 Katelyn P. Daniels1*
Katelyn P. Daniels1* Kim E. Nichols1
Kim E. Nichols1 Arti S. Pandey1
Arti S. Pandey1 Gabriela Gheorghe2Sara Helmig1Kevin Garrett3
Gabriela Gheorghe2Sara Helmig1Kevin Garrett3 Asim K. Bag3
Asim K. Bag3 Hiroto Inaba1
Hiroto Inaba1 Raul Ribeiro1
Raul Ribeiro1
- 1Department of Oncology, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 2Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 3Department of Radiology, St. Jude Children’s Research Hospital, Memphis, TN, United States
We describe a 5-year-old boy who initially presented with a large left renal mass, suspected to be Wilms tumor (WT). However, biopsy results revealed B-cell lymphoblastic lymphoma (B-LBL) manifesting as an isolated renal mass. Tumor transcriptome analysis identified an EWSR1::FLI1 fusion, with breakpoints distinct from those typically associated with Ewing's sarcoma. Other somatic pathogenic variants affecting WT1, ETV6, SETD2, ADD2, EZH2, PRDM2, and NF2 were identified. The patient also carried a germline CHEK2 variant of unknown significance, raising concerns for cancer predisposition. Given the unusual clinical presentation, somatic and germline genetic findings, and impossibility of measuring early response to therapy, the classical treatment of lymphoblastic lymphoma was modified. To minimize exposure to agents that increase DNA breakage, blinatumomab was used for consolidation. This approach led to significant tumor regression and the patient remains in remission for eight months post-diagnosis. This case underscores the importance of precise diagnosis, comprehensive somatic and germline genetic evaluation, and adapted treatment in pediatric oncology.
1 Introduction
This report describes a rare presentation of B-cell lymphoblastic lymphoma (B-LBL) as an isolated renal mass, initially mimicking a Wilms tumor (WT). Notably, molecular analysis of the tumor identified an atypical EWSR1::FLI1 rearrangement, a finding previously unreported in this malignancy, along with several pathogenic variants associated with hematologic malignancies. Additionally, the patient was found to carry a germline CHEK2 variant of uncertain significance (VUS). These findings posed a unique clinical challenge, necessitating a tailored therapeutic approach.
The atypical presentation and the inability to accurately assess early treatment response required deviation from conventional B-LBL treatment protocols. Instead of standard anthracycline- and cyclophosphamide-based intensification, which carries risks of DNA damage, the patient was treated with an antimetabolite-based regimen, with blinatumomab incorporated for consolidation. The kidney was preserved, and post-induction imaging and biopsy showed no residual tumor. The decision to use blinatumomab was influenced by emerging data from the Children's Oncology Group, demonstrating its potential to significantly improve disease-free survival in pediatric B-acute lymphoblastic leukemia (B-ALL) (1).
This case highlights the critical role of molecular diagnostics in guiding therapeutic decisions and underscores the importance of an individualized approach in managing complex and atypical pediatric malignancies.
2 Case description
A previously healthy 5-year-old boy presented to his primary care physician with an abdominal mass that was incidentally noticed by his mother. He was otherwise asymptomatic. Specifically, constitutional symptoms such as fever, weight loss, and night sweats were denied. He underwent abdominal ultrasound (US), which demonstrated a large left renal mass. Subsequently, a computed tomography (CT) scan revealed a large 12.7 cm×14.6 cm×12 cm left renal mass with encasement of the left renal artery. The patient was referred to the local hospital where he was noted to be hypertensive to 141/103 (>99th percentile for age) and required anti-hypertensive medication. On examination, he had an easily palpable, firm, non-tender, non-mobile mass that crossed midline and stretched from the left lower costal margin to the left lower quadrant. The family was counseled regarding a likely diagnosis of WT, and he was referred to St. Jude Children's Research Hospital for further evaluation and management.
