 Reeti Kumar1
Reeti Kumar1 Vahakn Keskinyan1
Vahakn Keskinyan1 Megan Chryst Stangl1
Megan Chryst Stangl1 Brandon M. Lane1Anne F. Buckley2Laura Barisoni2
Brandon M. Lane1Anne F. Buckley2Laura Barisoni2 David N. Howell2
David N. Howell2 Rasheed A. Gbadegesin1*
Rasheed A. Gbadegesin1*- 1Division of Nephrology, Department of Pediatrics, Duke University Medical Center, Durham, NC, United States
- 2Division of Renal Pathology, Department of Pathology, Duke University Medical Center, Durham, NC, United States
Glomerular diseases (GDs) are a major cause of chronic kidney disease in children. The conventional approach to diagnosis of GDs includes clinical evaluation and, in most cases, kidney biopsy to make a definitive diagnosis. However, in many cases, clinical presentations of different GDs can overlap, leading to uncertainty in diagnosis and management even after renal biopsy. In this report, we identify a family with clinical diagnoses of postinfectious glomerulonephritis and IgA nephropathy in a parent and two children. Renal biopsies were initially inconclusive; however, genetic testing showed that the two individuals diagnosed at different points with IgA nephropathy carried novel segregating pathogenic variants in COL4A5 gene. We were only able to make the final diagnoses in each of the family members after genetic testing and reverse phenotyping. This case highlights the utility of genetic testing and reverse phenotyping in resolving clinical diagnosis in families with unusual constellations of different glomerulopathies. We propose that clustering of different glomerular disease phenotypes in a family should be an indication for genetic testing followed by reverse phenotyping.
Introduction
Glomerular diseases (GDs) account for 8–14% of all cases of chronic kidney disease (CKD) in children (1, 2). The pathogenesis of different GDs is not completely known; however, recent advances in genomics and proteomics have thrown more light on disease mechanisms. For example, it is now known that GDs that are associated predominantly with proteinuria (nephrotic syndrome) are likely due to primary or secondary injuries to the visceral epithelial cell of the glomerulus, the podocyte, hence, the name podocytopathy (3). More than 60 genes that are known to cause monogenic nephrotic syndrome are enriched in the podocyte (4). However, most cases of hereditary nephritis are due to defects in collagen alpha genes (COL4A3, COL4A4, and COL4A5) (5).
While the majority of GDs are diagnosed by standard clinical evaluation and kidney histopathology, additional investigations, such as genetic testing, may be indicated to establish diagnosis. Even when genetic etiology is established, there is considerable overlap between different GDs. For example it has been reported that up to 10% of classical hereditary podocytopathies (focal and segmental glomerulosclerosis) are due to pathogenic variants in COL4A genes, and pathogenic variants in these same genes have also been reported in patients with presumed IgA nephropathy (6–10). This reported phenotypic pleiotropy in genes associated with GD emphasizes the importance of genetic diagnosis and careful scrutiny of phenotype data after genetic diagnosis to ensure accurate disease classification (10). In this report, we describe a family with unusual aggregation of different GDs that was clearly defined by a combination of genetic diagnosis and careful reverse phenotyping.
Case Description
YB is a 7-year-old male with no significant medical history who presented to the emergency department with hematuria in the context of 3-day history of fever, cough, and sore throat. He denied dysuria, epistaxis, hematochezia, or mucosal bleeding. Physical examination was notable for normal blood pressure, normal oropharyngeal appearance, and absence of peripheral edema. Urinalysis revealed 1+ protein, 3+ blood, and 1+ leukocytes. Urine protein/creatinine ratio was 1,167 mg/g. Serum creatinine was 0.5 mg/dl. Additional serum chemistries, complete blood count, C3/C4, anti-streptolysin O, and anti-DNase B were all within normal limits. Serologic testing was negative for anti-dsDNA, ANCA, and anti-GBM. Renal ultrasound revealed symmetrically large kidneys bilaterally without hydronephrosis, nephrolithiasis, or masses. He was treated for presumed UTI and referred to a pediatric nephrology clinic for follow up.
