 Hanan AlQudairy1,†
Hanan AlQudairy1,† Hesham AlDhalaan2,†
Hesham AlDhalaan2,† Sarah AlRuways1,3,†Nouf AlMutairi1,3,†Maha AlNakiyah1,3,†Reema AlGhofaili1,3,†
Sarah AlRuways1,3,†Nouf AlMutairi1,3,†Maha AlNakiyah1,3,†Reema AlGhofaili1,3,† Albandary AlBakheet2Adeeb Alomrani4Omar A. Alharbi2Ehab Tous2
Albandary AlBakheet2Adeeb Alomrani4Omar A. Alharbi2Ehab Tous2 Moeen AlSayed5
Moeen AlSayed5 Hamad AlZaidan5
Hamad AlZaidan5 Maha M. AlRasheed3,6Ali AlOdaib7
Maha M. AlRasheed3,6Ali AlOdaib7 Namik Kaya1*
Namik Kaya1*
- 1Translational Genomic Department, Center for Genomic Medicine, King Faisal Specialist Hospital, and Research Centre (KFSHRC), Riyadh, Saudi Arabia
- 2Department of Neurosciences, KFSHRC, Riyadh, Saudi Arabia
- 3College of Pharmacy, King Saud University (KSU), Riyadh, Saudi Arabia
- 4Dentistry Department, KFSHRC, Riyadh, Saudi Arabia
- 5Department of Medical Genomics, Center for Genomic Medicine, KFSHRC, Riyadh, Saudi Arabia
- 6Department of Clinical Pharmacy, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia
- 7Training and Education Department, Research Centre, KFSHRC, Riyadh, Saudi Arabia
Background: SLC13A5 (solute carrier family 13, member 5) encodes sodium/citrate cotransporter, which mainly localizes in cellular plasma membranes in the frontal cortex, retina, and liver. Pathogenic variants of the gene cause an autosomal recessive syndrome known as “developmental and epileptic encephalopathy 25 with amelogenesis imperfecta.”
Results: Here, we have investigated six patients from three different consanguineous Saudi families. The affected individuals presented with neonatal seizures, developmental delay, and significant defects in tooth development. Some patients showed other clinical features such as muscle weakness, motor difficulties, intellectual disability, microcephaly, and speech problems in addition to additional abnormalities revealed by electroencephalography (EEGs) and magnetic resonance imaging (MRI). One of the MRI findings was related to cortical thickening in the frontal lobe. To diagnose and study the genetic defects of the patients, whole exome sequencing (WES) coupled with confirmatory Sanger sequencing was utilized. Iterative filtering identified two variants of SLC13A5, one of which is novel, in the families. Families 1 and 2 had the same insertion (a previously reported mutation), leading to a frameshift and premature stop codon. The third family had a novel splice site variant. Confirmatory Sanger sequencing corroborated WES results and indicated full segregation of the variants in the corresponding families. The patients’ conditions were poorly controlled by multiple antiepileptics as they needed constant care.
Conclusion: Considering that recessive mutations are common in the Arab population, SLC13A5 screening should be prioritized in future patients harboring similar symptoms including defects in molar development.
Introduction
SLC13 genes (SLC13A2 and SLC13A3) encode transporter proteins that carry Na+-coupled dicarboxylate (NaDC) across the plasma membrane with different substrate preferences (1). Unlike NaDC, SLC13A5 encodes a Na+-coupled citrate (NaCT) transporter protein, a cytoplasmic sodium-dependent citrate carrier, which can carry dicarboxylate in addition to tricarboxylate substrates (2). The transporter is widely expressed in neurons, localized in the plasma membrane of various cell types, including hepatocytes in the liver, spermatozoa in the testis, and mostly astrocytes and neurons in the brain (3). Citrate is vital in cellular metabolism and neurotransmitter biogenesis (3). It is known to have an important role in the tricarboxylic acid cycle, where the molecule represents the starting point for generating reducing equivalents nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (reduced form) (FADH2), which in turn enter the electron transport chain to generate ATP (4). The brain cannot produce citrate independently; hence, it depends on citrate uptake via NaCT. Thus, the carrier has a pivotal role in mediating the uptake of circulating citrate for metabolism (4), preferably in the trivalent form rather than the divalent form.
SLC13A5 is located on chromosome 17 and consists of 12 coding exons. Biallelic mutations in the gene cause a rare genetic syndrome known as “developmental and epileptic encephalopathy 25 with amelogenesis imperfecta” (DEE25; phenotype MIM: 615905). The mutant NaCT molecules can lose their ability to bind to the sodium molecules, therefore failing to transport citrate from the extracellular matrix to cytosol (3, 5, 6). Relatedly, when citrate transport and metabolism are disrupted, intracellular citrate levels fall, resulting in neuronal energy failure, which is thought to be one of the explanations behind epileptic symptoms (3). In other words, a lack of cellular citrate results in energy deficiency in the brain, thus possibly contributing to the pathogenesis of epilepsy and delayed brain development (6, 7).
Here, in this study, we investigate pathogenic homozygous variants of SLC13A5 (including a novel splice site variant), causing mainly neonatal seizures and developmental delay in six patients from three different Saudi families. The affected individuals also showed auxiliary clinical features such as muscle weakness, motor difficulties, intellectual disability, microcephaly, and speech problems, in addition to abnormalities revealed by electroencephalography (EEG). The patients’ conditions were poorly controlled by multiple antiepileptics as they needed constant care. Additionally, we reviewed the current literature about SLC13A variants.
