 Amreen Mughal
Amreen Mughal Chengwen Sun
Chengwen Sun Stephen T. O’Rourke
Stephen T. O’Rourke- Department of Pharmaceutical Sciences, North Dakota State University, Fargo, ND, United States
Apelin-APJ receptor signaling regulates vascular tone in cerebral and peripheral arteries. We recently reported that apelin inhibits BKCa channel function in cerebral arteries, resulting in impaired endothelium-dependent relaxations. In contrast, apelin causes endothelium-dependent relaxation of coronary arteries. However, the effects of apelin on BKCa channel function in coronary arterial myocytes have not yet been explored. We hypothesized that apelin-APJ receptor signaling does not have an inhibitory effect on coronary arterial BKCa channels and hence does not alter nitric oxide (NO)-dependent relaxation of coronary arteries. Patch clamp recording was used to measure whole cell K+ currents in freshly isolated coronary smooth muscle cells. Apelin had no effect on the increases in current density in response to membrane depolarization or to NS1619 (a BKCa channel opener). Moreover, apelin did not inhibit NO/cGMP-dependent relaxations that required activation of BKCa channels in isolated coronary arteries. Apelin-APJ receptor signaling caused a marked increase in intracellular Ca2+ levels in coronary arterial smooth muscle cells, but failed to activate PI3-kinase to increase phosphorylation of Akt protein. Collectively, these data provide mechanistic evidence that apelin has no inhibitory effects on BKCa channel function in coronary arteries. The lack of inhibitory effect on BKCa channels makes it unlikely that activation of APJ receptors in coronary arteries would adversely affect coronary flow by creating a vasoconstrictive environment. It can be expected that apelin or other APJ receptor agonists in development will not interfere with the vasodilator effects of endogenous BKCa channel openers.
Introduction
Apelin is a vasoactive peptide found in many organs and tissues including, for example, adipose tissue (Boucher et al., 2005), atria (Földes et al., 2003), blood vessels (Kleinz and Davenport, 2004), lungs, kidneys, brain (Folino et al., 2015; Yan et al., 2020). Apelin acts via binding to G-protein-coupled-receptors known as APJ receptors, which are widely expressed throughout the cardiovascular system (Katugampola et al., 2001; Kleinz et al., 2005; Pitkin et al., 2010; Luo et al., 2018). An increasing body of evidence indicates that the apelin-APJ receptor signaling system regulates vascular function in mammalian arteries and veins (Gurzu et al., 2006; Jia et al., 2007; Salcedo et al., 2007; Wang et al., 2016; Nagano et al., 2019), including those from humans (Katugampola et al., 2001; Kleinz and Davenport, 2004; Maguire et al., 2009; Schinzari et al., 2017). Based on their generally favorable hemodynamic properties, apelin and apelin-based mimetics are currently in development for the treatment of several cardiovascular disorders, including heart failure, pulmonary hypertension, and ischemia/reperfusion injury (Wang et al., 2013; Brame et al., 2015; Gerbier et al., 2017; Mughal and O'Rourke, 2018).
BKCa channels play a central role in maintaining membrane potential and excitability in vascular smooth muscle cells (Tykocki et al., 2017; Dopico et al., 2018). Opening of BKCa channels leads to efflux of potassium ions, cell membrane hyperpolarization, and smooth muscle relaxation (Brayden and Nelson, 1992; Nelson and Quayle, 1995). Several endogenous vasodilator molecules, including NO and certain hyperpolarizing factors released from endothelial cells, exert their smooth muscle relaxant effects by targeting BKCa channels (Bolotina et al., 1994; Campbell et al., 1996; Mistry and Garland, 1998; Earley et al., 2005; Zhang et al., 2012). Similarly, pharmacologic agents that increase BKCa channel opening cause vascular smooth muscle relaxation and vasodilation (Ghatta et al., 2006; Jackson, 2017). By contrast, substances that interfere with BKCa channel opening create an environment that favors vasoconstriction (Tang et al., 2017; Dogan et al., 2019). Thus, modulation of BKCa channel activity is a critical mechanism for regulating blood vessel diameter and flow.
