 Pai-Jui Yeh
Pai-Jui Yeh Shiu-Feng Huang2,3
Shiu-Feng Huang2,3 Ming-Chou Chiang
Ming-Chou Chiang Chao-Jan Wang
Chao-Jan Wang- 1Division of Pediatric Gastroenterology, Department of Pediatrics, Chang Gung Memorial Hospital, Linkou Branch and Chang Gung University College of Medicine, Taoyuan, Taiwan
- 2Department of Pathology, Chang Gung Memorial Hospital, Linkou Branch and Chang Gung University College of Medicine, Taoyuan, Taiwan
- 3Institute of Molecular and Genomic Medicine, National Health Research Institutes, Zhunan, Taiwan
- 4Division of Neonatology, Department of Pediatrics, Chang Gung Memorial Hospital, Linkou Branch and Chang Gung University College of Medicine, Taoyuan, Taiwan
- 5Department of Medical Imaging and Intervention, Chang Gung Memorial Hospital, Linkou Branch and Chang Gung University College of Medicine, Taoyuan, Taiwan
- 6Liver Research Center, Chang Gung Memorial Hospital, Linkou Branch, Taoyuan, Taiwan
Background: Gestational alloimmune liver disease (GALD) is a rare but critical cause of neonatal liver failure. After discovering the maternal–fetal alloimmune mechanism, intravenous immunoglobulin (IVIG) with or without exchange transfusion (ET) has gradually replaced antioxidant cocktails as the first-line therapy. Whether such therapy changes the outcome of neonates with GALD is yet to be defined.
Method: We reported a pair of twins with discordant presentations, mild and self-limited in the older, whereas liver failure in the younger, who was successfully rescued by ET and IVIG. To investigate the outcome after therapeutic alteration, 39 cases between 2005 and 2020 from literature research were collected.
Results: Half of the collected cases (47.1%) were preterm. Common presentations were ascites, jaundice, respiratory distress, hepatomegaly, and edema. Leading laboratory abnormalities were coagulopathy, hypoalbuminemia, and elevated serum ferritin. Salivary gland biopsy and magnetic resonance imaging detected extrahepatic siderosis in 70% (14/20) and 56% (14/25), respectively. IVIG, ET, and liver transplantation were performed in 19 (48.7%), 15 (38.5%), and 8 (20.5%) patients, respectively. The overall survival (OS) rate and native liver survival (NLS) rate were 64.1% (25/39) and 43.6% (17/39), respectively. Although the compiled results did not support a significant benefit, the OS and NLS were higher in the IVIG with/without ET group compared with those treated with conventional therapy [OS (70 vs. 57.9%) and NLS (55 vs. 31.6%), respectively].
Conclusion: A high index of suspicion for GALD is crucial when facing a neonate with liver failure. Despite no significant influence on the outcome over conventional therapy in such a rare and detrimental disease, IVIG with or without ET can be worth trying before resorting to liver transplantation, which is resource-demanding and technique-challenging in small infants.
Introduction
Gestational alloimmune liver disease (GALD), currently regarded as a maternal–fetal alloimmune disorder, is a leading etiology of neonatal liver failure and neonatal hemochromatosis (NH) (1). Patients mostly present with congenital cirrhosis, whereas some develop hyperacute process, leading to stillbirth or demise (2). Antenatally, oligohydramnios and intrauterine growth retardation may be noted. Most patients develop clinical signs of liver failure within hours after birth. Laboratory examination may reveal hyperbilirubinemia, coagulopathy, disproportionally normal aminotransferase levels, elevated alpha-fetoprotein, and raised ferritin. Histopathologic and imaging studies are required to confirm extrahepatic siderosis, a fundamental finding for the diagnosis of NH (2). Asides from the classical NH, reports of non-iron-overload cases and novel pathologic features have expanded the spectrum of GALD (2, 3).
Although the causal antigen has not been identified, an in utero alloimmune pathway is attributed as early as the 12th week of gestation, inducing specific transplacental immunoglobulin G to activate the complement system and downstream cascades of fetal liver injury (4). High recurrence in the progenies of the affected mother also reflects the successive impact of maternal alloimmunization. In the past two decades, treatment evolution has been elaborated from the perspective of alloimmunization (4–7). Intravenous immunoglobulin (IVIG) and plasma exchange transfusion (ET) have replaced conventional cocktail therapy (1, 2). The application of gestational IVIG also potentially prevents its recurrence in the next pregnancy (2, 8, 9).