Approximately 2 months following his initial presentation, YB had recurrence of gross hematuria concomitantly following a viral syndrome. Rapid streptococcal testing was negative. Urinalysis revealed 3+ blood and persistent subnephrotic-range proteinuria. His symptoms resolved within a week. Given the concern for postinfectious glomerulonephritis vs. IgA nephropathy, close follow-up was arranged. Over the subsequent 9 months, he continued with microscopic hematuria and low-grade proteinuria. Upon further review of family history, it was discovered that his mother had microscopic hematuria and hypertension at 9 years of age, which was thought to be due to IgA nephropathy; she was managed conservatively, and his maternal great grandfather had a history of end-stage kidney disease (ESKD) of unclear etiology (Figure 1). Given persistent low-grade proteinuria and significant family history of kidney disease, kidney biopsy and genetic testing were pursued in YB. Notably, YB had no history of hearing loss.
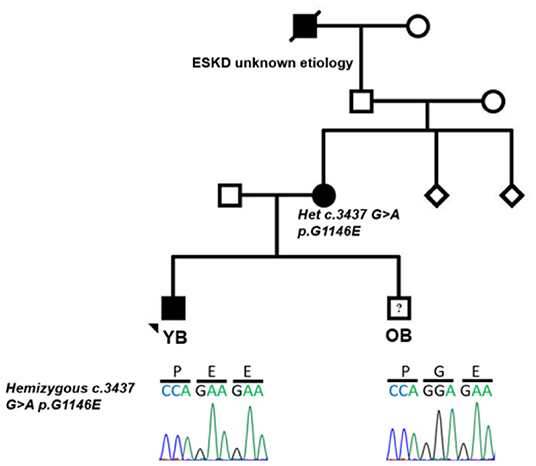
Figure 1. Pedigree of family with aggregation of different glomerular diseases showing segregation of c.3437 G>A p.G1146E in COL4A5. Filled circle and square represent affected individuals; unfilled circle, square, and diamond represent unaffected individuals.
Kidney biopsy revealed rare foci of extracapillary proliferation of epithelial cells without significant mesangial or endothelial cell proliferation, necrosis, or inflammation. Mild, focal, segmental staining of mesangial areas for IgA was identified by immunofluorescence. On electron microscopy, glomerular basement membranes appeared somewhat thinned, without thickening or lamellation; there was mild podocyte foot process effacement, but no unequivocal immune complex deposits were detected (Figures 2A,B). These findings were initially reported to be most consistent with IgA nephropathy. Genetic studies were carried out on a panel of COL4A and FSGS genes. Results revealed two variants of uncertain significance (VUS) in CD2AP (exon 7 heterozygous; c.730C>T p.Pro244Ser) and COL4A5 (exon 38 hemizygous; c.3437G>A genes (Figure 1). The COL4A5 variant is not present in over 250,000 exomes in the Genome Aggregation Database (gnomAD) database. The variant was predicted to be damaging by three in silico tools. The CD2AP variant was found in 14 individuals in the gnomAD database (minor allele frequency: 0.00006) and was also predicted to be benign by SIFT in silico tools. Based on these findings, immunofluorescent staining for COL4A was performed on the previously obtained kidney biopsy and demonstrated negative staining for collagen type IV alpha 3 and 5 in the glomerular basement membranes, negative staining for alpha 5 in Bowman's capsule, and positive staining for alpha 2 in the glomerular basement membranes and mesangium, all findings consistent with COL4A disease (Figures 2C,D).
