 Marc Gibaud1
Marc Gibaud1 Magalie Barth2
Magalie Barth2 Jérémie Lefranc3Karine Mention4Nathalie Villeneuve5Manuel Schiff6Hélène Maurey7Marie-Anne Barthez8Isabelle Caubel9
Jérémie Lefranc3Karine Mention4Nathalie Villeneuve5Manuel Schiff6Hélène Maurey7Marie-Anne Barthez8Isabelle Caubel9 Mondher Chouchane10Diane Doummar11
Mondher Chouchane10Diane Doummar11 Manoëlle Kossorotoff12Marie-Dominique Lamblin13
Manoëlle Kossorotoff12Marie-Dominique Lamblin13 Agathe Roubertie14
Agathe Roubertie14 Rima Nabbout12
Rima Nabbout12 Patrick Van Bogaert1,15*
Patrick Van Bogaert1,15*- 1Service de neuropédiatrie, CHU d'Angers, Angers, France
- 2Service de génétique médicale, CHU d'Angers, Angers, France
- 3Service de pédiatrie, CHU de Brest, Brest, France
- 4Centre de référence des Maladies Héréditaires du métabolisme, Hôpital Jeanne de Flandre CHRU Lille, Lille, France
- 5Service de neuropédiatrie, Hôpital de la Timone, APHM Marseille, Marseille, France
- 6Centre de référence maladies héréditaires du métabolisme Hôpital Robert Debré, APHP Paris, Paris, France
- 7Service de neuropédiatrie Hôpital Kremlin-Bicêtre APHP Paris, Paris, France
- 8Service de neuropédiatrie, CHU de Tours, Tours, France
- 9Service de pédiatrie, CH Lorient, Lorient, France
- 10Service de pédiatrie, CHU de Dijon, Dijon, France
- 11Service de neuropédiatrie, Hôpital d'Enfants Armand-Trousseau APHP Paris, Paris, France
- 12Service de neuropédiatrie et maladies métaboliques, Hôpital Necker-Enfants Malades APHP Paris, Paris, France
- 13Service de physiologie et explorations fonctionnelles, Hôpital Jeanne de Flandre CHRU Lille, Lille, France
- 14Service de neuropédiatrie, CHU de Montpellier, Montpellier, France
- 15Laboratoire Angevin de Recherche en Ingénierie des Systèmes (LARIS), Université d'Angers, Angers, France
Objective: To characterize the electro-clinical presentation of patients with pyridoxine-dependent epilepsy (PDE) and pyridoxal phosphate (PLP)-dependent epilepsy in order to determine whether some of them could be diagnosed as de novo West syndrome, i. e., West syndrome that starts after the age of 2 months without other types of seizures (focal seizures for instance) before the onset of epileptic spasms.
Methods: We analyzed data from an unpublished cohort of 28 genetically confirmed cases of PDE with antiquitine (ATQ) deficiency and performed a review of the literature looking for description of West syndrome in patients with either PDE with ATQ deficiency or PLP-dependent epilepsy with Pyridox(am)ine phosphate oxidase (PNPO) deficiency.
Results: Of the 28 cases from the ATQ deficiency French cohort, 5 had spasms. In four cases, spasms were associated with other types of seizures (myoclonus, focal seizures). In the last case, seizures started on the day of birth. None of these cases corresponded to de novo West syndrome. The review of the literature found only one case of PNPO deficiency presenting as de novo West syndrome and no case of ATQ deficiency.
Significance: The presentation of PDE- and PLP-dependent epilepsy as de novo West syndrome is so exceptional that it probably does not justify a systematic trial of pyridoxine or PLP. We propose considering a therapeutic trial with these vitamins in West syndrome if spasms are associated with other seizure types or start before the age of 2 months.
Introduction
West syndrome is an epilepsy syndrome arising between 2 months and 1 year, with an incidence estimated at 2–3/10,000 of live birth (1). Epileptic spasms are the hallmark of West syndrome. These epileptic seizures are characterized by contractions of the axial musculature of variable intensity, ranging from brief head nodding to massive abduction and flexion of the upper limbs, and occurring in clusters. A very abnormal interictal EEG pattern called hypsarrhythmia, i.e., high voltage, asynchronous, slow activity mixed with multifocal spikes and sharp waves, and arrest of psychomotor development complete the classical triad of West syndrome (2). Developmental arrest may be absent at the onset of spasms and is therefore not a mandatory feature to pose a diagnosis of West syndrome. In some patients, seizures occur before the age of 2 months, showing either a semiology of spasms or a different semiology (focal seizure, myoclonic seizure, and tonic seizures). When associated with a suppression-burst (SB) pattern on EEG, these early seizures are part of early epileptic encephalopathy with SB, which is a different epileptic syndrome than West syndrome but may evolve to West syndrome with age. Some patients may also start an epileptic disorder after the age of 2 months with other types of seizures (focal seizures for instance) before the onset of infantile spasms. In the other cases (de novo cases), epileptic spasms are the first epileptic manifestations of West syndrome.