Upon arrival, a repeat CT of the chest, abdomen, and pelvis with contrast showed no pulmonary or nodal metastases. The scan confirmed the presence of a large left renal mass replacing most of the parenchyma of the left kidney with extrarenal extension and potential retroperitoneal lymphadenopathy. Rupture of the renal capsule could not be excluded. (Figure 1A) The patient underwent an interventional radiology (IR)-guided biopsy that revealed immature lymphoid cells/blasts replacing renal tissue. Immunostaining showed the neoplastic cells to be positive for CD45 (dim), CD19, TdT, PAX5, CD43, CD20 and negative for CD3, CD123, CD34, ALK1, EBER, NKX2.2, PHOX2B, BCOR, and WT1. In conjunction with morphology, this immunophenotype was diagnostic of B-lymphoblastic leukemia/lymphoma. (Figures 2A–F) Further diagnostic workup, including bone marrow biopsy, lumbar puncture, and a positron emission tomography (PET) scan confirmed that disease was confined to the left kidney. Cerebrospinal fluid (CSF) and bone marrow examination showed no evidence of malignancy. Results of this staging evaluation indicated the presence of Stage III disease.
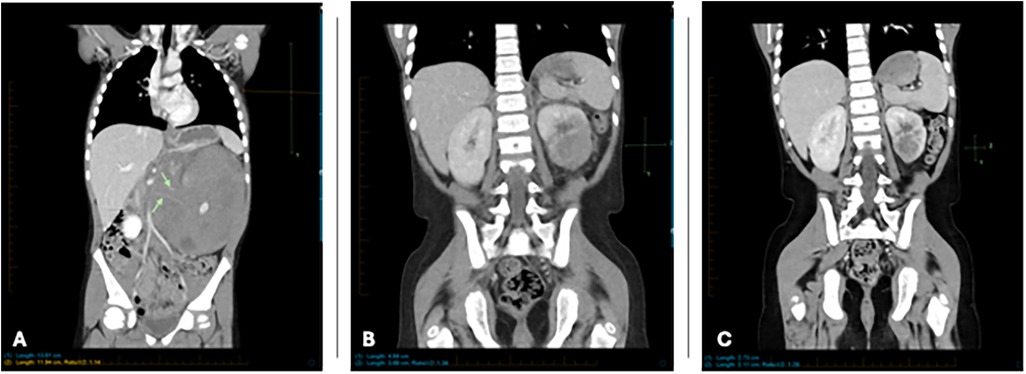
Figure 1. (A) Coronal reformatted CT at diagnosis showing large mass occupying and enlarging the entirety of the left kidney. Note encasement of renal arteries (green arrows). (B) Coronal reformatted CT 9 weeks after initial imaging shows significant improvement with smaller residual mass. (C) Coronal reformatted CT 19 weeks after initial imaging shows continued improvement with smaller residual mass.
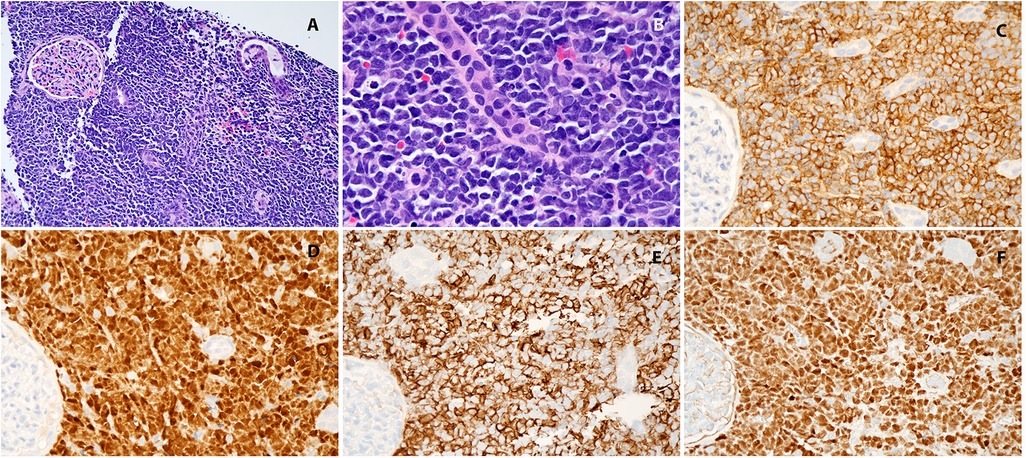
Figure 2. (A) Kidney, hematoxylin-eosin, 100×; (B) kidney, hematoxylin-eosin, 400×; (C) CD19, 400×; (D) TdT 400×; (E) CD20, 400×; (F) Pax5, 400×.
