 Mahmoud Zaki El-Readi1,2
Mahmoud Zaki El-Readi1,2 Ahmed M. Al-Abd3,4*Mohammad A. Althubiti1Riyad A. Almaimani1Hiba Saeed Al-Amoodi1Mohamed Lotfy Ashour5,6Michael Wink7Safaa Yehia Eid1
Ahmed M. Al-Abd3,4*Mohammad A. Althubiti1Riyad A. Almaimani1Hiba Saeed Al-Amoodi1Mohamed Lotfy Ashour5,6Michael Wink7Safaa Yehia Eid1- 1Department of Biochemistry, Faculty of Medicine, Umm Al-Qura University, Makkah, Saudi Arabia
- 2Department of Biochemistry, Faculty of Pharmacy, Al-Azhar University, Assiut, Egypt
- 3Department of Pharmaceutical Sciences, College of Pharmacy & Thumbay Research Institute for Precision Medicine, Gulf Medical University, Ajman, United Arab Emirates
- 4Pharmacology Department, Medical Division, National Research Centre, Cairo, Egypt
- 5Department of Pharmaceutical Sciences, Pharmacy Program, Batterjee Medical College, Jeddah, Saudi Arabia
- 6Department of Pharmacognosy, Faculty of Pharmacy, Ain Shams University, Cairo, Egypt
- 7Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, Heidelberg, Germany
Plant secondary metabolites (SMs) common natural occurrences and the significantly lower toxicities of many SM have led to the approaching development and use of these compounds as effective pharmaceutical agents; especially in cancer therapy. A combination of two or three of plant secondary metabolites together or of one SM with specific anticancer drugs, may synergistically decrease the doses needed, widen the chemotherapeutic window, mediate more effective cell growth inhibition, and avoid the side effects of high drug concentrations. In mixtures they can exert additive or even synergistic activities. Many SM can effectively increase the sensitivity of cancer cells to chemotherapy. In phytotherapy, secondary metabolites (SM) of medicinal plants can interact with single or multiple targets. The multi-molecular mechanisms of plant secondary metabolites to overcome multidrug resistance (MDR) are highlighted in this review. These mechanisms include interaction with membrane proteins such as P-glycoprotein (P-gp/MDR1); an ATP-binding cassette (ABC) transporter, nucleic acids (DNA, RNA), and induction of apoptosis. P-gp plays an important role in the development of MDR in cancer cells and is involved in potential chemotherapy failure. Therefore, the ingestion of dietary supplements, food or beverages containing secondary metabolites e.g., polyphenols or terpenoids may alter the bioavailability, therapeutic efficacy and safety of the drugs that are P-gp substrates.
Plant Secondary Metabolites Biosynthesis Pathways and Classification
All higher plants have the capacity to produce secondary metabolites (SM) Table 1.
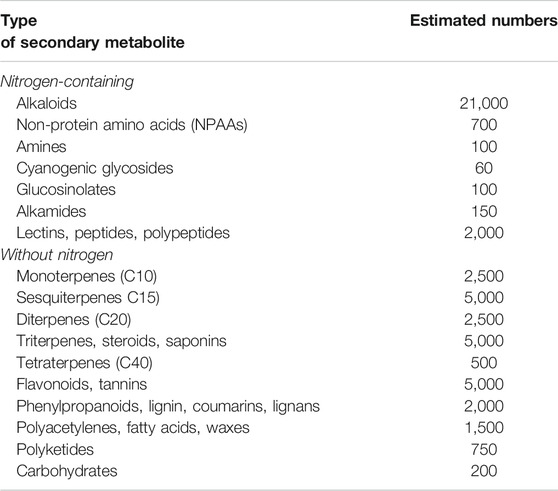
TABLE 1. Estimated number of plant secondary metabolites (Wink, 2010).
The great majority of these metabolites are derived from five precursor pathways, acetyl coenzymeA (polyketides such as anthraquinones, flavonoids), active isoprene (various terpenenoids), shikimic acid (aromatic amino acids, cinnamic acids, tannins, indole and isoquinoline alkaloids), glycolysis (sugars, gallic acid), and tricarboxylic acid cycle; krebs cycle (alkaloids) (Figure 1). At present, more than 140,000 structurally diverse SM have been produced and identified from these pathways (Wink, 2010).
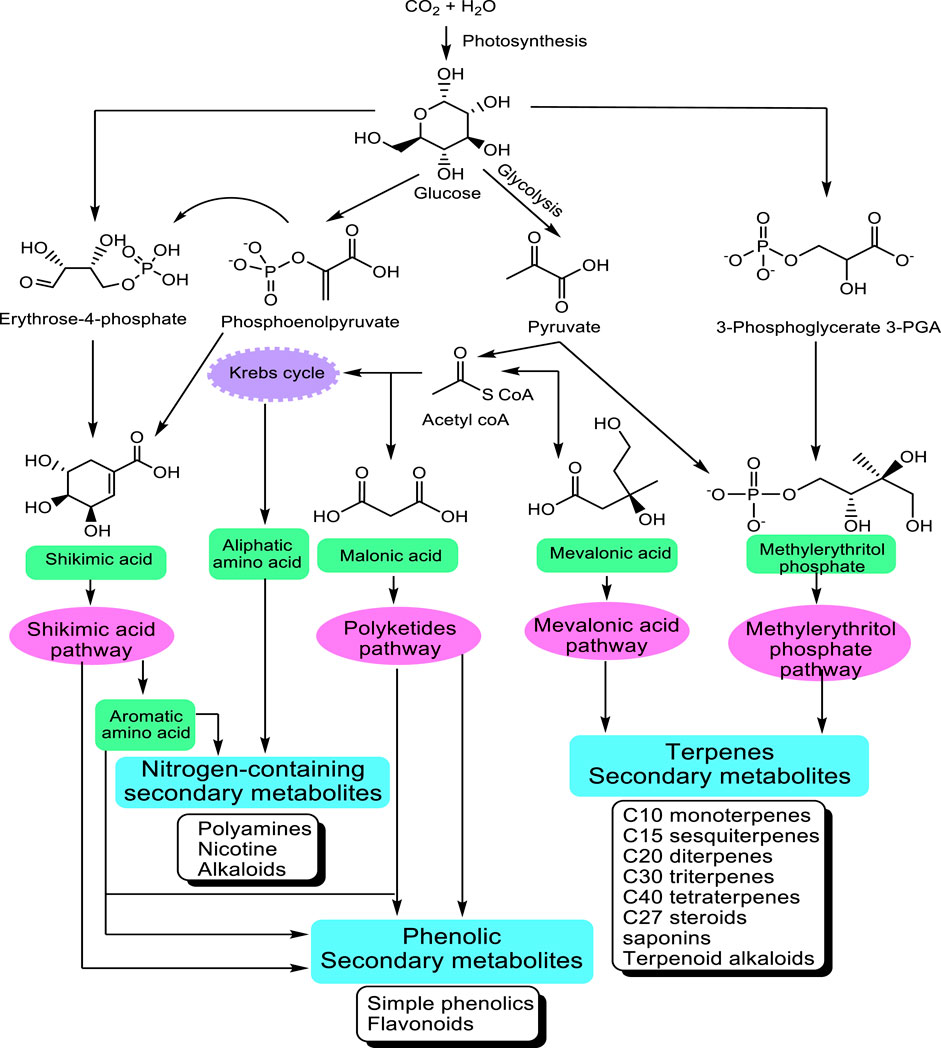
FIGURE 1. Main pathways leading to SM modified from Wink (2010). The non-mevalonate pathway or 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate pathway (MEP/DXP pathway) of isoprenoid biosynthesis is an alternative metabolic pathway leading to the formation of isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) and finally terpenes.
The structurally diverse SM can be classified based on the presence or absence of a nitrogen group in their structures into two large groups: SM with nitrogen in their structures and those without nitrogen. In Table 1, an estimate of the numbers of known SM is given in the table above.
Biosynthesis Site and Storage of Plant Secondary Metabolites
Most biosynthetic pathways completely, or at least partially, occur in the cytoplasm, in the endoplasmic reticulum or in organelles. Hydrophilic compounds are usually stored in the vacuole. The lipophilic substances are sequestered in laticifers, resin ducts, glandular hairs, or on the cuticle. Many terpenoids (such as monoterpenes, diterpenes, and carotenoids) are synthesized through the pyruvate/glyceraldehyde phosphate pathway, or in the chloroplast (Wink, 1999). The corresponding genes are apparently localized in the cell nucleus. Sesquiterpenes are formed mainly in the endoplasmic reticulum (ER) Figure 2.
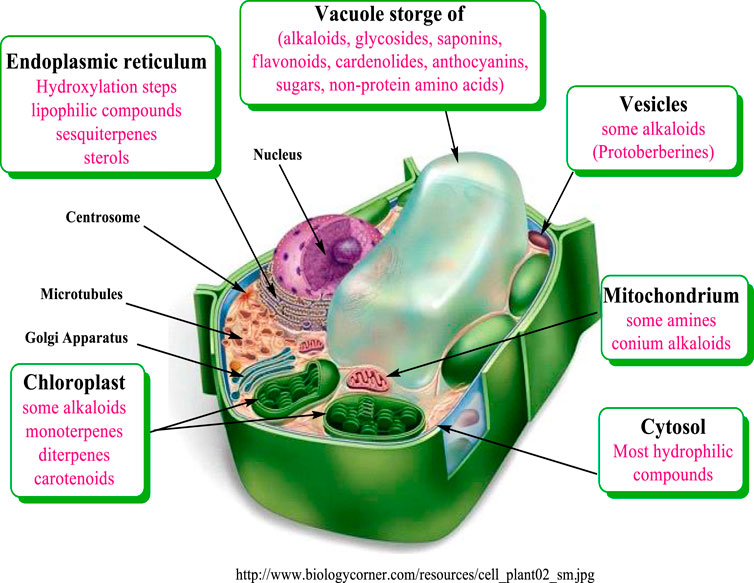
FIGURE 2. Compartments of biosynthesis and storage for plant secondary metabolites modified from Wink (1999).
Function of Secondary Metabolites for the Plants
SMs have vital functions, which are important for the fitness of their plants. Their main roles include a defense function against herbivores (insects, vertebrates), fungi, bacteria, viruses, and other plants. Some SM act as signal compounds to attract fertilizing and seed-dispersing animals. Others serve as communication-signals between the plants and symbiotic microorganisms (mycorrhizal fungi or N-fixing Rhizobia). Moreover, SMs have a protection role against UV light or other physical stress (Wink, 2003).
Our study focuses on a selection of SM which represents structures from each chemical class, such as glaucine, harmine, and sancguinarine for alkaloids and menthol, aromadendrene, β-sitosterol, β-carotene, crocin, retinoic acid, canthaxanthin, fucoxanthin, and digitonin (saponin) for terpenes and epigallcatechinegallate (EGCG) and thymol for polyphenols. In order to understand the role of this set of representative SM in cancer treatment, it is important to introduce notes about the cellular targets, mode of action and mechanisms of anticancer activity of SM classes.
Major Cellular Targets for Secondary Metabolites
SMs interfere with several important molecular targets present in a cell (Figure 3).
FIGURE 3. Molecular targets of secondary metabolites modified from (Wink, 2007).
SM can be classified into two major groups according to their biological activities (Wink, 2008):
(1) SM which are highly selective for cellular targets and developing strong biological activity.
(2) SM which are non-selective and show moderate or weak bioactivity. They can attack various cellular targets. This non-selective type includes broad spectrum phytochemicals, which may occur in multicomponent mixtures and can exert synergistic interactions.
There are four major cellular targets for SM including:
(1) Proteins are represented in different forms such as receptors, enzymes, ion channels, transporters, regulatory proteins(Alexander et al., 2019a; Alexander et al., 2019b; Alexander et al., 2019c; Alexander et al., 2019d; Alexander et al., 2019e; Alexander et al., 2019f; Alexander et al., 2019g), cytokines, chemokines (Khare et al., 2019), structural proteins, cytoskeletal proteins, microtubules forming the mitotic spindle, transcription factors, and hormones. MHC-B complex genes that encode for cell surface proteins, are activated and affect the tumor outcome (Khare et al., 2018). It has been reported that most SM apparently interact with proteins and change the protein conformation in an unselective manner (binding, complexing, and denaturing). SM with reactive functional groups, such as aldehyde, epoxide or terminal and/or exocyclic methylene, and cyclopropane groups (found in several terpenes), are able to undergo electrophilic or nucleophilic substitutions allowing the formation of covalent bonds with a protein by binding to free amino-, SH- or OH- groups. This covalent interaction might lead to changes in the 3D conformation and thus loss of protein activity; or it might alter the protein turnover due to the inability of proteases to break down the alkylated protein. Polyphenols are example of SM which change the three-dimensional structures of proteins (Wink, 2008; Wink, 2010; Wink and Schimmer, 2010).