The incidence of GALD-NH is around 4 per 100,000 live births in the United States (10). However, the research from Asia seems scanty. We encountered GALD-associated liver failure in a younger twin successfully rescued with ET and IVIG. Meanwhile, another patient whose diagnosis was made until liver transplantation was reported at a local pediatric gastroenterology meeting in Taiwan. Therefore, it is intriguing to understand the effect of IVIG on this rare disease based on a case series collected from a literature review.
Materials and Methods
Systematic Review of the Literature
A literature search in the PubMed database using keywords “gestational alloimmune liver disease” and “neonatal hemochromatosis” was conducted, retrieving case reports or case series published in English with full text from January 2005 to March 2020. Case series with only summarized or statistical results were excluded.
Extracted data of interest included countries, gestational ages (GAs) (prematurity defined as GA <37 weeks), sexes, birth bodyweights, modes of delivery, antenatal history (intrauterine growth retardation, oligohydramnios, family history, and other specific prenatal diagnoses), clinical presentations, laboratory results (aminotransferases, bilirubin, coagulation function, albumin, alpha-fetoprotein, and ferritin), magnetic resonance imaging (MRI), histopathologic findings (salivary gland or liver), treatments (ET, IVIG, and liver transplantation), outcomes, and the age of the last follow-up. Elevation of ferritin or alpha-fetoprotein was defined if the value exceeded 800 (ng/ml) and 100,000 (ng/ml), respectively (2). Coagulopathy was defined if the international normalized ratio (INR) exceeded 1.5. Hyperbilirubinemia was defined if total bilirubin exceeded 2 mg/dl. Patients with uncertain diagnoses, history of antenatal IVIG prevention, or fetal death were excluded. A proportion was used in some demographics or laboratory results with missing data. Depiction of differential diagnoses of each report was evaluated in detail. Cases with an evident argument for congenital infection, mitochondrial cytopathy, bile acid synthesis defect, transaldolase deficiency, or syndromic presentation would be excluded.
Statistical Analyses
Data collection and processing were conducted using Excel and SPSS v. 20 software. Continuous variables were expressed as means ± standard deviations or as medians with the interquartile range as appropriate. Categorical variables were expressed as percentages and were analyzed using the chi-square test. Comparative analyses of descriptive and inferential data were performed using the Student's t-test. A p-value < 0.05 was considered significant.
Results
Our Case
This female neonate was born to a G1P1 mother via Cesarean section due to twin pregnancy (dizygotic twin) at a local hospital with GA 37 + 3/7 weeks and a birth bodyweight of 2,115 g without any abnormality in the antenatal examination. The baby required oxygen support for respiratory distress. Abdominal distention was noted at 4 days old. Laboratory test revealed white blood cell 3,980/μl, hemoglobin 10.7 g/dl, platelet 74,000/μl, INR 2.53, albumin 1.9 g/dl, direct/total bilirubin 1.78/4.66 mg/dl, and aspartate transaminase/alanine aminotransferase: 55/22 U/L. She was transferred to our neonatal intensive care unit.
The abdominal sonography reported splenomegaly and profuse ascites. Congenital infections and surveys for an inborn error of metabolism were all negative except for a lower carnitine level. Blood, urine, and ascites culture yielded no pathogen. GALD was considered and confirmed by the findings of elevated ferritin (1,010 ng/ml), decreased transferrin (79.9 mg/dl), elevated alpha-fetoprotein (285,834.7 ng/ml), and siderosis of the liver and pancreas by MRI (Figure 1). Double volume ET followed by IVIG (1 g/kg) was completed on the 21st day of life. A liver biopsy performed a month later after the subsidence of coagulopathy revealed cirrhosis and siderosis (grade 1) (Figure 2) and positive immunohistochemical (IHC) stain of C5b-9 (Figure 2). During the subsequent follow-up, bilirubin gradually normalized in 4 months. Fibroscan at 1 year and 9 months old showed a liver stiffness measurement of 8.4 Kpa (F2).
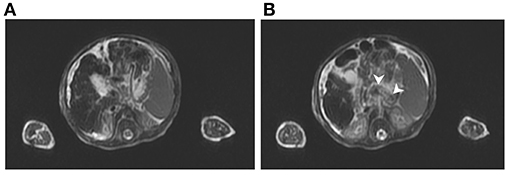
Figure 1. T2-weighted magnetic resonance imaging of case: (A) Siderosis and cirrhosis of liver and (B) metal-induced susceptibility artifact, indicating siderosis of pancreatic body (arrowhead).
