 Junghan Lee
Junghan Lee Sungji Ha
Sungji Ha Seung-Tae Lee3
Seung-Tae Lee3 Keun-Ah Cheon
Keun-Ah Cheon- 1Division of Child and Adolescent Psychiatry, Department of Psychiatry, Severance Hospital, Institute of Behavioral Science in Medicine, Yonsei University College of Medicine, Seoul, South Korea
- 2Department of Psychiatry, Institute of Behavioral Science in Medicine, Yonsei University College of Medicine, Seoul, South Korea
- 3Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, South Korea
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impairments in social communication and restricted and repetitive behaviors and interests. Identifying the genetic background may be one of the key features for the future diagnosis and treatment of ASD. With the tremendous development in genetic diagnosis techniques, next-generation sequencing (NGS) can be used to analyze multiple genes simultaneously with a single test in laboratory and clinical settings and is well suited for investigating autism genetics. According to previous studies, there are two types of genetic variants in ASD, rare variants and common variants, and both are important in explaining pathogenesis. In this study, NGS data from 137 participants with ASD were reviewed retrospectively with consideration for comorbid epilepsy. Diagnostic yield was 17.51% (24/137), and pathogenic/likely pathogenic variants were seen more frequently in female participants. Fourteen participants were diagnosed with comorbid epilepsy, six of them had pathogenic/likely pathogenic variants (43%). Genes with variants of unknown significance (VOUS) which have one or more evidence of pathogenicity following the American College of Medical Genetics (ACMG) criteria were also reviewed in both ASD and ASD with comorbid epilepsy groups. We found that most frequently found VOUS genes have previously been reported as genes related to ASD or other developmental disorders. These results suggest that when interpreting the NGS results in the clinical setting, careful observation of VOUS with some pathological evidence might contribute to the discovery of genetic pathogenesis of neurodevelopmental disorders such as ASD and epilepsy.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder with core symptoms of persistent deficits in social communication and restricted, repetitive patterns of behavior, interests, or activities (APA, 2013). According to recent reports, ASD no longer seems to be a rare disease; the overall prevalence is 16.8 per 1,000 children aged 8 years, with overall male-to-female ratio of 4:1 in the United States (Baio et al., 2018). Autism spectrum disorder is not simply a disorder but a significant social problem because the annual costs for ASD patients are tremendous (Lavelle et al., 2014). Costs may include medical and nonmedical costs and indirect costs such as parental productivity loss (Buescher et al., 2014). Accurate diagnosis and treatment guidance might substantially impact treatment of the disorder and reduce annual costs.
Autism spectrum disorder is associated with various coexisting factors including those related to genetics, the prenatal environmental, and the postnatal environmental (Lord et al., 2018). While knowledge about the neurobiological basis of ASD is still insufficient, genetic factors are regarded as crucial components according to previous studies (Rosenberg et al., 2009; Hallmayer et al., 2011). Studies of monozygotic twin concordance and sibling recurrence rates clearly reveal that genetic factors play important roles in the development of ASD (Geschwind, 2011; Sandin et al., 2014; Tick et al., 2016). In this context, identifying the genetic background of each ASD patient could be the ‘cornerstone’ of proper diagnosis and individualized treatment. In general, genetic diagnosis has the benefits of informing prognoses and preventing further superfluous invasive testing, leading to tailored treatment and family counseling (Han et al., 2018). In particular, genetic testing may be a key component in the development of precision medicine, in hope of predicting treatment outcomes on an individual basis. Tools for genetic analyses are rapidly developing and the collection of genetic information is accelerating tremendously (Geschwind and State, 2015; Loth et al., 2016).
However, there are inevitable obstacles to defining a genetic basis of ASD. First, the majority of ASD cases cannot be explained by a single gene mutation. Previous reports have indicated that only 10% of ASD cases originate from a rare variant in a single gene (Persico and Napolioni, 2013). Moreover, ASD can occur as a result of a combination of common variants (Gaugler et al., 2014). The fact that common variants are not causal for disease makes it harder for researchers to define their clinical importance (Geschwind, 2011; Persico and Napolioni, 2013). Second, several characteristics of autism genetics, such as an extremely heterogeneous genetic contribution, many different loci underlying disease, variable phenotypic expression, and lack of specificity make it difficult to understand the neuropathology of the condition (Persico and Napolioni, 2013). Solving heterogeneity may be the most important future task for ASD researchers.
It is clear that traditional candidate gene studies are not suited to investigate common gene variants. As next-generation sequencing (NGS) technologies developed, whole exome sequencing (WES) was used to identify diverse genetic variants, including common variants in ASD (Sanders et al., 2012; Persico and Napolioni, 2013). The use of NGS, with its ability to simultaneously analyze multiple genes (in case of WES) in a single test, is currently being established in clinics and laboratories (Yohe and Thyagarajan, 2017).
From a genetic perspective, an ASD patient should be monitored for comorbid epilepsy because ASD and epilepsy are known to share genetic backgrounds, which may be related to neuropathophysiology during brain development (Tuchman and Rapin, 2002; Tuchman and Cuccaro, 2011). Autism spectrum disorder patients with rare gene variants related to genetic syndromes such as Rett’s syndrome, are strongly suspected to have comorbid epilepsy (Canitano, 2007). Previous studies show that epilepsy in ASD is highly related to intellectual disability (ID) (Amiet et al., 2008) and associated with severity of ASD (Ko et al., 2016). To reveal the genetic background of neurodevelopment, examining the discriminative characteristics of genetic components of ASD with epilepsy and ID and comparing them with ASD with no comorbidities is essential. To investigate genetic variants associated with ASD and ASD with comorbid epilepsy, we planned a retrospective review of the medical records and NGS data of ASD patients.
Materials and Methods
Participants
We reviewed medical records of ASD patients who underwent NGS for genetic evaluation, who visited a specialist in an out-patient clinic for autism at the Severance Children’s Hospital, from January 1, 2016. In clinical settings, we recommend NGS to parents when patients show severe autistic symptoms, morphological problems, or other medical or neurological comorbidities. Data from 141 patients were collected, and four patients among them were excluded due to lack of clinical assessment and follow-ups which are vital for diagnosing ASD. One hundred and thirty-seven enrolled participants were clinically diagnosed as ASD by a specialized psychiatrist on the basis of diagnostic criteria suggested in DSM-5, and several clinical assessments (see Clinical Assessments) support the diagnosis. As data were reviewed retrospectively and it was impossible to fulfill assessments not performed in the clinical setting, there were missing scores for Intelligence-Quotient (IQ), the Social Responsiveness Scale (SRS), and the Social Communication Questionnaire (SCQ), in several participants. We examined not only NGS and clinical assessment data, but also checked for comorbid epilepsy and other comorbidities, history of seizure, and electroencephalogram (EEG) reports. For the cases with comorbid epilepsy, we followed the diagnostic decision of neurologists in the Severance Children’s Hospital. This study was approved by the applicable institutional Review Boards for research with human subjects at Severance Hospital, Yonsei University College of Medicine, where this study was performed.
Clinical Assessments
All participants had a previous clinical diagnosis of ASD by a specialized child psychiatrist. The diagnosis of ASD was established using Autism Diagnostic Interview-Revised (ADI-R) and Autism Diagnostic Observation Schedule (ADOS), the gold standard for ASD diagnosis. Clinical autistic characteristics of the participants were supplemented by the CARS, SRS and SCQ.
The Childhood Autism Rating Scale (CARS) is a 15-item behavioral rating scale, developed to distinguish ASD and other developmental disorders and to assess the severity of ASD. Each item is scored from one to four points, and midpoint scores are also possible. Higher scores indicate more severe ASD symptoms. The cut-off score, which distinguishes ASD from other developmental disorders, is 30 points (Amiet et al., 2008). The reliability and validity of the Korean version of CARS (K-CARS) have been verified (Shin and Kim, 1998).
The SRS is a 65-item questionnaire that asks parents and/or teachers about the characteristics of the social interactions shown by children over the past 6 months (Constantino et al., 2000). Each question is scored from zero to three points, depending on the frequency of the action described in each item. Higher scores mean a lower social function. We previously confirmed the clinical validity of the SRS in Korean children and provided the Korean T-score norm (Cheon et al., 2016). In the present study, we used total T-score of the participants.
The SCQ is a 40-item screening instrument that is based on ADI-R, a tool for more in-depth assessment of ASD symptoms, and selects key items that deviate from normal development (Chandler et al., 2007). The Korean version of the Social Communication Questionnaire (K-SCQ) was verified as a reliable and valid instrument for screening autistic symptoms in the Korean population (Kim et al., 2015). Each question is answered with yes or no. Higher scores indicate more severe symptoms associated with ASD.
