 Elkhansa Elgaali1
Elkhansa Elgaali1 Massimo Mezzavilla2
Massimo Mezzavilla2 Ikhlak Ahmed3
Ikhlak Ahmed3 Mohammed Elanbari3
Mohammed Elanbari3 Aesha Ali3
Aesha Ali3 Ghada Abdelaziz4
Ghada Abdelaziz4 Khalid A. Fakhro3
Khalid A. Fakhro3 Ayman Saleh1Tawfeg Ben-Omran5,6Naima Almulla1
Ayman Saleh1Tawfeg Ben-Omran5,6Naima Almulla1 Chiara Cugno1,3*
Chiara Cugno1,3*
- 1Pediatric Hematology and Oncology Department, Sidra Medicine, Doha, Qatar
- 2Department of Biology, University of Padua, Padua, Italy
- 3Research Department, Sidra Medicine, Doha, Qatar
- 4Division of Pediatrics, Sidra Medicine, Doha, Qatar
- 5Division of Genetic and Genomic Medicine, Sidra Medicine, Doha, Qatar
- 6Department of Medical Genetics, Hamad Medical Corporation, Doha, Qatar
Background: Familial hemophagocytic lymphohistiocytosis (FHLH) is an inherited life-threatening disease. Five types are identified, with the addition of congenital immunodeficiency syndromes in which HLH is a typical manifestation. The literature on this disease is very scarce in the Middle East, with only a few scattered reports.
Methods: We report detailed demographic, clinical, and genomic data from 28 patients diagnosed with primary and familial HLH over the last decade in Qatar. An evaluation was performed of allele frequencies of deleterious variants from 12 primary and familial HLH causative genes on the Qatar Genome Programme (QGP) cohort of 14,669 Qatari individuals.
Results: The genetic diagnosis was obtained in 15 patients, and four novel mutations in Perforin 1 (PRF1), UNC13D, LYST, and RAB27A genes were found. We identified 22,945 low/high/moderate/modifier impact variants significantly enriched in the QGP in those 12 genes. The variants rs1271079313 in PRF1 and rs753966933 in RAB27A found in our patient cohort were significantly more prevalent in the QGP compared to the Genome Aggregation Database (gnomAD) database, with a high carrier frequency in the Qatari population.
Conclusions: We established the first primary and familial HLH Registry in the Gulf Region and identified novel possibly pathogenic variants present at higher frequency in the Qatari population, which could be used for screening purposes. Raising awareness about primary and familial HLH and implementing screening activities in the Qatari highly inbred population could stem into more comprehensive premarital and prenatal evaluations and faster diagnosis.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) represents a hyperinflammatory syndrome characterized by reduced cytotoxic T-cell and natural killer (NK)-cell activity and uncontrolled aggressive proliferation of cytotoxic lymphocytes, leading to increasing levels of activated macrophages and inflammatory cytokines, which target multiple organs including the spleen, liver, bone marrow, and brain (1).
HLH is classified into primary, familial (FHLH), and secondary HLH. FHLH is an autosomal recessive rare immunodeficiency with onset in early childhood defined by mutations in FHL-causing loci, which render the cytotoxicity pathway ineffective in eradicating immune stimuli. Five types are identified, with four of the gene mutations described and discussed in the literature. Locus 9q21.3–22 has been associated with FHLH type 1 [Online Mendelian Inheritance in Man (OMIM) #267700]; however, the corresponding gene defect and protein have not been recognized so far. FHLH types 2–5 are caused by mutations in the perforin (PRF1, OMIM #603553), UNC13D (OMIM #608898), Syntaxin 11 (STX11, OMIM #603552), and syntaxin-binding protein-2 (STXBP2, OMIM #613101) genes (2, 3).
Additional genetic diseases in which HLH is a typical and common manifestation (primary HLH) include congenital immunodeficiency syndromes, such as Griscelli syndrome type 2 (GS2), caused by mutations of RAB27A, Chédiak–Higashi syndrome, X-linked lymphoproliferative disease, X-linked immunodeficiency with magnesium defect, interleukin-2-inducible T-cell kinase deficiency, and Hermansky–Pudlak syndrome. Acquired or secondary HLH can be triggered by infections, neoplasia, and autoimmune inflammatory syndromes, or can be found in the context of metabolic disorders and immune deficiencies, such as CD27 deficiency, lysinuric protein intolerance, chronic granulomatous disease, and so on (4).
The onset of the clinical manifestation of primary and familial HLH usually occurs in the first year of life, with the disease developing between 1 and 6 months of age or even in utero and manifesting at birth, and it is invariably fatal if left untreated. The common clinical findings include fever, hepatosplenomegaly, and bicytopenia (affecting at least two of three lineages in the peripheral blood: erythrocytes, leukocytes, and thrombocytes). Neurologic abnormalities can be isolated or indicate advanced primary and familial HLH. The diagnosis of primary and familial HLH is made by either fulfilling the HLH-2004 protocol's clinical and laboratory diagnostic criteria, or genetic testing that reveals an HLH-causing mutation (5). Early diagnosis and prompt and aggressive initiation of therapy can be life-saving (5).
To our knowledge, the literature on primary and familial HLH is very scarce in the Middle East and North Africa. Reports from Israel (6) and Oman (7) have described the clinical presentation and outcome of 11 and 13 children diagnosed with FHLH, respectively. The largest case series was recently published and reported 87 Saudi patients diagnosed with both familial and secondary HLH between 1995 and 2014: 36% of FHLH patients were found to have a STXBP2 mutation (8). In another case series, five patients with STX11 mutation and two patients with PRF1 mutation were described in a highly consanguineous setting (9).
GS2 (OMIM: 607624), caused by pathogenic variants in the RAB27A gene, is characterized by partial albinism, variable T lymphocyte and NK-cell deficiencies, and neurological impairment, i.e., seizures, strabismus, hemiparesis, ataxia, and cognitive disorders (10–12).