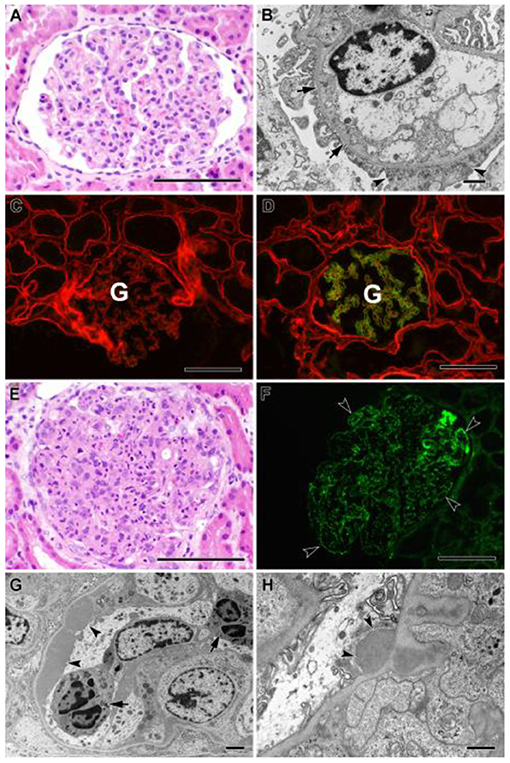
Figure 2. (A–D) Biopsy findings in patient YB. (A) Glomerulus from paraffin section showing minimal histologic abnormality (hematoxylin and eosin stain). Bar = 100 μm. (B) Electron micrograph showing mild, patchy podocyte foot process effacement (arrowheads), and mild thinning of capillary loop basement membrane without thickening or lamellation (arrows). No immune complex deposits were identified. Bar = 1 μm. (C,D) Two-color immunofluorescent stains of fresh frozen tissue for α2 (red) and α5 (green) chains of type IV collagen. (C) Patient's biopsy, showing total loss of staining for α5 chain in the glomerulus (G), consistent with Alport syndrome. (D) Positive control tissue showing normal pattern of staining for α5 chain confined to capillaries in the glomerulus (G), with more diffuse staining for α2 chain. Bars = 100 μm. (E–H) Biopsy findings in patient OB. (E) Glomerulus from paraffin section, showing global hypercellularity with abundant infiltration of neutrophils (hematoxylin and eosin stain). Bar = 100 μm. (F) Immunofluorescent stain of fresh frozen biopsy tissue for IgG showing extensive granular staining of capillary loops (arrowheads). Bar = 100 μm. (G) Electron micrograph showing large subendothelial immune complex deposit (arrowheads) and neutrophils within capillary lumen (arrows). Bar = 2 μm. (H) Electron micrograph showing subepithelial immune complex deposit (“hump,” arrowheads). Bar = 800 nm.
Based on the family history, genetic testing, and COL4A staining, YB was diagnosed with Alport syndrome due to a variant in the COL4A5 gene. YB has no history of hearing loss and passed his hearing test in the newborn period; however, his mother was recently diagnosed with bilateral hearing loss and ongoing microscopic hematuria. We carried out genetic testing in the mother and showed that she has the same COL4A5 variant detected in YB but not the CD2AP variant (Figure 1). We confirmed the COL4A5 variant by direct sequencing.
Shortly after YB was diagnosed with Alport syndrome, his 13-year-old brother (OB) presented to the emergency department (ED) with a 2-day history of fever, poor oral intake, emesis, abdominal pain, and oliguria. History was significant for Group A streptococcal pharyngitis diagnosed 1 month prior. Physical examination in the ED was notable for hypertension, petechial rash on the face and chest, and bilateral pitting pedal edema. Renal function panel revealed hyperkalemia (7.1 mmol/l) and stage three acute kidney injury (BUN 71 mg/dl, Cr 3.1 mg/dl). Urinalysis revealed 3+ blood, 2+ protein, urine protein/creatinine ratio of 388 mg/g, low C3 complement, and normal C4 complement. Serology and infectious disease work up were all negative. A presumptive diagnosis of postinfectious glomerulonephritis was made, but a renal biopsy was performed to rule out Alport syndrome in view of the family history. Histology of the kidney biopsy done on OB showed diffuse proliferative and exudative glomerulonephritis with global finely granular and focal segmental globular IgG and C3 deposits on immunofluorescence. Electron microscopy showed extensive subendothelial immune complex deposits as well as occasional subepithelial “hump-like” deposits, consistent with postinfectious glomerulonephritis (Figures 2E–H). Alport immunofluorescence staining showed no definitive loss of the alpha 3, and 5 chains of type IV collagen. OB's acute kidney injury was managed with acute dialysis, fluid restriction, and blood pressure control. His renal function normalized. He currently has no proteinuria, and his creatinine at the last follow up was 0.6. His genetic testing did not show the COL4A5 and CD2AP variant that was reported in his mother and brother (Figure 1).