Etiologies of West syndrome are variable, including structural, genetic, metabolic, and unknown etiologies. The structural group includes patients with malformations of cerebral cortical development, including tuberous sclerosis complex, and those with other types of prenatal, perinatal, or postnatal lesions of ischemic, infectious, traumatic, or even tumoral origin. The genetic etiologies include chromosomal aberrations and rearrangements, and mutations of various genes (CDKL5, ARX, etc.) that are usually associated with developmental delay prior to the onset of spasms. The metabolic etiologies were recently reviewed by Salar et al. (3). The authors conclude that West syndrome has a metabolic cause in about 3–5% of the cases in developed countries, but this proportion is higher in countries with high level of consanguinity (4). According to this paper, phenylcetonuria and related disorders are the most prevalent causes, and other metabolic causes include disorders of glucose metabolism and transport, Menkes disease, mitochondrial disorders, organic aciduria, biotinidase deficiency, congenital disorders of glycosylation, and vitamin dependencies: pyridoxine (vitamin B6) and pyridoxal phosphate (PLP) dependencies.
Pyridoxine and PLP dependencies are different diseases resulting in a deficit in PLP, which is the biologically active form of pyridoxine. It is a very important vitamin for brain functioning, as it is the cofactor for many enzymatic reactions, some of them being involved in epileptogenesis through GABA and glutamate metabolism (5). Pyridoxine-dependent epilepsy (PDE) is an epileptic syndrome with heterogeneous genetic background. Most cases are related to homozygous mutation of the ALDH7A1 gene encoding for antiquitine (ATQ), a key enzyme in the metabolism of lysine (5). This disease is not exceptional, with an incidence estimated at about 1:64,000 live births, and represents the most frequent form of PDE (6). In this disease as well as in hyperprolinemia type 2, metabolites such as pipecolic acid, alpha-aminoadipic semialdehyde (AASA), and Δ1-piperideine-6-carboxylate accumulate. The accumulation of these compounds will result in inactivation of PLP and possibly in neurotoxicity. Other cases of PDE are related to mutations of proline synthetase co-transcribed homolog (PROSC), a protein renamed as PLP-binding protein (PLPBP) and involved in the homeostasic regulation of PLP (7). PDE can also be related to impairment of PLP import into the brain, such as in hypophosphatasia and glycosylphosphatidylinositol (GPI) anchor synthesis defects (5). Finally, PDE has been described in molybdenum cofactor deficiency (8). PLP-dependent epilepsy is rarer than PDE and is related to Pyridox(am)ine phosphate oxidase (PNPO) deficiency, i.e., the enzyme that catalyzes the reaction from Pyridoxine-phosphate or Pyridoxamine-phosphate to PLP. Patients typically respond to PLP but not to pyridoxine, though partial response to pyridoxine has been described and some patients with PNPO deficiency for unknown reasons only respond to pyridoxine (9–11). In PDE as well as in PLP-dependent epilepsy, seizures typically start in the neonatal period and patients show an association of different types of seizures that may be either generalized (spasms, myoclonic, or tonic seizures) or focal (12, 13). They may lead to drug-resistant status epilepticus. The SB pattern or a combination of continuous and discontinuous patterns was considered as suggestive of B6 dependency (14). However, SB was found in only 21% of the patients in a more recent and larger series (15). Moreover, in that series, seizures had onset after the age of 1 month in 11% of the cases. PDE related to ATQ deficiency may be screened in searching for elevated AASA in urines, the test being highly sensitive and specific (15). However, measurement of AASA in urine is only available in specialized labs and, due to its instability, requires thorough cold chain handling during transport and storage. Pipecolic acid measurement is more readily available but less specific for ATQ deficiency. As these metabolic biomarkers of PDE are not available for rapid diagnosis, trials with pyridoxine (30 mg/kg/day over 1–3 days), followed by add-on of folinic acid (3–5 mg/kg/day) if ineffective, and then switch to PLP (30–60 mg/kg/day over 3 days) if ineffective, are recommended when PDE is suspected (5).
Guidelines for the treatment of West syndrome include hormonal therapy (ACTH, hydrocortisone, or prednisolone) and vigabatrin as first-line treatments. Recently, the association of hormonal therapy and vigabatrin was shown as more efficient than hormonal therapy alone in patients aged from 2 to 14 months of age with West syndrome of recent onset (16).