The patient began standard remission induction therapy according to guidelines of the St. Jude Total 17 trial while awaiting further genetic and cytogenetic test results. Rapid RNA sequencing revealed an EWSR1::FLI1 fusion transcript, an unusual finding for this diagnosis. Standard fusions such as ETV6::RUNX1 and TCF3::PBX1 were not detected. Whole exome sequencing (WES) identified somatic pathogenic or likely pathogenic variants in WT1 and PRDM2 with deletions within ETV6, SETD2, ADD2, EZH2, PRDM2, and NF2. Germline WES revealed a missense variant of uncertain significant (VUS) in CHEK2. Analysis of microsatellite instability, tumor mutational burden, and chromosomal breakage was not performed.
During induction therapy, the patient experienced a prolonged seizure (>1 hour), necessitating intensive care. Imaging revealed findings consistent with posterior reversible encephalopathy syndrome (PRES), likely related to his history of hypertension. Aggressive blood pressure management and anticonvulsant therapy were initiated, leading to full recovery. Subsequent imaging showed resolution of PRES-associated abnormalities. (Figure 3A–H) Treatment modifications were made to minimize anthracycline and cyclophosphamide exposure due to concerns about constitutional DNA instability. Blinatumomab was used during consolidation. A repeat PET scan after one cycle of blinatumomab showed significant regression of the left renal mass, with residual hypodense areas on CT. (Figure 1B) After a second cycle, the residual mass further decreased in size. (Figure 1C) IR-guided biopsy of the residual lesion revealed no evidence of active leukemia/lymphoma, only benign inflammatory changes and necrosis.
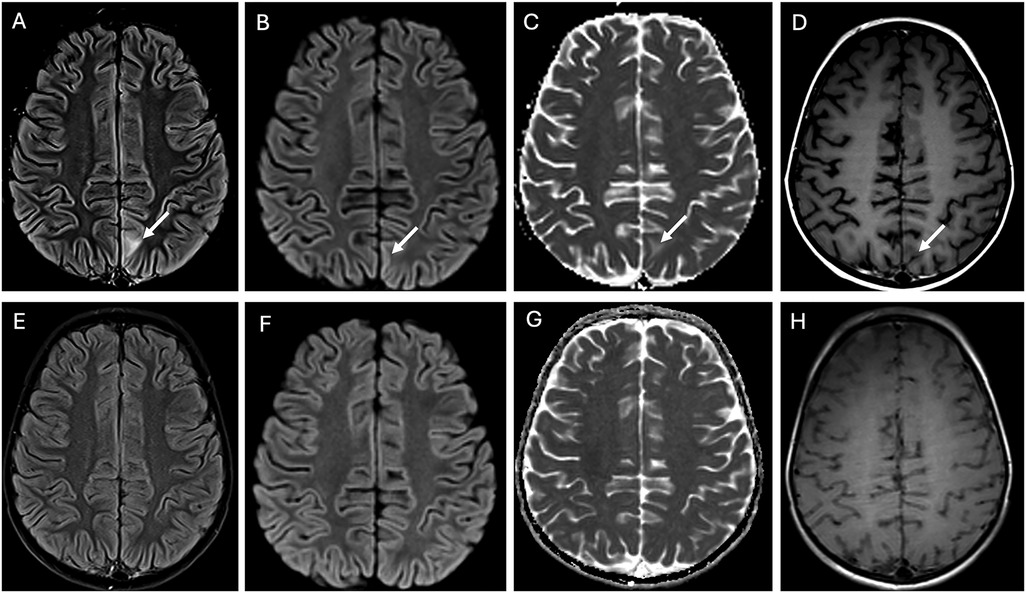
Figure 3. Posterior reversible encephalopathy syndrome (PRES). The top row of images at presentation shows an ill-defined area of hyperintensity in the juxtacortical region of the left paramedian parietal lobe on the FLAIR image arrow in (A). This is associated with facilitated diffusion, evidenced by subtle cortical hyperintensity on the diffusion trace image arrow in (B) and an increased apparent diffusion coefficient (ADC) in the juxtacortical region arrow in (C), consistent with vasogenic edema. Notably, there is no enhancement arrow in (D). At the 4-week follow-up (bottom row images), all findings on FLAIR (E) and diffusion imaging (F,G) had completely resolved, demonstrating full reversal of the condition.
