 Sinja Ohlsson1*
Sinja Ohlsson1* Alex Zaufel2
Alex Zaufel2 Natalia Qvartskhava2
Natalia Qvartskhava2 Friedrich Stock3Sophie Hinreiner4
Friedrich Stock3Sophie Hinreiner4 Hideo A. Baba5
Hideo A. Baba5 Benas Prusinskas6
Benas Prusinskas6 Ralf Kubitz7Boris Görg2Diran Herebian8
Ralf Kubitz7Boris Görg2Diran Herebian8 Elke Lainka1
Elke Lainka1 Simone Kathemann1
Simone Kathemann1
- 1Clinic for Pediatrics II, University Medicine Essen, Essen, Germany
- 2Department of Gastroenterology, Hepatology and Infectious Diseases, University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, Düsseldorf, Germany
- 3Institute of Human Genetics, University Medicine Essen, Essen, Germany
- 4Center for Human Genetics, Practice for Human Genetics Hehr, Regensburg, Germany
- 5Institute for Pathology, University Medicine Essen, Essen, Germany
- 6Department of Pediatrics, Vilnius University Hospital Santaros Clinics, Vilnius, Lithuania
- 7Department of Gastroenterology and Oncology, Bethanien Hospital, Moers, Germany
- 8Department of General Pediatrics, Neonatology and Pediatric Cardiology, University Hospital Düsseldorf, Heinrich Heine Universität Düsseldorf, Düsseldorf, Germany
We report on three children with novel variants in the lipolysis-stimulated lipoprotein receptor (LSR) gene with clinical presentation with early onset intrahepatic cholestasis and the main symptom being uncontrollable itching. Two patients showed dystrophy, short stature and microcephaly, whilst one patient had neurological developmental delay. LSR is one component of special tricellular tight junctions (tTJs) with expression in the liver and brain. We analyzed clinical data for all patients and performed multigene panel sequencing followed by Human Phenotype Ontology (HPO) based exome analysis, classifying the sequenced variants according to the American College of Medical Genetics and Genomics (ACMG) guidelines. We performed immunostaining on the liver cryosections. The lack of LSR expression in immunofluorescence of the patients’ liver tissue confirmed the pathogenicity of genetic variants. We analyzed bile acids (BA) and their derivatives by ultra-performance liquid chromatography-mass spectrometry (UPLC-MS) in two of the three patients, confirming disturbed bile salt secretion. We also describe the use of an ileal bile acid transport (IBAT) inhibitor in two patients with LSR-associated intrahepatic cholestasis for the first time. Both patients showed a good response to the therapy in terms of itch control. In conclusion, LSR-associated early onset intrahepatic cholestasis is a new and likely underdiagnosed disease. Patients with an unclear progressive familial intrahepatic cholestasis (PFIC)-like clinical picture should therefore undergo genetic testing of the LSR gene. Treatment with an IBAT inhibitor should be considered.
Introduction
The diagnosis of progressive familial intrahepatic cholestasis (PFIC) currently includes 13 heterogeneous subtypes (PFIC 1-13) of genetic, non-obstructive cholestatic liver disease due to impaired bile composition or secretion. PFIC 1-13 is caused by variants in the ATP8B1, ABCB11, ABCB4, TJP2, NR1H4, SLC51A, USP53, KIF12, ZFYVE19, MYO5B, SEMA7A, VPS33B and PSKH1 genes (1–6). The clinical symptoms vary depending on the type, with the main symptoms being cholestasis, itching and progressive liver fibrosis, through to end-stage liver disease in some types. Odevixibat (Bylvay®) received approval as a therapeutic option for symptom control in patients with PFIC in 2021, whilst Maralixibat (Livmarli®) has been approved since 2024. Odevixibat and Maralixibat inhibit ileal bile acid transport (IBAT), thus promoting bile acid (BA) excretion via the intestine (7).
In addition to classic PFIC, four cases of PFIC-like infantile intrahepatic cholestasis (IIC) caused by mutation in the lipolysis-stimulated lipoprotein receptor (LSR) gene (*616582) have been described to date (4, 8). The patients conformed to the clinical picture of PFIC with cholestasis and uncontrollable itching as the main symptom, with onset in early childhood. Two patients were described with neurological developmental delay and three of them with microcephaly (4, 8).