Materials and methods
Patient recruitment, sample collection, and nucleic acid isolation
Six patients and related family members were recruited under approvals provided by Institution's Research Advisory Council (Ethics Statement) at King Faisal Specialist Hospital and Research Center, Riyadh, KSA (KFSHRC, IRB-approved protocols, RAC#2120022). Peripheral blood samples (5 ml) were drawn into EDTA tubes. The Gentra Puregene DNA Purification Kit was utilized during DNA isolation (Gentra Systems, Inc. Minneapolis, MN, United States).
PCR and Sanger sequencing-based variant screening
Gene-specific primers were designed using the Primer 3 web tool. Primers were tested on human control DNA samples and optimized for successful PCR amplifications. After the PCR, samples were purified and sent to the sequencing core facility for Sanger sequencing. The direct sequencing was done on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, United States) according to the manufacturer's instructions.
Genome-wide SNP screening using GeneChip Axiom assays and autozygosity analysis
Genotyping was undertaken using GeneChip Human Axiom Arrays and other equipment such as a hybridization oven and a titan platform from Affymetrix (Thermo Fisher Inc., Santa Clara, CA, United States). Sample processing, labeling, hybridization, washing, and scanning were all done according to manuals and assay protocols provided by the manufacturers.
Whole exome sequencing
Exomes were captured using the Sure Select kit (Agilent Technologies, Santa Clara, CA, United States). Library construction was done using the same kits (Exons V5, 50 Mb capture kit, Agilent Technologies). After library preparation, captured fragments were run on an Illumina HiSeq 2500 Sequencer and mapped against UCSC hg19 (Illumina, Inc., San Diego, CA, United States). Comprehensive filtering of the detected variants was done as previously published (8–11). Especially, variants based on homozygosity, coding, and splicing features, being within the autozygome of the affected individuals in the families, were prioritized during filtering analysis. Additionally, publicly available databases and local resources, such as the program for Saudi Human Genome-based data outputs, were also screened during filtering. In silico pathogenicity was carried out as reported before (8–11).
Results
Clinical features
Patient 1 (family 1, IV:3)
The patient is a 17-year-old girl from a Saudi consanguineous family with a positive family history of a similar medical condition (Figure 1A). The patient developed a seizure on the first day of her life but was only seen in the clinic at the age of 8 years with a history of developmental delay and seizures mainly described in the form of eye fluttering. According to her parents, she started crawling, standing, and sitting at the age of 11 years but was unable to walk. On examination, all her growth parameters were below the third percentile with microcephaly. Microcephaly was detected in a local hospital when she was 4 months old. Neurological examination showed spastic quadriplegia, with contracture and increased deep tendon reflexes. She was on levetiracetam (equivalent to 66 mg/kg/day), phenobarbital (equivalent to 7 mg/kg/day), and topiramate (equivalent to 7 mg/kg/day) and was free of seizure for more than 2 years. At the age of 14, she suffered a femur fracture without any clear history of trauma and developed a seizure. The seizure was in the form of generalized tonic posturing with up-rolling of the eyes with involuntary movements. It lasted for 1 min and aborted spontaneously. Her GAL-1P levels were less than 49, and this result was consistent with the diagnosis of a very mild form of Duarte galactosemia. EEG was done twice at 8 and 14 years of age, and it showed intermittent slow activity, maximum in the left temporal, with excessive fast activity that could be due to phenobarbital (a chronic medication used to treat the patient during that time).
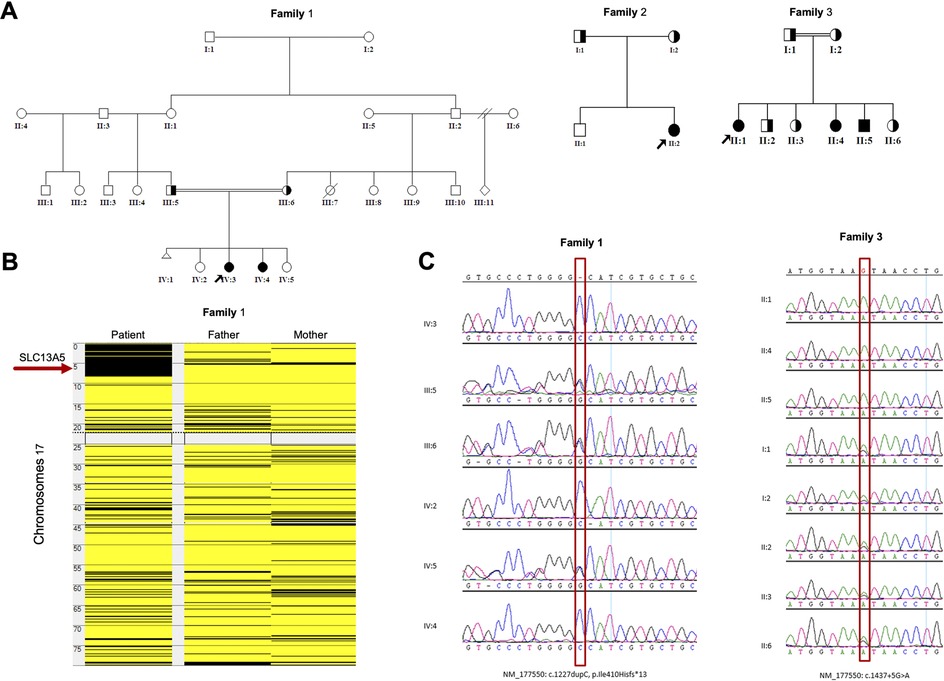
Figure 1. Genetic analyses of the families: (A) Pedigrees of the families of the study showing six affected members. (B) Homozygosity mapping showing the location of the SLC13A5 gene in major ROH in family 1. (C) Sanger sequence chromatograms of the tested individuals. SLC13A5, solute carrier family 13, member 5; ROH, runs of homozygosity.