Early response is a key point for the treatment of West syndrome, especially in patients of unknown etiology and normal psychomotor development prior to the onset of spasms. Indeed, early effective treatment with ACTH was associated with normal or only slightly impaired cognitive outcome in 70–100% in two reported series (17, 18). It is now well admitted that longer lead time to treatment is associated with poorer outcome (19, 20). Rapid recognition of vitamin-dependant epilepsy is also a key point for long-term outcome as early treatment with pyridoxine or PLP may lead to better psychomotor development (10, 21). Consensus guidelines for the management of PDE related to ATQ deficiency have recently been published and include the implementation of lysine restricted diet and arginine supplementation in order to improve the cognitive outcome (22). However, a trial with pyridoxine or PLP is only rarely proposed in patients with West syndrome of unknown etiology (23).
The purpose of this study is to characterize the electro-clinical features of patients with PDE and PLP-dependent epilepsy in order to determine whether some of them could be diagnosed as West syndrome, either de novo or following another type of epilepsy. This should prompt to propose a trial with pyridoxine or PLP in a subgroup of patients with West syndrome before starting a more classical treatment associating hormonal therapy and vigabatrin. To achieve this goal, electro-clinical data from an unpublished cohort of PDE related to ATQ deficiency from France were analyzed, and the current literature was reviewed.
Materials and Methods
French Cohort of Patients With ATQ Deficiency
A cohort of patients with PDE due to ATQ deficiency was recruited through an announcement diffused to the physicians participating to the Reference Center for Rares Epilepsies, based in Hôpital Necker-Enfants Malades (Paris), which constitutes a network covering almost all the country. Electro-clinical data were collected by the same physician (M.G.) from the patients' hospital medical files. Data collected focused on familial history of epilepsy, course of pregnancy, delivery, clinical signs preceding seizures, age at onset of seizures, type of first seizures, initial treatment, pyridoxine sensitivity, AED response, alternative treatments (folinic acid, lysine-restricted diet), clinical condition in the first months, seizure control, psychomotor development, EEG before and after pyridoxine administration, cerebral imaging, school performance, metabolic biomarkers, and genetics. It should be noted that there is no agreement between authors on the semiology features that allow distinction between epileptic spasms and tonic seizures in newborns. In describing the syndrome that bears his name, Ohtahara speaks about “tonic spams with or without clustering” (24). In this study, we decided to speak about epileptic spasms for seizures that occurred in clusters. Isolated events were called tonic or myoclonic seizures according to the duration of seizures and the ictal pattern on EEG if available.
Bibliographic Analysis
We investigated in the literature the presence of cases of vitamins dependent epilepsy, which presented epileptic spasms in the course of the disease. This search was done under PubMed for English language using the following associations of keywords:
Antiquitine + (infantile spasm or west syndrome)
ALDH7A1 + (infantile spasm or west syndrome)
PNPO + (infantile spasm or west syndrome)
Pyridoxine + (infantile spasm or west syndrome)
PLPBP + (infantile spasm or west syndrome): 0
MOSC + (infantile spasm or west syndrome): 0
Hyperprolinemia + (infantile spasm or west syndrome): 0
Hypophosphatasia + (infantile spasm or west syndrome): 3
GPI anchor + (infantile spasm or west syndrome): 8
Molybdenum cofactor + (infantile spasm or west syndrome): 4.
We considered here West syndrome when there was an association of clusters of spasms arising or persisting after the age of 2 months and an EEG pattern of hypsarrhythmia. In patients without genetic confirmation of either ATQ or PNPO deficiency, we considered that the diagnosis of PDE or PLP-dependent epilepsy was likely only in patients with epilepsy that responded to one of these two vitamins and either recurred after vitamin withdrawal or presented biological markers of these diseases. We did not include patients described only in abstracts or those with limited clinical information.
Results
Cohort of Patients With ATQ Deficiency
We collected a previously non-reported cohort of 28 patients with PDE due to ATQ deficiency with genetic mutations. Clinical characteristics are summarized in Table 1.