(2) Bio-membrane (fluidity, permeability). Saponins and other lipophilic terpenoids are examples of SM which influence the bio-membrane. Saponins are the glycosides of triterpenes or steroids, including steroidal alkaloids and cardiac glycosides. The bidesmosidicsaponins (inactive) cleave into the monodesmosidicsaponins (active) by β-glucosidase. Monodesmosidicsaponins, such as digitonin, have an amphipathic nature, their lipophilic terpenoid moiety is able to make a complex with cholesterol in biomembranes, while their sugar side chain binds to surface glycoproteins and glycolipids (Wink and Van Wyk, 2008). As a result, transient or permanent pores are generated in the membrane and make it leaky. This unspecific activity affects a wide set of cells and can easily be demonstrated with erythrocytes, which lose their haemoglobin when in contact with monodesmosidicsaponins (Wink, 2008; Wink and Van Wyk, 2008; Wink and Schimmer, 2010).
(3) Nucleic acids (DNA, RNA). Alkylating or intercalating SM affect a wider range of animals and microorganisms. The ability of SM to affect more than one molecular target is a typical SM behavior; therefore, additive and even synergistic activities can be expected. Generally, the SM that target membranes and DNA have cytotoxic activity and the affected cells usually undergo apoptosis (Wink, 2000; Wink, 2007). In addition, the covalent modifications are one major cause of point-mutations (deletion or exchange of one or more nucleic acid base) as well as failure of repair enzymes to exchange the modified bases (Wink, 2000).
(4) Inhibition of energy generation or cell division can lead to cell death, especially apoptosis (Pratt et al., 2020).
SM exert several effects on cancer cells in vitro, and on tumors in vivo (experimental animals), as well as interacting with anti-cancer drugs (affecting positively or negatively their efficacy), and protecting normal host organism tissues from the adverse effects of anti-cancer therapies (Wink, 2015; Seca and Pinto, 2018). These effects can be summarized as the follows:
DNA Alkylation
All genes are carried in DNA which encodes all proteins that are important for metabolism and normal cell development. Thus DNA is a main target of many SM (Roberts and Wink, 1998). Alkylating agents target N7 guanine in DNA. These compounds add methyl (anion, cation, or free radical) or other alkyl groups and form covalent bonds with the N7 guanine of DNA strands lead to strand breaks. Alkylguanine-DNA alkyl transferase (AGT) is a repair enzyme, which able to remove this modification. If the repair processes are failed, several types of mutation are arising such as nucleotide exchanges (transitions and transversions) or deletions. This leads to the inhibition of the correct utilization of guanine by base pairing and causes a miscoding of DNA and frame-shift mutation, translated gene into nonsense protein, and consequently, loss of protein function (Wink, 2007). Alkaloids e.g. aristolochic acids and pyrrolizidine alkaloids, lactones e.g. 3-propiolactone and parasorbic acid, glycosides e.g. cycasin and macrozamin are examples of classes of plant SM that exert alkylation of DNA (Wink, 2007).
DNA Intercalation
DNA intercalation is a non-covalent interaction between small molecules (mutagenic agents) and DNA (Persil and Hud, 2007). The intercalation is a result of the hydrophobic interactions between appropriate functional groups of SM and stacked base pairs of DNA. It is followed by ionic attraction, as expected between several cationic SM, such as alkaloids and nucleic acid (anionic). Intercalating SM is generally characterized as aromatic, planar, and hydrophobic. These features of SM lead to the hypothesis that they could very easily form π–π complexes with other planar aromatic molecules, such as nucleotide bases in DNA; adenine-thymine (AT) and guanine-cytosine (GC) (Wink and Schimmer, 2010; Hill et al., 2011). This interaction stabilizes the double-stranded DNA; therefore, DNA replication or transcription is stopped (Schmeller et al., 1997; Roberts and Wink, 1998).The intercalation also takes place in RNA double-stranded stem structures due to complementary base pairing (Wink, 2007). Frame-shift mutation is considered as a trigger of some lethal effects of intercalation on cells, which leads to changes in the sequence of corresponding amino acid (Wink, 2007). There is a positive correlation with the strongest being the intercalation and the inhibition of DNA and RNA processing enzymes, such as DNA polymerase I and reverse transcriptase (RT) (Roberts and Wink, 1998). Examples of intercalating agents are sanguinarine, harmine, glaucine, berberine, camptothecin, chelerythrine, doxorubicin, caffeine, theophylline, acridine orange, and ellipticine (Bathaie et al., 2007; Wink and Van Wyk, 2008; Hill et al., 2011). It has been reported that intercalating and alkylating SM induce apoptosis or inhibit carbohydrate-processing enzymes. Swainsonine, a Golgi a-mannosidase II inhibitor, is an example of alkaloids that intercalate with many glycosyltransferase enzymes leading to change in the produced carbohydrates, which participate in cell-cell and cell-substratum interactions affecting processes such as lymphocyte trafficking, immune cell stimulation, embryogenesis, and cancer metastasis (Goss et al., 1995).
DNA Topoisomerase
DNA replication, repair and recombination are essential processes for living cells. Topoisomerases (topo) are located in the nucleus of the cell; they alter the supercoiling and prevent entanglement of DNA double strands during DNA replication, transcription, exchanges of DNA segments between chromosomes (recombination) and elimination of erroneous DNA sequences (repair) (Wang et al., 2014). Therefore, topoisomerase inhibitors lead to cell death by blocking these essential processes. Mammalian cells contain two types of topoisomerases (topo) I and II. Topo I acts by transiently cutting single strands of DNA whereas topo II break double strands of DNA. Both types of topoisomerase break DNA via a catalytic tyrosine attack on a phosphodiester bond on the DNA backbone. The inhibition of topoisomerase occurs either directly by inhibiting this catalytic activity or by stabilizing the cleavable ternary complexes (CTC) by preventing strand resealing and converting CTC to lethal lesions that result when the cell tries to use the damaged DNA template for replication (Bailly, 2000; Ganguly et al., 2007; Wink, 2007; Alberts, 2008). Inhibition of one type of topoisomerase is enough to lead to cell-cycle arrest and cell death by apoptosis while synergistic cytotoxicity is observed when inhibiting both types of topoisomerase (Wink, 2007). Topoisomerase I is an important target of several SM in cancer cell; it can be targeted by several alkaloids such as quinazoline–quinoline, camptothecin, quinolones, and also by several flavones (Alison, 2002; Ma et al., 2004). A number of SM affect topo I and II enzymes such as sanguinarine, indenoquinolone, coralyne, acridine, and celastrol (triterpene) (Gatto et al., 1996; Makhey et al., 1996; Lin et al., 2005; Wink, 2007).
Telomeres and Telomerase
Human telomeres are tandem repeats of the hexameric sequence TTAGGG at the ends of linear chromosomes. The telomere sequence has a role in maintaining chromosomal integrity through its ability to prevent degradation, recombination, and be mistaken for DNA double-strand breaks (Shay and Wright, 2005). After finishing the replication many times, the telomeres are lost in subsequent cell divisions, leading to replicative senescence (Levy et al., 1992). Telomerase is a ribonucleoprotein enzyme responsible for the de novo synthesis of telomeres by adding telomeric DNA onto 3’ ends of chromosomes (Greider and Blackburn, 1985). Telomerase is an interesting target for anticancer therapy because it is necessary for the growth and survival of cancer cells. SM can trigger a DNA damage response, either via the “drug stacking” model of interaction with telomeres, or by inhibiting telomerase enzymatic activity. Inhibitor combinations (with different mechanism of actions) could accelerate telomere shortening and reduce the period of time required for killing cancer cells (Hathcock et al., 2004; Alberts, 2008). The combination of a major SM of green tea (EGCG and retinoic acid) inhibits telomerase activity (Yokoyama et al., 2008). Alkaloids including tetrandrine, fangchinoline, berbamine, ellipticine, cryptolepine, and neocryptolepine, terpenoids including genistein, ginsenoside, and ginseng saponin are examples of telomerase inhibitors from natural plant SM (Alberti et al., 2003; Guittat et al., 2003; Garbett and Graves, 2004; Liu et al., 2004; Ouchi et al., 2005; Zhang et al., 2005; Wang and Fang, 2006; Wink, 2007; Ji et al., 2011).
Cytoskeleton
The cytoskeleton (CSK) is structural protein classified into microfilaments (actin filaments), intermediate filaments, and microtubules. Microtubules play an important role in maintaining cell structure, providing platforms for intracellular transport, and assembling the spindle during mitosis (cell division); as well as, other cellular processes, e.g. apoptosis (Gierke et al., 2010). Microtubules are responsible for completing the mitosis process of cell division by separating the duplicated chromatids and pulling each part into daughter cells. Tubulin α- and β- dimers are polymerized into proto-filaments. A group of 13 proto-filaments laterally associate to form a long hollow cylindrical structure of microtubules. GTP molecules promote the polymerization (assembly) and bind it with both α- and β-tubulin dimer (Alberts, 2008). GTP-α-tubulin complex is stable while GTP-β-tubulin complex is unstable and it might be hydrolyzed to GDP soon after assembly. Therefore GDP-tubulin is prone to depolymerization and fall off (disassembly) (Wink, 2007). The dynamic instability of microtubules leads to cell cycle arrest (at mitosis) and subsequently apoptosis (Zhou and Giannakakou, 2005). The inhibition of the microtubule assembly or disassembly becomes a major target for anticancer agents because the cell division in cancer cells is faster than differentiated normal cells (Wink, 2007). It has been reported that almost all natural SM which have anticancer activity can interact with microtubules (Wink, 2007). Vinblastine and vincristine inhibit microtubule assembly while Taxol® (diterpene alkaloid) inhibits the microtubule disassembly and blocks cell division in the late G2 phase (Lobert et al., 1996; Wink, 2007). Chelidonine, colchicine, sanguinarine, phalloidin, and noscapine are examples of SM that interacting with microtubules (Wink, 2007).
Induction of Apoptosis
Apoptosis, or programmed cell death, is an intrinsic death process that plays an important role during development and in adult life (Kerr et al., 1972). Therefore, too little apoptosis or escape from apoptosis can promote carcinogenesis and progression (Bellamy et al., 1995). In addition, the inability of cancer cells to undergo apoptosis after treatment is one of the main reasons for the development MDR in cancer cells (Gimenez-Bonafe et al., 2009). Thus, induction of apoptosis is considered to be a novel approach to overcoming MDR in cancer treatment. The apoptosis pathways are the receptor pathway or extrinsic pathway that involve stimulation of the tumor necrosis factor (TNF) superfamily e.g. TNF-related apoptosis induces ligand (TRAIL) receptors or CD95 (APO-1/Fas) and the mitochondrial pathway (intrinsic). In the extrinsic pathway, the stimulation of death receptors by natural SM ligands or other stimuli, initiate the oligomerization of receptors followed by the recruitment of adaptor molecules (e.g. FADD procapase-8) to the stimulated death receptors, which leads to the induction of caspase-8 and finally an increase of caspase-3 (Sayers, 2011). Alternatively, caspases-8 may cleave Bid (BH3 protein of the Bcl-2 family) into tBid which activates the outer membrane permeabilization of mitochondria. Therefore, Bid is considered a link between the extrinsic and the intrinsic pathways (Walczak and Krammer, 2000). In the intrinsic pathway DNA damage and p53 stimulate the release of inter-membrane space proteins into the cytosol. These proteins include cytochrome c and/or second mitochondria-derived activators of caspase (Smac)/direct IAP binding protein with low pI (DIABLO), which trigger the activation of effector caspases (Tait and Green, 2010). Cytochrome c is involved in the apoptosome complex formation (cytochrome c + Apaf-1 + caspase-9) which leads to subsequent activation of the caspase-9, then induction of caspase-3 (Alberts, 2008; Tait and Green, 2010). However, Smac/DIABLO is able to bind and antagonize the inhibitor of apoptosis proteins (IAP) including survivin, XIAP, c-IAP2, c-IAP1, and livin/melanoma-IAP (ML-IAP) promoting the activation of caspases-3, -7 and -9 (LaCasse et al., 2008). There are several proteins that can control apoptosis pathways. Pro-apoptotic proteins include multi-domain proteins (e.g. Bak and Bax) and BH3 domain-proteins (e.g. Bid, Noxa, Bim, and Puma) and anti-apoptotic proteins such as the Bcl-2 family (e.g. Bcl-2, Mcl-1, and Bcl-xL), which significantly participate in intrinsic pathway regulation (Tait and Green, 2010). Examples of SM that induce apoptosis are sanguinarine, harmine, colchicine, vinblastine, berberine, emetine, and cinchonine(Rosenkranz and Wink, 2008; Cao et al., 2011; Chan, 2011; Hamsa and Kuttan, 2011; Lee et al., 2012).