The ADI-R is a semi-structured parent interview tool for parents of children aged 2 years and older (Lord et al., 1994). This is generally conducted in conjunction with ADOS, which directly monitors and assesses the child, and is used to complement the interpretation of results (Lord et al., 2000).
To assess the cognitive levels of participants, we used the Korean-Wechsler Intelligence Scale for Children-IV (K-WISC-IV) and the Korean Wechsler Preschool and Primary Scales of Intelligence-IV (K-WPPSI-IV). We also used Korean-Bayley-III for children who were unable to perform the Wechsler’s intelligence scales because of their age or development status.
Next-Generation Sequencing
The xGen Inherited Diseases Panel (Integrated DNA Technologies, Coralville, IA, USA) including 4,503 candidate genes was used for exome sequencing. Genes associated with various neurodevelopmental disorders such as ASD, epilepsy, seizure disorder, and X-linked ID are included in this panel.
Genomic DNA extracted from individuals’ samples was used for library preparation and target capture using a custom panel targeting candidate genes. Massively parallel sequencing was performed using the NextSeq 550Dx System (Illumina, San Diego, CA, USA). Quality control and sequence analysis was carried out using our custom analysis pipeline. Copy number analysis was carried out using our custom analysis pipeline (Kim et al., 2019). The GRCh37 (hg19) build was used as the reference sequence for mapping and variant calling while using Burrows-Wheeler alignment (BWA) tool (version 0.7.12). HaplotypeCaller and MuTect2 in the GATK package (3.8-0) and VarScan2 (2.4.0) were used to identify single nucleotide variations (SNV) and insertion and deletions (indels). Databases used for analyses and variant annotation include Online Mendelian Inheritance in Man (OMIM), the Human Gene Mutation Database (HGMD), Clinvar, dbSNP, 1000 Genomes, the Exome Aggregation Consortium (ExAC), the Exome Sequencing Project (ESP), and the Korean Reference Genome Database (KRGDB). Classification of variants followed the standards and guidelines established by the American College of Medical Genetics (ACMG) (Richards et al., 2015), with a scoring algorithm implemented in the DxSeq Analyzer (Dxome, Seoul, Korea). All pathogenic and likely pathogenic variants were further confirmed by Sanger sequencing.
Genetic variants that are not met for pathogenic/likely pathogenic nor benign/likely benign are classified as variants of unknown/uncertain significance (VOUS) according to the ACMG guideline. Benign and likely benign variants were excluded in our NGS clinical reports. If VOUS had one or more evidence of pathogenicity but unmet criteria for pathogenic/likely pathogenic, they were regarded as VOUS with a relatively high probability of pathogenicity. The VOUS with high probability of pathogenicity were selected by physicians in laboratory medicine referencing the criteria on evidence of pathogenicity in the ACMG guideline. Among VOUS with high probability of pathogenicity, we selected five or less variants for analysis.
Statistics
To compare demographic characteristics and results of clinical assessments between patients with and those without pathogenic/likely pathogenic variants, we used the Chi-squared test and the independent t-test. Statistical significance was defined at p < 0.05. Analyses were performed using the Statistical Package for the Social Sciences software (version 25.0; SPSS Inc., Chicago, IL, USA).
Results
Among 137 patients, only three patients showed no pathogenic/likely pathogenic variants nor VOUS according to our NGS clinical reports. Seven cases were identified with pathogenic variants, and 17 participants had likely pathogenic variants. The diagnostic yield acquired from the total NGS data was about 17.51% (24/137). Differences in demographic information and clinical assessment results are presented in Table 1. The proportion of females to males was significantly higher in the pathogenic/likely pathogenic variants group (62.5%, p = 0.006). The pathogenic/likely pathogenic variants group was associated with higher incidence of comorbid epilepsy (25%, p = 0.008). There were no between-group differences in age and clinical assessment scores (IQ, SRS T-score, SCQ, CARS). Characteristics of epilepsy and reports of electroencephalogram (EEG) in ASD with comorbid epilepsy were listed in Supplementary Material 1.
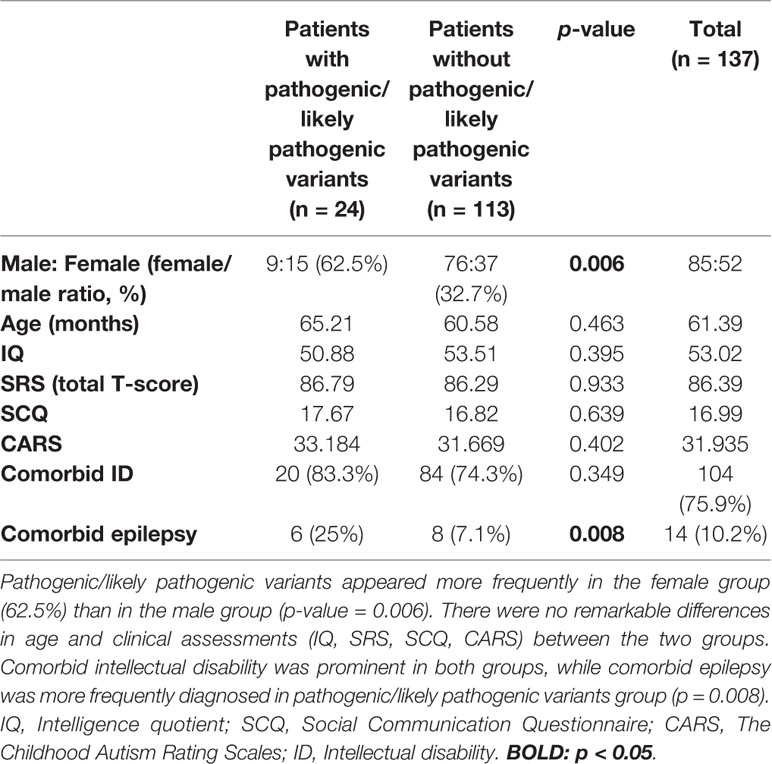
Table 1 Demographic data of participants.
By comparing males with females (Table 2), we found that females appear to have higher scores for the SRS total T-score (p = 0.024) as well as frequently detected pathogenic/likely pathogenic variants (p = 0.006). The CARS score was also slightly higher in females (p = 0.045), while age and other scores (IQ, SCQ) showed no significant statistical differences.
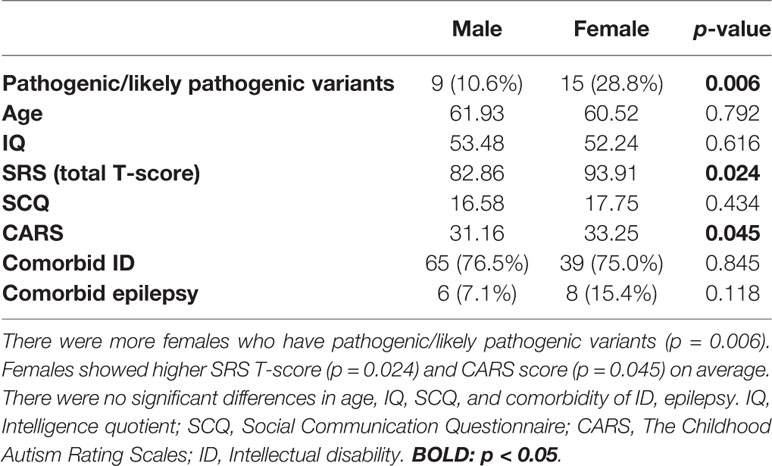
Table 2 Male–Female comparison.
Genes that harbored pathogenic variants included SHANK3, PTEN, NSD1, PAFAH1B1, and RAI1. Mutation types include exon deletion and nonsense mutations. We also identified copy number variants (CNV), chromosome 8p23.2 duplication, and chromosome 15q11.2–q13.2 duplication. These variants were expected to lead to loss of genetic function (Richards et al., 2015) which may play a role in pathogenesis of disease. Genetic information from OMIM were also described in Table 3. Among pathogenic variants, only PAFAH1B1 was not previously reported to be related to neurodevelopmental disorder including ASD, ID, and epilepsy.