Al-Sulaiman et al. recently described a founder RAB27A variant (NM_183235.3:c.244C>T) causing GS2 in highly consanguineous Qatari families (10). HLH was diagnosed in 33% of the patients and suspected in an additional 33% of patients who died. Overall, a relevant phenotypic heterogeneity was found, ranging from asymptomatic individuals to patients with isolated skin manifestations or neurological complications and/or HLH (10).
The aims of the present study were as follows: (1) to report on primary and familial HLH cases in Qatar in the last decade, while establishing the first Registry in the Gulf Region; and (2) to investigate the genetic background of the primary and familial HLH spectrum in the Qatari population, exploring the allele frequency of the variants in known HLH causative genes in the context of the Qatar Genome Programme (QGP), which is a population genome project aiming to sequence the genomes of the local population (13).
Methods
Detailed demographic, clinical, and genomic data were retrospectively collected from 28 patients diagnosed with primary and familial HLH between 2010 and 2022 at the Pediatric Oncology and Hematology, and Immunology Departments of Hamad General Hospital and Sidra Medicine (Doha), which is the only tertiary pediatric care hospital in Qatar. The clinical diagnosis was made in the presence of molecular confirmation or meeting five of the following eight criteria: fever; splenomegaly; cytopenia affecting at least two out of three peripheral blood lineages; hypertriglyceridemia and/or hypofibrinogenemia; hemophagocytosis in bone marrow, spleen, or lymph nodes; low or absent NK-cell activity; hyperferritinemia; or high soluble CD25/interleukin-2 receptor levels.
A total of 15 patients affected by GS2 were previously partially reported (10).
Clinical exome sequencing was performed on 24 patients at GeneDx, USA, as previously described (10). Four patients (patients 3, 4, 6, and 10) received diagnoses based solely on clinical criteria and did not undergo subsequent genetic testing confirmation, as detailed in Table 1.
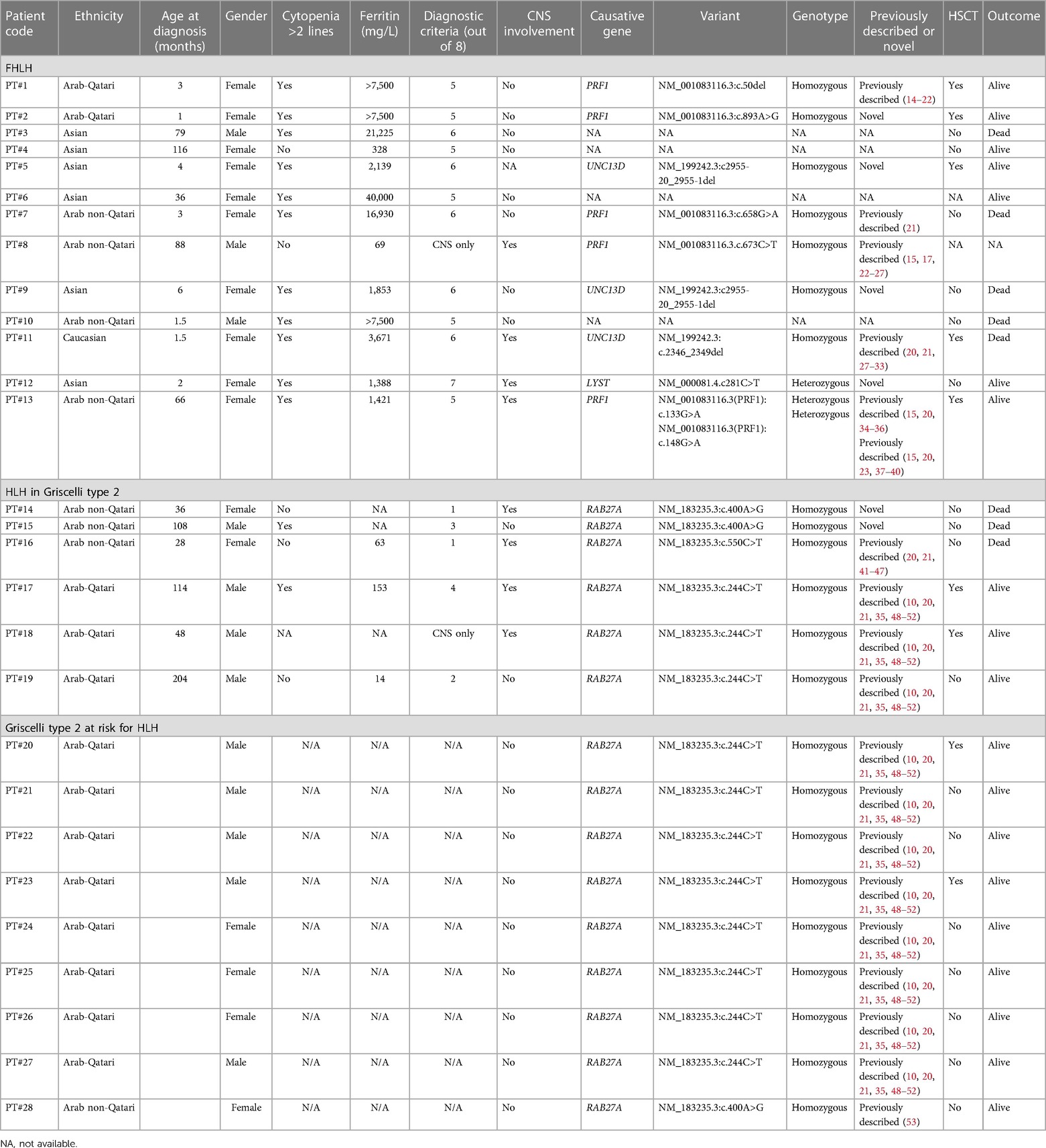
Table 1. Clinical features, laboratory characteristics, and genetic findings of the patients’ cohort.
