts KY, Baas AF, Boekholdt SM, et al. Genetics, clinical features, and long-term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. (2018) 71:711–22. doi: 10.1016/j.jacc.2017.12.019
30. Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, et al. Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2. Circ Res. (2007) 100:112–20. doi: 10.1161/01.RES.0000253095.44186.72
31. Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, et al. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol. (2010) 55:1127–35. doi: 10.1016/j.jacc.2009.11.016
32. Coll M, Pérez-Serra A, Mates J, Del Olmo B, Puigmulé M, Fernandez-Falgueras A, et al. Incomplete penetrance and variable expressivity: hallmarks in channelopathies associated with sudden cardiac death. Biology. (2017) 7:3. doi: 10.3390/biology7010003
33. Fan LL, Huang H, Jin JY, Li JJ, Chen YQ, Xiang R. Whole-exome sequencing identifies a novel mutation (p.L320R) of alpha-actinin 2 in a Chinese family with dilated cardiomyopathy and ventricular tachycardia. Cytogenet Genome Res. (2019) 157:148–52. doi: 10.1159/000496077
34. Murphy AC, Young PW. The actinin family of actin cross-linking proteins - a genetic perspective. Cell Biosci. (2015) 5:49. doi: 10.1186/s13578-015-0029-7
35. Lornage X, Romero NB, Grosgogeat CA, Malfatti E, Donkervoort S, Marchetti MM, et al. ACTN2 mutations cause “multiple structured core disease” (MsCD). Acta Neuropathol. (2019) 137:501–19. doi: 10.1007/s00401-019-01963-8
36. Savarese M, Palmio J, Poza JJ, Weinberg J, Olive M, Cobo AM, et al. Actininopathy: A new muscular dystrophy caused by ACTN2 dominant mutations. Ann Neurol. (2019) 85:899–906. doi: 10.1002/ana.25470
37. Jefferies JL, Wilkinson JD, Sleeper LA, Colan SD, Lu M, Pahl E, et al. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: results from the pediatric cardiomyopathy registry. J Card Fail. (2015) 21:877–84. doi: 10.1016/j.cardfail.2015.06.381
38. Deltas C. Digenic inheritance and genetic modifiers. Clin Genet. (2018) 93:429–38. doi: 10.1111/cge.13150
Keywords: left ventricular non-compaction, heterogeneous phenotype, ACTN2 variant, exome sequencing, cardiac magnetic resonance imaging
Citation: Park J, Cho YG, Park HW and Cho JS (2021) Case Report: Novel Likely Pathogenic ACTN2 Variant Causing Heterogeneous Phenotype in a Korean Family With Left Ventricular Non-compaction. Front. Pediatr. 9:609389. doi: 10.3389/fped.2021.609389
Received: 23 September 2020; Accepted: 08 March 2021;
Published: 30 March 2021.
Edited by:
Swarkar Sharma, Shri Mata Vaishno Devi University, IndiaReviewed by:
Marco Savarese, University of Helsinki, FinlandLiang-Liang Fan, Central South University, China
Arshia Angural, Shri Mata Vaishno Devi University, India
Copyright © 2021 Park, Cho, Park and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jung Sun Cho, dHdvcnVnaUBkYXVtLm5ldA==
†These authors have contributed equally to this work