Patient 2 (family 1, IV:4)
The patient is a 14-year-old girl, the sister of patient 1 (Figure 1A), diagnosed with developmental delay and seizure disorder. Her history of seizures began after 12 h of birth. She suffered from seizures many times per day. Her seizures were controlled by phenobarbitone for a few months only. Afterward, topiramate was added to her medication, which reduced the frequency of her seizures (3–20 times/day) per day. At 15 months, she was neither able to support herself nor was she attempting to stand. The last EEG done when the patient was 1 year old showed mild abnormality, probably secondary to a medication's effect.
Patient 3 (family 2, II:2)
The patient is a 7-year-old girl (Figure 1A). She started to have symptoms on her first day of life. The patient presented to the clinic at the age of 9 months and was diagnosed with severe infantile epileptic encephalopathy, global developmental delay (GDD), and intractable epilepsy disorder. Currently, the patient is unable to walk, crawl, or sit without support, and lacks control of her head and neck. Her seizures had multiple semiologies with left-sided facial twitches and secondary generalization with generalized tonic and clonic seizures. Another type of seizure was also noticed as right-sided tonic flexion of the extremities and extension of the left side accompanied by secondary generalization followed by postictal incontinence, both urinary and bowel, as well as sleepiness and confusion. At the age of 3, her seizures have been controlled for 1 year, though previously occurring on a daily basis. Since her birth and during the disease course, the patient was on multiple medications to control her seizures (e.g., phenobarbital, levetiracetam, carbamazepine, valproic acid, topiramate, clobazam) with poor response and sometimes no response. EEG done at the age of 9 months showed diffuse encephalopathy, which might be due to medications or postictal status. Subsequent EEG examinations at the age of 2 and 3 years were all normal. However, the last EEG at the age of 6 years was abnormal and suggestive of focal epileptogenicity from the left occipitotemporal head region. Lab tests done during the last visit revealed ammonia levels higher than normal.
Patient 4 (family 3, II:1)
The patient is a 28-year-old woman with a positive family history of two affected siblings with similar phenotypes, a 17-year-old sister with a more severe condition (patient 5) and a 12-year-old brother with a mild condition (patient 6) (Figure 1A). Her seizures started on the second day of life with abnormal EEG showing epileptiform discharges. Her seizures were controlled with valproic acid, and her last EEG was normal. She also has GDD. She can speak a few words. She started to walk at the age of 4 years. Her IQ was below 50. She is on risperidone for anxiety and aggressiveness. She is wearing eyeglasses for divergent squint. Her brain MRI (magnetic resonance imaging) and MRA (magnetic resonance angiography) are normal.
Patient 5 (family 3, II:4)
The patient is a 17-year-old woman (Figure 1A). Her first seizure started when she was a neonate (Table 1). She first presented in our hospital at the age of 4 years with infantile epileptic encephalopathy, severe global developmental delay, neonatal seizures, microcephaly, and hypertonia. She was noticed to have microcephaly when she was 1 month old. Her seizures started with left-sided body tonic posture and versive head deviation to the left, evolving to secondary generalization and myoclonic attacks. The first EEG was done at the age of 4, which showed severe abnormalities with the presence of a focal area of cortical neuro irritability on the right central and left parietal regions, indicating a predisposition for epileptic seizures and focal slowing on the right parasagittal region, which suggested an underlying structural abnormality. The last EEG was done at the age of 11 years. The finding was suggestive of mild cortical dysfunction and nonspecific encephalopathy. MRI done at the age of 3 years showed a mildly increased signal intensity on a T2-weighted image (Figure 2A). The patient is still having frequent seizures, is bedridden, is totally dependent on her family, and is taking multiple antiepileptic medications.