LSR is one component of special tricellular tight junctions (tTJs) that form tricellular cell contacts between epithelial cells. As an integral membrane protein, human LSR comprises 581 amino acids and contains an extracellular immunoglobulin G (IgG) domain, and transmembrane and cytoplasmatic domains. In their study, Masuda et al. (9) proposed LSR to act as a landmark to recruit other molecular components such as tricellulin to the tTJs. LSR is expressed in the fetal and adult liver as well as in ovaries, testis, the adrenal gland, lung, intestine, kidney and brain. Mesli et al. (10) also showed that mice with two inactivated LSR alleles (LSR-/-) displayed embryonic lethality, liver hypoplasia and reduced cellular density.
Methods
Patient data
We report on three children with variants in the LSR gene and a PFIC-like clinical picture. All patients receive, or have received, inpatient and outpatient care at Essen University Children's Hospital. The clinical data (medical history, examination results, laboratory parameters, and genetic and histopathological findings) have been collected since September 2023 from existing patient files after obtaining the consent of the legal guardians. All blood and tissue samples from patients used in the study were taken as a part of regular clinical diagnostics and according to clinical indications for unclear cholestatic liver disease. Consent forms for genetic testing and blood and tissue sampling were available. The parents gave their consent for publication of the data.
Massive parallel sequencing and sanger sequencing
Multigene panel sequencing followed by Human Phenotype Ontology (HPO) based exome analysis was carried out for all patients. Exome sequencing was performed followed by virtual panels. In cases where only the LSR gene was examined, virtual single-gene analysis was performed. Genomic deoxyribonucleic acid (DNA) was processed using the Nextera DNA Flex Exome enrichment protocol (Illumina, Inc., San Diego, California, USA) and sequenced on a NextSeqTM500 system (Illumina, Inc. San Diego, California, USA). All reads were aligned to the human reference genome (hg 19) and variant detection was performed by the SeqNext module (JSI Medical Systems GmbH, Ettenheim, Germany) software tool, which was used to filter and categorize all variants found. Novel sequence variants were further evaluated using AlamutVisualPlus (Interactive Biosoftware, Sofia Genetics, Saint-Sulpice-des-Rivoires, France) as well as public databases (gnomAD, ClinVar, HGMD and Decipher). After in silico characterization, the sequence variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (12). Sanger sequencing was performed on an ABI 3100Dx XL Avant Sequencer using the ABI Prism Big-DyeTM Terminator Cycle Sequencing Kit version 3.1 (Applied Biosystems, ThermoFisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer's recommendations. This method was carried out to confirm sequence variants identified by multigene panel sequencing and for parental carrier testing.
Immunofluorescence staining of liver cryosections
Liver biopsies were performed as ultrasound-guided percutaneous transhepatic core biopsies using MONOPTY Disposable Core Biopsy Instrument 16 G × 10 cm, 22 mm penetration, 1.7 cm length (BARD Biopsy Systems, Tempe, Arizona, USA). The histopathological sections were analyzed in our pathology department. Control pieces of normal human livers were surgically removed from patients with metastatic liver disease. Immunostaining of liver cryosections was performed as previously described (10). In brief, cryosections of human liver biopsies were immunostained with murine antibodies against multidrug resistance-associated protein 2 (MRP2; M2I-4, ALX-801-015), TJP1; 33-9100, Na+/K+ATPase (ATP1A1; clone C464.6, ZMS1029) and LSR; NBP1-89631 at a dilution of 1:50. Tissue samples were incubated with secondary antibodies conjugated to Alexa Fluor 488 (goat anti-mouse, A11029, green) or 546 (goat anti-rabbit, A11035, red) for 1 h at room temperature. Unless otherwise indicated, images were acquired with ZEN software (Ver.3.11) on an LSM 880 confocal laser scanning microscope (Zeiss, Jena, Germany) using Immersol 518 F (Zeiss) oil on a Zeiss Plan-APOCHROMAT 63 × oil/DIC objective at room temperature.
Quantification of serum BA levels
BA and their glycine and taurine derivatives were analyzed by ultraperformance liquid chromatography-mass spectrometry (UPLC-MS/MS). The method has already been described in detail elsewhere (10). The system consisted of an Acquity UPLC-H Class (Waters, UK) coupled to a Xevo-TQS tandem mass spectrometer (Waters, Milford, Massachusetts, USA) which is equipped with an ESI source in the negative ion mode. Data were collected in the multiple reaction monitoring (MRM) mode.