Discussion
This case highlights the utility of genetic testing and reverse phenotyping in resolving clinical diagnosis (11). While presumptive diagnoses of IgA nephropathy vs. postinfectious glomerulonephritis were considered in the proband (YB), these diagnoses were questioned in the setting of persistent proteinuria and suspicious family history; inconclusive kidney biopsy findings prompted genetic testing, which enabled the subsequent delineation of glomerulopathies in this family.
Alport syndrome (AS) is an inherited glomerulopathy characterized by hematuria, proteinuria, progressive renal failure, hearing loss, and ocular abnormalities (5). Alport syndrome is caused by mutations in type IV collagen genes (COL4A3, COL4A4, and COL4A5) (5, 12). 85% of known mutations are inherited in x-linked manner (COL4A5), and the remaining 15% of mutations are inherited in an autosomal recessive or autosomal dominant manner (COL4A3 or COL4A4 mutations) (5, 12), although it should be noted that autosomal dominant Alport syndrome is probably underdiagnosed because of its subtle phenotype in the majority of affected individuals.
A 2018 and, more recently, a 2021 consensus report suggests new classification for AS because of wide variability in its clinical phenotype and estimated that the lifetime risk for chronic kidney disease (CKD) is 100, 25, 100, and 20% for X-linked recessive, X-linked heterozygous, autosomal recessive, and autosomal dominant AS respectively (5, 13).
Our report adds to previously reported data on wide variability in the clinical manifestations of COL4A-associated nephropathy and significant overlap of this condition with other distinct glomerular pathologies. We and others reported that up to 10% of patients with diagnosis of familial FSGS have pathogenic variants in one of the COL4A genes; subsequent large studies showed that COL4A variants are the most common cause of CKD of unknown etiology in adults (6–8). Two studies also found pathogenic variants in COL4A3, COL4A4, and COL4A5 in 10–20% of patients with presumed IgA nephropathy (9, 10). One approach that investigators have used to ensure accurate diagnosis of kidney disease after genetic testing is by performing reverse phenotyping after genetic testing. Reverse phenotyping is a process whereby patients and their family are reevaluated after genetic testing with the goal of segregating identified genetic variants to previously unrecognized clinical symptoms and increase the sensitivity of the genetic analysis (11). In a recent study, Landini et al. showed that reverse phenotyping was able to identify previously unrecognized clinical signs of an unexpected underlying genetic nephropathy in 28% of patients in their cohort after whole-exome sequencing (11).
The COL4A5 variant reported in this family has not been found in patients with AS before and was initially reported as variant of unknown significance (VUS). However, further clinical evaluation and additional ancillary work-up clearly establish the pathogenicity of this variant. In addition, it also allows for accurate diagnosis and management of a sibling who presented with a completely different non-monogenic glomerular disease shortly after establishing diagnosis of AS in the proband. Our findings highlight the need for the future nephrologists to be trained in the use and interpretation of genetic testing to make accurate diagnosis of different kidney diseases. They also emphasize the need for integration of clinical, pathologic, and genomic data in the diagnosis of glomerular and other kidney diseases.
In conclusion, we propose that unusual aggregation of different glomerular diseases in a family is an indication for genetic testing followed by reverse phenotyping.