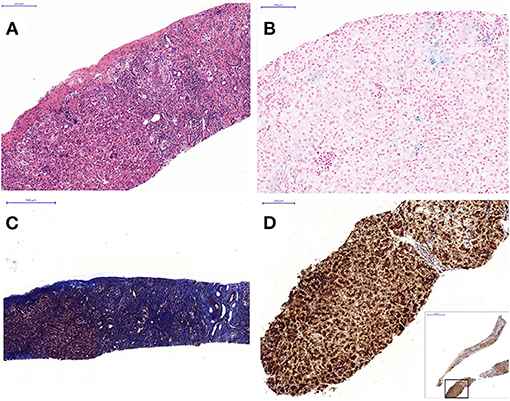
Figure 2. Liver histopathology: (A) hematoxylin and eosin stain shows heavy inflammatory cell infiltration including eosinophils and residual hepatic hematopoiesis (100×), (B) iron stain shows grade 1 siderosis (blue-stained area) (200×), (C) Masson Trichrome stain shows severe fibrosis with cirrhotic change (40×). (D) Immunohistochemistry stain with C5b-9 antibody shows strongly positive cells (brownish stained area) involving 90% of the tissue (100×).
Her dizygotic twin A was born with a birth bodyweight of 2,540 g. Laboratory examination disclosed INR 1.45, aspartate transaminase/alanine aminotransferase 272/43 U/L, direct/total bilirubin 0.95/1.65 mg/dl, albumin 2.2 g/dl, alpha-fetoprotein 97025.8 ng/ml, transferrin 92 mg/dl, and ferritin 762.4 ng/ml. After being transferred to our hospital, repeated laboratory tests did not fulfill the diagnostic levels (ferritin: 358.9 ng/ml, alpha-fetoprotein: 97,025.8 ng/ml). She was kept on an observation plan with continuous improvement.
Because the maternal factors may participate in the pathogenesis, we checked the asymptomatic mother with findings of an elevated serum immunoglobulin G level (2,610 mg/dl), a declined complement level (C3 70.7 mg/dl), positive antinuclear antibody, and positive anti-double-strand DNA antibody.
Case Series
General Demographics