3 Discussion
Lymphoblastic lymphomas (LBL) are immature malignancies arising from T-lymphoblasts (70%–80%) and B-lymphoblasts (20%–25%) (2). According to the current World Health Organization (WHO) classification, LBL and ALL exist on a spectrum, differentiated by the degree of bone marrow involvement (3). Consequently, LBL is typically treated with protocols designed for ALL. B-LBL is predominately a disease of childhood. Most patients are diagnosed before 6 years of age, with a slight male predominance. B-LBL tends to present at a lower stage than T-LBL, and there is a decreased propensity for bone marrow involvement (4, 5). Common presentation sites include bone, manifesting as osteolytic bone lesions (26%), and skin or subcutaneous tissues (23%) (4). Less common sites include the mediastinum or pleura (11%), lymph nodes (13%), or visceral organs (4%). Among the visceral sites, the gastrointestinal tract and kidneys each account for approximately 2% of cases (6).
Wilms tumor (WT), or nephroblastoma, is the most common pediatric malignancy of the kidney, with approximately 500 new cases diagnosed annually in the United States (7). Most children present asymptomatically when a caregiver or primary care physician notices a palpable abdominal mass in a child between three and five years of age. Associated symptoms such as hypertension, hematuria, and/or flank pain occur in 20%–25% of cases (8). Mutations involving WT1 are typically seen, and several genetic syndromes such as Wilms Tumor Aniridia Genitourinary Anomalies Syndrome (WAGR), Denys-Drash, Beckwith-Wiedemann, and Frasier Syndrome have been implicated in children presenting with bilateral WT (9). Treatment approaches for Wilms tumor differ by institution. Currently, the standard of care for WT within Children's Oncology Group (COG) institutions is to perform nephrectomy at the time of diagnosis (with few exceptions) while International Society of Pediatric Oncology (SIOP) recommends nephrectomy after preoperative chemotherapy (10). Biopsy is generally avoided, as it upstages the tumor to Stage III in both staging systems.
Proceeding with treatment without histologic confirmation may result in either exposing a patient to the morbidity associated with an unnecessary surgical procedure or inappropriate administration of chemotherapeutic agents delaying appropriate therapy. While most patients treated with COG strategies will undergo primary resection, about one-fourth of patients will have IR-guided biopsy followed by chemotherapy prior to surgery. However, patients treated with the SIOP approach may not have tissue obtained for histologic confirmation of diagnosis. Biopsy is only recommended for “atypical cases”. There are defined SIOP clinical, laboratory, and radiological criteria that would lead to biopsy rather than a presumed diagnosis of WT (11, 12). (Supplementary Table 1) Although up-front nephrectomy would have been reasonable in our patient with unilateral, localized disease below the age of six, normal renal parenchyma was not visualized on diagnostic imaging leading to appropriate concern for an alternative diagnosis.
Our patient's genetic findings added further complexity. An EWSR1::FLI1 fusion was detected, a hallmark cytogenetic abnormality in Ewing sarcoma (EWS), seen in 85% of cases. Interestingly, the fusion breakpoints in our patient differed from those typically associated with EWS (13). Two common EWSR1::FLI1 variants are seen in EWS: Type 1 and Type 2 fusions. Type 1 fusions consist of breakpoints in exon 7 of EWSR1 and exon 6 of FLI1, while Type 2 fusions consist of breakpoints in exon 7 of EWSR1 and exon 5 of FLI1. These specific breakpoints bring together the transcriptional activation domain of EWSR1 and the DNA-binding domain of FLI1 leading to the promotion of oncogenesis. Our patient's fusion consisted of breakpoints in exon 14 of EWSR1 and exon 4 of FLI1. Exon 14 of EWSR1 encodes a domain rich in glycine and arginine, and fusions at this exon have been reported with CREM in a novel myxoid mesenchymal tumor and in a malignant epithelioid neoplasm (14, 15) The breakpoint in exon 4 of FLI1 is part of a known breakpoint cluster region and is predicted to retain the ETS-type DNA-binding domain in its C-terminus (16). Although most common in EWS, EWSR1::FLI1 fusions have rarely been described in hematopoietic malignancies including acute leukemias, myeloid sarcoma, and in secondary leukemia after a diagnosis of EWS (17–19). In addition to this atypical EWSR1::FLI1 fusion, a heterozygous VUS in CHEK2 was discovered in our patient. Our patient's CHEK2 variant (CHEK2 c.1037G > A; p.Arg346His) was a missense mutation with frequency of about 0.002% in the population in gnomAD v2.2.1 (https://gnomad.broadinstitute.org/). Although classified as a VUS, the in-silico tool REVEL predicted a deleterious effect on protein function, and functional studies have supported this predicted deleterious impact by demonstrating DNA damage induced by alkylating agents (20, 21). This variant has been documented in individuals with breast cancer (22, 23).