Interaction With ABC Transporters
After heart diseases, cancer is the second-leading cause of death worldwide. Annually; more than 19.3 × 106 cancer cases and 10 × 106 cancer deaths are reported (Ferlay et al., 2010). The rate of cancer death is expected to increase worldwide, with an estimated 10 × 106 humans dying from cancer in 2020 and 28.4 × 106 dying in 2040 (Cho, 2007; Jemal et al., 2011). In addition, cancer is a multi-factorial disease resulting in abnormal and unlimited division of cells. Treatment strategies that inhibit growth of cancer cells cause some serious side effects on normal cells and cancer cells often develop resistance to drug treatment. Drug resistance develops not only to single drugs, but can also occur to several classes of drugs, even those with diverse chemical structures and modes of action. This is known as multidrug resistance (MDR). This might explain why treatment with cytotoxic agents or even multiple agent combinations with different targets is sometimes not effective. Several types of drug resistance have been identified (Fan et al., 1994). They have been classified into cellular and non-cellular mechanisms, as summarized in Figure 4.
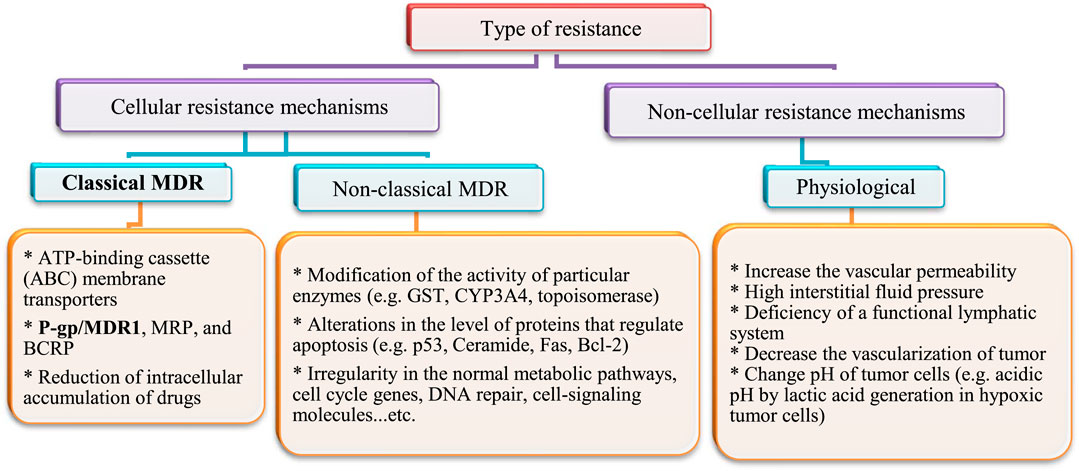
FIGURE 4. Mechanisms of multidrug resistance (MDR) in cancer cells.
This section will highlight the role of membrane transporters in the classical type of multi-drug resistance.
Membrane Transporters
Membrane transporters are classified into two main families: The ATP binding cassette (ABC) transporter family and the solute carrier (SLC) transporter family (Choudhuri and Klaassen, 2006).
The ATP-Binding Cassette (ABC) Transporters
Several substrates (lipids, bile acids, xenobiotic, and peptide antigens) are transported against a concentration gradient by ATP-binding cassette (ABC) transporters proteins. This active process is powered by ATP hydrolysis. ABC-transporters are expressed in different organs such as the small and large intestine, kidney, adrenal gland, placenta, liver, and capillary endothelial cells of brain and testis (Cordon-Cardo et al., 1989; Fricker et al., 1998; Fricker, 2008).The tissue localization of ABC-transporters suggests that they play a physiological function in the detoxification process. They transport and reduce the amount of exogenous and endogenous substances in the body and excrete the xenobiotics and metabolites into the gastrointestinal lumen, bile, and urine (Figure 5). Thus, ABC-transporters play an important role in the absorption and distribution of drugs; consequently, determining the pharmacokinetic properties and clinical response of several drugs (Gutmann et al., 1999; Lin and Yamazaki, 2003; Fricker and Miller, 2004; Mahringer et al., 2011). However, drug resistance can develop through this action when therapeutic agents are also effluxed from cells and eliminated from the body.
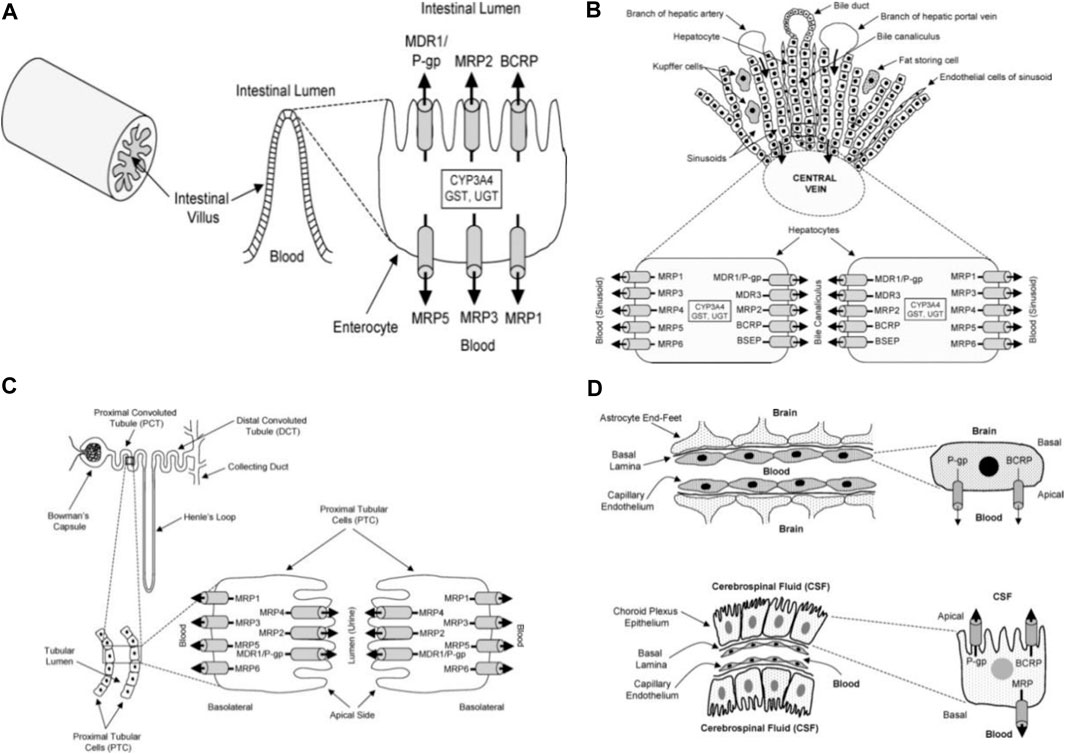
FIGURE 5. Diagrammatic section of the small intestine (A), liver (B), proximal tubular cells of kidney (C), choroid plexus epithelium and blood brain barrier (D) adapted from Choudhuri and Klaassen (2006).
Classification of (ABC) Transporters
Christopher Higgins coined the term “ABC transporter” in 1992 (Higgins, 1992). Today, 49 human ABC transporter family genes have been identified. They are classified into 7 subfamilies (number of members): ABCA (12), ABCB (11), ABCC (13), ABCD (4), ABCE (1), ABCF (3), and ABCG (5). There are three subfamilies which confer important drug resistance and they are expressed in several organs; ABCB1 (MDR1/P-glycoprotein of subfamily ABCB), subfamily ABCC (MRPs), and ABCG2 (BCRP of subfamily ABCG). Italics and numerical suffixes are used to designate genes e.g. ABCB/MDR (italicized) with a numerical suffix, such as ABCB1 (or MDR1), ABCC1 (or MRP1); whereas, a lack of italics indicates a protein as ABCB1 (or MDR1), ABCC1 (or MRP1) (Efferth, 2001; Gottesman et al., 2002).
P-glycoprotein (P-gp)
P-gp or MDR1 is a polypeptide dimer (1280-residue), which represents a pore forming membrane protein. It was the first studied member of ABC transporters. P-gp has long been of interest to molecular and cellular biologists because its overexpression is linked to MDR in human cancers. It transports many types of drugs such as cytotoxic agents, immuno-suppressants, protease inhibitors, statins, calcium channel blockers, steroids, beta-blockers, anticonvulsants, antihistamines, and antidepressants (Takara et al., 2006). Differing stimuli, including light ultraviolet (UV), retinoic acid, phorbol esters, butyrate, and several chemotherapeutics can induce P-gp gene expression. Additionally, environmental and/or genetic factors likely influence P-gp expression (Greiner et al., 1999).
Multidrug Resistance Protein (MRP)
It was originally believed that the overexpression of P-gp (170-kDa) alone led to multidrug resistance in cancer cells. Eight additional MRPs that potentially contribute to drug resistance have since been discovered. The multidrug resistance protein 1 (190-kDa; MRP1) is located in most organs in the basolateral membrane of epithelial cells (Fricker and Miller, 2002) (Figure 6). It appears to play an important role in protecting the cells from bilirubin toxicity. MRP1 is able to transport many anticancer agents, such asanthracycline antibiotics (e.g., doxorubicin, daunorubicin), epipodophyllotoxins (e.g. etoposide, teniposide), and Vinca alkaloids (e.g. vincristine, vinblastine) (Gottesman et al., 2002).
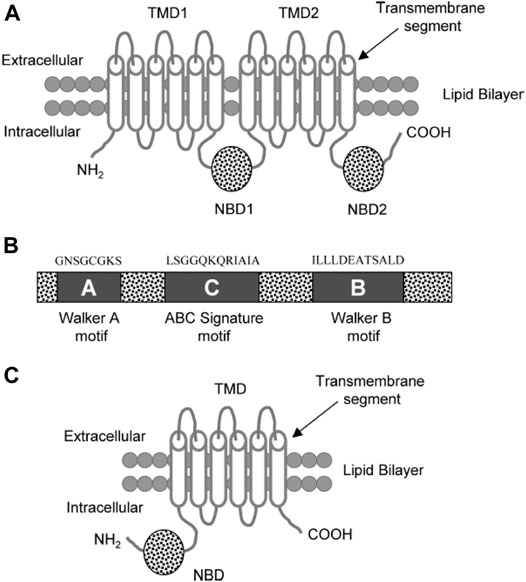
FIGURE 6. (A) Diagram of a typical ABC transporter protein inserted in the lipid bilayer. The trans-membrane protein consists of 2 trans-membrane domains (TMDs) and the two nucleotide ATP binding domains (NBDs) (B) The NBDs consists of walker A, walker B, and ABC signature C motifs with characteristic residues of the respective amino acid sequences above these motifs. (C) ABCG2 diagram showing a half transporter (6 trans-membrane segments and 1 NBDs) at the N-terminal end (Choudhuri and Klaassen, 2006).
Breast Cancer Resistance Protein (BCRP)
ABCG2 (also called BCRP/ABCP/MXR) is an ABC transporter that is about half the size of P gp (∼70-kDa). It contains six trans-membrane segments and one NBD at the N-terminal end in the cytoplasmic side. It is an efflux transporter found in most tissues and is clinically relevant as another mediator of drug resistance (Kühnle et al., 2009).