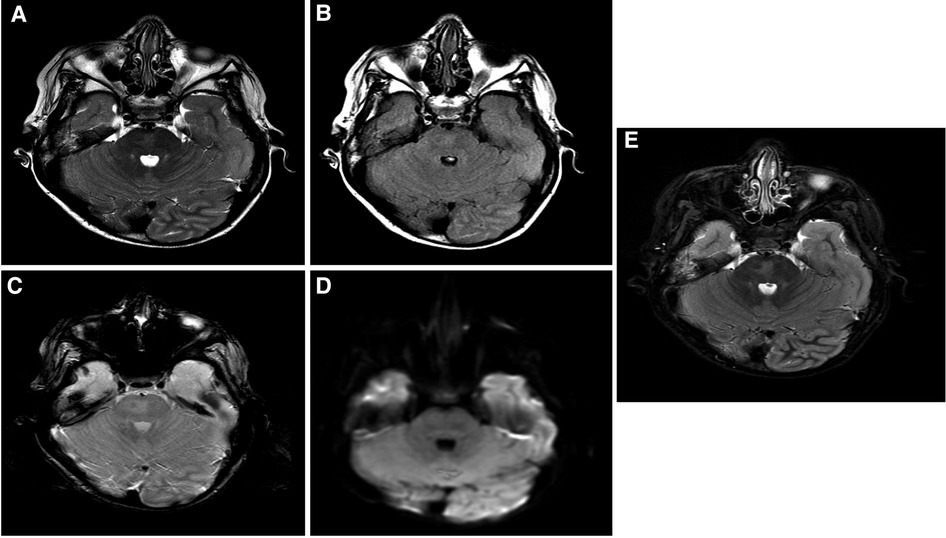
Figure 2. Brain MRI findings: selected images at the level of superior cerebellar peduncles showing a mildly increased signal intensity on a T2-weighted image (A), FLAIR (B), and inversion recovery (C) without evidence of blood staining on a T2 star image (D) or an increased diffusion signal (E). These findings are most suggestive of nonspecific pontine gliosis. MRI, magnetic resonance imaging; FLAIR, fluid-attenuated inversion recovery.
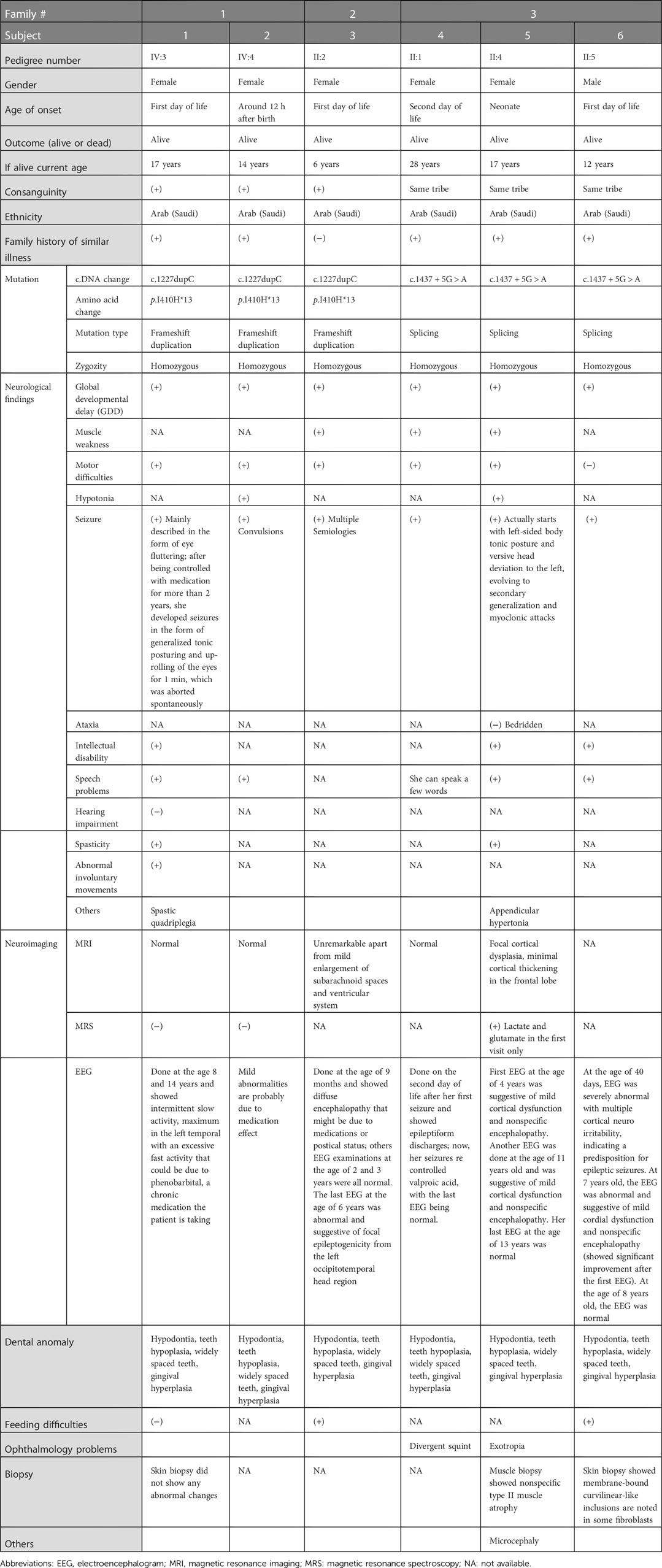
Table 1. Clinical features of subjects reported in this study.
Patient 6 (family 3, II:5)
The patient is a 12-year-old boy (Figure 1A). He was diagnosed with familial autosomal recessive epileptic encephalopathy, intractable seizures, and global developmental delay. His seizures started on the first day of his life. At the age of 40 days, his EEG was severely abnormal, with multiple cortical neuroirritability, indicating predisposition for epileptic seizures. At 7 years of age, the EEG was still abnormal and suggestive of mild cordial dysfunction and nonspecific encephalopathy but showed significant improvement compared to the first. The last EEG was performed at the age of 8 years and showed normal results. The patient is still having frequent seizures in addition to his global developmental delay, but from the developmental perspective, he is improving gradually.