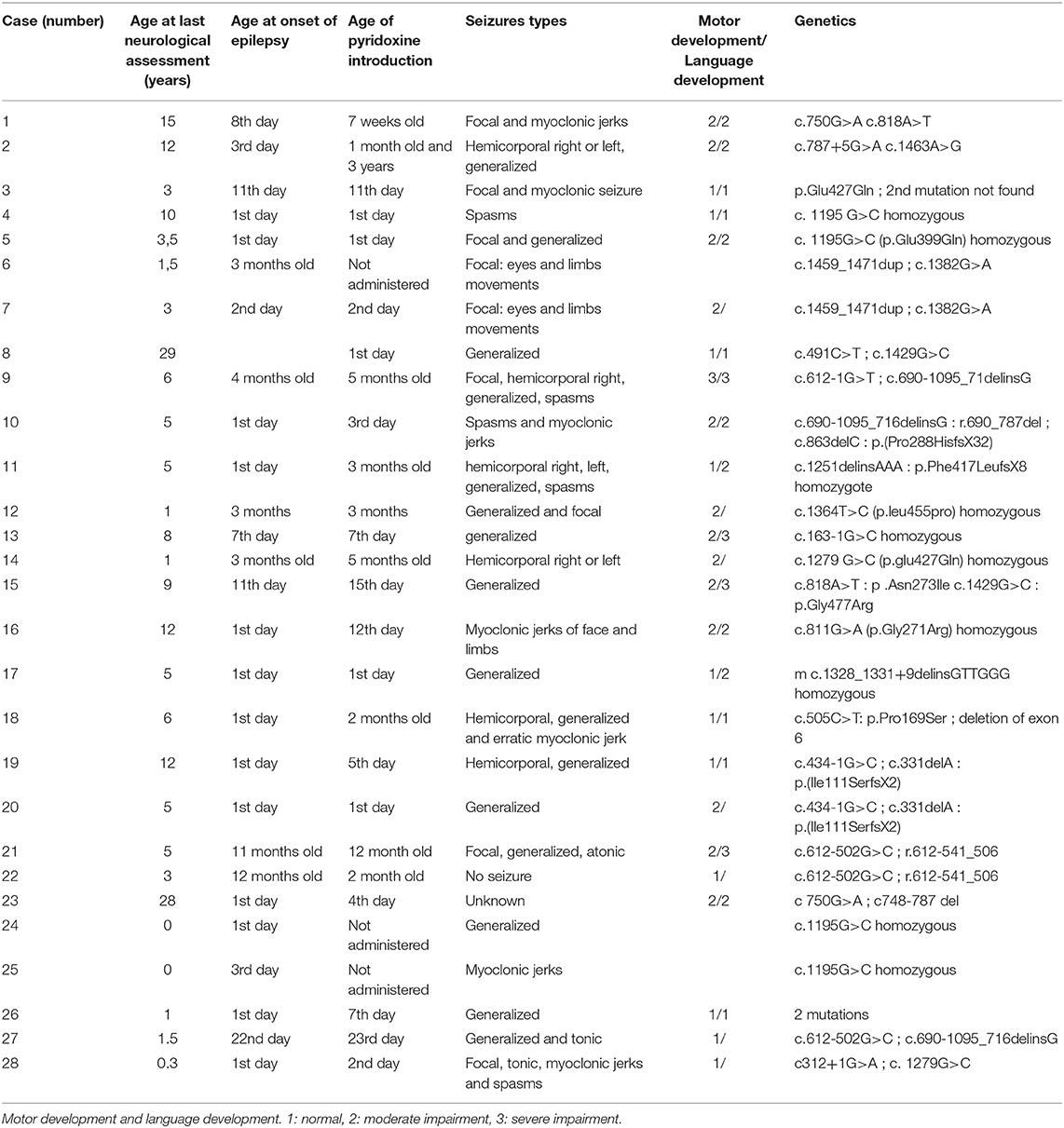
Table 1. Clinical description of the cases, and genetic mutations of ALDH7A1 gene.
All but one patient presented epileptic seizures, a patient diagnosed on the presence of familial history who received pyridoxine from the first day of life. The age at onset of seizures was within the 24 h after birth in 14/28 (50%), and in the first 2 months in 21/28 (75%). Pyridoxine was never started in three children, who died.
The children presented different types of seizures: focal seizures in 14 patients, myoclonic seizures in 7, generalized tonic–clonic seizures in 16, and epileptic spasms in 5. Fifteen children presented more than one type of seizure.
Epileptic spasms were associated with other types of seizures in 4/5 and started between 1 and 16 weeks of life. Only one patient showed spasms only (case # 4). In this patient, the spasms started on the first day of life, and the EEG showed a SB pattern. In the six patients with onset of seizures after the age of 2 months, only one had epileptic spasms. This patient (case # 9) started her epilepsy at the age of 4 months with focal seizures, and then presented epileptic spasms. Her psychomotor development was mildly impaired from birth, with delayed ocular fixation. Interictal EEG performed at the onset of epileptic spasms showed an atypical hypsarrhythmic pattern.
Neurological development was very often impaired. At an average age at last assessment of 7.3 years (standard deviation: 5.4), only 8 children had normal development, 12 had moderate impairment, 4 had severe impairment, and 3 had died. Data were missing in one patient.