General Structural of ABC Transporter (Mainly P-gp)
A typical ABC transport protein consists of two parts sharing a high level of sequence uniformity. One half is composed of two hydrophobic trans-membrane domains (TMDs) and the other of two hydrophilic nucleotide-binding domains (NBDs) found at the cytoplasmic face of the membrane. Each of the NBDs is associated with one of the TMDs; NBD1 with TMD1 and NBD2 with TMD2 (Figure 6). There are usually six trans-membrane segments in each TMD; therefore, 12 segments (α-helices) in total are located in the apical part of the cell membrane (Raviv et al., 1990; Rosenberg et al., 1997; Loo and Clarke, 1999).
Two of the TMDs of P-gp that are coupled with one another by a ∼75–amino acid linker region fold in a unique way (as known from Computer-assisted algorithms) (Rao and Nuti, 2003).The NBDs contain three characteristic motifs (Walker A, Walker B, and an ABC signature (C) motif). The ABC signature motif of NBDs is located across from the Walker A sequence of NBD2 in a sandwich configuration (Figure 6). Thus, ATPs are sandwiched between adjacent Walker A and ABC signature motifs (Akhtar et al., 2011). The three-dimensional structure with high resolution (25- Å) shows that there is a large central chamber in P-gp which is closed at the cytoplasmic side and opens to the lipid phase. This structure allows substrates to have free access from the cytoplasm, while denying access to substrates from the outer leaflet. A cross-sectional view of a P-gp molecule (4.5 Å) shows an overall clover-leaf shape and the central cavity is opened in the TMDs by conformational changes in the protein. These changes mediate substrate transportation (Rosenberg et al., 1997).
The Models and Mechanisms of P-gp Drug Efflux
The Classical Model
Simply, this model suggests that, P-gp actively expels substrate drugs from the cytoplasm to the extracellular locationthrough the two TMDs in a pore-forming arrangement (Borst and Schinkel, 1997).
Flippase Model
The substrate-binding site of P-gp is located on the inner face of the plasma membrane and in front of the cytoplasmic face. The hydrophobic portions of the substrate molecule are oriented toward the hydrophobic core of the membrane whereas the charged portion is oriented toward the polar cytosolic face of the membrane (Figure 7) (Homolya et al., 1993).
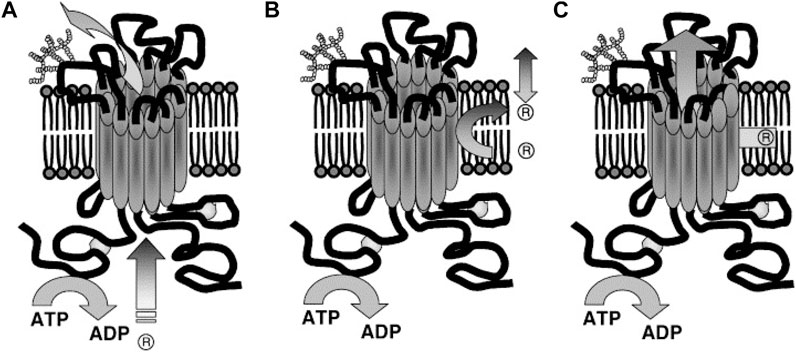
FIGURE 7. Suggested Models illustrating P-gp mechanisms of drug efflux. (A) Classical or pore, (B) flippase and (C) hydrophobic vacuum cleaner models. In the classical model: substrate effluxes out of the cell through a protein channel by interacting with P-gp in the cytoplasm. In the flippase model: substrate transports into the inner leaflet, binds to P-gp (within the membrane), translocates to the outer leaflet, and passively diffuses into the extracellular. In the hydrophobic vacuum cleaner model: the characteristics of ‘pore’ and ‘flippase’ models are mixed (Li et al., 2007).
The transport stages in this model are 1) the substrate is laterally diffused until it binds to the substrate-binding site in the inner leaflet of the lipid bilayer (pocket in the cytoplasmic leaflet). 2) The ATPase activity of P-gp hydrolyzes ATP and allows the protein to flip the substrate into the outer leaflet. 3) Then the substrate diffuses outside of the cell into the extracellular aqueous phase or else the outer leaflet (Higgins and Gottesman, 1992). It is believed the drug release from P-gp includes re-orientation of the drug binding site from the inner leaflet (cytosolic side) to the outer leaflet (extracellular side) of the membrane and is followed by a change from high to low drug binding affinity. Thus, it is hypothesized that ATP binding, rather than hydrolysis, is the driver behind the conformational changes which accompany the transport process (Rosenberg et al., 2001; Rosenberg et al., 2003). This flipping triggers the rearrangement of the P-gp structure, expelling the substrate. For example, in the lipopolysaccharide (sugar head and lipid tail) molecule as a substrate model, the hydrophilic heads are sequestered and then “flipped” in the internal chamber dragging the lipophilic tails through the bilayer as suggested by its “flipping” structure (Regev et al., 2005).
Hydrophobic Vacuum Cleaner Model
P-gp recognizes substrates as foreign to the cell membrane and pumps them from the intracellular compartment into the extracellular medium or from the lipid bilayer, thereby detecting and eliminating xenobiotic and hydrophobic drugs as they penetrate the lipid bilayer, much like a hydrophobic vacuum cleaner (Gottesman and Pastan, 1993). The vacuum cleaner model suggests that the lipid bilayer is integral to drug concentration, whereas P-gp itself has a low intrinsic substrate binding affinity (Sharom, 2014). This model suggests that substrates and modulators partition into membranes prior to interacting with the transporter (Figure 7C). P-gp substrates typically contain high lipid/water partition coefficients. They are concentrated at very high levels within the membrane (Regev et al., 2005).
P-Glycoprotein Substrates
P-gp can interact with hundreds of substrates, which are diverse in chemical structure and action, such as those found in natural products, anti-cancer drugs, cardiac glycosides, calcium channel blockers, analgesics, immunosuppressive agents, anthelminthic, HIV protease inhibitors (HPI), antibiotics, H2-receptor antagonists, fluorescent dyes, steroids, and peptides (linear and cyclic) (Amin, 2013).
Generally, P-gp substrates come in a variety of sizes, ranging from small (e.g. daunorubicin and doxorubicin) to large complex molecules (e.g. paclitaxel and vinblastine). Researchers have not yet fully defined the structural activity relationship of P-gp substrates; however, it is known that most substrates are hydrophobic organic cations, amphipathic, having a wide range of molecular weight (300‒2000 Da), and contain a minimum of two planar rings (Seelig, 1998; Cianchetta et al., 2005). Substrates may be anionic, cationic, or uncharged (Ueda et al., 1997). Structural characters required for the compound to interact with P-gp are made up of either two or three of electron donors (hydrogen bond acceptors) with a fixed spatial separation configuration (Seelig, 1998; Cianchetta et al., 2005). It seems to be a direct correlation between the affinity of a compound for P-gp and its number of hydrogen bonds plus its lipophilicity (Ecker et al., 1999). The three dimensions quantitative structure activity relationship (3DQSAR) study shows that the interaction between the substrate with the binding sites of P-gp is crucial to efflux (Xing et al., 2009).
TMDs of P-gp have a high number of amino acids in the side chains which are able to interact with substrates and act as hydrogen bond donors (Seelig, 1998; Cianchetta et al., 2005). In addition, the aromatic amino acid residues of substrates may have a role in the binding and transporting of these substrates (Pawagi et al., 1994). Therefore, the efficiency of a substrate to bind is dependent on how many points are simultaneously interacting with P-gp which in fact depends on the chemical structure of the drug. Van der Waals and hydrophobic interactions have a role in substrate binding because of the electrostatic interactions between charged amino acid in the side chains of P-gp and charged functional groups on the drug molecules (Watkins et al., 2001). Furthermore, a high concentration of the substrate in the membrane is important for enhancing the affinity interaction of the substrate with P-gp (Cascorbi, 2011). Additionally, the lipid environment surrounding P-gp can modulate the ATPase activity of P-gp and affect coupling of the drug binding site with NBDs.
P-Glycoprotein Modulators
Additionally, another class of compounds "modulators" interacts with P-gp. These are also known by other names such as MDR reversal agents or chemosensitizers. Modulators have similar structural properties to substrates which interact in the drug binding pocket and are transported by P-gp. Generally, substrates with a high affinity and high trans-bilayer diffusion rate are effective modulators. Effluxed compounds can re-enter the outer leaflet and then diffuse across the membrane (or "flip-flop" into the inner leaflet), where they once again interact with P-gp and are re-exported. Substrates re-enter the membrane slowly enough that the P-gp can keep up, establishing a drug gradient, and developing drug resistance (Li-Blatter et al., 2012). On the other hand, modulators re-enter so quickly that the P-gp cannot efflux them fast enough, causing a futile cycle to operate such as Rho123 which has a t1/2 of 3 min (Eytan et al., 1996). The performance of modulators has been shown to be related to their diffusion rate beyond the membrane (Eytan and Kuchel, 1999). Transporter turnover and ATP hydrolysis rates are high while no concentration gradient is generated, resulting in cells that are not resistant to modulators. MDR modulators can inhibit the activity of the P-gp efflux pump. Therefore, they have the ability to potentiate cytotoxicity, making them valuable alternatives for use in overcoming MDR (Robert and Jarry, 2003). Even though, cells are not resistant to modulators, they can be effectively affected by substrates combined with modulators (Eckford and Sharom, 2009; Sharom, 2011). Secondary metabolites (SM) of plants including phenolics, terpenoids, and alkaloids are intriguing candidate for use in chemosensitizing or reversing MDR in ABC transporters expressed in cancer cells (Wink et al., 2012). Most terpenes (mono-, di-, tri-, and sesquiterpenes) are very hydrophobic SM which can bind to bio-membranes. At higher doses, these SM are able to unselectively disturb membrane fluidity and permeability causing uncontrolled efflux of metabolites and ions and possibly even cell leakage. Terpenes can stimulate death receptors, induce apoptosis, and modulate proteins in the membrane (Wink and Van Wyk, 2008).The aforementioned modulators interfere with P-gp's ability to extrude drugs and exert a drug concentration gradient; thus, reversing MDR in cancer cells in vitro. It is clinically important because it can act as a selective blocker of the P-gp efflux pump and improve drug uptake and bioavailability (Fu and Arias, 2012).
Established P-Glycoprotein Inhibitors/Modulators
Some drug substrates of P-gp can inhibit P-gp mediated efflux or uptake of other P-gp substrates. Several P-gp inhibitors can inhibit P-gp function either by binding to drug-binding sites of the membrane transport proteins as competitive inhibitors or via indirect mechanisms by stopping the ATP hydrolysis process (non-competitive inhibitors). Indirect mechanisms are related to phosphorylation of the transport proteins or the expression of the P-gp gene (Sikic, 1999; Wink et al., 2012). Ca2+ channel blockers (e.g. verapamil, diltiazem, nifedipine, nicardipine, quinidine, quinine), antimicrobial agents (e.g., ketoconazole, itraconazole, erythromycin, cefoperazone, ceftriaxone, clarithromycin), HPIs (ritonavir, indinavir, saquinavir, nelfinavir), benzimidazole proton pump inhibitors (PPIs) or gastric H+/K+-ATPase inhibitors (e.g. omeprazole, lansoprazole, pantoprazole, rabeprazole), tamoxifen, propranolol, hydrocortisone, and progesterone have been shown to inhibit P-gp activity, leading to potentially relevant drug interactions (Pauli-Magnus et al., 2001; DuBuske, 2005; Fricker, 2008).
Additionally, several herbal constituents which are often used by cancer patients; as well as, dietary phytochemicals, are used as complementary and alternative medicines (CAMs). They can modulate or inhibit P-gp activity and/or expression (Fricker, 2008; Mahringer et al., 2010; Efferth and Greten, 2012; Eichhorn and Efferth, 2012). Recently, the effect of many natural occurring polyphenols, terpenoids, and alkaloids on P-gp expression and activity were reviewed (Wink et al., 2012). In resistant cancer cells, P-gp, MRP1, and BCRP can be competitively inhibited by some alkaloids (isoquinoline, protoberberine, quinoline, indole, monoterpeneindole, and steroidal alkaloids), lipophilic terpenoids (mono-, di-, tri-terpenes including saponins), tetraterpenes (including carotenoids) (Eid et al., 2020), and steroids (including cardiac glycosides) (Wink et al., 2012). Remarkably, initial observations also show that many more polar phenolic SM (phenolic acids, flavonoids, xanthones, chalcones, catechins, anthocyanins, tannins, stilbenes, anthraquinones, and naphthoquinones) (El-Readi et al., 2019) can directly disrupt the 3D structure of P-gp, thus inhibiting its activity in MDR cancer cells (Wink, 2008). There is a need for specific studies with ABC- transporters (mainly P-gp) with the end goal of gaining knowledge which leads to the development of new drugs and/or therapies to improve human quality of life. The next section will focus on the combination of plant secondary metabolites as an approach to overcoming MDR and exerting synergistic interactions.