Ethics statement
This study involving human participants was reviewed and approved by the Ethics Committee of the Medical faculty of the University Duisburg-Essen (vote 25-12327-Bo). Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with national legislation and the institutional requirements.
Results
Case series
Patient 1
Female patient of Indian origin from non-consanguineous parents had presented with clinically relevant itching since the age of 6 months (Table 1). The physical and neurological development was unremarkable. Laboratory chemistry showed a significant increase in BAs and a mild increase in aminotransaminases and gamma GT with normal bilirubin. The liver biopsy revealed grade 2–3 fibrosis, and the immunohistochemical examination for known transporter defects (BSEP, MDR3, MRP2 and CD10/CD13) was negative. Previous multi gene panel testing revealed one heterozygous variant each in the ABCB4 and ABCB11 genes, which were each assessed as unclear variants (ACMG class 3). In addition, a heterozygous variant was found in the CFTR gene, which was assessed as a likely pathogenic variant (ACMG class 4). In the case of autosomal recessive inheritance for CFTR, ABCB11 and ABCB4 related disorders a second variant would be necessary to confirm the diagnosis, but this could not be detected. A whole exome sequencing performed in India revealed two variants in the LSR gene. These two LSR variants were therefore analyzed in the patient and her parents by Sanger sequencing. Two compound-heterozygous variants were found in the LSR gene (c.257A>T, p.(Asp86Va); c.1274dup, p.(Glu426Glyfs*43)), which were classified as likely pathogenic (class 4) according to ACMG guidelines.
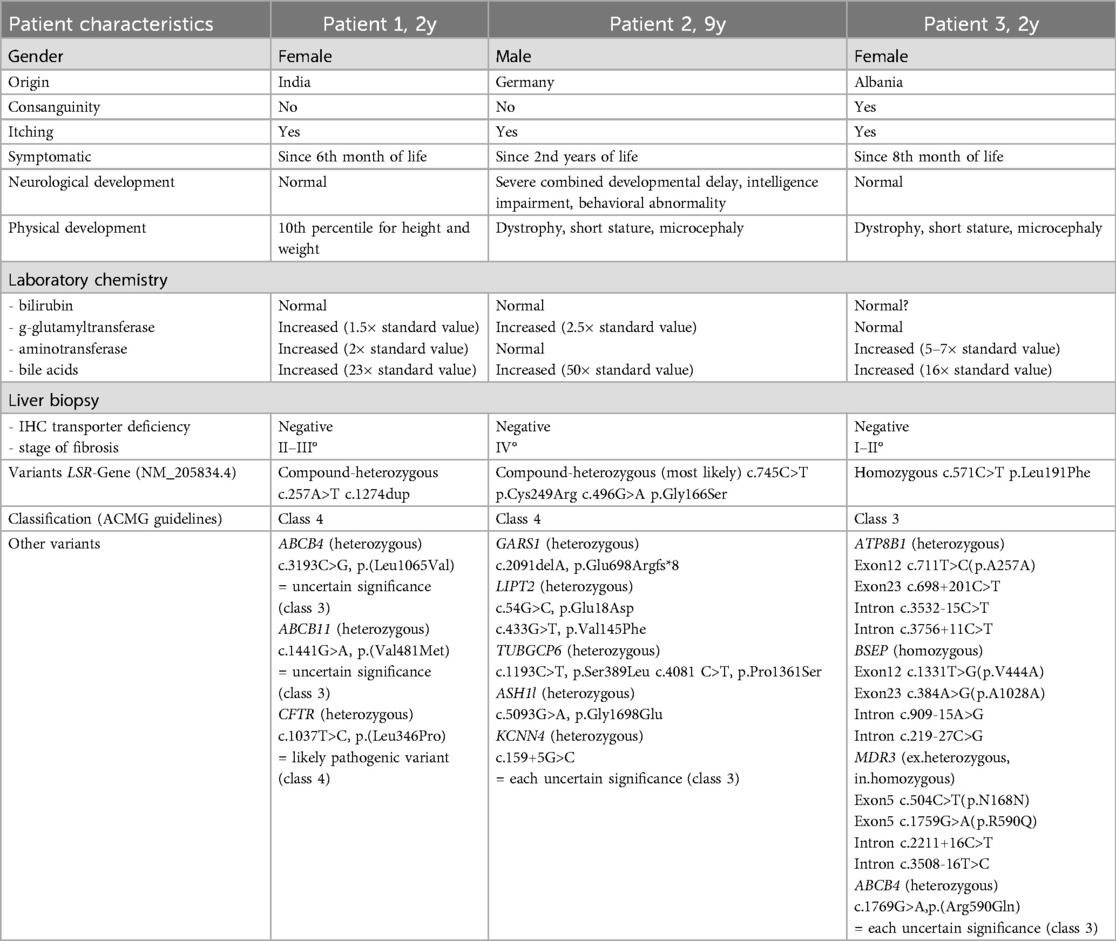
Table 1. Summary of the clinical features and genetic findings in our three patients with infantile intrahepatic cholestasis (IIC) with variants in the LSR gene.