A total of 45 cases from 41 articles were collected from 2005 to 2020. We excluded three cases with antenatal IVIG usage, one with an uncertain diagnosis, one with NH due to mitochondrial long-chain fatty acid oxidation defect, one with Martinez–Frias syndrome, and one fetal death. Together with our twin B, 39 cases were enrolled (Table 1). Most of the patients were reported from the United States. In contrast, Japan and Turkey were two leading countries in Asia.
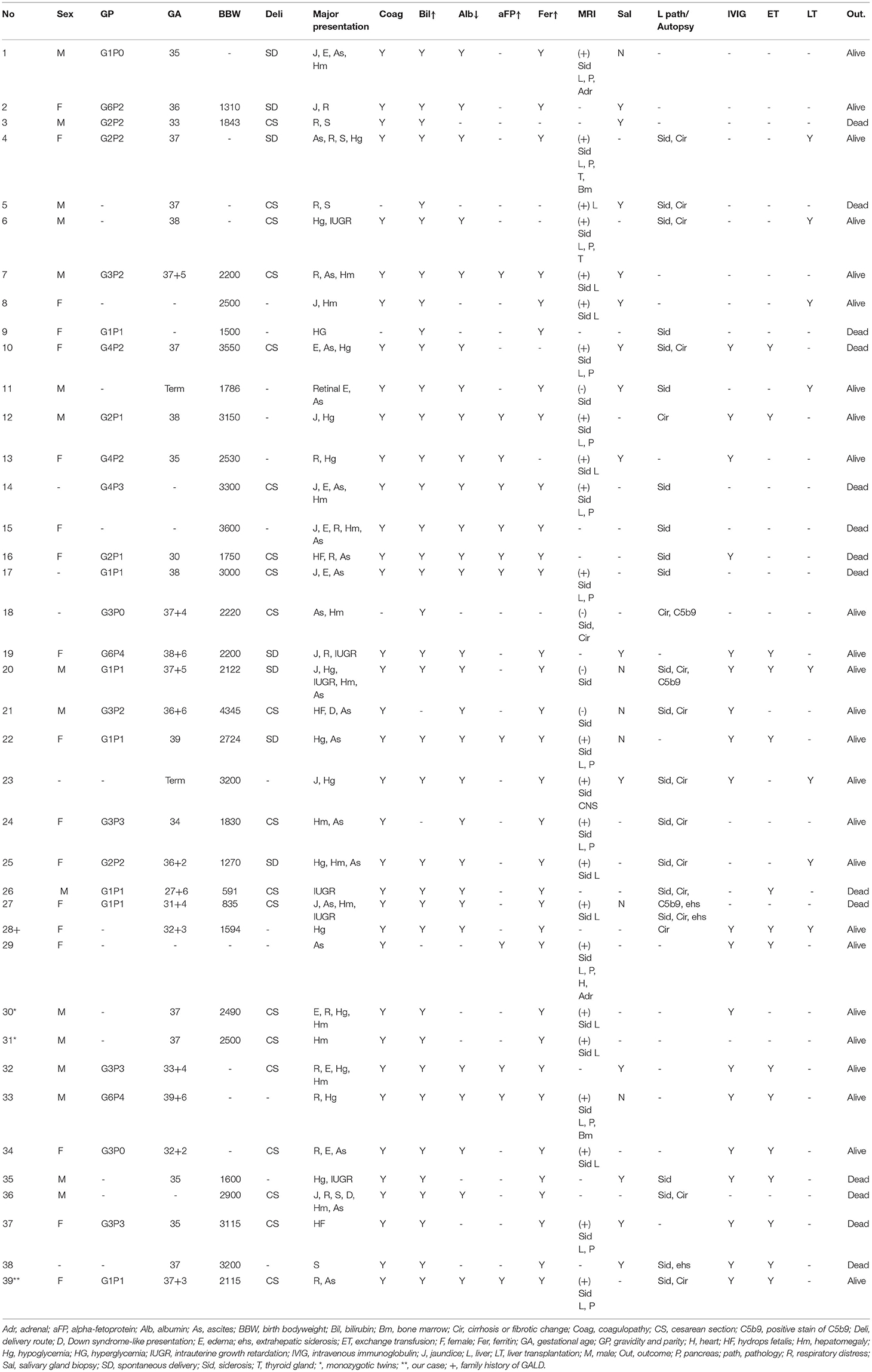
Table 1. Baseline data, presentations, examination results, treatments, and outcomes of the 39 cases.
The male-to-female ratio was close to 1. Prematurity comprised 47.1% of cases, mostly belonging to the late-preterm (35–37 weeks). Intrauterine growth retardation (IUGR) and oligohydramnios were documented in 10 (25.6%) and 5 (12.8%) cases, respectively. A pair of monozygotic twins (case numbers 30 and 31 in Table 1) was included.
Clinical Presentations
The most common presentations were ascites, jaundice, respiratory distress, hypoglycemia, hepatomegaly, and edema (including hydrops fetalis) (Table 2). Down syndrome was observed in three. Several cases developed rare organ dysfunction associated with extrahepatic siderosis, including one with pituitary dysfunction and secondary hypothyroidism and another with bilateral retinal edema. One case developed esophageal varices with hematemesis.
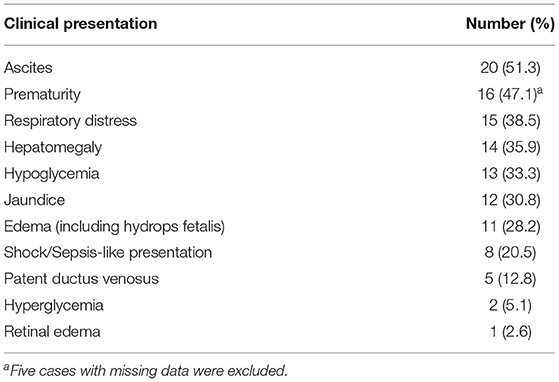
Table 2. Major clinical presentation.
Laboratory Examination and Imaging Evaluation
Coagulopathy, hyperbilirubinemia, hypoalbuminemia, and elevated ferritin were seen in 100% (36/36), 89.2% (33/37), 96.6% (28/29), and 94.3% (33/35) of patients, respectively. However, alpha-fetoprotein and aminotransferase levels were missed in 66.6 and 30.8% of the cases, respectively.