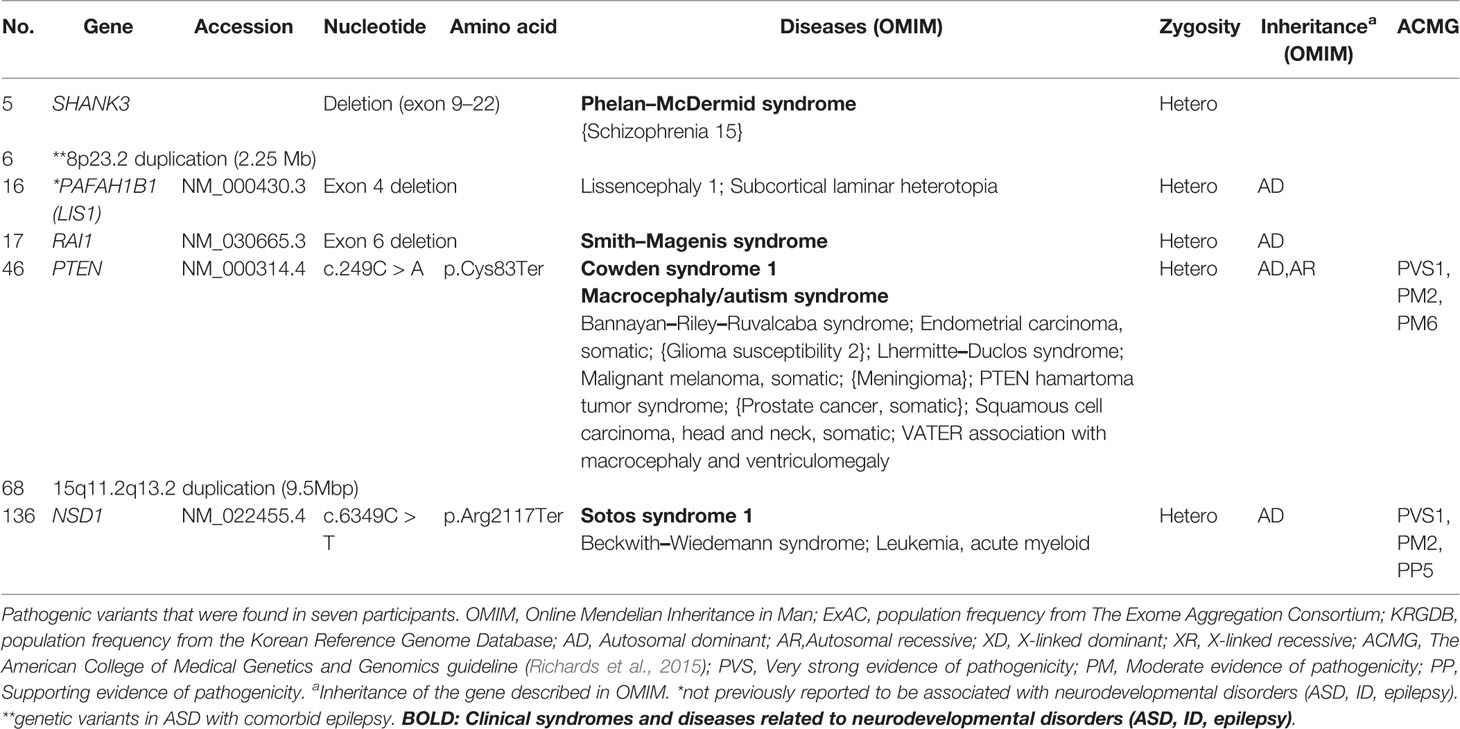
Table 3 Genetic characteristics: genes with pathogenic variants.
Likely pathogenic variants showed various types of mutations such as copy number variants, exon deletion, nonsense mutation and missense mutation (Table 4). Both patients with variants in TSC2 were diagnosed with tuberous sclerosis clinically. Likewise, both patients with variants in MECP2 were diagnosed with Rett’s syndrome in clinical setting. While most of genes are known to be related to ASD, ID or epilepsy, ABCC2, CCDC50 and SLC26A4 were not reported to be related to neurodevelopmental disorder according to OMIM. Likely pathogenic group showed significantly lower SRS T-score compared to pathogenic group (Supplementary Material 2).
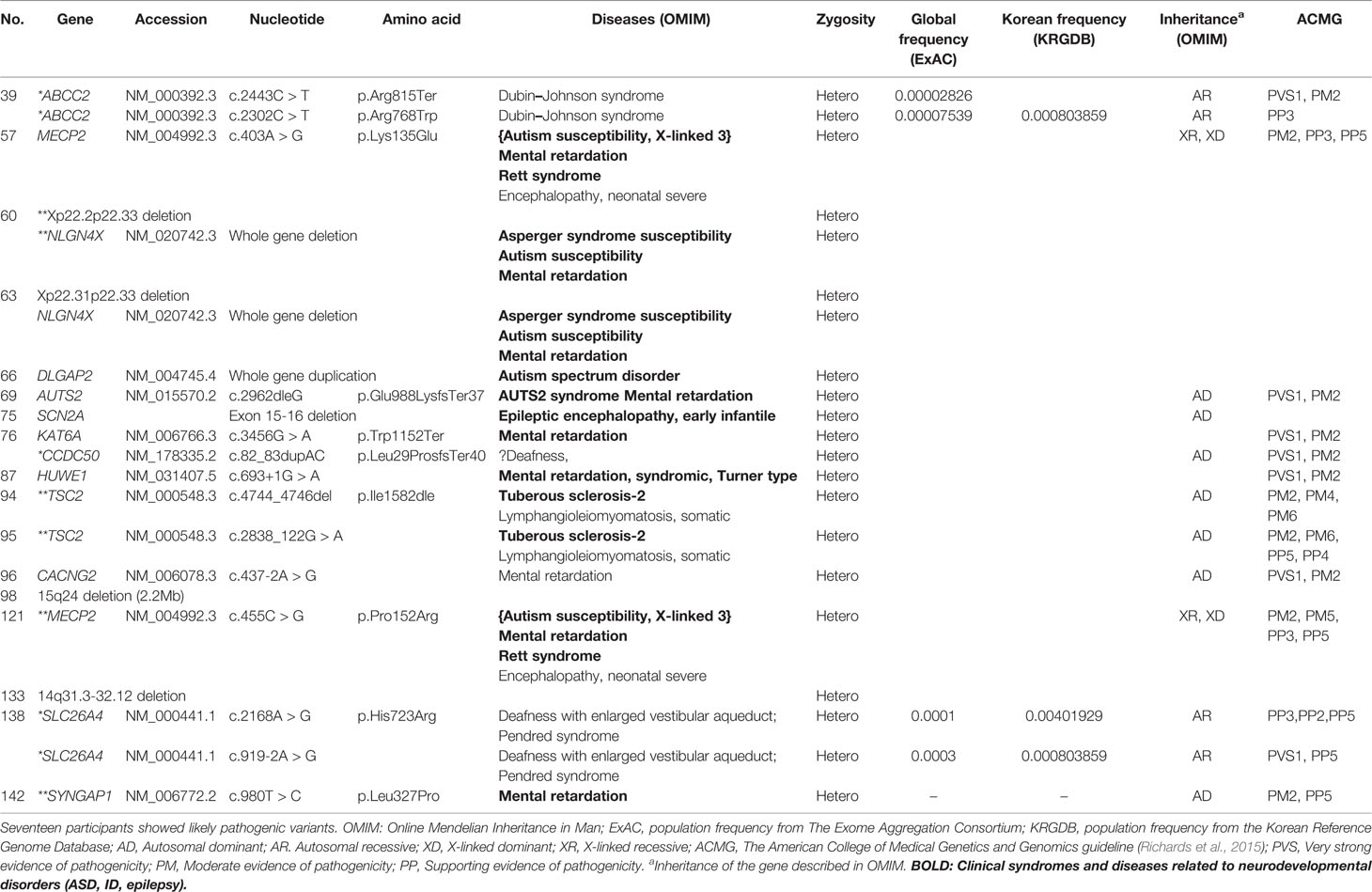
Table 4 Genetic characteristics: genes with likely pathogenic variants.
Importantly, pathogenic or likely pathogenic gene variants were found in approximately 43% (6/14) of participants with comorbid epilepsy. 8p23.2 duplication was the only pathogenic variant, and variations in Xp22.2p22.33 and the genes NLGN4X, TSC2, MECP2, SYNGAP1, were classified as likely pathogenic. Suspected genetic variants of each patient with comorbid epilepsy were shown in Table 5. There was no significant differences in IQ, SRS T-score, SCQ and CARS between patients with pathogenic/likely pathogenic variants and with VOUS (Supplementary Material 3).