Pedigrees are provided in Supplementary Figure S1. Conventional itching treatment for liver-related itching, such as colestyramine, ursodeoxycholic acid, rifampicin, naltrexone, phenobarbital and ondansetron, did not alleviate symptoms. Treatment with Odevixibat (Bylvay®) was initiated at the age of 2 years following the diagnosis of IIC. Follow-up was possible for 9 months until the patient moved back to India. A convincing response with a significant reduction of itching was seen after just 5 days and within the whole follow-up period. No relevant side effects were observed. As it is a genetic defect, the family planned a curative liver transplantation in India through a living donation from the father.
Patient 2
Male patient of German origin from non-consanguineous parents has presented with clinically relevant pruritus since the age of two. The patient has suffered from recurrent infection-associated seizures since the postnatal period. He has also displayed severe combined developmental delay, intellectual disability and behavioral problems, and in addition has dystrophy, short stature and microcephaly. Laboratory chemistry showed a significant increase in BA and a mild increase in gamma GT with normal bilirubin. The liver biopsy revealed liver cirrhosis but the immunohistochemical examination for known transporter defects was negative. Previous multi gene panel testing for genes associated with cholestasis and further testing revealed unclear heterozygous unclear variants. Based on the findings in patient 1, we also performed a targeted examination of the LSR gene. Two heterozygous variants were found in the LSR gene (c.745C>T p.Cys249Arg, c.496G>A p.Gly166Ser), which were classified as likely pathogenic (class 4) according to ACMG guidelines. Based on the clinical manifestation we suspect that both variants are compound heterozygous. However, as the father could not be examined, this cannot be stated conclusively. Pedigrees are provided in Supplementary Figure S1. Since the boy had refused all medication, pruritus therapy had not been possible until after the installation of a percutaneous endoscopic gastrostomy, after which medication could be administered for the first time. At age 9 years, the patient was treated with Odevixibat (Bylvay®) for 7 months and demonstrated a significant reduction in pruritus, with an accompanying improvement in quality of life, which continues to date. In addition, no relevant side effects were observed. Unfortunately, after 7 months, the gastric tube became blocked by Odevixibat, which dissolves only poorly, so that the patient was switched to the liquid drug Maralixibat (Livmarli®).
Patient 3
Female patient of Albanian origin from consanguineous parents has presented with clinically relevant itching and elevation of aminotransferases since the age of 8 months. The neurological development has so far been unremarkable, but the patient shows dystrophy, short stature and microcephaly. Laboratory chemistry shows a mild increase in BA but with normal bilirubin and gamma GT. The liver biopsy revealed grade 1–2 fibrosis and the immunohistochemical examination for known transporter defects was negative. Previous multi gene panel testing for genes associated with PFIC and congenital defect in bile acid synthesis revealed one heterozygous variant in the ATP8B1 gene, one heterozygous variant in the ABCB4 gene, one homozygous variant in the BSEP gene and a variant in MDR3 gene, which were each assessed as unclear variants (ACMG class 3). Based on the findings in patient 1, we also performed a targeted examination of the LSR gene. One homozygous variant (c.571C>T p.Leu191Phe) was found. Since the parents (both heterozygous for the variant) are consanguineous, the variant can only be classified as unclear variant (class 3) according to ACMG guidelines. Pedigrees are provided in Supplementary Figure S1. At present, the itching is only mild and barely affects the patient's quality of life. An IBAT-inhibitor has therefore not been used to date, with only ursodeoxycholic acid prescribed as a regular medication. Rifampicin is given on demand during rare episodes of extreme itching.