Despite the lack of definitive evidence to categorize our patient's CHEK2 variant as pathogenic, we remained concerned. Although family history was largely negative for malignancy, our patient's parents remain young. (Figure 4) The knowledge that this variant could produce deleterious effects including DNA damage resulting from exposure to alkylating agents, reports of this variant in other patients with malignancies, various coexisting somatic mutations present in the tumor sample, as well as our patient's unusual presentation left grounds for suspicion that cancer predisposition may be playing a role. Furthermore, significant limitations existed in measuring early treatment response and assessing residual disease in this case without involvement of the bone marrow. CHEK2 encodes a serine-threonine kinase involved in DNA repair and cell cycle regulation (24). Additional anthracycline and cyclophosphamide exposure would lead to further DNA damage that our patient, with a missense variant exacting a deleterious effect on CHEK2, may not be able to adequately repair potentially raising the risk of malignancy later in life. This concern led to treatment modifications and the choice to eliminate “early intensification” (a treatment phase consisting of cyclophosphamide, cytarabine, and mercaptopurine) in favor of two cycles of blinatumomab, which would not carry the same risk of DNA damage.
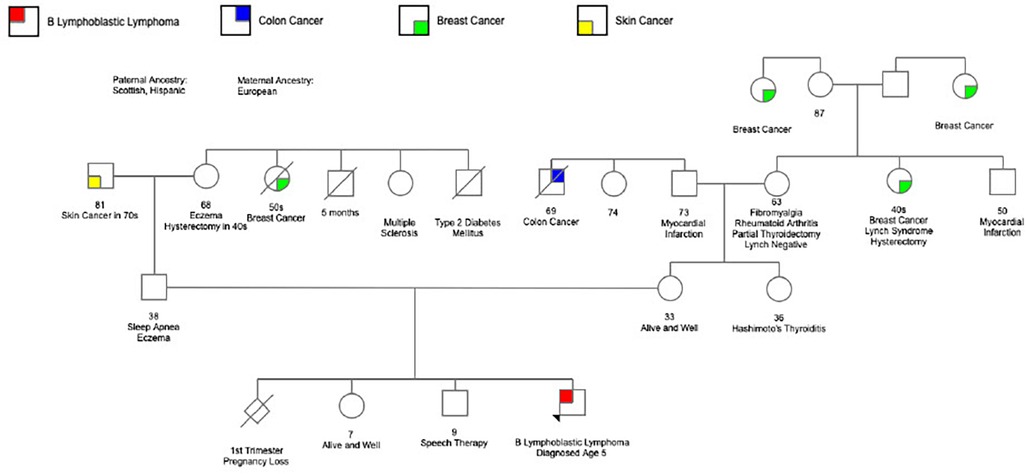
Figure 4. Family pedigree.