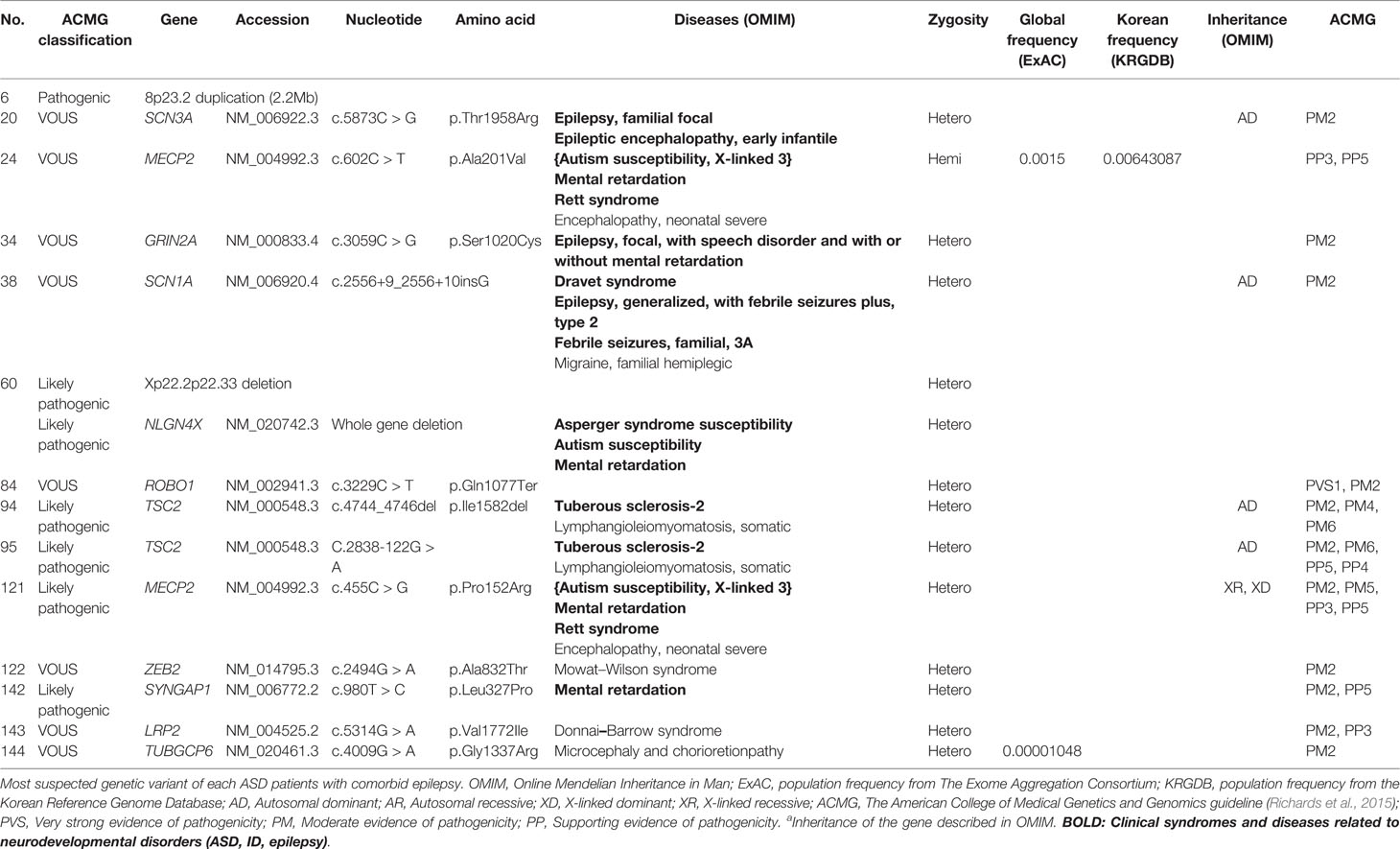
Table 5 Genetic characteristics: genes with most suspected variants to be related to ASD with epilepsy.
All patients had 0 to 37 VOUS genetic variants, 11.45 variants on average in our NGS clinical reports. There were several genes commonly observed with various variations. TSC2, ADGRV1, RAI1, CDH7, RELN, and NSD1 were the most commonly reported genes with variants of unknown origin regardless of mutation types. Genes with VOUS were repeatedly identified about 1.8 times on average in our data, with standard deviation of 1.79. Figure 1 shows the most frequently identified genes presenting VOUS in our patients without considering the variant type. More specifically, we also examined variants of unknown significance, including types of mutation and locations of the variants. As shown in Figure 2, an identical missense mutation in the FOXP1 gene was found three times among 137 patients, and other missense mutations were seen twice. These results suggest that large portion of genes with VOUS were restricted to missense mutation and have already been reported as genes related to ASD according to OMIM and SFARI database.
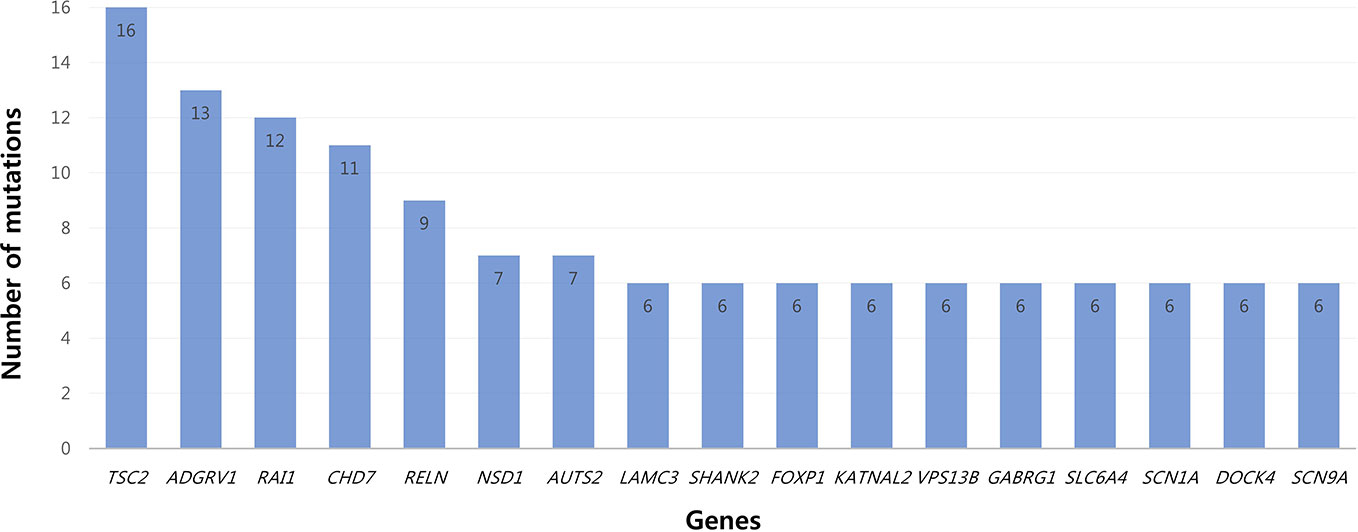
Figure 1 Most common genes with variants of unknown significance. Among genes with VOUS, TSC2 (16 times) was most frequently observed. After TSC2, common genes appeared in the order of ADGRV1 (13), RAI1 (12), CHD (11), RELN (9), NSD1/AUTS2 (7) and LAMC3/SHANK2/FOXP1/KATNAL2/VPS13B/GABRG1/SLC6A4/SCN1A/DOCK4/SCN9A (6).