Detection of LSR in human liver by immunofluorescence
To investigate the pathophysiological relevance of these newly discovered mutations, liver biopsies were co-stained with LSR and specific antibodies. In normal human liver tissue, LSR surrounds the bilirubin transporter MRP2 within the canalicular membrane and co-localizes with TJP1 at the canalicular tight junctions to display a distinct immunoreactivity. LSR was still detectable in the livers of all three patients (Figure 1), although to a greatly reduced extent. However, while the staining of MRP2 and TJP1 was mostly preserved and localized to the canalicular membrane and canalicular tight junctions, respectively, LSR was predominantly aberrantly located at the basolateral plasma membrane, as shown by co-staining with ATP1A1 in patients 2 and 3. The detection of aberrantly located LSR without gross disruption of the bile canaliculus therefore confirms that LSR is of central importance for the physiological functions of the bile canaliculus.
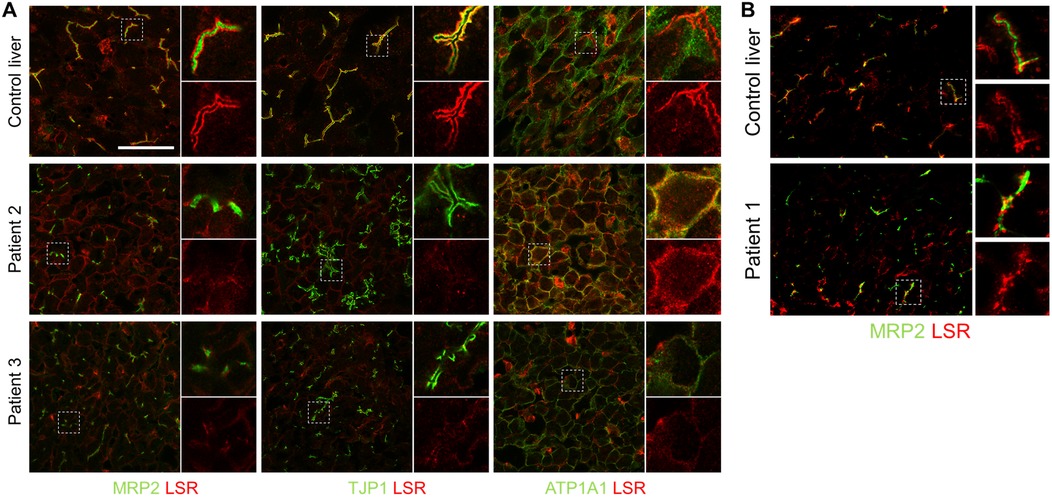
Figure 1. Immunofluorescence staining of LSR in liver slices. In normal human liver tissue (control liver), LSR (red), MRP2, TJP1, and ATP1A1 (green) were all strongly expressed and LSR colocalized at canalicular tight junctions with TJP1. (A) In the livers of both patients 2 and 3, TJP1 immunofluorescence (green) was largely preserved, while LSR immunoreactivity (red) was markedly reduced and colocalized aberrantly with ATP1A1 (green) at the basolateral plasma membrane. (B) In the liver of patient 1, MRP2 immunofluorescence (green) was preserved, while LSR immunoreactivity (red) was markedly reduced. There was insufficient patient material available for a complete set of immunofluorescence stains as shown in (A) Images in (B) were acquired with ZEN Blue 3.5.093 imaging software (ZEISS) on an Axio Observer.Z1 microscope with a C-Apochromat 63×/1.20 W Korr UV VIS IR objective. Scale bar, 50 μm. LSR, lipolysis-stimulated lipoprotein receptor; MRP2, multidrug resistance-associated protein 2; TJP1, tight junction protein 1; ATP1A1, ATPase Na(+)/K(+) transporting subunit alpha 1.