Combination Therapy to Overcome MDR
Drug combinations have been used for the treatment of a wide range of serious diseases including cancer, HIV, AIDS, TB, malaria, diabetes, hypertension, MRSA etc. Drug combination has a long history in herbal medicines such as traditional Chinese medicines (TCM) which is considered as poly-pharmacy with many multidrug regimens (Efferth, 2010).
Today, drug combinations are more established and developed because of the valuable advances in the technology of phytochemical isolation science and chemical synthetic capability. Over 90% of cancer deaths are related to MDR, and thus a failure in chemotherapy (Coley, 2010).
Finding a way to overcome MDR in cancer cells is the main focus of chemotherapy research which is mainly concerned with a combinational strategy. The use of many agents with different mechanisms of action may target several targets or diseases. The probable outcome advantages of drug combination include:
(1) Enhancing the efficacy of the therapy.
(2) Reducing the dose while enhancing or maintaining the same efficacy to prevent toxicity.
(3) Overcoming or reversing MDR (El-Readi et al., 2019; Abdalla et al., 2020; Abdallah et al., 2020).
(4) Achieving selective synergism against a target vs. host (Chou, 2006).
The effect of the drug combinations can be classified into three types: synergistic; the biological effects of the drugs in combinations are greater than the sum of individual drug effects, additive; the effects of the drug combinations are equal to additional effects and antagonistic; the biological activity of drugs in combination are lower than the sum of the individual drug effects. Efficient strategies as seen in “theoretical basis experimental design and computerized simulation in combinational drug studies” have been reported by Chou (2006). The combinational index, isobologram, and the correlation coefficient of linear regression depending on the medium effect equation are the most proper methods to apply when evaluating the drug combination.
Definition of Synergy
There are many definitions for the term “synergy”. The “isobole method” is considered as one of the most common and demonstrative methods to prove a proposed synergistic effect (Berenbaum, 1989). In an isobologram of isobole methods, the x and y axes reflect the dose of the single drugs A and B that exert certain effects (e.g. IC50) and the various doses in combinations are evaluated for the same effect. An isobole is represented by a line or curve between points of the same effect. According to the Berenbaumisobole method, the interaction results can be classified into 3 categories; additive, antagonism, and synergism (Berenbaum, 1989). The additive effect or zero (0) interaction means that the effect of two drugs (A and B) is a pure summation effect of both individual drugs as the follows:
The universal observed effect (E); DA and DB are the doses of drugs A and B.
Antagonistic interaction means the combination effect is less than a predictable effect from the summation of the individual drug effects and the data points of a combination will create a convex curve.
In a synergistic interaction, the overall combination effect of two drugs (A and B) is greater than would be expected by the summation of the individual drug effects. The result of the combination data points is then a concave curve.
In synergism, lower doses of drugs A and B are required to achieve the desired effect. The synergistic effect can be quantified as fold or factors as doubling or even greater multiplication of the expected effect. The advantage of dose reduction of combined drugs is a probable decrease in the side effects of potent cytotoxic drugs.
Mechanisms of Synergy Effects
The mechanisms of synergistic interactions are usually unknown. However, it is important to gain knowledge regarding the mechanism of action of combined drugs before managing drug combination studies. For example combining P-gp-substrate drugs (vinblastine, paclitaxel, and doxorubicin) with P-gp-reversing agents (e.g. ningalin, ardeemin, and limonin) (Chou, 2006; El-Readi et al., 2010). The synergistic mechanisms can be classified into at least 4 different possible mechanisms depending on the pharmacological, biochemical, molecular biological and clinical effects (Wink, 2008; Wagner, 2011):
1. Synergistic multi-target effects: Plant SM targets not only one site, but many targets and can simultaneously interact in a synergistic fashion. In addition, the SM can act as a synergistic partner or different molecules of the same signalling cascade (multi-target effects (Wink et al., 2012). A good example of such synergetic effect is seen in case of the activity of Urtica dioica roots towards prostate hyperplasia, which has been associated with a synergism of antiproliferative and anti-inflammatory effects by lectins and polysaccharides (Efferth and Koch, 2011).
2. Pharmacokinetic and physiochemical effects: Plant SM can improve solubility and/or the resorption rate and thereby the bioavailability of the synergistic partner. For example, enhancing the solubility or absorption rate of the active SM via inactive by-products (e.g. tannins). Furthermore, it can be supposed that bioavailability, interaction with ABC- transporter pathways, deactivation of active SM to inactive SM or activation of inactive pro-drugs, prevention of binding to target proteins (e.g. by tannins)....etc. may take place as a part of the mode of action in the synergistic process (Wink, 2008; Efferth and Koch, 2011).
3. Reverse the resistance mechanisms of cancer cells (against chemotherapy). SM can increase intracellular accumulation of pharmacologically active compounds by inhibiting the P-gp efflux pump. For example, the interaction of 5-methoxy-hydnocarpine (P-gp inhibitor) and berberine (P-gp substrate) in Berberis plants (Stermitz et al., 2000).
4. Neutralization or elimination of the toxic effects of SM via combination with synthetic drugs which that improve the overall efficacy results. Additionally, some SM may induce phase I metabolizing enzymes, e.g. CYP3A4, which are responsible for changing the pro-drugs (those with no pharmacological activity) into active metabolites (El-Readi et al., 2013; Eid et al., 2015).
Plant Secondary Metabolites and Combination Therapy
Plant secondary metabolites have important roles in chemotherapy (Eid et al., 2012a; Eid et al., 2012b; Eid et al., 2013). Over 60% of currently used chemotherapeutical agents are derived from natural sources such as plants, microorganisms, and marine organisms (Newman et al., 2003). More than 3350 plants species have been used in cancer treatment (Hartwell, 1982; Graham et al., 2000). However, most isolated plant compounds do not serve as drugs; rather, they provide lead structures for the development of potential novel agents. There are many examples of effective anticancer drugs or chemotherapeutical agents derived from plants, which are used for the treatment of many types of tumours including camptothecin, etoposide, epipodophyllotoxin such as paclitaxel (taxol), and Vincaalkolloids (vinblastine, vincristine) (Wink, 2007; Efferth, 2010). Ulrich-Merzenich et al., have mentioned several reasons for the interest in research into the development of a new generation of phytopharmaceuticals from natural sources by synergistic drug combinations (Ulrich-Merzenich et al., 2010).
Generally, hydrophobic SM, such as terpeneoids (including saponins), steroids (including cardiacglycosides) can modulate P-gp in cancer cells. Being lipophilic, they usually inhibit the active transporters by competing for binding to the active side, which present an excellent opportunity when administered as a chemosensitizer in combination with a cytotoxic agent. Limonin isolated from many Citrus species at 20 μM dose significantly enhanced doxorubicin cytotoxicity 2.98-fold and 2.2-fold in Caco2 and CEM/ADR5000 cells, respectively (El-Readi et al., 2010).
On the other hand, the alkaloids isolated from Chelidonium majus such as berberine chelidonine, and sanguinarine represent good examples for SMs that can synergistically act with many chemopreventive and epigenetic modifiers (HDACs inhibitors), which are widely used in cancer treatment. Concomitant treatment of chelidonine at 20 μM dose with doxorubicin dramatically decreased the IC50 of the known cytotoxic drug on Caco-2 cells to almost one tenth of the original (El-Readi et al., 2013). The other alkaloidal analogue sanguinarine reduced the IC50 value of doxorubicin in two-drug combinations (sanguinarine + doxorubicin) to 17.58-fold in the highly resistant Caco-2 cells (Eid et al., 2012; Eid et al., 2013). On the same context, berberine can also acts synergistically with vorinostat in SW480 colon cancer cells (Li et al., 2021).
Traditional systems of medicine, such as Chinese medicine, provide a lot of clinical prescriptions for many known diseases. The term “Reverse pharmacology” has been coined for traditional medicines and the biodiversity of medicinal plants has so far remained mainly unexplored and is still a valuable strategy for the finding of new drugs (Pieters and Vlietinck, 2005; Patwardhan and Mashelkar, 2009). However, the isolation and chemical identification of pure single SM has only resulted in moderate yield. For example, the National Cancer Institute, USA investigated the anticancer activity of 114,000 extracts (from about 35,000 plants) and only a few compounds from this large screening project have been clinically applied as chemotherapeutical agents such as paclitaxel and topotecan (Cragg and Boyd, 1996). Moreover, it is difficult to treat multifactorial diseases such as cancer, diabetes, or cardiovascular diseases with mono-target therapies. The synergy of multi-target therapies created by the combination of naturally-occurring phytochemicals is considered as a good approach to increase the efficacy of such treatment, and it might be cheaper than synthetic treatment (Vernon et al., 2010). In this study the pre-approval cost of new anticancer entities can be reduced to more than half of the total cost of two new anticancer agents in addition to the increased efficacy of such combinations. Hence, some combination therapies are currently in use in modern medical therapy for disease treatment. These are often used without documenting the original expected cumulative efficacy or adverse effects of both single treatments. Therefore, there is an increasing interest in the synergy concept (Ulrich-Merzenich et al., 2009).
In addition, the worldwide need for phytopharmaceuticals is steadily growing. The world-bank has reported that the trade in medicinal herbs, plant derived drug products, and crud plant materials is increasing annually by a growth rate between 5–15% (Patwardhan et al., 2005). For example, the herbal industry had a turnover of about 62 billion-dollars in 2005 (Patwardhan et al., 2005). Therefore, it is important to gain understanding about plant SM and their molecular and cellular targets to exert their cytotoxic effects on cancer cells.
Conclusion
Many studies provide important information regarding the molecular targets of plants secondary metabolites. The drug-herbal and drug-diet interactions associated with ABC-transporters, mainly P-gp is very important topic in cancer therapy. Understanding drug efflux and uptake, knowing the effects of other phytochemicals and natural products on multidrug resistance mediated transport, and the mechanism of modulating the drug transport proteins is essential for predicting clinical interactions in humans. Further research could provide knowledge which might prevent potentially harmful drug-drug interactions due to altered chemotherapeutic efficacy, when plant SM contained in (for example), an herbal medicine, is taken concurrently with prescribed medicines.
Author Contributions
All Authors have an equal contribution in research question, study design, relevant articles retrieve, articles selection, writing and revise the manuscripts.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdalla, A. N., Abdallah, M. E., Aslam, A., Bader, A., Vassallo, A., Tommasi, N., et al. (2020). Synergistic Anti Leukemia Effect of a Novel Hsp90 and a Pan Cyclin Dependent Kinase Inhibitors. Molecules 25, 2220. doi:10.3390/molecules25092220
Abdallah, M. E., El-Readi, M. Z., Althubiti, M. A., Almaimani, R. A., Ismail, A. M., Idris, S., et al. (2020). Tamoxifen and the PI3K Inhibitor: LY294002 Synergistically Induce Apoptosis and Cell Cycle Arrest in Breast Cancer MCF-7 Cells. Molecules 25, 3355. doi:10.3390/molecules25153355
Akhtar, N., Ahad, A., Khar, R. K., Jaggi, M., Aqil, M., Iqbal, Z., et al. (2011). The Emerging Role of P-Glycoprotein Inhibitors in Drug Delivery: a Patent Review. Expert Opin. Ther. Patents 21, 561–576. doi:10.1517/13543776.2011.561784
Alberti, P., Lacroix, L., Guittat, L., Helene, C., and Mergny, J.-L. (2003). Nucleic Acids as Targets for Antitelomerase Agents. Mini. Rev. Med. Chem. 3, 23–36. doi:10.2174/1389557033405485
Alberts, B. (2008). Molecular Biology of the Cell. 5th edition. New York: Extended version, Garland Science.