This Registry study was conducted under the ethical approval of Sidra Medicine Institutional Review Board (IRB) (protocol no. 1760852).
The QGP study population consisted of 14,669 apparently healthy adult Qatari individuals who gave consent and were recruited by the Qatar Biobank (QBB) (13), and who underwent whole-genome sequencing at Sidra Medicine.
In the QGP study, only anonymized datasets were accessed and used for the analysis after obtaining approval from the QBB IRB (QF-QBB-RES-ACC-00052).
Whole-genome sequencing data
The evaluation of allele frequency of primary and familial HLH causative genes (PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, RAB27A, LYST, AP3B1, ITK, MAGT1, and NLRC4) was performed on the QGP cohort: raw data generated from those samples were mapped to the human reference genome (genome reference consortium human build 38 patch, GRCh38) using a Burrow-Wheeler Aligner (BWA). Joint variant calling was performed using the Genome Analysis Toolkit (GATK). Quality control (QC) was performed at different stages of the pipeline to ensure data quality and consistency. Poor quality samples were removed based on the low call rate due to poor DNA quality, outlying heterozygosity due to sample contamination or inbreeding, duplication or relatedness of samples based on identity by state (IBS), mismatches with external information, and outlying population ancestry due to population structure. Variant quality control was performed as previously described (54). The specific set of variants encompassing primary and familial HLH causative genes was extracted from the multi-sample vcf file using bcftools (55) for interrogation of their pathogenic effects in our population. Genetic variations from those genes were extracted from the sequencing data and annotated using SnpEff/SnpSift (v4.3t) (https://pcingola.github.io/SnpEff/), categorizing their impact on gene function as “high,” “moderate,” “low,” and “modifier.”
Statistical analysis
Fisher's exact test was applied to identify variants in the 12 known primary and familial HLH causative genes that showed a statistically different enrichment of the alternate allele frequency between QGP and the Genome Aggregation Database (gnomAD) release 3.0.
We corrected for multiple testing using the Benjamini–Hochberg method for controlling the false discovery rate (FDR). All variants with an adjusted p-value (adjusted) <0.05 were considered to have differential enrichment.
Analyses were run using R software version 4.0.4.
In addition, we analyzed the level of genic constraints for each primary and familial HLH gene using two metrics: (1) the loss of function intolerance score (pLi) (56), measuring from gnomAD how much a gene is prone to accumulate loss of function variants; and (2) the residual variation intolerance score (RVIS) (57), indicating whether a gene has more or less common functional genetic variation compared to the genome-wide expectation. A relatively essential or fundamental gene should have a high intolerance of both loss of function and functional variation (high pLi, low RVIS). Finally, we explored the possible protein–protein interactions (PPIs) between primary and familial HLH genes using the data obtained from the BioGRID database (https://thebiogrid.org/) (58).
Results
Based on HLH-2004 criteria, a total of 19 out of 28 (67.8%) patients were diagnosed with primary and familial HLH, 6 (21.4%) of them in the context of GS2. The detailed clinical data of the patients are reported in Table 1.
The patients (63.2% female; 36.8% male) had a mean age at the time of diagnosis of 31.3 and 89.7 months, respectively for isolated FHLH and primary HLH with underlying GS2. Different ethnicities were represented: Qatari (n = 5, 26.3%), other non-Qatari Arabs (n = 7, 36.8%), Asian (n = 6, 31.7%), and Caucasian (n = 1, 5.2%).
Primary and familial HLH were localized only to the central nervous system (CNS) in five (26.3%) patients (four in the GS2 group). Overall, CNS involvement (clinical symptoms and/or radiological findings and/or cerebrospinal fluid pleocytosis/hemophagocytosis) was identified in eight (44.4%) patients.
Other clinical diagnostic criteria were fever (83.3%); splenomegaly (74%; spleen >3 cm below costal margin); cytopenia >2 lineages (72.2%; hemoglobin <9 g/dl, absolute neutrophil count <100/µl, platelets < 100,000/µl); hypertriglyceridemia and/or hypofibrinogenemia (68.7%); hemophagocytosis in bone marrow, spleen, or lymph nodes (43.7%); hyperferritinemia (68.7%; >500 mg/L); and high soluble CD25/interleukin-2 receptor levels (100%; available only for eight patients). An NK cytotoxicity test was not performed due to the lack of testing availability in Qatar.
Genetic studies were performed in 15 patients and the following genes were identified: PRF1 (33.3%), UNC13D (20%), and RAB27A (40%). LYST was detected in only one (6.6%) patient. We found four novel mutations not previously described in PRF1, UNC13D, LYST, and RAB27A (Table 1).
Two patients (patients 8 and 18) presented with isolated CNS HLH, exhibiting suggestive radiological findings alongside a positive molecular diagnosis. Isolated CNS HLH is increasingly recognized as a distinct clinical entity, characterized by chronic inflammation confined to the CNS and germline mutations in known primary HLH-associated genes (59).
We also report on 15 GS2 patients: 6 developed primary HLH, 2 of them are alive after hematopoietic stem cell transplantation (HSCT), and 1 is awaiting the transplant procedure. A total of 11 children carried the founder mutation (NM_183235.3:c.244C > T) described by Al-Sulaiman et al. (10). Three patients with different RAB27A mutations died before having access to HSCT. Out of the 11 GS2 patients with RAB27A Qatari founder mutation, 2 were pre-emptively transplanted before developing any signs or symptoms of HLH while the others are currently being closely followed.
The patients were mostly treated with HLH protocols 1994 and 2004, and 7 out of 17 underwent HSCT.