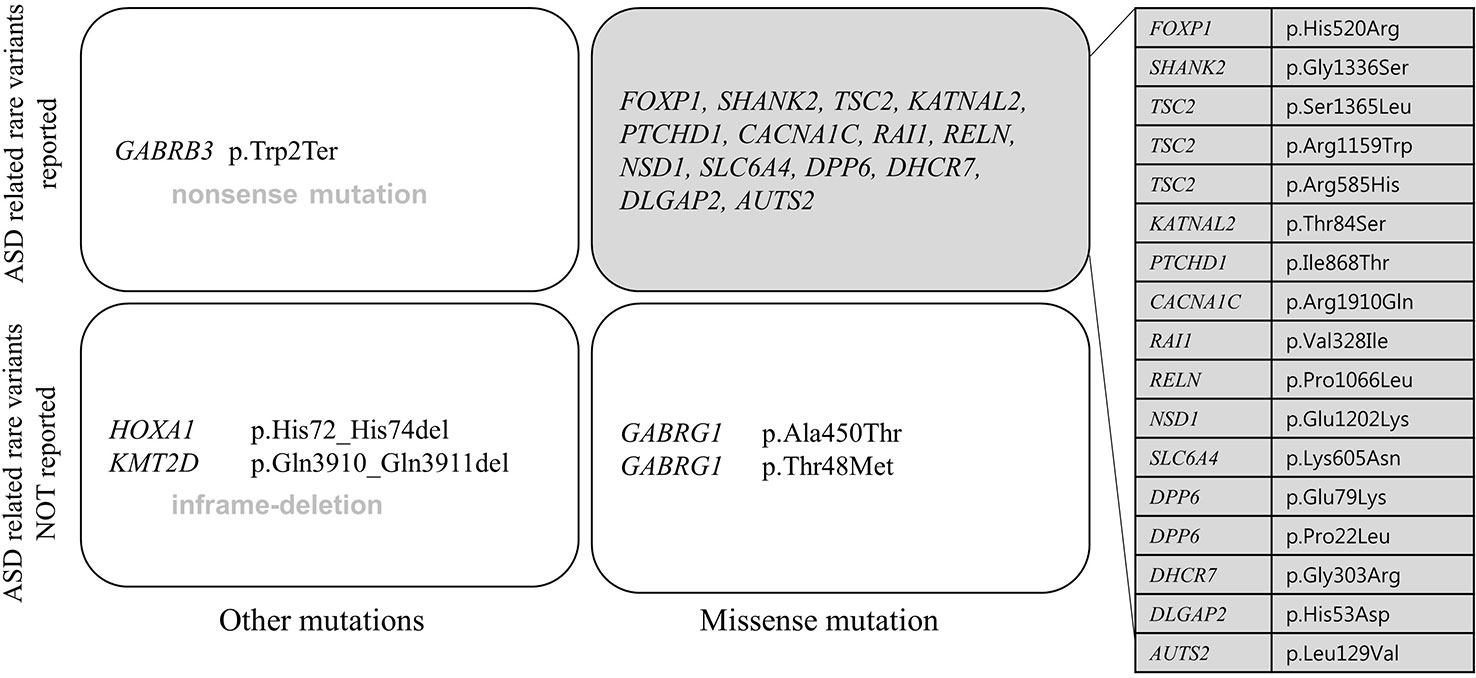
Figure 2 Repeatedly reported variations of unknown significance. A missense mutation (p.His520Arg) of FOXP1 gene was most commonly found. Only FOXP1 gene variation appeared three times in 137 patients, the rest of missense mutations were found twice each. In perspective of mutation type, one nonsense mutation (p.Trp2Ter in GABRB3 gene), two in-frame deletions (p.Gln3910_Gln3911del in KMT2D gene, p.His72_His74del in HOXA1 gene), 21 missense mutations were related to each gene. Most genes have already been reported to be related to ASD according to OMIM and SFARI database.
Discussion
Among 137 patients, only three patients showed no pathogenic, likely pathogenic variants and VOUS. This might be because patients who had severe symptoms or signs suggesting a genetic etiology in the clinical setting underwent NGS. Severe symptoms are usually related to genetic burden in ASD (Pizzo et al., 2019).
According to previous studies, diagnostic yields vary case by case (Yang et al., 2014; Tammimies et al., 2015; Rossi et al., 2017). Our yield of 17.51% may be within the predicted range, but the remarkable differences between males and females are concerning. The diagnostic yield of the female group was 28.8% which was significantly higher than that of the males. Higher SRS T-scores and CARS scores that indicate severity of autism symptoms were also significantly high in females. Though females are less prevalent in ASD (Baio et al., 2018), genetic burden and symptom severity can be higher than males. Females with ASD are known to have more genetic load than males (Lai et al., 2015), and severe clinical conditions also tend to be related with genetic variants (Lovato et al., 2019). Such reports support our results which highlight the importance of genetic evaluation in females with ASD.
Rare Genetic Variants in Autism Spectrum Disorder
Most pathogenic variants were found in genes such as SHANK3, PTEN, NSD1, and the 8p23.2 duplications that have already been reported to be associated with ASD. Most pathogenic variants are related to specific neurodevelopmental syndromes. Variants in SHANK3 can accompany Phelan–McDermid syndrome (Berg et al., 2018); PTEN, Cowden syndrome (Goffin et al., 2001); NSD1, Sotos syndrome (Kurotaki et al., 2002); and RAI1, Smith–Magenis syndrome (Slager et al., 2003; Laje et al., 2010). These syndromes are often reported to be related with ASD (Goffin et al., 2001; De Rubeis et al., 2014; Connolly et al., 2017). In the case of the 15q11.2–q13.2 duplication, a previously reported duplication in 15q11–13 was associated with ASD, and if variants are inherited from the father, Prader–Willi syndrome should also be considered (Bolton et al., 2004; Veltman et al., 2005). Autism spectrum disorder with these syndromes related genes should be monitored with caution, regarding comorbidities. However, unlike other genes, variants of PAFAH1B1 have not been previously reported as rare variants that affect ASD development. An animal study demonstrated that mutation in the murine ortholog of this gene contributes to diminished social interaction in mice (Sudarov et al., 2013). As the gene plays a role in synaptogenesis and nervous system development (Wall et al., 2009; Sudarov et al., 2013), the possibility of ASD pathogenicity should not be neglected. The 8p23.2 duplication is described in Rare Genetic Variants in Autism Spectrum Disorder With Comorbid Epilepsy.
Most genes containing likely pathogenic variants were reported to be associated with neurodevelopmental disorders. The copy number variants, Xp22.2p22.33 deletion, 15q24 deletion (Adam et al., 2018), and 14q31.3–32.12 deletion (Crkvenac Gornik et al., 2019) were also reported to be related to developmental delay. Otherwise, some genes which had not been considered as ASD related genes were discovered. ABCC2 is known to trigger Dubin–Johnson syndrome, which causes an increase in conjugated bilirubin levels (Keitel et al., 2003). The condition is characterized by black pigment in the liver. Mutations in SLC26A4 have been related to Pendred syndrome, leading to sensorineuronal hearing loss (Landa et al., 2013). The participant with history of comorbid hearing loss might be due to genetic mutation in high probability, but whether this genetic mutation is also responsible for ASD development or not is unclear due to lack of evidence. CCDC50 with a duplication in exon 11 was reported to be associated with progressive hearing loss in the Spanish group (Modamio-Høybjør et al., 2007), but to our knowledge, no reports were found to be related to CCDC50 with neurodevelopmental disorders. Further studies are needed to understand the relationship between these genes and ASD.
Rare Genetic Variants in Autism Spectrum Disorder With Comorbid Epilepsy
In our study, two cases with TSC2 variation and one case with MECP2 variation were diagnosed as tuberous sclerosis and Rett’s syndrome among ASD patients with comorbid epilepsy, upon evidence of general appearance, clinical manifestation, and brain magnetic resonance imaging. Heterozygous SYNGAP1 gene mutations have been associated with ASD, ID, several forms of idiopathic generalized epilepsy, and delay in psychomotor development (Pinto et al., 2010; Klitten et al., 2011). Though some variants (8p23.2 duplication, Xp22.2p22.33 deletion, and NLGN4X) classified as likely pathogenic, the association with epilepsy has not been reported so far.
We confirmed a 2.25 Mb duplication in the short arm of chromosome 8 (8p23.2), including the CSMD1 gene, by additional microarray examination. Though various size duplications of 8p23.2 are known to be associated with ASD and developmental delay, such as speech delay and learning difficulties (Glancy et al., 2009; Fisch et al., 2011), there was no evidence of association with epilepsy. The complement pathway is tightly controlled in the brain and disruption of microglia-specific complement receptor 3(CR3)/C3 signaling results in sustained deficits in synaptic connectivity. It is believed that deregulation of complement activity could induce aberrant synaptic elimination, which may influence susceptibility to both neurodegenerative and psychiatric disorders (Schafer et al., 2013). Although the mechanism of pathogenesis of epilepsy is not established, recent studies have reported that synaptic connectivity is associated with the development of epilepsy (Chu et al., 2010; Karoly et al., 2018). To date, there is insufficient evidence to explain the direct relationship between duplication of CSMD1 and epilepsy. However, we suggest that the overexpression of CSMD1 due to 8p23.2 duplication leads to abnormal synaptic connectivity and it may contribute to the occurrence of epilepsy.
It is known that deletion of chromosome Xp is associated with ID and ASD (Shinawi et al., 2009; Willemsen et al., 2012). More than 100 genes are known to be located on Xp22.2-p22.33. Males with deletions encompassing Xp22 exhibit a phenotype consistent with the loss of one or more of the genes located in this region (Melichar et al., 2007). In females, there are only a few reports that show de novo chromosomal deletions of Xp22 are associated with ASD and developmental delay (Thomas et al., 1999; Chocholska et al., 2006). In addition to the loss of genes located in Xp22, unfavorable X-inactivation of the intact chromosome would be another mechanism for the expressed phenotype in females (Shinawi et al., 2009). Additional tests to confirm the exact location and extent of the Xp deletion were recommended, but not performed in our case. NLGN4X is a gene located in the short arm of the X chromosome and is also known to be related to ASD and ID (Jamain et al., 2003; Macarov et al., 2007), but not with epilepsy. In an animal study, Nlgn4 knock-out (KO) mice showed decreased network response and increased protein expression of synaptic proteins, such as N-methyl-D-aspartate receptor (Nmdar) subunit 1 (Nr1), and metabotropic glutamate receptor 5 (mGluR5), which are involved in synaptic plasticity and excitatory circuit rewiring (Delattre et al., 2013). Imbalance between excitatory and inhibitory synapses is one of the main hypotheses explaining the pathogenesis of epilepsy (Matsumoto and Ajmonemarsan, 1964). Furthermore, excitation/inhibition imbalances resulting from neurodevelopmental deficits have been suggested as pathogenic mechanisms for both ASD and epilepsy (Bozzi et al., 2018).