Twenty-five cases (64.1%, 25/39) received an abdominal MRI. Hepatic siderosis was found in 21 (84%, 21/25), and extrahepatic siderosis in 14 (56%, 14/25), involving the pancreas, the heart, and the adrenal gland. A case with adenohypophysis siderosis had a reversal of iron overload after liver transplantation. Via imaging, liver fibrosis or cirrhosis was documented in six (15.4%, 6/39).
Histopathological Findings
Lip or buccal mucosa biopsy was performed in 20 cases (51%, 20/39), with 14 (70%, 14/20) positive for iron deposition. Liver tissue was obtained in 24 patients (62%, 24/39, including autopsy in 8) with iron deposition in 21 (87.5%, 21/24). Regarding the three patients without hepatic siderosis, one demonstrated strong C5b-9 IHC staining, another received biopsy 6 months post-ET and IVIG, and the other presented with marked hepatocytes loss and fibrosis. Only three cases applied C5b-9 IHC staining as a diagnostic modality. Via histopathology, liver fibrosis or cirrhosis was documented in 16 patients (66.7%, 16/24). If combining MRI and histopathology, siderosis was documented in 37 (94.9%, 37/39) and liver fibrosis/cirrhosis in 18 (46.2%, 18/39).
Treatment and Outcome
Conventional cocktail therapy comprising antioxidants, chelating agents, and blood transfusion was applied for the management in the earlier cases. IVIG or ET application became more prevalent after 2010. Seven patients reported before 2010 were all treated “exclusively” with conventional therapy with or without liver transplantation.
IVIG, ET, and liver transplantation were performed in 19 (48.7%), 15 (38.5%), and 8 (20.5%) patients, respectively. Among them, 14 cases (35.9%) received both ET and IVIG. The IVIG dose was 1 g/kg, whereas a few cases received multiple doses up to 5 g/kg in 2 weeks. The administration timing varied as early as the second day of life. As to ET, around 25% of cases underwent multiple courses (33 courses in one). The ET timing usually accompanied or preceded IVIG.
In this series, the overall survival and native liver survival (NLS) rates were 64.1% (25/39) and 43.6% (17/39), respectively. On survival analysis of IVIG with/without ET vs. the conventional cocktail therapy groups, the overall survival and NLS rates were higher in the IVIG with/without ET group, although not statistically significant (Table 3). Hence, a significant benefit was not supported by the current compiled results.
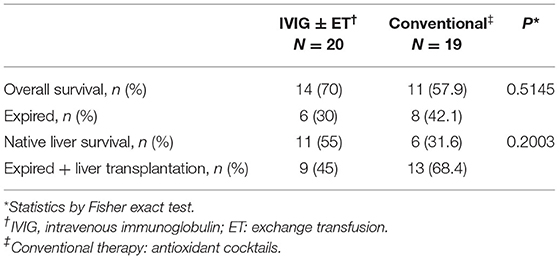
Table 3. Survival rates in GALD cases receiving IVIG with/without ET or conventional therapy.
Discussion
We shared a successful experience of a neonate (the younger twin) with GALD-related liver failure, managed with ET followed by IVIG with rapid clinical improvement, gradual resolution of stigmata of portal hypertension, and regression of liver fibrosis from cirrhosis (F4) to F2 (Fibroscan). Complete regression of cirrhosis has been reported in some cases approximately 2 to 3.5 years later (11).
Interestingly, the twin A of our case presented minor liver dysfunction, echoing the discordant presentations in reported GALD twins (12–14). Similar to different hemolytic severity between twins born to rhesus-sensitized mothers, attributed to alloantigen polymorphism or dissimilar antigen presentation by HLA variants (15), a parallel mechanism may apply to GALD-NH. Dizygosity of our twin may well-explain the different manifestations in siblings born to the same mother.
NH may arise from non-GALD etiologies, including perinatal infection, trisomy 21, mitochondrial DNA depletion due to deoxyguanosine kinase deficiency (DGUOK gene mutations), bile acid synthetic defect (SRD5B1 mutations), GRACILE syndrome (BCS1L mutation), myofibromatosis, tricho-hepato-enteric syndrome, and Martinez–Frias syndrome (2). Although Down syndrome (DS)-associated NH may be explained by transient megakaryocytic leukemia (TML), in some DS with NH, TML was not observed in the pathology or autopsy (16). Maternal antinuclear antibodies were documented at pregnancy in another reported DS-NH case without TML, which supported an immune-mediated mechanism (17). This series included three NH cases associated with DS without TML. Two of them received IVIG therapy with survival in one.