In conclusion, our patient's case highlights the complex diagnostic processes and decision making often involved in the care of pediatric cancer patients. What initially appeared to be a classic presentation of WT turned out to be a rare and unexpected case of B-LBL, with an isolated renal mass as the presenting manifestation. This unusual presentation underscores the importance of considering alternative diagnoses, even when clinical and imaging findings align with more common conditions. The patient's genetic findings, including the presence of an atypical EWSR1::FLI1 fusion and a possible cancer predisposition due to a CHEK2 variant, further complicate the clinical picture, necessitating a more nuanced treatment approach. Ultimately, the choice to forgo traditional anthracycline-based intensification in favor of blinatumomab was influenced by the need to minimize DNA damage, given the possibility of susceptibility to future malignancies. This case emphasizes the importance of personalized medicine, genetic testing, and careful consideration of treatment strategies for patients with pediatric malignancies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
KPD: Writing – original draft, Writing – review & editing. KN: Writing – review & editing. AP: Writing – review & editing. GG: Writing – review & editing. SH: Writing – review & editing. KG: Writing – review & editing. AB: Writing – review & editing. HI: Writing – review & editing. RR: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants CA-21765 and CA-71907 from the National Institutes of Health (U.S. Department of Health and Human Services), by a Center of Excellence grant from the State of Tennessee, and by the American Lebanese Syrian Associated Charities (ALSAC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1569506/full#supplementary-material
References
1. Gupta S, Rau RE, Kairalla JA, Rabin KR, Wang C, Angiolillo AL, et al. Blinatumomab in standard-risk B-cell acute lymphoblastic leukemia in children. N Engl J Med. (2025) 392(9):875–91. doi: 10.1056/NEJMoa2411680
2. Burkhardt B, Zimmermann M, Oschlies I, Niggli F, Mann G, Parwaresch R, et al. The impact of age and gender on biology, clinical features and treatment outcome of non-hodgkin lymphoma in childhood and adolescence. Br J Haematol. (2005) 131(1):39–49. doi: 10.1111/j.1365-2141.2005.05735.x
3. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. (2022) 36(7):1720–48. doi: 10.1038/s41375-022-01620-2
4. Ducassou S, Ferlay C, Bergeron C, Girard S, Laureys G, Pacquement H, et al. Clinical presentation, evolution, and prognosis of precursor B-cell lymphoblastic lymphoma in trials LMT96, EORTC 58881, and EORTC 58951. Br J Haematol. (2011) 152(4):441–51. doi: 10.1111/j.1365-2141.2010.08541.x
5. Lin P, Jones D, Dorfman DM, Medeiros LJ. Precursor B-cell lymphoblastic lymphoma: a predominantly extranodal tumor with low propensity for leukemic involvement. Am J Surg Pathol. (2000) 24(11):1480–90. doi: 10.1097/00000478-200011000-00003
6. Kroeze E, Arias Padilla L, Bakker M, Boer JM, Hagleitner MM, Burkhardt B, et al. Pediatric precursor B-cell lymphoblastic malignancies: from extramedullary to medullary involvement. Cancers (Basel). (2022) 14(16):3895. doi: 10.3390/cancers14163895
7. Breslow N, Olshan A, Beckwith JB, Green DM. Epidemiology of Wilms tumor. Med Pediatr Oncol. (1993) 21(3):172–81. doi: 10.1002/mpo.2950210305
8. Irtan S, Ehrlich PF, Pritchard-Jones K. Wilms tumor: “state-of-the-art” update, 2016. Semin Pediatr Surg. (2016) 25(5):250–6. doi: 10.1053/j.sempedsurg.2016.09.003
9. Huff V. Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer. (2011) 11(2):111–21. doi: 10.1038/nrc3002
10. Pater L, Melchior P, Cooper RC, McAleer BT, Kalapurakal MF, A J, et al. Wilms tumor. Pediatr Blood Cancer. (2021) 68(Suppl 2):e28257. doi: 10.1002/pbc.28257