Interestingly, though estimated as VOUS, some previously reported epilepsy genes (SCN1A, SCN3A, MECP2, and GRIN2A) were also detected. Genes may have different mutation types or location, which leads to different effect sizes on ASD or epilepsy development, but they still have an important impact on disease occurrence.
Variants of Unknown Origin
In the development of ASD, multiple loci tend to show relatively weak genotype–phenotype correlations and act additively (Persico and Napolioni, 2013). This means that common variants with low effect sizes should not be ignored considering the heterogeneity of ASD. Unfortunately until now, studies such as genome-wide association studies (GWAS), which focus on the contribution of common variants to disease, have not yield consistent results (Geschwind, 2011). According to the ACMG guideline, the VOUS variant can also have one or more evidence of pathogenicity even it was classified as VOUS. So the NGS results should be interpreted carefully, as it is possible to suggest new common variants relevant to ASD pathogenesis.
Except two genes (ADGRV1 and GABRG1), 15 genes frequently classified as VOUS were also reported multiple times with rare variants in ASD (Geschwind, 2011; Persico and Napolioni, 2013; Sener et al., 2016). As shown in Figure 2, except one nonsense mutation and two in-frame deletions, most VOUS were missense mutations. That is, the usual types of VOUS mutation are less likely to disrupt function of gene. For this reason, even if a mutation occurs in the same gene, the effect on the development of ASD might be different depending on the mutation type.
Likewise, ROBO1 which was identified in ASD with comorbid epilepsy was implicated in developmental dyslexia and dysfunction of language acquisition system (Hannula-Jouppi et al., 2005; Bates et al., 2011). In addition to the roles in guiding axons and the Slit/Robo signaling pathway, ROBO1 is also involved in cellular processes such as cell migration and immune cell activation during neuroinflammatory responses (Mirakaj and Rosenberger, 2017). Recently, it has been suggested that inflammation and autoimmunity play important roles in childhood seizures and epilepsies (Korff and Dale, 2017). In our case, the patient with a nonsense mutation in ROBO1 was diagnosed with ASD and had clinical manifestations of focal seizure. This case suggests that although a genetic variation does not satisfy the criteria of pathogenic/likely pathogenic variants, it might affect an individual’s phenotype.
Through these results, it is possible to surmise that genes with known pathogenic variants may often appear with VOUS also. As variants affect genetic functions such as synaptic and neuronal plasticity (Ben-David and Shifman, 2012), the influence on ASD would be exerted when loss of function occurred, even though the effect may vary by location and type of mutation. It is necessary not to overlook genes with VOUS if the gene has been previously reported with pathogenic variants in neurodevelopmental disorders, including ASD.
Heterogeneity has been a great challenge for developing tailored treatment of ASD as there are a large number of genes related to ASD, and loss of function differs according to each type of mutations. Through the advancement of genetic analysis technology, NGS results are being used in clinical fields, but it is still difficult to interpret and identify the clinical significance. To provide proper management to ASD individually, discrimination of the pathogenic variant among multiple variants should be achieved. Our results show that it is necessary to notice genes with VOUS although their function is not clearly defined yet. Especially in ASD presenting heterogeneous clinical manifestation and frequent comorbid disorders, results of genetic analysis should be performed with caution. The VOUS in ASD related genes involved with unclear mutation, or non-ASD related genes with clinically relevant phenotype may be of primary importance in investigating genetic data (Lovato et al., 2019). Efforts to identify the function of genes with VOUS will lead to discovering genetic pathogenesis of neurodevelopment disorder in the future.
There are several limitations to this study. First, as this study reviewed medical records retrospectively, clinical assessment could not be performed without bias. This may have influenced the statistical results of the demographic data. Second, in cases of age under 4 years, we could estimate intellectual disability only by Bayley Scales of Infant Development. Third, as our participant group mostly showed severe phenotypes (SRS T-score 86.39 on average), further studies are needed to compare differences in genetic components according to severity of ASD phenotype. Furthermore, as we analyzed the clinical NGS reports retrospectively, we could not show data from typically developing control group. To define the pathogenicity of variants of genes, comparing the result with that of normal population might be helpful. Finally, as medical records were reviewed cross-sectionally, we could not evaluate the development of comorbidities including epilepsy.
Despite the limitations mentioned above, there are several strengths in our study. First, to our knowledge, this is the first NGS study in ASD patients with or without comorbid epilepsy in Korea. As all patients are Korean, our results are not confounded by population genetic heterogeneity. Second, because NGS was carried out only in ASD patients who had severe phenotypes, comorbid disorders, or suspicious general appearance in a clinical setting, genetic variants thought to impact ASD development were able to be easily obtained. Third, we found some genes that have not previously been reported but are possibly pathogenic in ASD. Finally, by considering comorbid epilepsy, we confirmed genetic overlaps in ASD and epilepsy, even though genetic variations are currently known to be related just with either ASD or epilepsy.
In conclusion, we suggest that rare variants (pathogenic/likely pathogenic) and common variants (VOUS) are both necessary in investigating individuals’ genetic characteristics in ASD and epilepsy. Pathogenic/likely pathogenic variants might be useful in confirming genetic syndrome, predicting comorbidity, and treatment planning. The VOUS might also influence the phenotype characteristics of ASD and epilepsy, even though the evidence and possibility are not strong enough. Careful efforts in interpreting the VOUS might contribute to understand the genetic cause of ASD and epilepsy.
Data Availability Statement
The datasets generated for this study will not be made publicly available because it includes the patient’s genetic data for clinical purpose.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Boards at Severance Hospital, Yonsei University College of Medicine. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
Conceived and designed the experiments: JL, SH, K-AC. Performed and analyzed NGS: S-TL, S-GP, SS, JC. Analyzed data: JL, SH, S-GP, SS. Wrote the manuscript: JL, SH, S-TL, S-GP, SS, JC, K-AC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by a faculty research grant of Yonsei University College of Medicine for 2014(6-2014-0143), a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI12C0021-A120029) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2016R1A2B4006737).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00585/full#supplementary-material
References
Adam, M. P., Ardinger, H. H., Pagon, R. A., Wallace, S. E., Bean, L. J. H., Stephens, K., et al. (2018). GeneReviews® [Internet].