To sum up, none of the patients from this large cohort of PDE were diagnosed as de novo West syndrome.
Review of the Literature
The different combinations of searches made it possible to find 37 articles. Ten of them reported relevant cases.
In the North American cohort of PDE published by Bennett et al. (25), four patients presented with epileptic spasms. Two of them were published as K30031 and two by Battaglioli et al. (26). They are two siblings who both had epileptic spasms at 6 months of age. Results of EEG at diagnosis of epileptic spams are not provided. The boy did not develop severe epilepsy later, and his EEG was normal at age 4. Pyridoxine had a beneficial effect on language and cognition at age 8 years. His sister was treated with pyridoxine at the age of 6 months, allowing a resolution of spasms in 3 days. There was no mention of stopping pyridoxine supplementation to assert PDE. Neither mutation nor deletion in the ALDH7A1 gene was found and the level of pipecolic acid was normal in the second child. The third case (K3009) was published by Bennett et al. (27). This girl experienced epileptic spasms at the age of 3 months, resistant to ACTH and vigabatrin, but with a sustained response to pyridoxine. The search for mutation or deletion in the ALDH7A1 gene was negative and the blood level of pipecolic acid was normal (28). The fourth case is a boy having started spasms with hypsarrhythmia at 5 months that were drug-resistant but rapidly responded to pyridoxine. No mutation or deletion in the ALDH7A1 gene was found, and the level of pipecolic acid was normal (28). To sum up, these four patients presented as West syndrome that responded to pyridoxine, but genetic investigations were negative and biological markers for ATQ deficiency were absent. Therefore, we cannot conclude that these four patients had PDE presenting as West syndrome, even if PDE related to another gene, that ALDH7A1 gene remains a possibility.
The article published by Mills et al. (29) reports a child (patient 7, see additional data of this article) who started his epilepsy at 5 months of life with epileptic spasms, without other types of seizures and hypsarrhythmia on EEG. Her epilepsy was rapidly controlled by vigabatrin and PLP. When PLP was removed, the seizures recurred. Evolution was favorable for epilepsy and development with PLP monotherapy. The molecular diagnosis of PNPO deficiency was confirmed (homozygous R116Q mutation). So, the clinical presentation for this patient is de novo West syndrome.
In a recent article published by Salar et al. (3), the authors list the metabolic causes of West syndrome. It is mentioned in this article that the deficit in ATQ can be a cause, citing Liu et al. (30) who reported a patient with clinical response to pyridoxine but without PDE as seizures did not recur when pyridoxine was stopped.
The study published in 2018 by Xue et al. (31) describes characteristics of 744 patients with epileptic spasms. Eleven of them had a response to pyridoxine alone or had hypsarrhythmic EEG, and spasms were not preceded by other types of seizures. However, no mutation was found for the ALDH7A1 or PNPO genes, and other genes of PDE were not researched.
In a genetic study of epileptic spasms (32), there was 1 case who presented a mutation of PNPO. His epilepsy began on the first day of life, with myoclonus and focal seizures, and SB pattern on EEG. A trial of IV pyridoxine followed by oral pyridoxine was unsuccessful. PLP was not tried. He developed flexion spasms at 3.5 months of age, followed by recurrent status epilepticus. The spasms were drug-resistant. So, this patient had early epileptic encephalopathy with SB that evolved to West syndrome.
Finally, a case of West syndrome related to hypophosphatasia responding remarkably to PLP after failure of many anti-epileptic drugs was reported (33). This patient had intractable tonic seizures and a SB pattern from day 2, and epileptic spasms associated with hypsarrhythmia occurred at the age of 2 months. So, this patient did not present as de novo West syndrome, and had in addition clinical and radiological features of skeletal dysplasia.
Discussion
We report here a new cohort of 28 patients with PDE and ALDH7A1 mutation. This is one of the larger series reported so far. The analysis of this French cohort did not identify any cases presenting as typical de novo West syndrome, i.e., onset of epilepsy with epileptic spasms after the age of 2 months and hypsarrhythmia on EEG. Indeed, the five patients who presented epileptic spasms had either early neonatal epilepsy, or other types of seizures before the onset of spasms. In addition, none of these five patients did show a typical hypsarrhythmia on EEG. The review of the literature identified a single patient with PLP-dependent epilepsy due to PNPO deficiency who presented as a typical de novo West syndrome (29). To our knowledge, this is the only published case, and we did not find any case of PDE presenting as de novo West syndrome.