It is widely recognized that the effects of vasoactive agents may vary depending on the anatomic location of the blood vessel under investigation and/or the experimental conditions. Previous work from our laboratory demonstrates that APJ receptors are expressed in coronary and cerebral arteries (Mughal et al., 2018a; Mughal et al., 2018b). In coronary arteries, apelin acts on endothelial APJ receptors to cause nitric oxide (NO) release and endothelium-dependent relaxation (Mughal et al., 2018a). By contrast, apelin causes neither vasoconstriction nor vasorelaxation in cerebral arteries, despite the presence of APJ receptors on both endothelial cells and myocytes (Mughal et al., 2018b). However, activation of cerebral arterial smooth muscle APJ receptors by apelin inhibits BKCa channel function, thereby impairing arterial relaxation induced by NO and an endothelium-dependent hyperpolarization (EDH) pathway (Mughal et al., 2018b; Mughal et al., 2020).
Direct effects of apelin on BKCa channels in coronary arterial smooth muscle cells, and on the functional response to endogenous mediators that act via BKCa channel activation, are presently unknown. Thus, the present study was designed to increase our understanding of the apelin-APJ receptor signaling pathway in coronary arteries. Here, we tested the hypothesis that apelin-APJ receptor signaling does not have an inhibitory effect on coronary arterial BKCa channels and hence does not alter nitric oxide (NO)-dependent relaxation of coronary arteries.
Materials and Methods
Animals and Tissue Preparation
Twelve-week-old male Sprague-Dawley rats were used for all experiments (Envigo RMS, Indianapolis, IN, United States). Rats were housed at 22 ± 2°C (32% humidity) on a 12–12 h light-dark cycle and provided with food and water ad libitum. All animal protocols were approved by the North Dakota State University Institutional Animal Care and Use Committee. Rat hearts were isolated from animals anesthetized with isoflurane and placed into ice-cold physiological salt solution (PSS). Both left and right epicardial coronary arteries and associated branches were dissected and cleaned of surrounding tissues.
Smooth Muscle Cell Isolation
Smooth muscle cells were isolated by enzymatic digestion as described previously (Mughal et al., 2018b). Briefly, cell isolation was performed in isolation solution of the following composition (in mM): 60 NaCl, 80 Na-glutamate, 5 KCl, 2 MgCl2, 10 glucose, and 10 HEPES (pH 7.2). Arterial segments were first incubated in 1.2 mg/ml papain (Worthington, Lakewood, NJ, United States) and 2.0 mg/ml dithioerythritol (Sigma Aldrich, St. Louis, MO, United States) for 17 min at 37°C, and then in 0.8 mg/ml type II collagenase (Worthington) for 12 min at 37°C. After digestion, the segments were washed with ice-cold isolation solution and triturated with a glass pipette to liberate single smooth muscle cells. Isolated cells were kept on ice and were used within 6–8 h following isolation for electrophysiology.
Electrophysiological Recording
Freshly isolated arterial myocytes were placed into a recording chamber (Warner Instruments, Hamden, CT, United States) and whole cell current recordings were performed using our previously described protocol (Mughal et al., 2018a). Briefly, cells were perfused with a bath solution containing (in mM) 145 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, 10 glucose; pH 7.4 (NaOH). The internal pipette solution contained (in mM) 145 KCl, 5 NaCl, 0.37 CaCl2 (estimated free Ca2+ = 172 nM), 2 MgCl2, 10 HEPES, 1 EGTA, 7.5 glucose; pH 7.2 (KOH). Whole cell K+ currents were recorded at the room temperature (25°C) by stepping in 10 mV increments from a holding potential of −60 to +80 mV using an AxoPatch 200B amplifier equipped with an Axon CV 203BU headstage (Molecular Devices). The patch electrodes (3–4 MΩ) were pulled from 1.5 mm borosilicate glass capillaries. Voltage-activated currents were filtered at 2 kHz and digitized at 10 kHz, and capacitative and leakage currents were subtracted digitally. Series resistance and total cell capacitance were calculated from uncompensated capacitive transients in response to 10 ms hyperpolarizing step pulses (5 mV) or obtained by adjusting series resistance and whole-cell capacitance using the Axopatch 200B amplifier control system. All drugs were diluted in fresh bath solution and perfused into the experimental chamber. Data were collected and analyzed with pCLAMP 10.0 software (Molecular Devices, Sunnyvale, CA, United States). All currents were expressed as current density (current at the end of each voltage step divided by cell membrane capacitance).