The mortality rate was 42% (one patient was lost to follow-up). Seven out of eight patients died early after the onset of the disease, while one died from HSCT-related complications. The mortality rate for GS2 patients with primary HLH was 50%.
In the QGP cohort, only three variants (rs147035858, rs1271079313, and rs753966933), all identified in Qatari patients, were found. The variants rs1271079313 and rs753966933 were significantly more frequent in QGP when compared to the gnomAD database. A total of 19 heterozygous carriers for rs1271079313, and 145 heterozygous carriers and 7 homozygous individuals for rs753966933 were identified in the QGP cohort (Table 2).
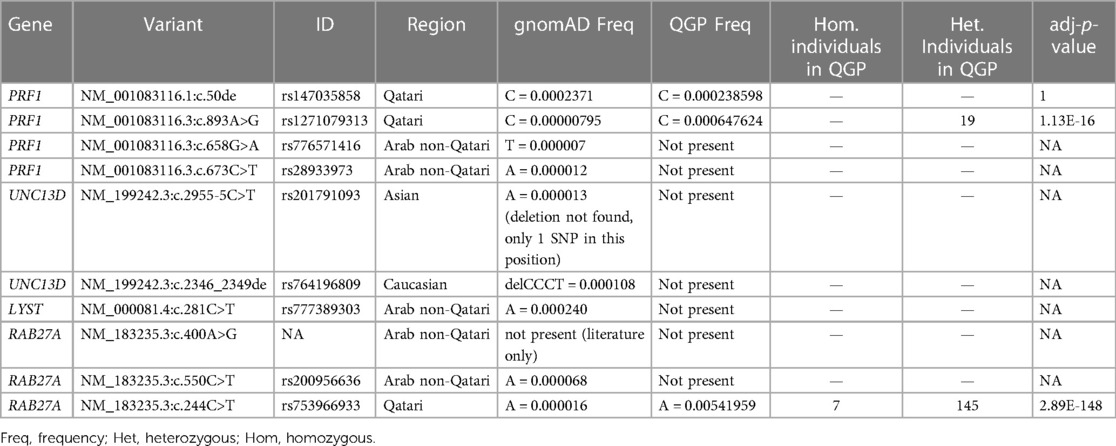
Table 2. Allele frequencies of patients’ variants in Qatar Genome Programme and gnomAD.
When restricting the analysis to unrelated samples (individuals not sharing a first- or second-degree relationship with any other individual in the QGP cohort), 18 primary and familial HLH gene variants were found at a higher frequency in QGP samples than in gnomAD samples (OR >1; 95% confidence interval (CI); adj-p-value <0.05); none of these variants was identified in our patient cohort (Supplementary Table 1).
The low/high/moderate impact variants significantly enriched in the QGP vs. gnomAD (adj-p-value <0.05) were 22,945, with higher values for STXBP2 and LYST (Figure 1).
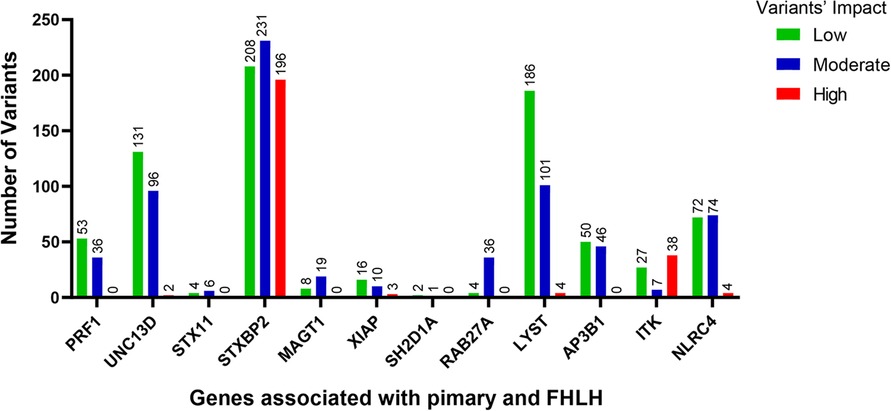
Figure 1. The number of variants with low (green bars), moderate (blue bars) and high (red bars) impact significantly enriched in the QGP cohort vs. gnomAD database. Fisher's exact test was carried out for comparisons between QGP and gnomAD to identify variants in the 12 known primary and FHLH causative genes. Adj-p-value <0.05.
In addition, we observed that three out of four genes (PRF1, LYST, and RAB27A) present in our cohort harbored a high level of genic intolerance (low RVIS, <25%), whereas none of them showed evidence of pLi (Table 3).
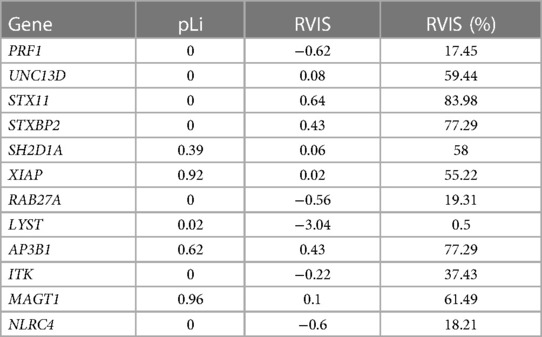
Table 3. Genic intolerance scores of the 12 primary and FHLH causative genes.
All primary and familial HLH genes found in our cohort did not display any evidence of physical interaction (PPI) between each other except for RAB27A with UNC13D (data not shown).
Discussion
The present study reports all cases of primary and familial HLH diagnosed in Qatar over the last decade and establishes the first HLH Registry in the Gulf Region.
We found that the majority (72%) of the patients had been diagnosed in the last 3 years, indicating both increased awareness of the clinical team and meaningful use of next-generation sequencing techniques for the routine diagnostic workup. Signs and symptoms often appear sequentially or late during the disease course, rendering the diagnosis difficult; molecular testing has the advantage of allowing early diagnosis in suspected cases where not all required clinical criteria are yet fulfilled.