There is abundant but discordant literature on the use of pyridoxine and PLP in the treatment of West syndrome whatever the underlying etiology, suggesting a non-specific anti-epileptic effect. Pyridoxine and PSP were considered as very important treatment options in a literature of the 80s−90s, mainly issued from Japan (34, 35). More recent studies are less enthusiastic. Debus et al. (36) did not find any efficacy in 37 patients, and Heiskala et al. (37) reported only one success out of 30 patients. Kunnanayaka et al. (38) compared the combination of pyridoxine–prednisone vs. prednisolone alone in a population of 62 infants and did not find any significant difference. In the study published by Kaushik et al. (39), 135 out of 144 patients with epileptic spasms received pyridoxine, with partial response (>50% improvement) in 12 cases (8.9%) and complete spasm cessation in only 1 patient (0.7%). Thus, high-dose Vitamin B6 might have some antiepileptic action in West syndrome, but the level of evidence is very low.
In conclusion, data from the literature and data analysis of this cohort of patients suggest that de novo West syndrome is not a typical feature of PDE or PNPO dependency. Spasms are among the types of seizures presented by patients with PDE but, unlike de novo West syndrome, these patients usually present other types of seizures, have an earlier epilepsy onset (predominantly in their first month of life), and do not present hypsarrhythmia but other types of EEG abnormalities, like the SB pattern. We think that it is not legitimate to propose a systematic pyridoxine or PLP trial in classical de novo West syndrome as it is likely to delay the use of other therapeutic strategies, such as hormonal therapy and vigabatrin, which could have a strong negative impact on the long-term outcome (19, 20). This is in line with the recommendations of the American Academy of Neurology and of the Practice Committee of the Child Neurology Society for treatment of West syndrome (40). On the other hand, vitamin trials and metabolic screening should be considered when the epileptic spasms have an onset before the age of 2 months, are associated with a SB aspect on EEG, are preceded by other types of seizures, or do not respond to first-line treatments.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
MG and MB: study design, data collection, data analysis, data interpretation, drafting and revising the manuscript. JL, KM, NV, MS, HM, M-AB, IC, MC, DD, MK, M-DL, AR, and RN: data collection, data analysis, data interpretation, drafting and revising the manuscript. PV: study design, data analysis, data interpretation, drafting and revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Association pour le développement de la neuropéditrie—ADN Anjou.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Trevathan E, Murphy CC, Yeargin-Allsopp M. The descriptive epidemiology of infantile spasms among atlanta children. Epilepsia. (1999) 40:748–51. doi: 10.1111/j.1528-1157.1999.tb00773.x
2. Hrachovy RA, Frost JD. Infantile spasms. Handb Clin Neurol. (2013) 111:611–8. doi: 10.1016/B978-0-444-52891-9.00063-4
3. Salar S, Moshé SL, Galanopoulou AS. Metabolic etiologies in West syndrome. Epilepsia Open. (2018) 3:134–66. doi: 10.1002/epi4.12102
4. Alrifai MT, AlShaya MA, Abulaban A, Alfadhel M. Hereditary neurometabolic causes of infantile spasms in 80 children presenting to a tertiary care center. Pediatr Neurol. (2014) 51:390–7. doi: 10.1016/j.pediatrneurol.2014.05.015
5. Wilson MP, Plecko B, Mills PB, Clayton PT. Disorders affecting vitamin B 6 metabolism. J Inherit Metab Dis. (2019) 42:629–46. doi: 10.1002/jimd.12060
6. Coughlin CR II, van Karnebeek CDM, Al-Hertani W, Shuen AY, Jaggumantri S, Jack RM, et al. Triple therapy with pyridoxine, arginine supplementation and dietary lysine restriction in pyridoxine-dependent epilepsy: neurodevelopmental outcome. Mol Genet Metab. (2015) 116:35–43. doi: 10.1016/j.ymgme.2015.05.011