Alexander, S. P. H., Armstrong, J. F., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., et al. (2019a). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and Other Protein Targets. Br. J. Pharmacol. 176 (Suppl. 1), S1–S20. doi:10.1111/bph.14747
Alexander, S. P. H., Cidlowski, J. A., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., et al. (2019b). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Nuclear Hormone Receptors. Br. J. Pharmacol. 176 (Suppl. 1), S397–S493. doi:10.1111/bph.14750
Alexander, S. P. H., Christopoulos, A., and Davenport, A. P., Kelly, E., Mathie, A., Peters, J. A., et al. (2019c). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 176 (Suppl. 1), S21–S141. doi:10.1111/bph.14748
Alexander, S. P. H., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., Armstrong, J. F., et al. (2019d). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters. Br. J. Pharmacol. 176 (Suppl. 1), S397–S493. doi:10.1111/bph.14753
Alexander, S. P. H., Fabbro, D., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., et al. (2019e). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic Receptors. Br. J. Pharmacol. 176 (Suppl. 1), S247–S296. doi:10.1111/bph.14751
Alexander, S. P. H., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., Striessnig, J., et al. (2019f). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Br. J. Pharmacol. 176 (Suppl. 1), S142–S228. doi:10.1111/bph.14749
Alexander, S. P. H., and Fabbro, D., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., et al. (2019g). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. Br. J. Pharmacol. 176 (Suppl. 1), S297–S396. doi:10.1111/bph.14752
Amin, M. L. (2013). P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target. Insights 7, 27–34. doi:10.4137/dti.s12519
Bailly, C. (2000). Topoisomerase I Poisons and Suppressors as Anticancer Drugs. Curr. Med. Chem. 7, 39–58. doi:10.2174/0929867003375489
Bathaie, S. Z., Bolhasani, A., Hoshyar, R., Ranjbar, B., Sabouni, F., and Moosavi-Movahedi, A. A., (2007), Interaction of Saffron Carotenoids as Anticancer Compounds with ctDNA, Oligo (dG.dC)15, and Oligo (dA.dT)15, DNA Cell Biol. 26, 533–540. doi:10.1089/dna.2007.0598
Bellamy, C. O. C., Malcolmson, R. D. G., Harrison, D. J., and Wyllie, A. H. (1995). Cell Death in Health and Disease: the Biology and Regulation of Apoptosis. Semin. Cancer Biol. 6, 3–16. doi:10.1006/scbi.1995.0002
Borst, P., and Schinkel, A. H. (1997). Genetic Dissection of the Function of Mammalian P-Glycoproteins. Trends Genet. 13, 217–222. doi:10.1016/s0168-9525(97)01112-8
Cao, M.-R., Li, Q., Liu, Z.-L., Liu, H.-H., Wang, W., Liao, X.-L., et al. (2011). Harmine Induces Apoptosis in HepG2 Cells via Mitochondrial Signaling Pathway. Hepatobiliary Pancreat. Dis. Int. 10, 599–604. doi:10.1016/s1499-3872(11)60102-1
Cascorbi, I. (2011). P-glycoprotein: Tissue Distribution, Substrates, and Functional Consequences of Genetic Variations. Handbook Exp. Pharmacol. 201, 261–283. doi:10.1007/978-3-642-14541-4_6
Chan, W.-H. (2011). Embryonic Toxicity of Sanguinarine through Apoptotic Processes in Mouse Blastocysts. Toxicol. Lett. 205, 285–292. doi:10.1016/j.toxlet.2011.06.018
Cho, W. C. (2007). Contribution of Oncoproteomics to Cancer Biomarker Discovery. Mol. Cancer 6, 25. doi:10.1186/1476-4598-6-25
Chou, T.-C. (2006). Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 58, 621–681. doi:10.1124/pr.58.3.10
Choudhuri, S., and Klaassen, C. D. (2006). Structure, Function, Expression, Genomic Organization, and Single Nucleotide Polymorphisms of Human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) Efflux Transporters. Int. J. Toxicol. 25, 231–259. doi:10.1080/10915810600746023
Cianchetta, G., Singleton, R. W., Zhang, M., Wildgoose, M., Giesing, D., Fravolini, A., et al. (2005). A Pharmacophore Hypothesis for P-Glycoprotein Substrate Recognition Using GRIND-Based 3D-QSAR. J. Med. Chem. 48, 2927–2935. doi:10.1021/jm0491851
Coley, H. M. (2010). Overcoming Multidrug Resistance in Cancer: Clinical Studies of P-Glycoprotein Inhibitors. Methods Mol. Biol. 596, 341–358. doi:10.1007/978-1-60761-416-6_15
Cordon-Cardo, C., O'Brien, J. P., Casals, D., Rittman-Grauer, L., Biedler, J. L., Melamed, M. R., et al. (1989). Multidrug-resistance Gene (P-Glycoprotein) Is Expressed by Endothelial Cells at Blood-Brain Barrier Sites. Proc. Natl. Acad. Sci. 86, 695–698. doi:10.1073/pnas.86.2.695
Cragg, G. M., and Boyd, M. (1996). “Drug Discovery and Development at the National Cancer Institute,” in Medicinal Resources of the Tropical Forest : Biodiversity and its Importance to Human Health. Editors M. J. Balick, E. Elisabetsky, and S. A. Laird (New York: Columbia University Press), 101.
DuBuske, L. M. (2005). The Role of P-Glycoprotein and Organic Anion-Transporting Polypeptides in Drug Interactions. Drug Saf. 28, 789–801. doi:10.2165/00002018-200528090-00004
Ecker, G., Huber, M., Schmid, D., and Chiba, P. (1999). The Importance of a Nitrogen Atom in Modulators of Multidrug Resistance. Mol. Pharmacol. 56, 791–796.
Eckford, P. D. W., and Sharom, F. J. (2009). ABC Efflux Pump-Based Resistance to Chemotherapy Drugs. Chem. Rev. 109, 2989–3011. doi:10.1021/cr9000226
Efferth, T., and Greten, H. J. (2012). The European Directive on Traditional Herbal Medicinal Products: Friend or Foe for Plant-Based Therapies? J. Chin. Integr. Med. 10, 357–361. doi:10.3736/jcim20120401
Efferth, T., and Koch, E. (2011). Complex Interactions between Phytochemicals. The Multi-Target Therapeutic Concept of Phytotherapy. Curr. Drug Targets 12, 122–132. doi:10.2174/138945011793591626
Efferth, T. (2001). The Human ATP-Binding Cassette Transporter Genes From the Bench to the Bedside. Curr. Mol. Med. 1, 45–65. doi:10.2174/1566524013364194
Efferth, T. (2010). Cancer Therapy with Natural Products and Medicinal Plants. Planta Med. 76, 1035–1036. doi:10.1055/s-0030-1250062
Eichhorn, T., and Efferth, T. (2012). P-glycoprotein and its Inhibition in Tumors by Phytochemicals Derived from Chinese Herbs. J. Ethnopharmacology 141, 557–570. doi:10.1016/j.jep.2011.08.053
Eid, S. Y., El-Readi, M. Z., and Wink, M. (2012a). Carotenoids Reverse Multidrug Resistance in Cancer Cells by Interfering with ABC-Transporters. Phytomedicine 19, 977–987. doi:10.1016/j.phymed.2012.05.010
Eid, S. Y., El-Readi, M. Z., and Wink, M. (2012b). Synergism of Three-Drug Combinations of Sanguinarine and Other Plant Secondary Metabolites with Digitonin and Doxorubicin in Multi-Drug Resistant Cancer Cells. Phytomedicine 19, 1288–1297. doi:10.1016/j.phymed.2012.08.010
Eid, S. Y., El-Readi, M. Z., Eldin, E. E. M. N., Fatani, S. H., and Wink, M. (2013). Influence of Combinations of Digitonin with Selected Phenolics, Terpenoids, and Alkaloids on the Expression and Activity of P-Glycoprotein in Leukaemia and Colon Cancer Cells. Phytomedicine 21, 47–61. doi:10.1016/j.phymed.2013.07.019
Eid, S. Y., El-Readi, M. Z., Ashour, M. L., and Wink, M. (2015). Fallopia Japonica, a Natural Modulator, Can Overcome Multidrug Resistance in Cancer Cells. Evidence-Based Complementary and Alternative Medicine. eCAM 2015, 868424. doi:10.1155/2015/868424
Eid, S. Y., Althubiti, M. A., Abdallah, M. E., Wink, M., and El-Readi, M. Z. (2020). The Carotenoid Fucoxanthin Can Sensitize Multidrug Resistant Cancer Cells to Doxorubicin via Induction of Apoptosis, Inhibition of Multidrug Resistance Proteins and Metabolic Enzymes. Phytomedicine 77, 153280. doi:10.1016/j.phymed.2020.153280
El-Readi, M. Z., Hamdan, D., Farrag, N., El-Shazly, A., and Wink, M. (2010). Inhibition of P-Glycoprotein Activity by Limonin and Other Secondary Metabolites from Citrus Species in Human Colon and Leukaemia Cell Lines. Eur. J. Pharmacol. 626, 139–145. doi:10.1016/j.ejphar.2009.09.040
El-Readi, M. Z., Eid, S., Ashour, M. L., Tahrani, A., and Wink, M. (2013). Modulation of Multidrug Resistance in Cancer Cells by Chelidonine and Chelidonium Majus Alkaloids. Phytomedicine 20, 282–294. doi:10.1016/j.phymed.2012.11.005
El-Readi, M. Z., Eid, S., Abdelghany, A. A., Al-Amoudi, H. S., Efferth, T., and Wink, M. (2019). Resveratrol Mediated Cancer Cell Apoptosis, and Modulation of Multidrug Resistance Proteins and Metabolic Enzymes. Phytomedicine 55, 269–281. doi:10.1016/j.phymed.2018.06.046
Eytan, G. D., and Kuchel, P. W. (1999). Mechanism of Action of P-Glycoprotein in Relation to Passive Membrane Permeation. Int. Rev. Cytol. - a Surv. Cell Biol. 190, 175–250. doi:10.1016/s0074-7696(08)62148-8
Eytan, G. D., Regev, R., Oren, G., and Assaraf, Y. G. (1996). The Role of Passive Transbilayer Drug Movement in Multidrug Resistance and its Modulation. J. Biol. Chem. 271, 12897–12902. doi:10.1074/jbc.271.22.12897
Fan, D., Beltran, P. J., and O’Brien, C. A. (1994). Reversal of Multidrug Resistance (Boca Raton, FL: CRC Press).
Ferlay, J., Shin, H.-R., Bray, F., Forman, D., Mathers, C., and Parkin, D. M. (2010). Estimates of Worldwide Burden of Cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 127, 2893–2917. doi:10.1002/ijc.25516
Fricker, G., and Miller, D. S. (2002). Relevance of Multidrug Resistance Proteins for Intestinal Drug Absorption In Vitro and In Vivo. Pharmacol. Toxicol. 90, 5–13. doi:10.1034/j.1600-0773.2002.900103.x
Fricker, G., and Miller, D. S. (2004). Modulation of Drug Transporters at the Blood-Brain Barrier. Pharmacology 70, 169–176. doi:10.1159/000075545
Fricker, G., Drewe, J., and Miller, D. S. (1998). Excretion of a Fluorescent Rapamycin-Derivative in Proximal Kidney Tubules Is Mediated by P-Glycoprotein. Int. J. Clin. Pharmacol. Ther. 36, 67–69.
Fricker, G. (2008). Drug Interactions with Natural Products at the Blood Brain Barrier. Curr. Drug. Metab. 9, 1019–1026. doi:10.2174/138920008786927758
Fu, D., and Arias, I. M. (2012). Intracellular Trafficking of P-Glycoprotein. Int. J. Biochem. Cell Biol. 44, 461–464. doi:10.1016/j.biocel.2011.12.009
Ganguly, A., Das, B., Roy, A., Sen, N., Dasgupta, S. B., Mukhopadhayay, S., et al. (2007). Betulinic Acid, a Catalytic Inhibitor of Topoisomerase I, Inhibits Reactive Oxygen Species Mediated Apoptotic Topoisomerase I DNA Cleavable Complex Formation in Prostate Cancer Cells but Does Not Affect the Process of Cell Death. Cancer Res. 67, 11848–11858. doi:10.1158/0008-5472.can-07-1615
Garbett, N., and Graves, D. (2004). Extending Natures Leads: The Anticancer Agent Ellipticine. Cmcaca 4, 149–172. doi:10.2174/1568011043482070
Gatto, B., Sanders, M. M., Yu, C., Wu, H. Y., Makhey, D., LaVoie, E. J., et al. (1996). Identification of Topoisomerase I as the Cytotoxic Target of the Protoberberine Alkaloid Coralyne. Cancer Res. 56, 2795–2800.