Vascular Function Studies
Coronary arterial segments (120–150 μm; 1.2 mm in length) were suspended in wire myograph chambers (DMT, Aarhus, Denmark) for isometric tension recording, as described previously (Mughal et al., 2018a). Briefly, the tissues were bathed in physiologic salt solution (PSS) of the following composition [(in mM): 119 NaCl, 15 NaHCO3, 4.6 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 1.5 CaCl2 and 5.5 glucose] at 37°C. Vessel reactivity was confirmed using KCl (60 mM) and presence of endothelium was tested using the endothelium-dependent vasodilator, acetylcholine (ACh; 10−6 M). Vessels that showed >90% relaxation to ACh were considered endothelium-intact vessels. Responses to the vasodilators used in this study were obtained in arterial rings contracted with 5-hydroxytryptamine (5-HT; 10−7 M) to a sub-maximal level [∼30% of maximum contraction induced by KCl (60 mM)]. Paired experiments with inhibitors were performed in control rings isolated from the same animals.
Intracellular Ca2+ Imaging
Smooth muscle cells were plated in eight-well borosilicate cover glass chambers (ThermoFisher Scientific, Waltham, MA, United States) and allowed to adhere to the glass surface (at 37°C) in endothelial growth media with 5% FBS (Promocells, Heidelberg, Germany) for 60 min. After incubation, cells were thoroughly washed with Hanks’ balanced salt solution and incubated for 60 min with fluo-4 AM (5 μM). At the end of incubation, cells were washed again and perfused with Hanks’ balanced salt solution. Real-time Ca2+ imaging was performed using a Zeiss confocal laser-scanning microscope (Carl Zeiss, Germany) equipped with a 40× numerical aperture oil immersion lens (with excitation at 488 nm and emission at 515 nm). All experiments were carried out at room temperature in Hanks’ balanced salt solution within 6–8 h following isolation. The effect of apelin (10−7 M) on intracellular Ca2+ levels, in the absence and presence of the APJ receptor antagonist (Lee et al., 2005), F13A (10−7 M × 5 min), was determined in paired experiments. Smooth muscle cells were freshly isolated from three animals (each animal on different day) and three to four cells were analyzed for each treatment from each animal.
Western Immunoblotting
Protein expression was performed according to our previously published method for coronary arteries with minor modification (Mughal et al., 2018a). Briefly, isolated coronary arteries were treated with apelin (10−7 M) for different durations (5, 10, 15 and 30 min) at 37°C. Then arterial segments were snap frozen and protein was isolated. Protein estimation was performed using a Pierce BCA protein estimation kit (ThermoFisher Scientific, Waltham, MA, United States). Aliquots of supernatant containing equal amounts of protein (40 μg) were separated on 7.5–10% polyacrylamide gel by SDS-PAGE, and electroblotted onto a PVDF membrane (Bio-Rad Laboratories, Hercules, CA, United States). Blots were blocked with 5% nonfat dry milk in Tris-buffered saline (TBS, pH 7.4) and incubated with primary antibodies, followed by secondary antibody linked to horseradish peroxidase (1:100; Cell Signaling). The blots were first probed with Akt antibody (1:500 dilution; Cell Signaling, Denver, MA, United States), followed by chemical stripping and reprobing with phospho-Akt (p-Akt) antibody (1:500 dilution; Cell Signaling, Denver, MA, United States). To ensure equal loading, the blots were probed for β-actin using an anti-actin antibody (1:500; Santa Cruz Biotechnology Inc.). Immuno-detection was performed using an enhanced chemiluminescence light detection kit (ThermoFisher Scientific).