Managing isolated CNS HLH presents significant challenges due to its elevated mortality and morbidity rates. This may be due to distinct neuroinflammatory processes triggering isolated disease manifestation without systemic activation, potentially influenced by specific genetic factors (60). The patients listed in Table 1 who had only one diagnostic criterion or exhibited CNS involvement were the ones who benefited the most from the early molecular workup. This enabled a timely diagnosis before the clinical presentation could manifest in its full severity.
In our cohort of mostly Arabic and Asian patients, we described four novel mutations, previously categorized as of uncertain significance, in PRF1 (NM_001083116.3:c.893A > G), UNC13D (NM_199242.3:c2955-20_2955-1del), LYST (NM_000081.4.c281C > T), and RAB27A (NM_183235.3:c.400A > G), whose recognition as disease causative will surely help to drive faster diagnosis and treatment.
In our study, we observed that certain genes related to primary and familial HLH within our patient cohort showed lower scores in a measurement called RVIS compared to other genes linked to the same disease. This suggests a high level of purifying selection among these genes, except for UNC13D, and explains why the deleterious variants were found mainly in RAB27A, PRF1, and LYST. Our findings indicate that three distinct genes within the studied cohort, namely PRF1, LYST, and RAB27A, manifest a heightened degree of genic intolerance (the degree to which a gene can tolerate genetic variation without adversely affecting its function), as denoted by their low RVIS (which falls under the 25th percentile of the genomic distribution of all RVIS). This should be interpreted as a reduced tolerance to genetic variations. However, noteworthy is the absence of discernible evidence denoting pronounced intolerance to loss-of-function mutations, as reflected by the lack of pLi values higher than 0.9 (61) in these three genes.
We can summarize that these genes, classified as highly sensitive to mutations and genetic changes or “mutation and/or genic intolerant” by these measures, should be enriched for variants that lead to genetic diseases compared to genes that are more tolerant to mutations and classified as “ mutation and genic tolerant” (62, 63). More precisely, these genes show evidence of intolerance to functional variation (RVIS); however, they appear to be tolerant to putative loss of function variants. An interpretation is that there are few deleterious effects if these genes are not expressed due to the presence of loss of function variants, but the presence of missense variants that alter the function of these genes is less tolerated in the population due to their possible pathogenic effects.
Although all the genes are required for the assembly, exocytosis, and function of cytotoxic granules (64), the majority of them act independently at different steps of the cytotoxic degranulation pathway, as indicated by the absence of evident protein–protein interactions in our data. The lack of interactions should rule out any hypothesis of epistatic interplay between these genes. The only exception is represented by RAB27A with UNC13D, which in fact are both involved in the docking and priming of the cytotoxic granules (64, 65).
We performed a comprehensive analysis of the variants annotated that were significantly different between the Qatari population (14,669 whole genomes) and other world populations represented in the gnomAD v3 dataset (76,156 whole genomes): 22,945 variants predicted as low/high/moderate impact in the 12 known primary and familial HLH causative genes had higher allele frequencies in the Qatari population (Figure 1), confirming recent publications showing that several rare deleterious variants are more common in the Qatari population, in line with the high consanguinity rate (54).
Two of our variants of interest (rs1271079313 and rs753966933) were found to be considerably enriched in the Qatari population. The RAB27A variant (rs753966933) has already been described as a founder mutation for Qatar in a recent publication (10), while the PRF1 variant (rs1271079313) has not reported before and can be considered a population-enriched variant that drifted to an elevated allele frequency in Qatar.
The RAB27A variant (rs753966933) was identified in 7 homozygous Qatari individuals and 145 carriers. This possibly pathogenic variant is considered highly deleterious according to its combined annotation-dependent depletion (CADD) score (CADD = 33) (66); to our knowledge, no homozygous individuals were found in other databases, such as gnomAD and the 1000 Genomes Project (1KG). This observation, coupled with the level of constraint found in this gene, could highlight a possible increased burden of deleterious variants, due to endogamy and founder effects, which are common characteristics of the Qatar population in RAB27A (54), which ultimately could lead to an increased prevalence of primary HLH in this population.
No homozygous individuals were identified for the PRF1 variant (rs1271079313) in the QGP, while 19 carriers were observed.
Our data suggest that these inherited possibly pathogenic variants should be taken into consideration for premarital or early diagnostic screenings, which are currently limited in Qatar. They might be included in the newly developed “QChip1,” a genotyping microarray including the most common single gene disease pathogenic variants identified in Qatar that could become a valuable screening tool for newborns, premarital couples, and patients (67).
We corroborated recently published data describing additional patients affected by GS2 and characterized by an extremely wide spectrum of clinical manifestations, ranging from asymptomatic individuals to life-threatening HLH (10). The penetrance of the RAB27A mutation (NM_183235.3:c.244C>T) remains unclear, especially considering the identification of seven homozygous, apparently healthy, individuals in the QGP. There is increasing evidence that partial and combined genetic defects of the degranulation pathway can predispose to HLH together with environmental stimulus and/or genetic modifiers (64) and that several digenic combinations for HLH are also present at some frequency in healthy populations (68). Further studies are needed to identify those individuals at high risk of developing HLH, hence benefitting from preventative treatment. The use of whole exome/genome sequencing instead of targeted sequencing/genotyping could help to better understand the combined role of different genes involved in the granulation pathway and their clinical impact.
We believe that raising awareness about this disease as well as early consideration of signs and symptoms could lead to a faster diagnosis, shorten the diagnostic odyssey, prevent fatal outcomes, and facilitate a rational therapeutic approach, especially with the advent of gene therapy and gene editing methods. It will also lead to the identification of new variants, enabling the offering of various preventative measures and genetic counseling in the premarital, preconceptional, or prenatal period.