Global incidence, geographic distribution, and ethnic influence remain unknown. Large-scale studies and case series (over five cases) are mostly reported from the United States and Europe, whereas the reports in the Asian population are exiguous (5, 8, 9, 17–23). Underdiagnosis and the lack of a reporting system may lead to scarcity.
The clinical manifestation can be non-specific, resembling neonatal sepsis, respiratory failure, or metabolic disorders. Some presented with mild disease, whereas some suffered from complications of liver cirrhosis or fatal multiorgan failure. The challenge emphasizes the high suspicion of GALD when encountering neonates with acute severe liver disease (9, 17–19).
Prenatal clues of GALD-NH are not common. Sonogram or MRI may reveal oligohydramnios, IUGR, hydrops fetalis, atrophic liver, splenomegaly, iron deposition in liver or pancreas, edematous placenta, or cholelithiasis (24, 25). IUGR (25.6%) and oligohydramnios (12.8%) in this series were in the low end compared with previous studies, ranging from 20.6 to 100% and from 8 to 100%, respectively (17, 18, 20, 23, 24, 26). Still, these two findings are not exclusive to NH.
Extrahepatic siderosis is the fundamental criterion for the diagnosis of NH. However, diagnosis of GALD may still be justified if recurrence in siblings without evidence of extrahepatic siderosis (27). Another substantial pathological evidence is the demonstration of C5b-9 IHC staining, indicating complement-mediated liver injury. This neoantigen produced during the terminal complement cascade activation has been recognized as a defining feature for GALD (28). Cases with intense hepatic C5b-9 staining and supportive clinical presentation but an absence of extrahepatic siderosis have been reported (29). However, the specificity of C5b-9 staining is still arguable. A study of 44 cases with neonatal liver failure (22 NHs, and 22 non-NHs) that were found only six in the NH group were definite GALD if based on C5b-9 IHC (20). Dubruc et al. reported C5b-9 that was expressed in 100% of GALD cases, yet also in 27% of non-GALD cases (enterovirus hepatitis, bile acid synthetic defect, DGUOK mutation, Gaucher disease, cystic fibrosis, and giant-cell hepatitis with autoimmune hemolytic anemia) (30). Regarding these controversial findings, a careful application and interpretation of the C5b-9 IHC are indispensable for an accurate diagnosis.
The diagnosis rate of siderosis by MRI and salivary gland biopsy varied. Isa et al. reported 100 and 37.5% of hepatic and pancreatic siderosis by MRI, respectively; Feldman et al. suggested 60% of sensitivity by either MRI or salivary gland biopsy, whereas the combination of both tools yielded 80% (2, 19). This series showed hepatic siderosis by MRI in 84% of cases. As for extrahepatic siderosis, MRI and salivary gland biopsy detection rates were 56 and 70%, respectively.
Based on the alloimmune mechanism, treatment of GALD has switched from conventional cocktail therapy to the combination of ET and IVIG while saving liver transplantation for refractory cases. ET acts to remove existing reactive antibody, and IVIG blocks antibody-induced complement activation (2). The historically high mortality rate, reaching 80–90%, has been reduced with this revolutionary therapy (2, 4). Nevertheless, the survival rate differed in several reports. A significant difference in NLS between the IVIG/ET groups (75–79%) and the historical conventional therapy group (17%) was documented (9). However, Taylor et al. found a lower NLS of 45% in the IVIG with/without ET group in a different cohort (8). The NLS with IVIG with/without ET in this series (55%) was amid the two reported cohorts. The different outcomes may be multifactorial, including possible alternative diagnoses with NH, disease severity, the timing of application, doses, and single or combo use of ET and IVIG.