Gierke, S., Kumar, P., and Wittmann, T. (2010). Analysis of Microtubule Polymerization Dynamics in Live Cells. Methods Cell Biol 97, 15–33. doi:10.1016/s0091-679x(10)97002-7
Gimenez-Bonafe, P., Tortosa, A., and Perez-Tomas, R. (2009). Overcoming Drug Resistance by Enhancing Apoptosis of Tumor Cells. Curr. Cancer Drug Targets 9, 320–340. doi:10.2174/156800909788166600
Goss, P. E., Baker, M. A., Carver, J. P., and Dennis, J. W. (1995). Inhibitors of Carbohydrate Processing: A New Class of Anticancer Agents. Clin. Cancer Res. 1, 935–944.
Gottesman, M. M., and Pastan, I. (1993). Biochemistry of Multidrug Resistance Mediated by the Multidrug Transporter. Annu. Rev. Biochem. 62, 385–427. doi:10.1146/annurev.bi.62.070193.002125
Gottesman, M. M., Fojo, T., and Bates, S. E. (2002). Multidrug Resistance in Cancer: Role of ATP-dependent Transporters. Nat. Rev. Cancer 2, 48–58. doi:10.1038/nrc706
Graham, J. G., Quinn, M. L., Fabricant, D. S., and Farnsworth, N. R. (2000). Plants Used against Cancer - an Extension of the Work of Jonathan Hartwell. J. Ethnopharmacology 73, 347–377. doi:10.1016/s0378-8741(00)00341-x
Greider, C. W., and Blackburn, E. H. (1985). Identification of a Specific Telomere Terminal Transferase Activity in Tetrahymena Extracts. Cell 43, 405–413. doi:10.1016/0092-8674(85)90170-9
Greiner, B., Eichelbaum, M., Fritz, P., Kreichgauer, H.-P., Von Richter, O., Zundler, J., et al. (1999). The Role of Intestinal P-Glycoprotein in the Interaction of Digoxin and Rifampin. J. Clin. Invest. 104, 147–153. doi:10.1172/jci6663
Guittat, L., Alberti, P., Rosu, F., Van Miert, S., Thetiot, E., Pieters, L., et al. (2003). Interactions of Cryptolepine and Neocryptolepine with Unusual DNA Structures. Biochimie 85, 535–547. doi:10.1016/s0300-9084(03)00035-x
Gutmann, H., Fricker, G., Török, M., Michael, S., Beglinger, C., and Drewe, J. (1999). Evidence for Different ABC-Transporters in Caco-2 Cells Modulating Drug Uptake. Pharm. Res. 16, 402–407. doi:10.1023/a:1018825819249
Hamsa, T., and Kuttan, G. (2011). Harmine Activates Intrinsic and Extrinsic Pathways of Apoptosis in B16F-10 Melanoma. Chin. Med. 6, 11. doi:10.1186/1749-8546-6-11
Hartwell, J. L. (1982). Plants Used against Cancer : A Survey. Lawrence, Mass: Quarterman Publications.
Hathcock, K. S., Hodes, R. J., and Weng, N. P. (2004). Analysis of Telomere Length and Telomerase Activity. Curr. Protoc. Immunol. Chapter 10, Unit 10.30. doi:10.1002/0471142735.im1030s62
Higgins, C. F., and Gottesman, M. M. (1992). Is the Multidrug Transporter a Flippase? Trends Biochem. Sci. 17, 18–21. doi:10.1016/0968-0004(92)90419-a
Higgins, C. F. (1992). ABC Transporters: From Microorganisms to Man. Annu. Rev. Cell. Biol. 8, 67–113. doi:10.1146/annurev.cb.08.110192.000435
Hill, G. M., Moriarity, D. M., and Setzer, W. N. (2011). Attenuation of Cytotoxic Natural Product DNA Intercalating Agents by Caffeine. Sci. Pharm. 79, 729–747. doi:10.3797/scipharm.1107-19
Homolya, L., Holló, Z., Germann, U. A., Pastan, I., Gottesman, M. M., and Sarkadi, B. (1993). Fluorescent Cellular Indicators Are Extruded by the Multidrug Resistance Protein. J. Biol. Chem. 268, 21493–21496. doi:10.1016/s0021-9258(20)80566-3
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., and Forman, D. (2011). Global Cancer Statistics. CA: a Cancer J. clinicians 61, 69–90. doi:10.3322/caac.20107
Ji, X., Chen, J., Sun, H., Zhou, H., Xiang, J., Peng, A., et al. (2011). The Interaction of Telomere DNA G-Quadruplex with Threebis-Benzyltetrahydroisoquinoline Alkaloids. Nucleic acid Ther. 21, 415–422. doi:10.1089/nat.2011.0311
Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 26, 239–257. doi:10.1038/bjc.1972.33
Khare, V. M., Saxena, V. K., Tomar, A., Singh, K. P., Singh, K. B., and Tiwari, A. K. (2018). MHC-B Haplotypes Impact Susceptibility and Resistance to RSV-A Infection. Front. Biosci. 10, 506–519. doi:10.2741/e837
Khare, V. M., Saxena, V. K., Tomar, A., Nyinawabera, A., Singh, K. B., Ashby, C. R., et al. (2019). Cytokine Gene Expression Following RSV-A Infection. Front. Biosci. 24, 463–481. doi:10.2741/4729
Kühnle, M., Egger, M., Müller, C., Mahringer, A., Bernhardt, G., Fricker, G., et al. (2009). Potent and Selective Inhibitors of Breast Cancer Resistance Protein (ABCG2) Derived from the P-Glycoprotein (ABCB1) Modulator Tariquidar. J. Med. Chem. 52, 1190–1197. doi:10.1021/jm8013822
LaCasse, E. C., Mahoney, D. J., Cheung, H. H., Plenchette, S., Baird, S., and Korneluk, R. G. (2008). IAP-targeted Therapies for Cancer. Oncogene 27, 6252–6275. doi:10.1038/onc.2008.302
Lee, J. S., Jung, W.-K., Jeong, M. H., Yoon, T. R., and Kim, H. K. (2012). Sanguinarine Induces Apoptosis of HT-29 Human Colon Cancer Cells via the Regulation of Bax/Bcl-2 Ratio and Caspase-9-dependent Pathway. Int. J. Toxicol. 31, 70–77. doi:10.1177/1091581811423845
Levy, M. Z., Allsopp, R. C., Futcher, A. B., Greider, C. W., and Harley, C. B. (1992). Telomere End-Replication Problem and Cell Aging. J. Mol. Biol. 225, 951–960. doi:10.1016/0022-2836(92)90096-3
Li, X., Li, J.-P., Yuan, H.-Y., Gao, X., Qu, X.-J., Xu, W.-F., et al. (2007). Recent Advances in P-Gp-Mediated MDR Reversal Mechanisms. Methods Find Exp. Clin. Pharmacol. 29, 607–617. doi:10.1358/mf.2007.29.9.1139054
Li, G., Zhang, C., Liang, W., Zhang, Y., Shen, Y., and Tian, X. (2021). Berberine Regulates the Notch1/PTEN/PI3K/AKT/mTOR Pathway and Acts Synergistically with 17-AAG and SAHA in SW480 Colon Cancer Cells. Pharm. Biol. 59, 21–30. doi:10.1080/13880209.2020.1865407
Li-Blatter, X., Beck, A., and Seelig, A. (2012). P-glycoprotein-ATPase Modulation: the Molecular Mechanisms. Biophysical J. 102, 1383–1393. doi:10.1016/j.bpj.2012.02.018
Lin, J. H., and Yamazaki, M. (2003). Clinical Relevance of P-Glycoprotein in Drug Therapy. Drug Metab. Rev. 35, 417–454. doi:10.1081/dmr-120026871
Lin, J. P., Lu, H. F., Lee, J. H., Lin, J. G., Hsia, T. C., Wu, L. T., et al. (2005). (-)-Menthol Inhibits DNA Topoisomerases I, II Alpha and Beta and Promotes NF-kappaB Expression in Human Gastric Cancer SNU-5 Cells. Anticancer Res. 25, 2069–2074.
Liu, L., Berletch, J. B., Green, J. G., Pate, M. S., Andrews, L. G., and Tollefsbol, T. O. (2004). Telomerase Inhibition by Retinoids Precedes Cytodifferentiation of Leukemia Cells and May Contribute to Terminal Differentiation. Mol. Cancer Ther. 3, 1003–1009.
Lobert, S., Vulevic, B., and Correia, J. J. (1996). Interaction of Vinca Alkaloids with Tubulin: A Comparison of Vinblastine, Vincristine, and Vinorelbine. Biochemistry 35, 6806–6814. doi:10.1021/bi953037i
Loo, T. W., and Clarke, D. M. (1999). The Transmembrane Domains of the Human Multidrug Resistance P-Glycoprotein Are Sufficient to Mediate Drug Binding and Trafficking to the Cell Surface. J. Biol. Chem. 274, 24759–24765. doi:10.1074/jbc.274.35.24759
Ma, Z., Hano, Y., Nomura, T., and Chen, Y. (2004). Novel Quinazoline-Quinoline Alkaloids with Cytotoxic and DNA Topoisomerase II Inhibitory Activities. Bioorg. Med. Chem. Lett. 14, 1193–1196. doi:10.1016/j.bmcl.2003.12.048
Mahringer, A., Karamustafa, S., Klotz, D., Kahl, S., Konkimalla, V. B., Wang, Y., et al. (2010). Inhibition of P-Glycoprotein at the Blood-Brain Barrier by Phytochemicals Derived from Traditional Chinese Medicine. Cancer Genomics Proteomics 7, 191–205.
Mahringer, A., Ott, M., Reimold, I., Reichel, V., and Fricker, G. (2011). The ABC of the Blood-Brain Barrier - Regulation of Drug Efflux Pumps. Cpd 17, 2762–2770. doi:10.2174/138161211797440221
Makhey, D., Gatto, B., Yu, C., Liu, A., Liu, L. F., and LaVoie, E. J. (1996). Coralyne and Related Compounds as Mammalian Topoisomerase I and Topoisomerase II Poisons. Bioorg. Med. Chem. 4, 781–791. doi:10.1016/0968-0896(96)00054-5
Newman, D. J., Cragg, G. M., and Snader, K. M. (2003). Natural Products as Sources of New Drugs over the Period 1981−2002. J. Nat. Prod. 66, 1022–1037. doi:10.1021/np030096l
Ouchi, H., Ishiguro, H., Ikeda, N., Hori, M., Kubota, Y., and Uemura, H. (2005). Genistein Induces Cell Growth Inhibition in Prostate Cancer through the Suppression of Telomerase Activity. Int. J. Urol. 12, 73–80. doi:10.1111/j.1442-2042.2004.00973.x
Patwardhan, B., and Mashelkar, R. A. (2009). Traditional Medicine-Inspired Approaches to Drug Discovery: Can Ayurveda Show the Way Forward? Drug Discov. Today 14, 804–811. doi:10.1016/j.drudis.2009.05.009
Patwardhan, B., Warude, D., Pushpangadan, P., and Bhatt, N. (2005). Ayurveda and Traditional Chinese Medicine: a Comparative Overview. Evidence-Based Complement. Altern. Med. 2, 465–473. doi:10.1093/ecam/neh140
Pauli-Magnus, C., Rekersbrink, S., Klotz, U., and Fromm, M. F. (2001). Interaction of Omeprazole, Lansoprazole and Pantoprazole with P-Glycoprotein. Naunyn-Schmied Arch. Pharmacol. 364, 551–557. doi:10.1007/s00210-001-0489-7
Pawagi, A. B., Wang, J., Silverman, M., Reithmeier, R. A. F., and Deber, C. M. (1994). Transmembrane Aromatic Amino Acid Distribution in P-Glycoprotein. J. Mol. Biol. 235, 554–564. doi:10.1006/jmbi.1994.1013
Persil, Ö., and Hud, N. V. (2007). Harnessing DNA Intercalation. Trends Biotechnology 25, 433–436. doi:10.1016/j.tibtech.2007.08.003
Pieters, L., and Vlietinck, A. J. (2005). Bioguided Isolation of Pharmacologically Active Plant Components, Still a Valuable Strategy for the Finding of New Lead Compounds? J. Ethnopharmacology 100, 57–60. doi:10.1016/j.jep.2005.05.029
Pratt, S. J., Hernández-Ochoa, E., and Martin, S. S. (2020). Calcium Signaling: Breast Cancer's Approach to Manipulation of Cellular Circuitry. Biophys. Rev. 12, 1343–1359. doi:10.1007/s12551-020-00771-9
Rao, U. S., and Nuti, S. L. (2003). Identification of Two Different States of P-Glycoprotein in its Catalytic Cycle. J. Biol. Chem. 278, 46576–46582. doi:10.1074/jbc.m308078200
Raviv, Y., Pollard, H. B., Bruggemann, E. P., Pastan, I., and Gottesman, M. M. (1990). Photosensitized Labeling of a Functional Multidrug Transporter in Living Drug-Resistant Tumor Cells. J. Biol. Chem. 265, 3975–3980. doi:10.1016/s0021-9258(19)39690-5
Regev, R., Yeheskely-Hayon, D., Katzir, H., and Eytan, G. D. (2005). Transport of Anthracyclines and Mitoxantrone across Membranes by a Flip-Flop Mechanism. Biochem. Pharmacol. 70, 161–169. doi:10.1016/j.bcp.2005.03.032
Robert, J., and Jarry, C. (2003). Multidrug Resistance Reversal Agents. J. Med. Chem. 46, 4805–4817. doi:10.1021/jm030183a
Roberts, M. F., and Wink, M. (1998). Alkaloids: Biochemistry, Ecology, and Medicinal Applications. New York: Plenum Press.