Dental and oral manifestation findings
The clinical features of our six patients include not only early-onset epilepsy and delayed neurological development but also significant defects in tooth development such as hypodontia, teeth hypoplasia, widely spaced teeth, and gingival hyperplasia, as we see in amelogenesis imperfecta cases (i.e., the enamel is abnormally thin, soft, fragile, pitted, and discolored), causing severe embarrassment, eating difficulties, and pain in patients. These dental deformities are very difficult to treat as they involve both dental and skeletal deformities. Interestingly, we see teeth defect and malformation in both groups of young patients with primary teeth and older patients with permanent teeth, as well as open anterior-skeletal bite and overgrowth of gingiva (mostly due to poor oral hygiene and the side effect of antiepileptic medications) (Figure 3).
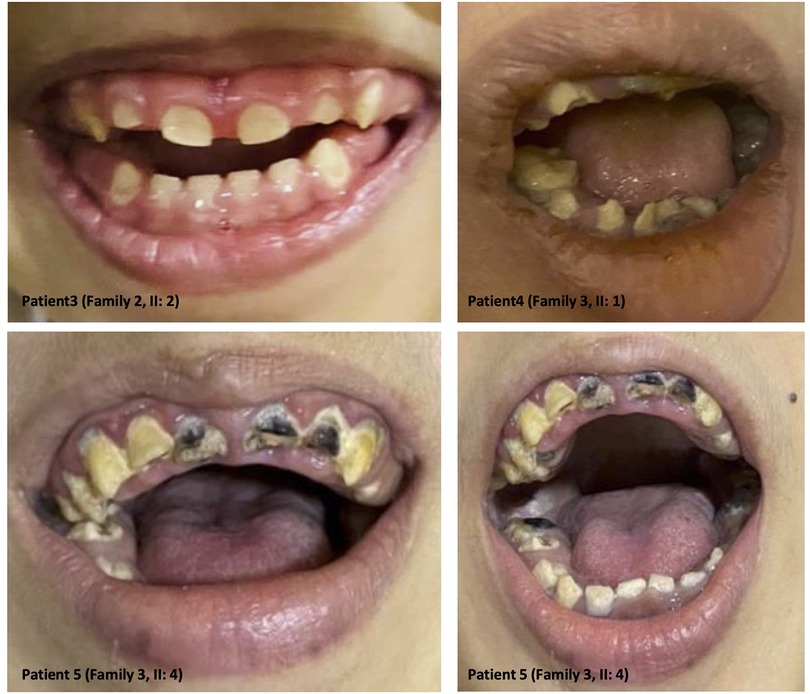
Figure 3. Dental anomalies: illustrative images from the selected patients showing teeth defects and malformations in both groups of young patients with primary teeth and older patients with permanent teeth, as well as open anterior-skeletal bite and overgrowth of the gingiva.
Genetic findings
Whole exome sequencing (WES) combined with single nucleotide polymorphism (SNP) analysis revealed two homozygous variants of SLC13A5 as the only strong candidates underlying the disorder. The gene is in a major run of homozygosity (ROH) block detected on the short arm of chromosome 17 in family 1 (Figure 1B). Patients 1, 2, and 3 harbor a previously known single nucleotide insertion (c.1227dupC) (12). The c.1227dupC produces a frameshift, leading to a premature stop codon (p.Ile410Hisfs*13). The other variant, a novel donor splice site variant of SLC13A5 (c.1437 + 5G > A), was identified in the remaining three patients 4, 5, and 6 (family 3) by WES. This variant was absent in local and international databases. Segregation of the variants in the families was performed using Sanger sequencing (Figure 1C).
MutationTaster and varSEAK were used to predict the pathogenicity of c.1227dupC (p.Ile410Hisfs*13) and c.1437 + 5G > A, respectively. MutationTaster predicted the variant to be deleterious, leading to a premature stop codon. Moreover, this variant is already reported in ClinVar as a disease-causing variant. On the other hand, varSEAK computed that c.1437 + 5G > A will have a class 5 splicing effect (an exon skipping), resulting in loss of function due to aberrant splicing. Details of these predictions and analysis results are presented in Supplementary File S1.
Discussion
Epileptic encephalopathies are broad and highly heterogeneous epileptic disorders characterized by seizures and abnormal EEG coupled with cognitive and developmental impairment. Biallelic mutations in SLC13A5 cause one such disorder called as “developmental and epileptic encephalopathy 25 with amelogenesis imperfecta” (DEE25; phenotype MIM: 615905). In this study, we focused on pathogenic variants of SLC13A5 in our population and identified six patients from three different Saudi families harboring two homozygous variants (a previously reported insertion and a novel splice site variant). Previously reported patients exceeded 100 cases with approximately 36 different variants (Supplementary Table S1). All these appear to be autosomal recessive, with several families having more than one affected child. Most of the variants are missense (n = 24) (13). Additionally, six splicing mutations (13–21), four small deletions (18, 20, 22–26), two small insertions (6, 12, 13, 16, 27, 28), and four gross deletions (13, 20, 29) have been reported.