Conclusion
GALD-NH is an important etiology of neonatal liver failure with the insult beginning in the fetal stage. Evidence of extrahepatic siderosis and complement-mediated hepatic injury are supportive features, but more research is required to define the specificity of different diagnostic modalities. Discordant presentations in twins suggest an independent in utero alloimmune process. Currently, IVIG with or without ET is recommended as the first-line therapy. Although IVIG/ET therapy did not significantly improve NLS in this series, it is worth trying in a neonate with liver failure before resorting to liver transplantation, which is resource-demanding and technique-challenging in small infants.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
M-WL, P-JY, S-FH, M-CC, and C-JW: substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data. M-WL and P-JY: drafting the article. M-WL: critical revision. All authors: final approval.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.680730/full#supplementary-material
References
1. Ibrahim SH, Jonas MM, Taylor SA, Gutierrez Sanchez LH, Wolf JL, Sundaram SS. Liver Diseases in the perinatal period: interactions between Mother and Infant. Hepatology. (2020) 71:1474–85. doi: 10.1002/hep.31109
2. Feldman AG, Whitington PF. Neonatal hemochromatosis. J Clin Exp Hepatol. (2013) 3:313–20. doi: 10.1016/j.jceh.2013.10.004
3. Wu H, Ferguson W, Castro E, Kearney D, Finegold M, Patel K. Extrahepatic nonreticuloendothelial siderosis is not specific to gestational alloimmune liver disease. Pediatr Dev Pathol. (2019) 22:356–64. doi: 10.1177/1093526619826429
4. Pan X, Kelly S, Melin-Aldana H, Malladi P, Whitington PF. Novel mechanism of fetal hepatocyte injury in congenital alloimmune hepatitis involves the terminal complement cascade. Hepatology. (2010) 51:2061–8. doi: 10.1002/hep.23581
5. Whitington PF, Kelly S. Outcome of pregnancies at risk for neonatal hemochromatosis is improved by treatment with high-dose intravenous immunoglobulin. Pediatrics. (2008) 121:e1615–21. doi: 10.1542/peds.2007-3107
6. Whitington PF, Hibbard JU. High-dose immunoglobulin during pregnancy for recurrent neonatal haemochromatosis. Lancet. (2004) 364:1690–8. doi: 10.1016/S0140-6736(04)17356-X
7. Baruteau J, Heissat S, Broue P, Collardeau-Frachon S, Bouvier R, Fabre M, et al. Transient neonatal liver disease after maternal antenatal intravenous Ig infusions in gestational alloimmune liver disease associated with neonatal haemochromatosis. J Pediatr Gastroenterol Nutr. (2014) 59:629–35. doi: 10.1097/MPG.0000000000000514
8. Taylor SA, Kelly S, Alonso EM, Whitington PF. The effects of gestational alloimmune liver disease on fetal and infant morbidity and mortality. J Pediatr. (2018) 196:123–8. e1. doi: 10.1016/j.jpeds.2017.12.054
9. Rand EB, Karpen SJ, Kelly S, Mack CL, Malatack JJ, Sokol RJ, et al. Treatment of neonatal hemochromatosis with exchange transfusion and intravenous immunoglobulin. J Pediatr. (2009) 155:566–71. doi: 10.1016/j.jpeds.2009.04.012
10. Kasko O, Klose E, Rama G, Newberry D, Jnah A. Gestational alloimmune liver disease: a case study. Neonatal Netw. (2018) 37:271–80. doi: 10.1891/0730-0832.37.5.271
11. Ekong UD, Melin-Aldana H, Whitington PF. Regression of severe fibrotic liver disease in 2 children with neonatal hemochromatosis. J Pediatr Gastroenterol Nutr. (2008) 46:329–33. doi: 10.1097/MPG.0b013e318046772f
12. Ekong UD, Kelly S, Whitington PF. Disparate clinical presentation of neonatal hemochromatosis in twins. Pediatrics. (2005) 116:e880–4. doi: 10.1542/peds.2005-0784
13. Midorikawa H, Mizuochi T, Okada JI, Hisano T. Disparate clinical findings in monochorionic twins with neonatal hemochromatosis. Pediatr Int. (2017) 59:1215–6. doi: 10.1111/ped.13421
14. Chee YY, Wong SCM, Wong MSR. Heterogeneous presentation of neonatal hemochromatosis in dichorionic twins. AJP Rep. (2018) 8:e332–e4. doi: 10.1055/s-0038-1675335