Data availability statement
Whole-genome sequence data related to 14,669 Qatari individuals can be accessed through application to the Qatar Biobank (https://www.qatarbiobank.org.qa/) by submitting an online request, subject to institutional review board approval by the Qatar Biobank.
Ethics statement
This registry study was conducted under the ethical approval of Sidra Medicine Institutional Review Board (IRB) (protocol no. 1760852). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
EE: Writing – original draft, Investigation, Resources. MM: Formal Analysis, Writing – review & editing, Data curation. IA: Formal Analysis, Writing – review & editing, Data curation. ME: Formal Analysis, Writing – review & editing. AA: Validation, Visualization, Writing – original draft, Writing – review & editing. GA: Writing – review & editing. KF: Formal Analysis, Writing – review & editing. AS: Writing – review & editing. TB: Writing – review & editing, Investigation, Resources. NA: Conceptualization, Writing – review & editing. CC: Formal Analysis, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing, Conceptualization, Resources.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We acknowledge QBB for providing QGP data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1326489/full#supplementary-material
Abbreviations
CI, confidence interval; CNS, central nervous system; FDR, false discovery rate; FHLH, familial hemophagocytic lymphohistiocytosis; GATK, the Genome Analysis Toolkit; gnomAD, Genome Aggregation Database; GRCh38, genome reference consortium human build 38 patch; GS2, Griscelli syndrome type 2; IBS, identity by state; IRB, institutional review board; QBB, Qatar Biobank; 1KG, 1000 Genomes Project; pLi, loss of function intolerance score; NK, natural killer; OMIM, Online Mendelian Inheritance in Man; QC, quality control; QGP, Qatar Genome Programme; RVIS, residual variation intolerance score; PRF1, Perforin 1; STXBP2, syntaxin-binding protein-2.
References
1. Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. (2002) 100(7):2367–73. doi: 10.1182/blood-2002-01-0172
2. Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program. (2011) 2011:178–83. doi: 10.1182/asheducation-2011.1.178
3. Esteban YM, de Jong JLO, Tesher MS. An overview of hemophagocytic lymphohistiocytosis. Pediatr Ann. (2017) 46(8):e309–13. doi: 10.3928/19382359-20170717-01
4. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. (2017) 49:20–6. doi: 10.1016/j.coi.2017.08.004
5. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48(2):124–31. doi: 10.1002/pbc.21039
6. Stark B, Hershko C, Rosen N, Cividalli G, Karsai H, Soffer D. Familial hemophagocytic lymphohistiocytosis (FHLH) in Israel. I. Description of 11 patients of Iranian-Iraqi origin and review of the literature. Cancer. (1984) 54(10):2109–21. doi: 10.1002/1097-0142(19841115)54:10%3C2109::AID-CNCR2820541010%3E3.0.CO;2-M
7. Al-Lamki Z, Wali YA, Pathare A, Ericson KG, Henter JI. Clinical and genetic studies of familial hemophagocytic lymphohistiocytosis in Oman: need for early treatment. Pediatr Hematol Oncol. (2003) 20(8):603–9. doi: 10.1080/0888001039024301
8. Ahmari AA, Alsmadi O, Sheereen A, Elamin T, Jabr A, El-Baik L, et al. Genetic and clinical characteristics of pediatric patients with familial hemophagocytic lymphohistiocytosis. Blood Res. (2021) 56(2):86–101. doi: 10.5045/br.2021.2020308
9. Elyamany G, Alzahrani A, Elfaraidi H, Alsuhaibani O, Othman N, Al Mussaed E, et al. Hemophagocytic lymphohistiocytosis: single-center series of 12 cases from Saudi Arabia. Clin Med Insights Pediatr. (2016) 10:21–6. doi: 10.4137/CMPed.S35853
10. Al-Sulaiman R, Othman A, El-Akouri K, Fareed S, AlMulla H, Sukik A, et al. A founder RAB27A variant causes Griscelli syndrome type 2 with phenotypic heterogeneity in Qatari families. Am J Med Genet A. (2020) 182(11):2570–80. doi: 10.1002/ajmg.a.61829
11. Anikster Y, Huizing M, Anderson PD, Fitzpatrick DL, Klar A, Gross-Kieselstein E, et al. Evidence that Griscelli syndrome with neurological involvement is caused by mutations in RAB27A, not MYO5A. Am J Hum Genet. (2002) 71(2):407–14. doi: 10.1086/341606
12. Rajadhyax M, Neti G, Crow Y, Tyagi A. Neurological presentation of Griscelli syndrome: obstructive hydrocephalus without haematological abnormalities or organomegaly. Brain Dev. (2007) 29(4):247–50. doi: 10.1016/j.braindev.2006.09.007
13. Al Thani A, Fthenou E, Paparrodopoulos S, Al Marri A, Shi Z, Qafoud F, et al. Qatar Biobank cohort study: study design and first results. Am J Epidemiol. (2019) 188(8):1420–33. doi: 10.1093/aje/kwz084
14. Roberston IM, Jordan JM, Whitlock FA. Emotions and skin (II)—the conditioning of scratch responses in cases of lichen simplex. Br J Dermatol. (1975) 92(4):407–12. doi: 10.1111/j.1365-2133.1975.tb03101.x
15. Molleran Lee S, Villanueva J, Sumegi J, Zhang K, Kogawa K, Davis J, et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. (2004) 41(2):137–44. doi: 10.1136/jmg.2003.011528