These pathogenic variants have been linked to various phenotypes, especially seizure-causing syndromes such as Kohlschütter–Tönz and West syndromes (19, 26). However, the current understanding of these syndromes, as well as dissecting the genetic and clinical heterogeneity of developmental and epileptic encephalopathies, revealed that different genes are involved in these syndromes. For example, mutations in ROGDI lead to Kohlschütter–Tönz syndrome (MIM: 226750; OMIM: 614574). In children SLC13A5-related early-onset epilepsy, which usually occurs within the first few weeks after birth, seizures continue as the children grow (3, 4). This is thought to be related to loss-of-function mutations. Such mutations cause an inability to transport citrate across the plasma membrane, leading to distinctive symptoms. Affected patients may exhibit physical and cognitive impairment, major delays in speech development, and characteristic dental problems (3, 4, 20). Of note, SLC13A5 is expressed in a wide range of tissues (22); most of the phenotypes observed in various affected individuals seem related to its expression in the brain. The patients presented with encephalopathy, GDD, and seizures, which appeared during early infancy and mostly began in the first 24 h of life. The electroencephalogram (EEG) recordings also showed multiple abnormalities. Such anomalies were also seen in previously reported patients. For example, Klotz et al. (18) reported nine patients from six different families; all had seizures from the first week of life except for one who experienced a delay in her first seizure at the age of months. Moreover, all of them were noticed to have motor and language delays.
Interestingly, some studies have reported defects in tooth development, such as hypodontia. Such symptoms are considered one of the key characteristics of “developmental and epileptic encephalopathy-25 (DEE25) disorder” (3, 6, 18, 29). It is noteworthy that the pioneering study by Thevenon et al. (3) did not report any major defects of tooth development except for widely spaced teeth. SLC13A5 has been found to have a crucial role in tooth development, where a gene expression analysis showed SLC13A5 to be one of the top 15 genes dysregulated during mouse molar development (6). Such involvement caused confusion due to the overlapping clinical features and manifestations between Kohlschütter–Tönz syndrome and DEE25. Moreover, clinical heterogeneity appeared in the disease expression, even between siblings with identical genotypes. For example, a brother and sister carrying the same compound heterozygous variants of SLC13A5 presented with varying intractable seizures and focal brain lesions; one of the patients had focal cortical dysplasia and was more developmentally delayed than the other sibling. Nevertheless, both had seizures within the first day of life, hypotonia, and developmental delay (30).
The earliest study identified SLC13A5 variants as the disease-causing variants that reported overlapping clinical features such as early-onset seizures, which appeared within any day of the first week of life, with severe developmental delay. In addition to seizures that may even appear as early as on the first day of life, there were other phenotypes, such as pediatric movement disorders and intellectual disability, linked to pathogenic variants of SLC13A5 (6, 14, 31).
The six affected patients in our study have the characteristics mentioned before, with predominant GDD and epilepsy. The other features observed in our patients are as follows: patients 1 and 5 also presented with microcephaly at early stages of life; patients 1 and 3 had spasticity; patients 3 and 5 had speech problems; patients 1, 2, 3, and 5 presented with motor difficulties; and finally patient 5 showed muscle atrophy, recurrent skin abscess, and exotropia.
Until now, there is no specific treatment for SLC13A5-epileptic encephalopathy. Considering that the seizures are refractory, all our patients are on multiple anticonvulsant medications to control their seizures. Some are on three anticonvulsant medications (levetiracetam, topiramate, and phenobarbital), while others were on topiramate along with valproic acid and diazepam. However, none of the patients have been reported to be effectively weaned off their medications. One of the proposed mechanisms that can help to control seizures in SLC13A5-epileptic encephalopathy is using medications that can increase the cytoplasmic level of citrate. This could lead to a decrease in the frequency of seizures and help control the patient's symptoms. Triheptanoinone acts by increasing the metabolism of odd-chain fatty acids in neuronal mitochondria; as a result, it increases the levels of succinyl-CoA, which leads to the increase of citrate concentration. Benzodiazepines are another example. These classes of medications share an augmentation mechanism by enhancing the inhibitory action of gamma aminobutyric acid (GABA) (28). Moreover, other antiepileptic medications can also be used, such as phenytoin or lamotrigine, as these drugs target sodium channels. One of the new-generation drugs that have multiple mechanisms of action to control seizures is stiripentol. One of its action mechanisms is based on enhancing GABAergic transmission. Stiripentol can also be used as an adjunctive therapy to valproate and clobazam. Moreover, acetazolamide, a carbonic anhydrase inhibitor and an atypical medication, can also suppress seizures by an unknown mechanism of action. On the other hand, nonpharmacological treatments such as ketogenic diet have been reported to reduce seizure frequency, although their efficiency is controversial (28). Still, there is a need to investigate new therapeutic targets and more effective treatments for this disorder in the future.
In conclusion, we reported a novel homozygous variant of SLC13A5 mutation in a previously unstudied population for this disease. Given the growing number of epilepsy genes linked to early-onset epileptic encephalopathies, WES and gene panels can improve the identifying known and unknown pathogenic variants in the gene. Although there is currently no curative treatment available for the disease, early detection of the deleterious variants may provide an advantage in terms of premarital screening, genetic counseling, and avoiding unnecessary and redundant diagnostic tests and treatments. Improving seizure control should also be one of the components of future therapeutic trials and research.