7. Darin N, Reid E, Prunetti L, Samuelsson L, Husain RA, Wilson M, et al. Mutations in PROSC disrupt cellular pyridoxal phosphate homeostasis and cause vitamin-B 6 -dependent epilepsy. Am J Hum Genet. (2016) 99:1325–37. doi: 10.1016/j.ajhg.2016.10.011
8. Struys EA, Nota B, Bakkali A, Al Shahwan S, Salomons GS, Tabarki B. Pyridoxine-dependent epilepsy with elevated urinary α-amino adipic semialdehyde in molybdenum cofactor deficiency. Pediatrics. (2012) 130:e1716-1719. doi: 10.1542/peds.2012-1094
9. Plecko B, Paul K, Mills P, Clayton P, Paschke E, Maier O, et al. Pyridoxine responsiveness in novel mutations of the PNPO gene. Neurology. (2014) 82:1425–33. doi: 10.1212/WNL.0000000000000344
10. Mills PB, Camuzeaux SSM, Footitt EJ, Mills KA, Gissen P, Fisher L, et al. Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome. Brain J. Neurol. (2014) 137(Pt 5):1350–60. doi: 10.1093/brain/awu051
11. Pearl PL, Hyland K, Chiles J, McGavin CL, Yu Y, Taylor D. Partial pyridoxine responsiveness in PNPO deficiency. JIMD Rep. (2013) 9:139–42. doi: 10.1007/8904_2012_194
12. Bok LA, Halbertsma FJ, Houterman S, Wevers RA, Vreeswijk C, Jakobs C, et al. Long-term outcome in pyridoxine-dependent epilepsy: long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol. (2012) 54:849–54. doi: 10.1111/j.1469-8749.2012.04347.x
13. van Karnebeek CDM, Tiebout SA, Niermeijer J, Poll-The BT, Ghani A, Coughlin CR II, et al. Pyridoxine-dependent epilepsy: an expanding clinical spectrum. Pediatr Neurol. (2016) 59:6–12. doi: 10.1016/j.pediatrneurol.2015.12.013
14. Nabbout R, Soufflet C, Plouin P, Dulac O. Pyridoxine dependent epilepsy: a suggestive electroclinical pattern. Arch Dis Child Fetal Neonatal Ed. (1999) 81:F125–9. doi: 10.1136/fn.81.2.F125
15. Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, Varadkar S, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain. (2010) 133:2148–59. doi: 10.1093/brain/awq143
16. O'Callaghan FJK, Edwards SW, Alber FD, Hancock E, Johnson AL, Kennedy CR, et al. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. Lancet Neurol. (2017) 16:33–42. doi: 10.1016/S1474-4422(16)30294-0
17. Riikonen R. ACTH therapy of West syndrome: finnish views. Brain Dev. (2001) 23:642–6. doi: 10.1016/S0387-7604(01)00306-0
18. Kivity S, Lerman P, Ariel R, Danziger Y, Mimouni M, Shinnar S, et al. Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia. (2004) 45:255–62. doi: 10.1111/j.0013-9580.2004.30503.x
19. O'Callaghan FJK, Edwards SW, Alber FD, Borja MC, Hancock E, Johnson AL, et al. Vigabatrin with hormonal treatment versus hormonal treatment alone (ICISS) for infantile spasms: 18-month outcomes of an open-label, randomised controlled trial. Lancet Child Adolesc Health. (2018) 2:715–25. doi: 10.1016/S2352-4642(18)30244-X
20. O'Callaghan FJK, Lux AL, Darke K, Edwards SW, Hancock E, Johnson AL, et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study: Lead Time and Age in Infantile Spasms. Epilepsia. (2011) 52:1359–64. doi: 10.1111/j.1528-1167.2011.03127.x
21. Basura GJ, Hagland SP, Wiltse AM, Gospe SM. Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr. (2008) 168:697–704. doi: 10.1007/s00431-008-0823-x
22. Coughlin CR, Tseng LA, Abdenur JE, Ashmore C, Boemer F, Bok LA, et al. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. (2021) 44:178–92. doi: 10.1002/jimd.12332
23. Knupp KG, Leister E, Coryell J, Nickels KC, Ryan N, Juarez-Colunga E, et al. Response to second treatment after initial failed treatment in a multicenter prospective infantile spasms cohort. Epilepsia. (2016) 57:1834–42. doi: 10.1111/epi.13557
24. Ohtahara S, Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res. (2006) 70:58–67. doi: 10.1016/j.eplepsyres.2005.11.021
25. Bennett CL, Chen Y, Hahn S, Glass IA, Gospe SM. Prevalence of ALDH7A1 mutations in 18 North American pyridoxine-dependent seizure (PDS) patients. Epilepsia. (2009) 50:1167–75. doi: 10.1111/j.1528-1167.2008.01816.x
26. Battaglioli G, Rosen DR, Gospe SM, Martin DL. Glutamate decarboxylase is not genetically linked to pyridoxine-dependent seizures. Neurology. (2000) 55:309–11. doi: 10.1212/WNL.55.2.309