Amiet, C., Gourfinkel-An, I., Bouzamondo, A., Tordjman, S., Baulac, M., Lechat, P., et al. (2008). Epilepsy in autism is associated with intellectual disability and gender: evidence from a meta-analysis. Biol. Psychiatry 64 (7), 577–582. doi: 10.1016/j.biopsych.2008.04.030
APA (2013). Diagnostic and statistical manual of mental disorders (DSM-5®). (Washington, DC, Arlington: American Psychiatric Publishing)
Baio, J., Wiggins, L., Christensen, D. L., Maenner, M. J., Daniels, J., Warren, Z., et al. (2018). Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States 2014. MMWR Surveillance Summaries 67 (6), 1. doi: 10.15585/mmwr.ss6706a1
Bates, T. C., Luciano, M., Medland, S. E., Montgomery, G. W., Wright, M. J., Martin, N. G. (2011). Genetic Variance in a Component of the Language Acquisition Device: ROBO1 Polymorphisms Associated with Phonological Buffer Deficits. Behav. Genet. 41 (1), 50–57. doi: 10.1007/s10519-010-9402-9
Ben-David, E., Shifman, S. (2012). Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PloS Genet. 8 (3), e1002556. doi: 10.1371/journal.pgen.1002556
Berg, E. L., Copping, N. A., Rivera, J. K., Pride, M. C., Careaga, M., Bauman, M. D., et al. (2018). Developmental social communication deficits in the Shank3 rat model of phelan-mcdermid syndrome and autism spectrum disorder. Autism Res. 11 (4), 587–601. doi: 10.1002/aur.1925
Bolton, P. F., Veltman, M. W., Weisblatt, E., Holmes, J. R., Thomas, N. S., Youings, S. A., et al. (2004). Chromosome 15q11-13 abnormalities and other medical conditions in individuals with autism spectrum disorders. Psychiatr. Genet. 14 (3), 131–137. doi: 10.1097/00041444-200409000-00002
Bozzi, Y., Provenzano, G., Casarosa, S. (2018). Neurobiological bases of autism-epilepsy comorbidity: a focus on excitation/inhibition imbalance. Eur. J. Neurosci. 47 (6), 534–548. doi: 10.1111/ejn.13595
Buescher, A. V., Cidav, Z., Knapp, M., Mandell, D. S. (2014). Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 168 (8), 721–728. doi: 10.1001/jamapediatrics.2014.210
Canitano, R. (2007). Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry 16 (1), 61–66. doi: 10.1007/s00787-006-0563-2
Chandler, S., Charman, T., Baird, G., Simonoff, E., Loucas, T., Meldrum, D., et al. (2007). Validation of the social communication questionnaire in a population cohort of children with autism spectrum disorders. J. Am. Acad. Child Adolesc. Psychiatry 46 (10), 1324–1332. doi: 10.1097/chi.0b013e31812f7d8d
Cheon, K. A., Park, J. I., Koh, Y. J., Song, J., Hong, H. J., Kim, Y. K., et al. (2016). The social responsiveness scale in relation to DSM IV and DSM5 ASD in Korean children. Autism Res. 9 (9), 970–980. doi: 10.1002/aur.1671
Chocholska, S., Rossier, E., Barbi, G., Kehrer-Sawatzki, H. (2006). Molecular cytogenetic analysis of a familial interstitial deletion Xp22.2-22.3 with a highly variable phenotype in female carriers. Am. J. Med. Genet. A 140 (6), 604–610. doi: 10.1002/ajmg.a.31145
Chu, Y. X., Jin, X. M., Parada, I., Pesic, A., Stevens, B., Barres, B., et al. (2010). Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. U. S. A. 107 (17), 7975–7980. doi: 10.1073/pnas.0913449107
Connolly, S., Anney, R., Gallagher, L., Heron, E. A. (2017). A genome-wide investigation into parent-of-origin effects in autism spectrum disorder identifies previously associated genes including SHANK3. Eur. J. Hum. Genet. 25 (2), 234. doi: 10.1038/ejhg.2016.153
Constantino, J. N., Przybeck, T., Friesen, D., Todd, R. D. (2000). Reciprocal social behavior in children with and without pervasive developmental disorders. J. Dev. Behav. Pediatr. 21 (1), 2–11. doi: 10.1097/00004703-200002000-00002
Crkvenac Gornik, K., Tonkovic Durisevic, I., Pokupec Bilic, A., Huljev Frkovic, S. (2019). Interstitial 14Q31. 3-Q32. 13 deletion: The role of molecular karyotyping in clarifying the etiology of developmental delay. Mol. Exp. Biol. Med. 1, 48–51.
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515 (7526), 209. doi: 10.1038/nature13772
Delattre, V., La Mendola, D., Meystre, J., Markram, H., Markram, K. (2013). Nlgn4 knockout induces network hypo-excitability in juvenile mouse somatosensory cortex in vitro. Sci. Rep. 3. doi: 10.1038/srep02897
Fisch, G. S., Davis, R., Youngblom, J., Gregg, J. (2011). Genotype-Phenotype Association Studies of Chromosome 8p Inverted Duplication Deletion Syndrome. Behav. Genet. 41 (3), 373–380. doi: 10.1007/s10519-011-9447-4
Gaugler, T., Klei, L., Sanders, S. J., Bodea, C. A., Goldberg, A. P., Lee, A. B., et al. (2014). Most genetic risk for autism resides with common variation. Nat. Genet. 46 (8), 881. doi: 10.1038/ng.3039
Geschwind, D. H., State, M. W. (2015). Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 14 (11), 1109–1120. doi: 10.1016/S1474-4422(15)00044-7
Geschwind, D. H. (2011). Genetics of autism spectrum disorders. Trends Cogn. Sci. 15 (9), 409–416. doi: 10.1016/j.tics.2011.07.003
Glancy, M., Barnicoat, A., Vijeratnam, R., de Souza, S., Gilmore, J., Huang, S., et al. (2009). Transmitted duplication of 8p23.1-8p23.2 associated with speech delay, autism and learning difficulties. Eur. J. Hum. Genet. 17 (1), 37–43. doi: 10.1038/ejhg.2008.133
Goffin, A., Hoefsloot, L. H., Bosgoed, E., Swillen, A., Fryns, J. P. (2001). PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 105 (6), 521–524. doi: 10.1002/ajmg.1477
Hallmayer, J., Cleveland, S., Torres, A., Phillips, J., Cohen, B., Torigoe, T., et al. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68 (11), 1095–1102. doi: 10.1001/archgenpsychiatry.2011.76
Han, J. Y., Jang, J. H., Park, J., Lee, I. G. (2018). Targeted next-generation sequencing of Korean patients with developmental delay and/or intellectual disability. Front. Pediatr. 6, 391. doi: 10.3389/fped.2018.00391
Hannula-Jouppi, K., Kaminen-Ahola, N., Taipale, M., Eklund, R., Nopola-Hemmi, J., Kaariainen, H., et al. (2005). The axon guidance receptor gene ROBO1 is a candidate gene for developmental dyslexia. PloS Genet. 1 (4), 467–474. doi: 10.1371/journal.pgen.0010050.
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34 (1), 27–29. doi: 10.1038/ng1136
Karoly, P. J., Kuhlmann, L., Soudry, D., Grayden, D. B., Cook, M. J., Freestone, D. R. (2018). Seizure pathways: A model-based investigation. PloS Comput. Biol. 14 (10). doi: 10.1371/journal.pcbi.1006403
Keitel, V., Nies, A. T., Brom, M., Hummel-Eisenbeiss, J., Spring, H., Keppler, D. (2003). A common Dubin-Johnson syndrome mutation impairs protein maturation and transport activity of MRP2 (ABCC2). Am. J. Physiol. Gastrointestinal Liver Physiol. 284 (1), G165–G174. doi: 10.1152/ajpgi.00362.2002
Kim, J. H., Sunwoo, H. J., Park, S. B., Noh, D. H., Jung, Y. K., Cho, I. H., et al. (2015). A validation study of the Korean version of Social Communication Questionnaire. J. Korean Acad. Child Adolesc. Psychiatry 26 (3), 197.
Kim, B., Lee, H., Jang, J., Kim, S. J., Lee, S. T., Cheong, J. W., et al. (2019). Targeted next generation sequencing can serve as an alternative to conventional tests in myeloid neoplasms. PloS One 14 (3), e0212228. doi: 10.1371/journal.pone.0212228
Klitten, L. L., Moller, R. S., Nikanorova, M., Silahtaroglu, A., Hjalgrim, H., Tommerup, N. (2011). A balanced translocation disrupts SYNGAP1 in a patient with intellectual disability, speech impairment, and epilepsy with myoclonic absences (EMA). Epilepsia 52 (12), e190–e193. doi: 10.1111/j.1528-1167.2011.03304.x
Ko, C., Kim, N., Kim, E., Song, D. H., Cheon, K.-A. (2016). The effect of epilepsy on autistic symptom severity assessed by the social responsiveness scale in children with autism spectrum disorder. Behav. Brain Functions 12 (1), 20. doi: 10.1186/s12993-016-0105-0
Korff, C. M., Dale, R. C. (2017). The Immune System in Pediatric Seizures and Epilepsies. Pediatrics 140 (3). doi: 10.1542/peds.2016-3534
Kurotaki, N., Imaizumi, K., Harada, N., Masuno, M., Kondoh, T., Nagai, T., et al. (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 30 (4), 365. doi: 10.1038/ng863
Lai, M.-C., Lombardo, M. V., Auyeung, B., Chakrabarti, B., Baron-Cohen, S. (2015). Sex/gender differences and autism: setting the scene for future research. J. Am. Acad. Child Adolesc. Psychiatry 54 (1), 11–24. doi: 10.1016/j.jaac.2014.10.003