16. Lee SM, Sumegi J, Villanueva J, Tabata Y, Zhang K, Chakraborty R, et al. Patients of African ancestry with hemophagocytic lymphohistiocytosis share a common haplotype of PRF1 with a 50delT mutation. J Pediatr. (2006) 149(1):134–7. doi: 10.1016/j.jpeds.2006.03.003
17. Trizzino A, zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet. (2008) 45(1):15–21. doi: 10.1136/jmg.2007.052670
18. Sánchez IP, Leal-Esteban LC, Álvarez-Álvarez JA, Pérez-Romero CA, Orrego JC, Serna ML, et al. Analyses of the PRF1 gene in individuals with hemophagocytic lymphohystiocytosis reveal the common haplotype R54C/A91V in Colombian unrelated families associated with late onset disease. J Clin Immunol. (2012) 32(4):670–80. doi: 10.1007/s10875-012-9680-5
19. Higa B, Velankar M. Familial haemophagocytic lymphohistiocytosis in twin infants. Pathology. (2013) 45(1):83–5. doi: 10.1097/PAT.0b013e32835b5db2
20. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. (2017) 19(10):1105–17. doi: 10.1038/gim.2017.37
21. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
22. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. (1999) 286(5446):1957–9. doi: 10.1126/science.286.5446.1957
23. Risma KA, Frayer RW, Filipovich AH, Sumegi J. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J Clin Invest. (2006) 116(1):182–92. doi: 10.1172/JCI26217
24. Blincoe A, Heeg M, Campbell PK, Hines M, Khojah A, Klein-Gitelman M, et al. Neuroinflammatory disease as an isolated manifestation of hemophagocytic lymphohistiocytosis. J Clin Immunol. (2020) 40(6):901–16. doi: 10.1007/s10875-020-00814-6
25. Clementi R, zur Stadt U, Savoldi G, Varoitto S, Conter V, De Fusco C, et al. Six novel mutations in the PRF1 gene in children with haemophagocytic lymphohistiocytosis. J Med Genet. (2001) 38(9):643–6. doi: 10.1136/jmg.38.9.643
26. Steinberg O, Yacobovich J, Dgany O, Kodman Y, Livni G, Rachmel A, et al. Prolonged course of familial hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. (2006) 28(12):831–3. doi: 10.1097/MPH.0b013e31802d3a96
27. Voskoboinik I, Thia MC, De Bono A, Browne K, Cretney E, Jackson JT, et al. The functional basis for hemophagocytic lymphohistiocytosis in a patient with co-inherited missense mutations in the perforin (PFN1) gene. J Exp Med. (2004) 200(6):811–6. doi: 10.1084/jem.20040776
28. Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. (2006) 27(1):62–8. doi: 10.1002/humu.20274
29. Santoro A, Cannella S, Trizzino A, Bruno G, De Fusco C, Notarangelo LD, et al. Mutations affecting mRNA splicing are the most common molecular defect in patients with familial hemophagocytic lymphohistiocytosis type 3. Haematologica. (2008) 93(7):1086–90. doi: 10.3324/haematol.12622
30. Angalena R, Naushad S, Devi A, Dalal B. Single gene disorders. Genomic Med. (2008) 2(3–4):241–52. doi: 10.1007/s11568-009-9118-1
31. Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. (2010) 95(12):2080–7. doi: 10.3324/haematol.2010.029389
32. Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet. (2011) 48(5):343–52. doi: 10.1136/jmg.2010.085456
33. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137(1):188–96.e4. doi: 10.1016/j.jaci.2015.06.048
34. Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. (2002) 99(1):61–6. doi: 10.1182/blood.V99.1.61
35. Jin Z, Wang Y, Wang J, Zhang J, Wu L, Gao Z, et al. Primary hemophagocytic lymphohistiocytosis in adults: the utility of family surveys in a single-center study from China. Orphanet J Rare Dis. (2018) 13(1):17. doi: 10.1186/s13023-017-0753-7
36. Al-Herz W, Chou J, Delmonte OM, Massaad MJ, Bainter W, Castagnoli R, et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front Immunol. (2018) 9:3146. doi: 10.3389/fimmu.2018.03146
37. Göransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, Söderhäll C, Samuelsson A, Janka G, et al. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet. (2001) 68(3):590–7. doi: 10.1086/318796
38. Okur H, Balta G, Akarsu N, Oner A, Patiroglu T, Bay A, et al. Clinical and molecular aspects of Turkish familial hemophagocytic lymphohistiocytosis patients with perforin mutations. Leuk Res. (2008) 32(6):972–5. doi: 10.1016/j.leukres.2007.11.033
39. Voskoboinik I, Thia MC, Trapani JA. A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood. (2005) 105(12):4700–6. doi: 10.1182/blood-2004-12-4935
40. Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci U S A. (2009) 106(24):9809–14. doi: 10.1073/pnas.0903815106
41. Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. (2000) 25(2):173–6. doi: 10.1038/76024
42. Sheela SR, Latha M, Injody SJ. Griscelli syndrome: Rab 27a mutation. Indian Pediatr. (2004) 41(9):944–7.15475639
43. Westbroek W, Tuchman M, Tinloy B, De Wever O, Vilboux T, Hertz JM, et al. A novel missense mutation (G43S) in the switch I region of Rab27A causing Griscelli syndrome. Mol Genet Metab. (2008) 94(2):248–54. doi: 10.1016/j.ymgme.2008.02.009
44. Meschede IP, Santos TO, Izidoro-Toledo TC, Gurgel-Gianetti J, Espreafico EM. Griscelli syndrome-type 2 in twin siblings: case report and update on RAB27A human mutations and gene structure. Braz J Med Biol Res. (2008) 41(10):839–48. doi: 10.1590/S0100-879X2008001000002
45. Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, Ramme K, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer. (2010) 54(4):563–72. doi: 10.1002/pbc.22357
46. Singh A, Garg A, Kapoor S, Khurana N, Entesarian M, Tesi B. An Indian boy with Griscelli syndrome type 2: case report and review of literature. Indian J Dermatol. (2014) 59(4):394–7. doi: 10.4103/0019-5154.135494
47. Ariffin H, Geikowski A, Chin TF, Chau D, Arshad A, Abu Bakar K, et al. Griscelli syndrome. Med J Malaysia. (2014) 69(4):193–4. doi: 10.1007/springerreference_41278
48. Netter P, Chan SK, Banerjee PP, Monaco-Shawver L, Noroski LM, Hanson IC, et al. A novel Rab27a mutation binds melanophilin, but not Munc13–4, causing immunodeficiency without albinism. J Allergy Clin Immunol. (2016) 138(2):599–601.e3. doi: 10.1016/j.jaci.2015.12.1337
49. Gadoury-Levesque V, Dong L, Su R, Chen J, Zhang K, Risma KA, et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. (2020) 4(12):2578–94. doi: 10.1182/bloodadvances.2020001605
50. Platt CD, Zaman F, Bainter W, Stafstrom K, Almutairi A, Reigle M, et al. Efficacy and economics of targeted panel versus whole-exome sequencing in 878 patients with suspected primary immunodeficiency. J Allergy Clin Immunol. (2021) 147(2):723–6. doi: 10.1016/j.jaci.2020.08.022
51. Forbes LR, Eckstein OS, Gulati N, Peckham-Gregory EC, Ozuah NW, Lubega J, et al. Genetic errors of immunity distinguish pediatric nonmalignant lymphoproliferative disorders. J Allergy Clin Immunol. (2022) 149(2):758–66. doi: 10.1016/j.jaci.2021.07.015
52. Zhang J, Sun Y, Shi X, Zhang R, Wang Y, Xiao J, et al. Genotype characteristics and immunological indicator evaluation of 311 hemophagocytic lymphohistiocytosis cases in China. Orphanet J Rare Dis. (2020) 15(1):112. doi: 10.1186/s13023-020-01390-z
53. Masri A, Bakri FG, Al-Hussaini M, Al-Hadidy A, Hirzallah R, de Saint Basile G, et al. Griscelli syndrome type 2: a rare and lethal disorder. J Child Neurol. (2008) 23(8):964–7. doi: 10.1177/0883073808315409
54. Mbarek H, Devadoss Gandhi G, Selvaraj S, Al-Muftah W, Badji R, Al-Sarraj Y, et al. Qatar genome: insights on genomics from the Middle East. Hum Mutat. (2022) 43(4):499–510. doi: 10.1002/humu.24336
55. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. (2011) 27(21):2987–93. doi: 10.1093/bioinformatics/btr509
56. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
57. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. (2013) 9(8):e1003709. doi: 10.1371/journal.pgen.1003709
58. Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. (2021) 30(1):187–200. doi: 10.1002/pro.3978
59. Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. (2019) 6(3):e560. doi: 10.1212/NXI.0000000000000560
60. Aytac S, Balta G, Kuskonmaz B, Aksu T, Okur FV, Unal S, et al. Isolated CNS hemophagocytic lymphohistiocytosis in children: what we know, what we don't know? Blood. (2021) 138(1):4190. doi: 10.1182/blood-2021-153509
61. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536(7616):285–91. doi: 10.1038/nature19057
62. Beck DB, Petracovici A, He C, Moore HW, Louie RJ, Ansar M, et al. Delineation of a human Mendelian disorder of the DNA demethylation machinery: TET3 deficiency. Am J Hum Genet. (2020) 106(2):234–45. doi: 10.1016/j.ajhg.2019.12.007
63. Oved JH, Babushok DV, Lambert MP, Wolfset N, Kowalska MA, Poncz M, et al. Human mutational constraint as a tool to understand biology of rare and emerging bone marrow failure syndromes. Blood Advances. (2020) 4(20):5232–45. doi: 10.1182/bloodadvances.2020002687
64. Steen EA, Hermiston ML, Nichols KE, Meyer LK. Digenic inheritance: evidence and gaps in hemophagocytic lymphohistiocytosis. Front Immunol. (2021) 12:777851. doi: 10.3389/fimmu.2021.777851
65. Kalinichenko A, Perinetti Casoni G, Dupré L, Trotta L, Huemer J, Galgano D, et al. RhoG deficiency abrogates cytotoxicity of human lymphocytes and causes hemophagocytic lymphohistiocytosis. Blood. (2021) 137(15):2033–45. doi: 10.1182/blood.2020008738
66. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46(3):310–5. doi: 10.1038/ng.2892
67. Rodriguez-Flores JL, Messai-Badji R, Robay A, Temanni R, Syed N, Markovic M, et al. The QChip1 knowledgebase and microarray for precision medicine in Qatar. NPJ Genom Med. (2022) 7(1):3. doi: 10.1038/s41525-021-00270-0
Keywords: familial hemophagocytic lymphohistiocytosis (FHLH), genetics, novel variants, QGP, hemophagocytic lymphohistiocytosis (HLH)
Citation: Elgaali E, Mezzavilla M, Ahmed I, Elanbari M, Ali A, Abdelaziz G, Fakhro KA, Saleh A, Ben-Omran T, Almulla N and Cugno C (2024) Genetic background of primary and familial HLH in Qatar: registry data and population study. Front. Pediatr. 12:1326489. doi: 10.3389/fped.2024.1326489
Received: 23 October 2023; Accepted: 8 April 2024;
Published: 9 May 2024.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Taher M. Mohiuddin, Umm al-Qura University, Saudi ArabiaDeniz N. Cagdas Ayvaz, Hacettepe University Medical School, Türkiye
© 2024 Elgaali, Mezzavilla, Ahmed, Elanbari, Ali, Abdelaziz, Fakhro, Saleh, Ben-Omran, Almulla and Cugno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Cugno, Y2N1Z25vQHNpZHJhLm9yZw==