Data availability statement
The original contributions presented in the study are included in the article and Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by King Faisal Specialist Hospital and Research Centre, Office of Research Affairs, Research Advisory Council, (RAC#2120022). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
NK conceptualized, designed, and supervised the project and involved in drafting the manuscript. HA, SA, NA, MA, RA, and AA performed the experiments, reviewed patients’ charts, and involved in writing the manuscript. HA, AA, ET, MA, and HA performed patient care, performed clinical evaluations, acquired clinical data, reviewed the patients’ charts, supervised drafting of the clinical descriptions, and involved in revising the manuscript. MMA and AA involved in drafting and revising the manuscript, supervised the students, and involved in data analysis. All authors contributed to the article and approved the submitted version.
Funding
Namik Kaya is supported by King Salman Center for Disability Research (RAC, 2180004).
Acknowledgments
The authors are grateful to the patients and their families, Saudi Genome Project team members, Core Facilities at the Center for Genomic Medicine, KFSHRC Research Advisory Council Committees, and KFSHRC Purchasing Department for facilitating and expediting our requests. The authors highly thank and appreciate King Salman Center for Disability Research for its generous support of our project (KSCDR-RAC, 2180004, Namik Kaya)
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.1051534/full#supplementary-material.
References
1. Pajor AM. Molecular properties of sodium/dicarboxylate cotransporters. J Membr Biol. (2000) 175(1):1–8. doi: 10.1007/s002320001049
2. Inoue K, Zhuang L, Ganapathy V. Human Na+-coupled citrate transporter: primary structure, genomic organization, and transport function. Biochem Biophys Res Commun. (2002) 299(3):465–71. doi: 10.1016/S0006-291X(02)02669-4
3. Thevenon J, Milh M, Feillet F, St-Onge J, Duffourd Y, Juge C, et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am J Hum Genet. (2014) 95(1):113–20. doi: 10.1016/j.ajhg.2014.06.006
4. Bhutia YD, Kopel JJ, Lawrence JJ, Neugebauer V, Ganapathy V. Plasma membrane Na(+)-coupled citrate transporter (SLC13A5) and neonatal epileptic encephalopathy. Molecules. (2017) 22(3. doi: 10.3390/molecules22030378
5. Inoue K, Zhuang L, Maddox DM, Smith SB, Ganapathy V. Human sodium-coupled citrate transporter, the orthologue of Drosophila Indy, as a novel target for lithium action. Biochem J. (2003) 374(Pt 1):21–6. doi: 10.1042/bj20030827
6. Hardies K, de Kovel CG, Weckhuysen S, Asselbergh B, Geuens T, Deconinck T, et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain. (2015) 138(Pt 11):3238–50. doi: 10.1093/brain/awv263
7. Weckhuysen S, Korff CM. Epilepsy: old syndromes, new genes. Curr Neurol Neurosci Rep. (2014) 14(6):447. doi: 10.1007/s11910-014-0447-7
8. Aldhalaan H, AlBakheet A, AlRuways S, AlMutairi N, AlNakiyah M, AlGhofaili R, et al.. A novel GEMIN4 variant in a consanguineous family leads to neurodevelopmental impairment with severe microcephaly, spastic quadriplegia, epilepsy, and cataracts. Genes. (2021) 13(1). doi: 10.3390/genes13010092
9. AlMuhaizea M, Dabbagh O, AlQudairy H, AlHargan A, Alotaibi W, Sami R, et al. Phenotypic variability of MEGF10 variants causing congenital myopathy: report of two unrelated patients from a highly consanguineous population. Genes. (2021) 12(11). doi: 10.3390/genes12111783
10. Sanderson LE, Lanko K, Alsagob M, Almass R, Al-Ahmadi N, Najafi M, et al. Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain. (2021) 144(3):769–80. doi: 10.1093/brain/awaa459
11. Al-Muhaizea MA, Aldeeb H, Almass R, Jaber H, Binhumaid F, Alquait L, et al. Genetics of ataxia telangiectasia in a highly consanguineous population. Ann Hum Genet. (2022) 86(1):34–44. doi: 10.1111/ahg.12445
12. Shamseldin HE, Maddirevula S, Faqeih E, Ibrahim N, Hashem M, Shaheen R, et al. Increasing the sensitivity of clinical exome sequencing through improved filtration strategy. Genet Med. (2017) 19(5):593–8. doi: 10.1038/gim.2016.155
13. Matricardi S, De Liso P, Freri E, Costa P, Castellotti B, Magri S, et al. Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia. (2020) 61(11):2474–85. doi: 10.1111/epi.16699
14. Cordeiro D, Bullivant G, Siriwardena K, Evans A, Kobayashi J, Cohn RD, et al. Genetic landscape of pediatric movement disorders and management implications. Neurol Genet. (2018) 4(5):e265. doi: 10.1212/NXG.0000000000000265