11. Vujanic GM, Kelsey A, Mitchell C, Shannon RS, Gornall P. The role of biopsy in the diagnosis of renal tumors of childhood: results of the UKCCSG Wilms tumor study 3. Med Pediatr Oncol. (2003) 40(1):18–22. doi: 10.1002/mpo.10216
12. de Kraker J, Graf N, van Tinteren H, Pein F, Sandstedt B, Godzinski J, et al. Reduction of postoperative chemotherapy in children with stage I intermediate-risk and anaplastic Wilms’ tumour (SIOP 93-01 trial): a randomised controlled trial. Lancet. (2004) 364(9441):1229–35. doi: 10.1016/S0140-6736(04)17139-0
13. Riggi N, Stamenkovic I. The biology of ewing sarcoma. Cancer Lett. (2007) 254(1):1–10. doi: 10.1016/j.canlet.2006.12.009
14. Argani P, Harvey I, Nielsen GP, Takano A, Suurmeijer AJH, Voltaggio L, et al. EWSR1/FUS-CREB Fusions define a distinctive malignant epithelioid neoplasm with predilection for mesothelial-lined cavities. Mod Pathol. (2020) 33(11):2233–43. doi: 10.1038/s41379-020-0646-5
15. Plougastel B, Zucman J, Peter M, Thomas G, Delattre O. Genomic structure of the EWS gene and its relationship to EWSR1, a site of tumor-associated chromosome translocation. Genomics. (1993) 18(3):609–15. doi: 10.1016/S0888-7543(05)80363-5
16. Berger M, Dirksen U, Braeuninger A, Koehler G, Juergens H, Krumbholz M, et al. Genomic EWS-FLI1 fusion sequences in ewing sarcoma resemble breakpoint characteristics of immature lymphoid malignancies. PLoS One. (2013) 8(2):e56408. doi: 10.1371/journal.pone.0056408
17. Martini A, La Starza R, Janssen H, Bilhou-Nabera C, Corveleyn A, Somers R, et al. Recurrent rearrangement of the Ewing’s sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res. (2002) 62(19):5408–12.12359745
18. Hawkins JM, Craig JM, Secker-Walker LM, Prentice HG, Mehta AB. Ewing’s sarcoma t(11;22) in a case of acute nonlymphocytic leukemia. Cancer Genet Cytogenet. (1991) 55(2):157–62. doi: 10.1016/0165-4608(91)90072-3
19. Kim GE, Beach B, Gastier-Foster JM, Murata-Collins JL, Rowland JM, Donnell O, et al. Ewing sarcoma as a second malignant neoplasm after acute lymphoblastic leukemia. Pediatr Blood Cancer. (2005) 45(1):57–9. doi: 10.1002/pbc.20238
20. Delimitsou A, Fostira F, Kalfakakou D, Apostolou P, Konstantopoulou I, Kroupis C, et al. Functional characterization of CHEK2 variants in a Saccharomyces cerevisiae system. Hum Mutat. (2019) 40(5):631–48. doi: 10.1002/humu.23728
21. Boonen R, Wiegant WW, Celosse N, Vroling B, Heijl S, Kote-Jarai Z, et al. Functional analysis identifies damaging CHEK2 missense variants associated with increased cancer risk. Cancer Res. (2022) 82(4):615–31. doi: 10.1158/0008-5472.CAN-21-1845
22. Le Calvez-Kelm F, Lesueur F, Damiola F, Vallee M, Voegele C, Babikyan D, et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. (2011) 13(1):R6. doi: 10.1186/bcr2810
23. Lhota F, Zemankova P, Kleiblova P, Soukupova J, Vocka M, Stranecky V, et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin Genet. (2016) 90(4):324–33. doi: 10.1111/cge.12748
Keywords: B lymphoblastic lymphoma, renal mass, EWSR1::FLI1 translocation, CHEK2, Wilms tumor
Citation: Daniels KP, Nichols KE, Pandey AS, Gheorghe G, Helmig S, Garrett K, Bag AK, Inaba H and Ribeiro R (2025) Case Report: A rare pediatric case of B-cell lymphoblastic lymphoma presenting as an isolated renal mass with EWSR1::FLI1 translocation and germline CHEK2 variant. Front. Pediatr. 13:1569506. doi: 10.3389/fped.2025.1569506
Received: 31 January 2025; Accepted: 9 June 2025;
Published: 24 June 2025.
Edited by:
Tomasz Szczepanski, Medical University of Silesia, PolandReviewed by:
Joseph Louis Lasky, Cure 4 The Kids, United StatesJessica Heath, University of Vermont Children’s Hospital, United States
Copyright: © 2025 Daniels, Nichols, Pandey, Gheorghe, Helmig, Garrett, Bag, Inaba and Ribeiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katelyn P. Daniels, a2F0ZWx5bi5wdXJ2aXNAc3RqdWRlLm9yZw==