15. Lewin S, Bussel JB. Review of fetal and neonatal immune cytopenias. Clin Adv Hematol Oncol. (2015) 13:35–43.
16. Ruchelli ED, Uri A, Dimmick JE, Bove KE, Huff DS, Duncan LM, et al. Severe perinatal liver disease and Down syndrome: an apparent relationship. Hum Pathol. (1991) 22:1274–80. doi: 10.1016/0046-8177(91)90111-2
17. Collardeau-Frachon S, Heissat S, Bouvier R, Fabre M, Baruteau J, Broue P, et al. French retrospective multicentric study of neonatal hemochromatosis: importance of autopsy and autoimmune maternal manifestations. Pediatr Dev Pathol. (2012) 15:450–70. doi: 10.2350/12-02-1155-OA.1
18. Fischer HS, Staufner C, Sallmon H, Henning S, Buhrer C. Early exchange transfusion to treat neonates with gestational alloimmune liver disease—an 11-year cohort study. J Pediatr Gastroenterol Nutr. (2020) 70:444–9. doi: 10.1097/MPG.0000000000002593
19. Isa HM, Mohamed AM. Neonatal hemochromatosis. Case series from Bahrain. Saudi Med J. (2013) 34:1274–80.
20. Heissat S, Collardeau-Frachon S, Baruteau J, Dubruc E, Bouvier R, Fabre M, et al. Neonatal hemochromatosis: diagnostic work-up based on a series of 56 cases of fetal death and neonatal liver failure. J Pediatr. (2015) 166:66–73. doi: 10.1016/j.jpeds.2014.09.030
21. Rodrigues F, Kallas M, Nash R, Cheeseman P, D'Antiga L, Rela M, et al. Neonatal hemochromatosis–medical treatment vs. transplantation: the king's experience. Liver Transpl. (2005) 11:1417–24. doi: 10.1002/lt.20497
22. Sheflin-Findling S, Annunziato RA, Chu J, Arvelakis A, Mahon D, Arnon R. Liver transplantation for neonatal hemochromatosis: analysis of the UNOS database. Pediatr Transplant. (2015) 19:164–9. doi: 10.1111/petr.12418
23. Heffron T, Pillen T, Welch D, Asolati M, Smallwood G, Hagedorn P, et al. Medical and surgical treatment of neonatal hemochromatosis: single center experience. Pediatr Transplant. (2007) 11:374–8. doi: 10.1111/j.1399-3046.2006.00675.x
24. Sciard C, Collardeau-Frachon S, Atallah A, Combourieu D, Massardier J, Heissat S, et al. Prenatal imaging features suggestive of liver gestational allo immune disease. J Gynecol Obstet Hum Reprod. (2019) 48:61–4. doi: 10.1016/j.jogoh.2018.11.005
25. Darouich S, Boujelbene N, Amraoui J, Amraoui N, Masmoudi A. Hemochromatosis associated with cholelithiasis as a cause of hydrops fetalis and stillbirth: Prenatal diagnosis. J Clin Ultrasound. (2019) 47:47–50. doi: 10.1002/jcu.22653
26. Alenezi K, Kamath BM, Siddiqui I, Tomlinson C, Chavhan GB. Magnetic resonance imaging findings in neonatal hemochromatosis. J Pediatr Gastroenterol Nutr. (2018) 66:581–7. doi: 10.1097/MPG.0000000000001880
27. Whitington PF. Neonatal hemochromatosis: a congenital alloimmune hepatitis. Semin Liver Dis. (2007) 27:243–50. doi: 10.1055/s-2007-985069
28. Whitington PF. Gestational alloimmune liver disease and neonatal hemochromatosis. Semin Liver Dis. (2012) 32:325–32. doi: 10.1055/s-0032-1329901
29. Tsunoda T, Inui A, Kawamoto M, Sogo T, Komatsu H, Kasahara M, et al. Neonatal liver failure owing to gestational alloimmune liver disease without iron overload. Hepatol Res. (2015) 45:601–5. doi: 10.1111/hepr.12381
Keywords: gestational alloimmune liver disease, intravenous immunoglobulin, neonatal hemochromatosis, hyperbilirubinemia, neonatal liver failure, exchange transfusion
Citation: Yeh P-J, Huang S-F, Chiang M-C, Wang C-J and Lai M-W (2021) Efficacy of Intravenous Immunoglobulin/Exchange Transfusion Therapy on Gestational Alloimmune Liver Disease. Front. Pediatr. 9:680730. doi: 10.3389/fped.2021.680730
Received: 15 March 2021; Accepted: 17 May 2021;
Published: 21 June 2021.
Edited by:
Pietro Vajro, University of Salerno, ItalyReviewed by:
Helen Savoia, Royal Children's Hospital, AustraliaChristoph Bührer, Charité - Universitätsmedizin Berlin, Germany
Sophie Collardeau-Frachon, Hospices Civils de Lyon, France
Copyright © 2021 Yeh, Huang, Chiang, Wang and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Wei Lai, YTIyMTQxQGNnbWgub3JnLnR3