Author Contributions
RG design the studies, participate in the recruitment of participants in the study, genetic studies, and deep phenotyping, and writing of the manuscript. RK participate in the recruitment of participants in the study, genetic studies, and deep phenotyping and writing of the manuscript. VK participate in the recruitment of participants in the study, deep phenotyping and editing of the manuscript. MS performed the genetic studies and was involved in writing of the manuscript. BL performed the genetic studies and was involved in writing of the manuscript. AB, LB, and DH generated and interpreted the renal biopsy. All three were involved in writing and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The expert assistance of Steven Conlon with preparation of the biopsy figure is greatly appreciated.
References
1. Harambat J, van Stralen KJ, Kim JJ, Tizard EJ. Epidemiology of chronic kidney disease in children. Pediatr Nephrol. (2012) 27:363–73. doi: 10.1007/s00467-011-1939-1
2. Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol. (2016) 12:133–46. doi: 10.1038/nrneph.2015.205
3. Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. (2007) 71:1205–14. doi: 10.1038/sj.ki.5002222
4. Kopp JB, Anders HJ, Susztak K, Podestà MA, Remuzzi G, Hildebrandt F, et al. Podocytopathies. Nat Rev Dis Primers. (2020) 6:68. doi: 10.1038/s41572-020-0196-7
5. Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport syndrome classification working group. Kidney Int. (2018) 93:1045–51. doi: 10.1016/j.kint.2017.12.018
6. Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. (2014) 86:1253–9. doi: 10.1038/ki.2014.305
7. Groopman EE, Marasa M, Cameron-Christie S, Christie SC, Petrovski S, Aggarwal VS, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. (2019) 380:142–51. doi: 10.1056/NEJMoa1806891
8. de Haan A, Eijgelsheim M, Vogt L, Knoers NVAM, de Borst MH. Diagnostic yield of next-generation sequencing in patients with chronic kidney disease of unknown etiology. Front Genet. (2019) 10:1264. doi: 10.3389/fgene.2019.01264
9. Li Y, Groopman EE, D'Agati V, Prakash S, Zhang J, Mizerska-Wasiak M, et al. Type IV collagen mutations in familial IgA nephropathy. Kidney Int Rep. (2020) 5:1075–8. doi: 10.1016/j.ekir.2020.04.011
10. Cambier A, Robert T, Hogan J, Rabant M, Peuchmaur M, Boyer O, et al. Rare collagenous heterozygote variants in children with IgA nephropathy. Kidney Int Rep. (20213) 6:1326–35. doi: 10.1016/j.ekir.2021.02.022
11. Landini S, Mazzinghi B, Becherucci F, Allinovi M, Provenzano A, Palazzo V, et al. Reverse phenotyping after whole-exome sequencing in steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2020) 15:89–100. doi: 10.2215/CJN.06060519
12. Quinlan C, Rheault MN. Genetic basis of type IV collagen disorders of the kidney. Clin J Am Soc Nephrol. (2021) 16:1101–9. doi: 10.2215/CJN.19171220
Keywords: glomerular disease, reverse phenotype, COL4A5 gene, IgA nephropathy, acute glomerulonephritis
Citation: Kumar R, Keskinyan V, Stangl MC, Lane BM, Buckley AF, Barisoni L, Howell DN and Gbadegesin RA (2022) Case Report: Unusual Aggregation of Different Glomerulopathies in a Family Resolved by Genetic Testing and Reverse Phenotyping. Front. Pediatr. 10:826330. doi: 10.3389/fped.2022.826330
Received: 30 November 2021; Accepted: 20 January 2022;
Published: 28 February 2022.
Edited by:
Nikoleta Printza, Aristotle University of Thessaloniki, GreeceReviewed by:
Vasiliki Karava, Aristotle University of Thessaloniki, GreeceDespoina Tramma, Aristotle University of Thessaloniki, Greece
Copyright © 2022 Kumar, Keskinyan, Stangl, Lane, Buckley, Barisoni, Howell and Gbadegesin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rasheed A. Gbadegesin, cmFzaGVlZC5nYmFkZWdlc2luQGR1a2UuZWR1