15. Costain G, Cordeiro D, Matviychuk D, Mercimek-Andrews S. Clinical application of targeted next-generation sequencing panels and whole exome sequencing in childhood epilepsy. Neuroscience. (2019) 418:291–310. doi: 10.1016/j.neuroscience.2019.08.016
16. Maddirevula S, Kuwahara H, Ewida N, Shamseldin HE, Patel N, Alzahrani F, et al. Analysis of transcript-deleterious variants in Mendelian disorders: implications for RNA-based diagnostics. Genome Biol. (2020) 21(1):145. doi: 10.1186/s13059-020-02053-9
17. Sallevelt S, Stegmann APA, de Koning B, Velter C, Steyls A, van Esch M, et al. Diagnostic exome-based preconception carrier testing in consanguineous couples: results from the first 100 couples in clinical practice. Genet Med. (2021) 23(6):1125–36. doi: 10.1038/s41436-021-01116-x
18. Klotz J, Porter BE, Colas C, Schlessinger A, Pajor AM. Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol Med. (2016) 22. doi: 10.2119/molmed.2016.00077
19. Schossig A, Bloch-Zupan A, Lussi A, Wolf NI, Raskin S, Cohen M, et al. SLC13A5 Is the second gene associated with Kohlschutter–Tonz syndrome. J Med Genet. (2017) 54(1):54–62. doi: 10.1136/jmedgenet-2016-103988
20. Yang QZ, Spelbrink EM, Nye KL, Hsu ER, Porter BE. Epilepsy and EEG phenotype of SLC13A5 citrate transporter disorder. Child Neurol Open. (2020) 7:2329048X20931361. doi: 10.1177/2329048X20931361
21. Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. (2015) 10(2):148–61. doi: 10.1016/j.celrep.2014.12.015
22. Bainbridge MN, Cooney E, Miller M, Kennedy AD, Wulff JE, Donti T, et al. Analyses of SLC13A5-epilepsy patients reveal perturbations of TCA cycle. Mol Genet Metab. (2017) 121(4):314–9. doi: 10.1016/j.ymgme.2017.06.009
23. Jiang T, Gao J, Jiang L, Xu L, Zhao C, Su X, et al. Application of trio-whole exome sequencing in genetic diagnosis and therapy in Chinese children with epilepsy. Front Mol Neurosci. (2021) 14:699574. doi: 10.3389/fnmol.2021.699574
24. Nolan D, Carlson M. Whole exome sequencing in pediatric neurology patients: clinical implications and estimated cost analysis. J Child Neurol. (2016) 31(7):887–94. doi: 10.1177/0883073815627880
25. Turro E, Astle WJ, Megy K, Graf S, Greene D, Shamardina O, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. (2020) 583(7814):96–102. doi: 10.1038/s41586-020-2434-2
26. Fernandez-Marmiesse A, Roca I, Diaz-Flores F, Cantarin V, Perez-Poyato MS, Fontalba A, et al. Rare variants in 48 genes account for 42% of cases of epilepsy with or without neurodevelopmental delay in 246 pediatric patients. Front Neurosci. (2019) 13:1135. doi: 10.3389/fnins.2019.01135
27. Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. (2017) 136(8):921–39. doi: 10.1007/s00439-017-1821-8
28. Alhakeem A, Alshibani F, Tabarki B. Extending the use of stiripentol to SLC13A5-related epileptic encephalopathy. Brain Dev. (2018) 40(9):827–9. doi: 10.1016/j.braindev.2018.05.020
29. Duan R, Saadi NW, Grochowski CM, Bhadila G, Faridoun A, Mitani T, et al. A novel homozygous SLC13A5 whole-gene deletion generated by Alu/Alu-mediated rearrangement in an Iraqi family with epileptic encephalopathy. Am J Med Genet A. (2021) 185(7):1972–80. doi: 10.1002/ajmg.a.62192
30. Anselm I, MacCuaig M, Prabhu SB, Berry GT. Disease heterogeneity in Na(+)/citrate cotransporter deficiency. JIMD Rep. (2017) 31:107–11. doi: 10.1007/8904_2016_546
31. Harripaul R, Vasli N, Mikhailov A, Rafiq MA, Mittal K, Windpassinger C, et al. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry. (2018) 23(4):973–84. doi: 10.1038/mp.2017.60
Keywords: SLC13A, novel variant, epileptic encephalopathy, tooth abnormalities, intellectual disability, whole exome sequencing (WES), Sanger sequencing
Citation: AlQudairy H, AlDhalaan H, AlRuways S, AlMutairi N, AlNakiyah M, AlGhofaili R, AlBakheet A, Alomrani A, Alharbi OA, Tous E, AlSayed M, AlZaidan H, AlRasheed MM, AlOdaib A and Kaya N (2023) Clinical, radiological, and genetic characterization of SLC13A5 variants in Saudi families: Genotype phenotype correlation and brief review of the literature. Front. Pediatr. 10:1051534. doi: 10.3389/fped.2022.1051534
Received: 22 September 2022; Accepted: 14 December 2022;
Published: 27 February 2023.
Edited by:
Mariasavina Severino, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Charles Marques Lourenco, Faculdade de Medicina de São José do Rio Preto, BrazilZehra Agha, COMSATS University, Pakistan
© 2023 Alqudairy, Aldhalaan, Alruways, Almutairi, Alnakiyah, Alghofaili, Albakheet, Alomrani, Alharbi, Tous, Alsayed, Alzaidan, AlRasheed, Alodaib and Kaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Namik Kaya bmtheWFAa2ZzaHJjLmVkdS5zYQ== bmFtaWtrYXlhQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Pediatric Neurology, a section of the journal Frontiers in Pediatrics