Bile salt analysis confirms disturbed bile salt secretion
Primary and secondary BA and their taurine and glycine conjugates were analyzed from the sera of patient 2, patient 3 and 40 controls by high performance liquid chromatography-mass spectrometry (HPLC-MS/MS) (Figure 2). Although both patients showed no or only slight elevations of serum bilirubin and gamma GT, total serum BA were significantly elevated to 71.0 µM in patient 2 and 22.3 µM in patient 3 (normal median: 0.94 µM, n = 40). Unconjugated and secondary BA, which are produced by bacterial deconjugation and dihydroxylation in the large intestine, were still present in both patients. However, the increased total serum BA concentrations were mainly due to a disproportionate increase in conjugated and primary BA concentrations, as indicated by the increased ratios between conjugated/unconjugated and primary/secondary BA. The hydrophobicity index tended to be decreased in both patients, suggesting that BA overload rather than toxic BA composition contributed to liver injury in patients 2 and 3. Thus, these data indicate that both patients excrete insufficient amounts of BA, but without complete disruption of the enterohepatic circulation.
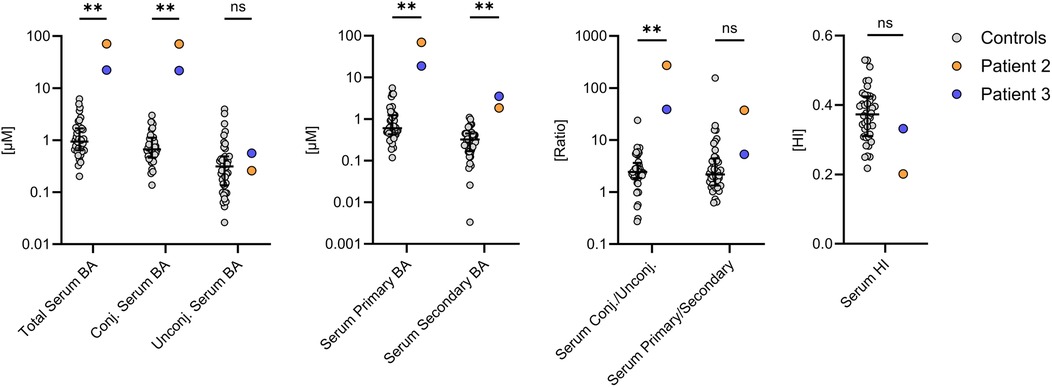
Figure 2. Serum bile acid (BA) composition in patients 2 and 3. Both patients had significantly increased serum BA concentrations, which was mainly due to a disproportionate increase in primary and conjugated BA. The BA profiles were compared with the measurements of 40 control subjects (grey). *P < 0.05, **P < 0.01. Statistical significance was calculated using an unpaired Mann–Whitney test. Data are depicted as median with interquartile range. BA, bile acids; HI, hydrophobicity index.
Discussion
We report on three children with early onset intrahepatic cholestasis caused by novel variants in the LSR gene. Based on the ACMG guidelines, the genetic findings of our patients were classified as likely pathogenic (patient 1 and 2) and variant of unknown significance (patient 3), respectively. The clinical characteristics of these children and the lack of LSR expression in the immunofluorescence of the patients’ liver tissue confirm the pathogenicity of the genetic variants. All patients had additional variants in other genes. Although no variants were described as primary causes for cholestasis and itching, they are potential confounders. Only four patients with LSR-associated early onset intrahepatic cholestasis have been reported to date (4, 8). These patients showed PFIC-like disease presentation, with itching being the main symptom. As with the other PFIC diseases, our patients showed a sub-individually variable clinical picture ranging from mild symptoms without neurological impairment to the full clinical picture with therapy-refractory itching, liver cirrhosis and severe neurodevelopmental delay. Previous data report on onset of symptoms in infancy (4, 8). Our case series shows that the obvious onset of symptoms does not necessarily have to be in infancy, but is rather a diagnosis of the early childhood with variable courses. As in the cases described above, one of our patients had severe neurological symptoms (combined neurodevelopmental delay, intellectual disability, behavioral problems, seizures) and two had microcephaly. Since expression of the LSR protein has also been detected in the brain, a neurological manifestation of the diseases is plausible. However, further research is required as to whether the variants in the LSR gene are the exclusive the cause of the neurological symptoms. Nevertheless, the neurological development of patients with LSR variants should receive close monitoring, whilst long-term observation is necessary to better assess any prognosis. Further biochemical investigations into the function of the LSR protein in the liver - but also in other tissues such as the brain - should be performed for a better understanding of the pathogenesis of this disease. Masuda et al. (9) previously described LSR as an integral component of tTJs to form tricellular cell contacts between epithelial cells. The pathogenesis may be similar to that of PFIC 4, in which the cell contacts in the canalicular membrane and between the cholangiocytes are disrupted due to deficiency of TJP2. As with LSR, TJP2 is also expressed in multiple tissues. In the pathomechanism of PFIC 4, it is assumed that the BA themselves additionally stress the cell-cell contacts, so that they are even less stable than in other tissues (11). This explanation would also be reasonable be for LSR-associated early onset intrahepatic cholestasis.