27. Bennett CL, Huynh HM, Chance PF, Glass IA, Gospe SM. Genetic heterogeneity for autosomal recessive pyridoxine-dependent seizures. Neurogenetics. (2005) 6:143–9. doi: 10.1007/s10048-005-0221-8
28. Mefford HC, Zemel M, Geraghty E, Cook J, Clayton PT, Paul K, et al. Intragenic deletions of ALDH7A1 in pyridoxine-dependent epilepsy caused by Alu-Alu recombination. Neurology. (2015) 85:756–62. doi: 10.1212/WNL.0000000000001883
29. Mills PB. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum Mol Genet. (2005) 14:1077–86. doi: 10.1093/hmg/ddi120
30. Liu X-M, Li R, Chen S-Z, Sang Y, Chen J, Fan C-H, et al. Screening of inherited metabolic disorders in infants with infantile Spasms. Cell Biochem Biophys. (2015) 72:61–5. doi: 10.1007/s12013-014-0404-8
31. Xue J, Qian P, Li H, Wu Y, Xiong H, Zhang Y-H, et al. Clinical characteristics of two cohorts of infantile spasms: response to pyridoxine or topiramate monotherapy. World J Pediatr WJP. (2018) 14:290–7. doi: 10.1007/s12519-018-0127-9
32. Michaud JL, Lachance M, Hamdan FF, Carmant L, Lortie A, Diadori P, et al. The genetic landscape of infantile spasms. Hum. Mol. Genet. (2014) 23:4846–58. doi: 10.1093/hmg/ddu199
33. Yamamoto H, Sasamoto Y, Miyamoto Y, Murakami H, Kamiyama N. A successful treatment with pyridoxal phosphate for West syndrome in hypophosphatasia. Pediatr Neurol. (2004) 30:216–8. doi: 10.1016/j.pediatrneurol.2003.08.003
34. Ohtsuka Y, Matsuda M, Ogino T, Kobayashi K, Ohtahara S. Treatment of the West syndrome with high-dose pyridoxal phosphate. Brain Dev. (1987) 9:418–21. doi: 10.1016/S0387-7604(87)80116-X
35. Pietz J, Benninger C, Schäfer H, Sontheimer D, Mittermaier G, Rating D. Treatment of infantile spasms with high-dosage vitamin B6. Epilepsia. (1993) 34:757–63. doi: 10.1111/j.1528-1157.1993.tb00458.x
36. Debus OM, Kurlemann G, Study group. Sulthiame in the primary therapy of West syndrome: a randomized double-blind placebo-controlled add-on trial on baseline pyridoxine medication. Epilepsia. (2004) 45:103–8. doi: 10.1111/j.0013-9580.2004.19003.x
37. Heiskala H, Riikonen R, Santavuori P, Simell O, Airaksinen E, Nuutila A, et al. West syndrome: individualized ACTH therapy. Brain Dev. (1996) 18:456–60. doi: 10.1016/S0387-7604(96)00024-1
38. Kunnanayaka V, Jain P, Sharma S, Seth A, Aneja S. Addition of pyridoxine to prednisolone in the treatment of infantile spasms: a pilot, randomized controlled trial. Neurol India. (2018) 66:385–90. doi: 10.4103/0028-3886.227281
39. Kaushik JS, Patra B, Sharma S, Yadav D, Aneja S. Clinical spectrum and treatment outcome of West Syndrome in children from Northern India. Seizure. (2013) 22:617–21. doi: 10.1016/j.seizure.2013.04.014
40. Go CY, Mackay MT, Weiss SK, Stephens D, Adams-Webber T, Ashwal S, et al. Evidence-based guideline update: medical treatment of infantile spasms. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. (2012) 78:1974–80. doi: 10.1212/WNL.0b013e318259e2cf
Keywords: epilepsy, West syndrome, infantile, spasms, pyridoxine, pyridoxal phosphate, antiquitine, B6
Citation: Gibaud M, Barth M, Lefranc J, Mention K, Villeneuve N, Schiff M, Maurey H, Barthez M-A, Caubel I, Chouchane M, Doummar D, Kossorotoff M, Lamblin M-D, Roubertie A, Nabbout R and Van Bogaert P (2021) West Syndrome Is an Exceptional Presentation of Pyridoxine- and Pyridoxal Phosphate-Dependent Epilepsy: Data From a French Cohort and Review of the Literature. Front. Pediatr. 9:621200. doi: 10.3389/fped.2021.621200
Received: 25 October 2020; Accepted: 04 January 2021;
Published: 05 March 2021.
Edited by:
Pasquale Striano, University of Genoa, ItalyReviewed by:
Raffaele Falsaperla, University Hospital Polyclinic Vittorio Emanuele, ItalyLucia Fusco, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2021 Gibaud, Barth, Lefranc, Mention, Villeneuve, Schiff, Maurey, Barthez, Caubel, Chouchane, Doummar, Kossorotoff, Lamblin, Roubertie, Nabbout and Van Bogaert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Van Bogaert, cGF0cmljay52YW5ib2dhZXJ0QGNodS1hbmdlcnMuZnI=