Laje, G., Morse, R., Richter, W., Ball, J., Smith, A. C. (2010). Autism spectrum features in Smith-Magenis syndrome. American journal of medical genetics part C: Seminars in medical genetics (Hoboken: Wiley Subscription Services, Inc., A Wiley Company.) 154 (4) 456–662 . doi: 10.1002/ajmg.c.30275
Landa, P., Differ, A.-M., Rajput, K., Jenkins, L., Bitner-Glindzicz, M. (2013). Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in Pendred syndrome/enlarged vestibular aqueducts. BMC Med. Genet. 14 (1), 85. doi: 10.1186/1471-2350-14-85
Lavelle, T. A., Weinstein, M. C., Newhouse, J. P., Munir, K., Kuhlthau, K. A., Prosser, L. A. (2014). Economic burden of childhood autism spectrum disorders. Pediatrics 133 (3), e520–e529. doi: 10.1542/peds.2013-0763
Lord, C., Rutter, M., Le Couteur, A. (1994). Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 24 (5), 659–685. doi: 10.1007/BF02172145
Lord, C., Risi, S., Lambrecht, L., Cook, E. H., Leventhal, B. L., DiLavore, P. C., et al. (2000). The Autism Diagnostic Observation Schedule—Generic: A standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 30 (3), 205–223. doi: 10.1023/A:1005592401947
Lord, C., Elsabbagh, M., Baird, G., Veenstra-Vanderweele, J. (2018). Autism spectrum disorder. Lancet 392 (10146), 508–520. doi: 10.1016/S0140-6736(18)31129-2
Loth, E., Murphy, D. G., Spooren, W. (2016). Defining precision medicine approaches to autism spectrum disorders: concepts and challenges. Front. Psychiatry 7, 188. doi: 10.3389/fpsyt.2016.00188
Lovato, D. V., Herai, R. R., Pignatari, G. C., Beltrao-Braga, P. C. B. (2019). The Relevance of Variants With Unknown Significance for Autism Spectrum Disorder Considering the Genotype-Phenotype Interrelationship. Front. Psychiatry 10, 409. doi: 10.3389/fpsyt.2019.00409
Macarov, M., Zeigler, M., Newman, J. P., Strich, D., Sury, V., Tennenbaum, A., et al. (2007). Deletions of VCX-A and NLGN4: a variable phenotype including normal intellect. J. Intellect Disabil. Res. 51 (Pt 5), 329–333. doi: 10.1111/j.1365-2788.2006.00880.x
Matsumoto, H., Ajmonemarsan, C. (1964). Cellular Mechanisms in Experimental Epileptic Seizures. Science 144 (3615), 193–194. doi: 10.1126/science.144.3615.193
Melichar, V. O., Guth, S., Hellebrand, H., Meindl, A., von der Hardt, K., Kraus, C., et al. (2007). A male infant with a 9.6 Mb terminal Xp deletion including the OA1 locus: Limit of viability of Xp deletions in males. Am. J. Med. Genet. Part A 143A (2), 135–141. doi: 10.1002/ajmg.a.31451
Mirakaj, V., Rosenberger, P. (2017). Immunomodulatory Functions of Neuronal Guidance Proteins. Trends Immunol. 38 (6), 444–456. doi: 10.1016/j.it.2017.03.007
Modamio-Høybjør, S., Mencía, Á., Goodyear, R., del Castillo, I., Richardson, G., Moreno, F., et al. (2007). A Mutation in CCDC50, a Gene Encoding an Effector of Epidermal Growth Factor–Mediated Cell Signaling, Causes Progressive Hearing Loss. Am. J. Hum. Genet. 80 (6), 1076–1089. doi: 10.1086/518311
Persico, A. M., Napolioni, V. (2013). Autism genetics. Behav. Brain Res. 251, 95–112. doi: 10.1016/j.bbr.2013.06.012
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466 (7304), 368–372. doi: 10.1038/nature09146
Pizzo, L., Jensen, M., Polyak, A., Rosenfeld, J. A., Mannik, K., Krishnan, A., et al. (2019). Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet. Med. 21 (4), 816. doi: 10.1038/s41436-018-0266-3
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405. doi: 10.1038/gim.2015.30
Rosenberg, R. E., Law, J. K., Yenokyan, G., McGready, J., Kaufmann, W. E., Law, P. A. (2009). Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch. Pediatr. Adolesc. Med. 163 (10), 907–914. doi: 10.1001/archpediatrics.2009.98
Rossi, M., El-Khechen, D., Black, M. H., Hagman, K. D. F., Tang, S., Powis, Z. (2017). Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr. Neurol. 70, 34–43. e32. doi: 10.1016/j.pediatrneurol.2017.01.033
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485 (7397), 237. doi: 10.1038/nature10945
Sandin, S., Lichtenstein, P., Kuja-Halkola, R., Larsson, H., Hultman, C. M., Reichenberg, A. (2014). The familial risk of autism. Jama 311 (17), 1770–1777. doi: 10.1001/jama.2014.4144
Schafer, D. P., Lehrman, E. K., Stevens, B. (2013). The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 61 (1), 24–36. doi: 10.1002/glia.22389
Sener, E. F., Canatan, H., Ozkul, Y. (2016). Recent advances in autism spectrum disorders: applications of whole exome sequencing technology. Psychiatry Invest. 13 (3), 255. doi: 10.4306/pi.2016.13.3.255
Shin, M. S., Kim, Y.-H. (1998). Standardization study for the Korean version of the Childhood Autism Rating Scale: Reliability, validity and cut-off score. Korean J. Clin. Psychol. 17 (1), 1–15.
Shinawi, M., Patel, A., Panichkul, P., Zascavage, R., Peters, S. U., Scaglia, F. (2009). The Xp Contiguous Deletion Syndrome and Autism. Am. J. Med. Genet. Part A 149A (6), 1138–1148. doi: 10.1002/ajmg.a.32833
Slager, R. E., Newton, T. L., Vlangos, C. N., Finucane, B., Elsea, S. H. (2003). Mutations in RAI1 associated with Smith–Magenis syndrome. Nat. Genet. 33 (4), 466. doi: 10.1038/ng1126
Sudarov, A., Gooden, F., Tseng, D., Gan, W. B., Ross, M. E. (2013). Lis1 controls dynamics of neuronal filopodia and spines to impact synaptogenesis and social behaviour. EMBO Mol. Med. 5 (4), 591–607. doi: 10.1002/emmm.201202106
Tammimies, K., Marshall, C. R., Walker, S., Kaur, G., Thiruvahindrapuram, B., Lionel, A. C., et al. (2015). Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. Jama 314 (9), 895–903. doi: 10.1001/jama.2015.10078
Thomas, N. S., Sharp, A. J., Browne, C. E., Skuse, D., Hardie, C., Dennis, N. R. (1999). Xp deletions associated with autism in three females. Hum. Genet. 104 (1), 43–48. doi: 10.1007/s004390050908
Tick, B., Bolton, P., Happé, F., Rutter, M., Rijsdijk, F. (2016). Heritability of autism spectrum disorders: a meta-analysis of twin studies. J. Child Psychol. Psychiatry 57 (5), 585–595. doi: 10.1111/jcpp.12499
Tuchman, R., Cuccaro, M. (2011). Epilepsy and autism: neurodevelopmental perspective. Curr. Neurol. Neurosci. Rep. 11 (4), 428–434. doi: 10.1007/s11910-011-0195-x
Tuchman, R., Rapin, I. (2002). Epilepsy in autism. Lancet Neurol. 1 (6), 352–358. doi: 10.1016/S1474-4422(02)00160-6
Veltman, M. W., Thompson, R. J., Craig, E. E., Dennis, N. R., Roberts, S. E., Moore, V., et al. (2005). A paternally inherited duplication in the Prader-Willi/Angelman syndrome critical region: a case and family study. J. Autism Dev. Disord. 35 (1), 117–127. doi: 10.1007/s10803-004-1039-1
Wall, D., Esteban, F., Deluca, T., Huyck, M., Monaghan, T., de Mendizabal, N. V., et al. (2009). Comparative analysis of neurological disorders focuses genome-wide search for autism genes. Genomics 93 (2), 120–129. doi: 10.1016/j.ygeno.2008.09.015
Willemsen, M. H., de Leeuw, N., de Brouwer, A. P., Pfundt, R., Hehir-Kwa, J. Y., Yntema, H. G., et al. (2012). Interpretation of clinical relevance of X-chromosome copy number variations identified in a large cohort of individuals with cognitive disorders and/or congenital anomalies. Eur. J. Med. Genet. 55 (11), 586–598. doi: 10.1016/j.ejmg.2012.05.001
Yang, Y., Muzny, D. M., Xia, F., Niu, Z., Person, R., Ding, Y., et al. (2014). Molecular findings among patients referred for clinical whole-exome sequencing. Jama 312 (18), 1870–1879. doi: 10.1001/jama.2014.14601
Keywords: autism spectrum disorder, epilepsy, next-generation sequencing, clinical exome sequencing, autism genetics
Citation: Lee J, Ha S, Lee S-T, Park S-G, Shin S, Choi JR and Cheon K-A (2020) Next-Generation Sequencing in Korean Children With Autism Spectrum Disorder and Comorbid Epilepsy. Front. Pharmacol. 11:585. doi: 10.3389/fphar.2020.00585
Received: 11 September 2019; Accepted: 16 April 2020;
Published: 14 May 2020.
Edited by:
Mohd Farooq Shaikh, Monash University, MalaysiaReviewed by:
Vinod RMT Balasubramaniam, Monash University Malaysia, MalaysiaKing-Hwa Ling, Universiti Putra Malaysia, Malaysia
Copyright © 2020 Lee, Ha, Lee, Park, Shin, Choi and Cheon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keun-Ah Cheon, a2FjaGVvbkB5dWhzLmFj