In our study, we describe the use of an IBAT inhibitor in two patients with LSR-associated early onset intrahepatic cholestasis for the first time. Both patients showed a good response to the therapy in terms of itch control. No side effects were observed. Although the long-term results of its use remain to be seen, and the number of cases is small, the use of an IBAT inhibitor appears to be promising. Odevixibat and Maralixibat may delay or prevent liver transplantation and be a conservative treatment for pruritus.
Conclusion
In summary, LSR-associated early onset intrahepatic cholestasis a new and probably underdiagnosed disease. Patients with an unclear PFIC-like clinical picture should therefore undergo genetic testing of the LSR gene. More genetic testing will expand the etiology and understanding of PFIC-related diseases and bile transport.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethic Committee of the Medical faculty of the University Duisburg-Essen (vote 25-12327-Bo). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
SO: Data curation, Formal analysis, Writing – original draft. AZ: Methodology, Resources, Visualization, Writing – review & editing, Investigation. NQ: Investigation, Methodology, Resources, Visualization, Writing – review & editing. FS: Data curation, Formal analysis, Software, Validation, Writing – review & editing. SH: Data curation, Formal analysis, Validation, Writing – review & editing. HB: Investigation, Writing – review & editing. BP: Writing – review & editing. RK: Investigation, Visualization, Writing – review & editing. BG: Investigation, Software, Visualization, Writing – review & editing. DH: Formal analysis, Investigation, Validation, Visualization, Writing – review & editing. EL: Conceptualization, Data curation, Formal analysis, Supervision, Writing – original draft, Writing – review & editing. SK: Conceptualization, Data curation, Formal analysis, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. AZ was supported in part by the MODS project funded from the program “Profilbildung 2020” (grant no. PROFILNRW-2020-107-A), an initiative of the Ministry of Culture and Science of the State of North Rhine-Westphalia.
Acknowledgments
Fluorescence microscopy was performed at the Advanced Light Microscopy Core Facility (Ad-Light), Medical Faculty of the Heinrich-Heine-University, Düsseldorf.
The authors gratefully acknowledge Lars Pape for critical reading of and commenting on this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1582769/full#supplementary-material
Supplementary Figure S1 | Immunofluorescence staining of LSR in liver slices of patient 1.
Abbreviations
ABCB, ATP binding cassette subfamily B; ACMG, American college of medical genetics and genomics; ATP, adenosine triphosphate; ATP1A1, ATPase Na(+)/K(+) transporting subunit alpha 1; BA, bile acids; BSEP, bile salt export pump; CD, cluster of differentiation; DNA, deoxyribonucleic acid; HI, hydrophobicity index; HPLC-MS, high performance liquid chromatography-mass spectrometry; HPO, human phenotype ontology; IBAT, ileal bile acid transport; IgG, immunoglobulin G; IIC, infantile intrahepatic cholestasis; LSR, lipolysis-stimulated lipoprotein receptor; MDR3, multidrug resistance gene 3; MRM, multiple reaction monitoring; MRP2, multidrug resistance-associated protein 2; PFIC, progressive familial intrahepatic cholestasis; TJP, tight junction protein; tTJs, tricellular tight junctions; UPLC-MS, ultra-performance liquid chromatography-mass spectrometry.