Rosenberg, M. F., Callaghan, R., Ford, R. C., and Higgins, C. F. (1997). Structure of the Multidrug Resistance P-Glycoprotein to 2.5 Nm Resolution Determined by Electron Microscopy and Image Analysis. J. Biol. Chem. 272, 10685–10694. doi:10.1074/jbc.272.16.10685
Rosenberg, M. F., Velarde, G., Ford, R. C., Martin, C., Berridge, G., Kerr, I. D., et al. (2001). Repacking of the Transmembrane Domains of P-Glycoprotein during the Transport ATPase Cycle. EMBO J. 20, 5615–5625. doi:10.1093/emboj/20.20.5615
Rosenberg, M. F., Kamis, A. B., Callaghan, R., Higgins, C. F., and Ford, R. C. (2003). Three-dimensional Structures of the Mammalian Multidrug Resistance P-Glycoprotein Demonstrate Major Conformational Changes in the Transmembrane Domains upon Nucleotide Binding. J. Biol. Chem. 278, 8294–8299. doi:10.1074/jbc.m211758200
Rosenkranz, V., and Wink, M. (2008). Alkaloids Induce Programmed Cell Death in Bloodstream Forms of Trypanosomes (Trypanosoma B. Brucei). Molecules 13, 2462–2473. doi:10.3390/molecules13102462
Sayers, T. J. (2011). Targeting the Extrinsic Apoptosis Signaling Pathway for Cancer Therapy. Cancer Immunol. Immunother. 60, 1173–1180. doi:10.1007/s00262-011-1008-4
Schmeller, T., Latz-Brüning, B., and Wink, M. (1997). Biochemical Activities of Berberine, Palmatine and Sanguinarine Mediating Chemical Defence against Microorganisms and Herbivores. Phytochemistry 44, 257–266. doi:10.1016/s0031-9422(96)00545-6
Seca, A. M. L., and Pinto, D. (2018). Plant Secondary Metabolites as Anticancer Agents: Successes in Clinical Trials and Therapeutic Application. Int. J. Mol. Sci. 19, 263. doi:10.3390/ijms19010263
Seelig, A. (1998). A General Pattern for Substrate Recognition by P-Glycoprotein. Eur. J. Biochem. 251, 252–261. doi:10.1046/j.1432-1327.1998.2510252.x
Sharom, F. J. (2011). The P-Glycoprotein Multidrug Transporter. Essays Biochem. 50, 161–178. doi:10.1042/bse0500161
Sharom, F. J. (2014). Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 4, 41. doi:10.3389/fonc.2014.00041
Shay, J. W., and Wright, W. E. (2005). Senescence and Immortalization: Role of Telomeres and Telomerase. Carcinogenesis 26, 867–874. doi:10.1093/carcin/bgh296
Sikic, B. I. (1999). New Approaches in Cancer Treatment. Ann. Oncol. 10, 149–153. doi:10.1023/a:1008355801988
Stermitz, F. R., Lorenz, P., Tawara, J. N., Zenewicz, L. A., and Lewis, K. (2000). Synergy in a Medicinal Plant: Antimicrobial Action of Berberine Potentiated by 5'-methoxyhydnocarpin, a Multidrug Pump Inhibitor. Proc. Natl. Acad. Sci. 97, 1433–1437. doi:10.1073/pnas.030540597
Tait, S. W. G., and Green, D. R. (2010). Mitochondria and Cell Death: Outer Membrane Permeabilization and beyond. Nat. Rev. Mol. Cell Biol 11, 621–632. doi:10.1038/nrm2952
Takara, K., Sakaeda, T., and Okumura, K. (2006). An Update on Overcoming MDR1-Mediated Multidrug Resistance in Cancer Chemotherapy. Cpd 12, 273–286. doi:10.2174/138161206775201965
Ueda, K., Taguchi, Y., and Morishima, M. (1997). How Does P-Glycoprotein Recognize its Substrates? Semin. Cancer Biol. 8, 151–159. doi:10.1006/scbi.1997.0066
Ulrich-Merzenich, G., Panek, D., Zeitler, H., Wagner, H., and Vetter, H. (2009). New Perspectives for Synergy Research with the "Omic"-Technologies. Phytomedicine 16, 495–508. doi:10.1016/j.phymed.2009.04.001
Ulrich-Merzenich, G., Panek, D., Zeitler, H., Vetter, H., and Wagner, H. (2010). Drug Development from Natural Products: Exploiting Synergistic Effects. Indian J. Exp. Biol. 48, 208–219.
Vernon, J. A., Golec, J. H., and Dimasi, J. A. (2010). Drug Development Costs when Financial Risk Is Measured Using the Fama-French Three-Factor Model. Health Econ. 19, 1002–1005. doi:10.1002/hec.1538
Wagner, H. (2011). Synergy Research: Approaching a New Generation of Phytopharmaceuticals. Fitoterapia 82, 34–37. doi:10.1016/j.fitote.2010.11.016
Walczak, H., and Krammer, P. H. (2000). The CD95 (APO-1/Fas) and the TRAIL (APO-2L) Apoptosis Systems. Exp. Cell Res. 256, 58–66. doi:10.1006/excr.2000.4840
Wang, Y., and Fang, M. Y. (2006). Effect of Ginseng Saponin, Arsenic Trioxide, Beta-Elemene Combined with CTX on Telomere-Telomerase System in K562 Cell Line. Zhongguo Shi Yan Xue Ye Xue Za Zhi 14, 1089–1095.
Wang, F., Lu, C.-H., and Willner, I. (2014). From Cascaded Catalytic Nucleic Acids to Enzyme-DNA Nanostructures: Controlling Reactivity, Sensing, Logic Operations, and Assembly of Complex Structures. Chem. Rev. 114, 2881–2941. doi:10.1021/cr400354z
Watkins, R. E., Wisely, G. B., Moore, L. B., Collins, J. L., Lambert, M. H., Williams, S. P., et al. (2001). The Human Nuclear Xenobiotic Receptor PXR: Structural Determinants of Directed Promiscuity. Science 292, 2329–2333. doi:10.1126/science.1060762
Wink, M., and Schimmer, O. (2010). “Molecular Modes of Action of Defensive Secondary Metabolites,” in Annual Plant Reviews Volume 39: Functions and Biotechnology of Plant Secondary Metabolites (Wiley-Blackwell), 21–161. doi:10.1002/9781444318876.ch2
Wink, M., and Van Wyk, B.-E. (2008). Mind-altering and Poisonous Plants of the World. Portland, OR: Timber Press.
Wink, M., Ashour, M. L., and El-Readi, M. Z. (2012). Secondary Metabolites from Plants Inhibiting ABC Transporters and Reversing Resistance of Cancer Cells and Microbes to Cytotoxic and Antimicrobial Agents. Front. Microbiol. 3, 130. doi:10.3389/fmicb.2012.00130
Wink, M. (1999). Biochemistry of Plant Secondary Metabolism. Sheffield, England, Boca Raton, FL: Sheffield Academic Press; CRC Press.
Wink, M. (2000). “Interference of Alkaloids with Neuroreceptors and Ion Channels,” in Studies in Natural Products Chemistry. Editor R. Atta ur (Amsterdam: Elsevier), 3–122. doi:10.1016/s1572-5995(00)80004-6
Wink, M. (2003). Evolution of Secondary Metabolites from an Ecological and Molecular Phylogenetic Perspective. Phytochemistry 64, 3–19. doi:10.1016/s0031-9422(03)00300-5
Wink, M. (2007). “Chapter 1 Molecular Modes of Action of Cytotoxic Alkaloids: From DNA Intercalation, Spindle Poisoning, Topoisomerase Inhibition to Apoptosis and Multiple Drug Resistance,” in The Alkaloids: Chemistry and Biology. Editor G. A. Cordell, 1–47. doi:10.1016/s1099-4831(07)64001-2
Wink, M. (2008). Evolutionary Advantage and Molecular Modes of Action of Multi-Component Mixtures Used in Phytomedicine. Cdm 9, 996–1009. doi:10.2174/138920008786927794
Wink, M. (2010). Introduction: Biochemistry, Physiology and Ecological Functions of Secondary Metabolites. Hoboken, NJ: Wiley-Blackwell.
Wink, M. (2015). Modes of Action of Herbal Medicines and Plant Secondary Metabolites. Medicines 2, 251–286. doi:10.3390/medicines2030251
Xing, L., Hu, Y., and Lai, Y. (2009). Advancement of Structure-Activity Relationship of Multidrug Resistance-Associated Protein 2 Interactions. AAPS J. 11, 406–413. doi:10.1208/s12248-009-9117-0
Yokoyama, M., Noguchi, M., Nakao, Y., Ysunaga, M., Yamasaki, F., and Iwasaka, T. (2008). Antiproliferative Effects of the Major Tea Polyphenol, (−)-epigallocatechin Gallate and Retinoic Acid in Cervical Adenocarcinoma. Gynecol. Oncol. 108, 326–331. doi:10.1016/j.ygyno.2007.10.013
Zhang, J., Tu, Y., and Smith-Schneider, S. (2005). Activation of P53, Inhibition of Telomerase Activity and Induction of Estrogen Receptor Beta Are Associated with the Anti-growth Effects of Combination of Ovarian Hormones and Retinoids in Immortalized Human Mammary Epithelial Cells. Cancer Cell Int 5, 6. doi:10.1186/1475-2867-5-6
Keywords: multidrug resistance, apoptosis, secondary metaabolites, cancer, molecular mechanism
Citation: El-Readi MZ, Al-Abd AM, Althubiti MA, Almaimani RA, Al-Amoodi HS, Ashour ML, Wink M and Eid SY (2021) Multiple Molecular Mechanisms to Overcome Multidrug Resistance in Cancer by Natural Secondary Metabolites. Front. Pharmacol. 12:658513. doi: 10.3389/fphar.2021.658513
Received: 25 January 2021; Accepted: 12 April 2021;
Published: 21 May 2021.
Edited by:
Nor Eddine Sounni, University of Liège, BelgiumReviewed by:
Nesrine Affara, Carnegie Mellon University in Qatar, QatarVishwa Khare, Eurofins Viracor, United States
Copyright © 2021 El-Readi, Al-Abd, Althubiti, Almaimani, Al-Amoodi, Ashour, Wink and Eid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed M. Al-Abd, YWhtZWRtYWxhYmRAcGhhcm1hLmFzdS5lZHUuZWc=