References
1. Qiu YL, Liu T, Abuduxikuer K, Hao CZ, Gong JY, Zhang MH, et al. Novel missense mutation in VPS33B is associated with isolated low gamma-glutamyltransferase cholestasis: attenuated, incomplete phenotype of arthrogryposis, renal dysfunction, and cholestasis syndrome. Hum Mutat. (2019) 40(12):2247–57. doi: 10.1002/humu.23770
2. Amirneni S, Haep N, Gad MA, Soto-Gutierrez A, Squires JE, Florentino RM. Molecular overview of progressive familial intrahepatic cholestasis. World J Gastroenterol. (2020) 26(47):7470–84. doi: 10.3748/wjg.v26.i47.7470
3. Gao E, Cheema H, Waheed N, Mushtaq I, Erden N, Nelson-Williams C, et al. Organic solute transporter alpha deficiency: a disorder with cholestasis, liver fibrosis, and congenital diarrhea. Hepatology. (2020) 71(5):1879–82. doi: 10.1002/hep.31087
4. Maddirevula S, Alhebbi H, Alqahtani A, Algoufi T, Alsaif HS, Ibrahim N, et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet Med. (2019) 21(5):1164–72. doi: 10.1038/s41436-018-0288-x
5. Luan W, Hao CZ, Li JQ, Wei Q, Gong JY, Qiu YL, et al. Biallelic loss-of-function ZFYVE19 mutations are associated with congenital hepatic fibrosis, sclerosing cholangiopathy and high-GGT cholestasis. J Med Genet. (2021) 58(8):514–25. doi: 10.1136/jmedgenet-2019-106706
6. Pan Q, Luo G, Qu J, Chen S, Zhang X, Zhao N, et al. A homozygous R148W mutation in semaphorin 7A causes progressive familial intrahepatic cholestasis. EMBO Mol Med. (2021) 13(11):e14563. doi: 10.15252/emmm.202114563
7. Thompson RJ, Arnell H, Artan R, Baumann U, Calvo PL, Czubkowski P, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. (2022) 7(9):830–42. doi: 10.1016/S2468-1253(22)00093-0
8. Uehara T, Yamada M, Umetsu S, Nittono H, Suzuki H, Fujisawa T, et al. Biallelic mutations in the LSR gene cause a novel type of infantile intrahepatic cholestasis. J Pediatr. (2020) 221:251–4. doi: 10.1016/j.jpeds.2020.01.064
9. Masuda S, Oda Y, Sasaki H, Ikenouchi J, Higashi T, Akashi M, et al. LSR defines cell corners for tricellular tight junction formation in epithelial cells. J Cell Sci. (2011) 124(Pt 4):548–55. doi: 10.1242/jcs.072058
10. Mesli S, Javorschi S, Berard AM, Landry M, Priddle H, Kivlichan D, et al. Distribution of the lipolysis stimulated receptor in adult and embryonic murine tissues and lethality of LSR-/- embryos at 12.5 to 14.5 days of gestation. Eur J Biochem. (2004) 271(15):3103–14. doi: 10.1111/j.1432-1033.2004.04223.x
11. Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. (2014) 46(4):326–8. doi: 10.1038/ng.2918
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24.25741868
Keywords: infantile intrahepatic cholestasis, early onset intrahepatic cholestasis, LSR gene, pruritus, itching, microcephaly, progressive familiar intrahepatic cholestasis, IBAT inhibitor
Citation: Ohlsson S, Zaufel A, Qvartskhava N, Stock F, Hinreiner S, Baba HA, Prusinskas B, Kubitz R, Görg B, Herebian D, Lainka E and Kathemann S (2025) The role of LSR gene variants in early onset intrahepatic cholestasis: a case series with treatment options. Front. Pediatr. 13:1582769. doi: 10.3389/fped.2025.1582769
Received: 24 February 2025; Accepted: 31 July 2025;
Published: 4 September 2025.
Edited by:
Patryk Lipiński, Uczelnia Warszawska im. Marii Skłodowskiej-Curie, PolandReviewed by:
Sateesh Maddirevula, King Faisal Specialist Hospital and Research Centre, Saudi ArabiaCharina Ramirez, University of Texas Southwestern Medical Center, United States
Copyright: © 2025 Ohlsson, Zaufel, Qvartskhava, Stock, Hinreiner, Baba, Prusinskas, Kubitz, Görg, Herebian, Lainka and Kathemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sinja Ohlsson, c2luamEub2hsc3NvbkB1ay1lc3Nlbi5kZQ==