Drugs
The following drugs were used: acetylcholine (ACh), diethylamine NONOate (DEA NONOate), 5-HT, and NS1619 (Sigma Chemical, St. Louis, MO, United States); iberiotoxin (IBTx) and eight-Bromo-cGMP (8-Br-cGMP) (Tocris, Ellisville, MO); apelin-13 and F13A (H-Gln-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Ala-OH trifluoroacetate salt) (Bachem, Torrance, CA, United States). Drug solutions were prepared fresh daily, kept on ice, and protected from light until used. All drugs were dissolved initially in double-distilled water. Drugs were added to the myograph chambers in volumes not greater than 0.02 ml. Drug concentrations are reported as final molar concentrations in the myograph chamber.
Data Analysis
Relaxation responses are expressed as a percent of the initial tension induced by 5-HT (10−7 M). EC50 values (drug concentration that produced 50% of its own maximal response) were determined, converted to their negative logarithm, and expressed as -log molar EC50 (pD2). For Ca2+ imaging studies, the amplitude of Fluo-4 AM fluorescence was calculated by peak fluorescence intensity (F) divided by baseline intensity (F0). Results are expressed as means ± SEM, and n refers to the number of animals from which blood vessels and/or smooth muscle cells were taken, unless otherwise stated. All data sets were tested for normal distribution using the Shapiro–Wilk test. Values were compared by Student’s t-test or one-way ANOVA using Tukey’s test as post-hoc analysis for paired or unpaired parametric distributed observations, as appropriate, to determine significance between groups. Mann-Whitney test was applied for non-parametric distributed observations. Values were considered significantly different when p < 0.05.
Results
Electrophysiology Studies (Whole Cell K+ Currents)
Whole cell K+ currents were recorded in freshly isolated smooth muscle cells from coronary arteries. Superfusion of apelin (10−7 M, 5 min) had no inhibitory effect on whole cell K+ currents, but addition of the selective BKCa channel blocker, iberiotoxin (10−7 M, 5 min), significantly reduced the current density (Figure 1). Incubation of coronary myocytes with NS1619 (10−5 M, 3 min), a selective BKCa channel opener, caused a significant increase in the current density (Figures 2A,D). The NS1619-induced increase in current density was markedly attenuated by iberiotoxin (Figures 2B,E), whereas apelin had no inhibitory effect (Figures 2C,F).
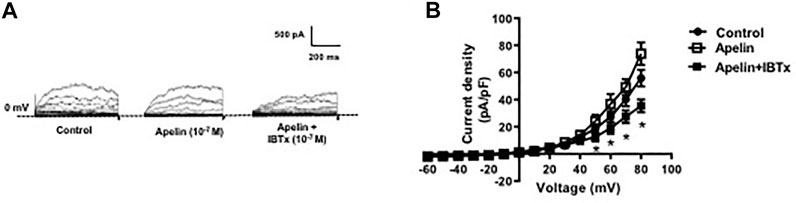
FIGURE 1. Effect of apelin on whole cell K+ currents in coronary arterial myocytes. Whole cell K+ currents were recorded in freshly isolated myocytes in response to successive voltage pulses of 800 ms duration, increasing in 10 mV increments from −60 to +80 mV with or without apelin (10−7 M) or apelin plus IBTx (10-7 M). (A) Representative traces showing the current recordings from smooth muscle cell in the absence and presence of apelin alone or apelin plus IBTx. (B) Summary I–V curves of whole cell K+ currents at baseline (control) and after treatment with apelin (10−7 M) alone or in the presence of IBTx (10−7 M). Dotted line represents zero current level (at 0 mV). Values are presented as the mean ± S. E. M. (n = 6). *p < 0.05 vs. control current density.
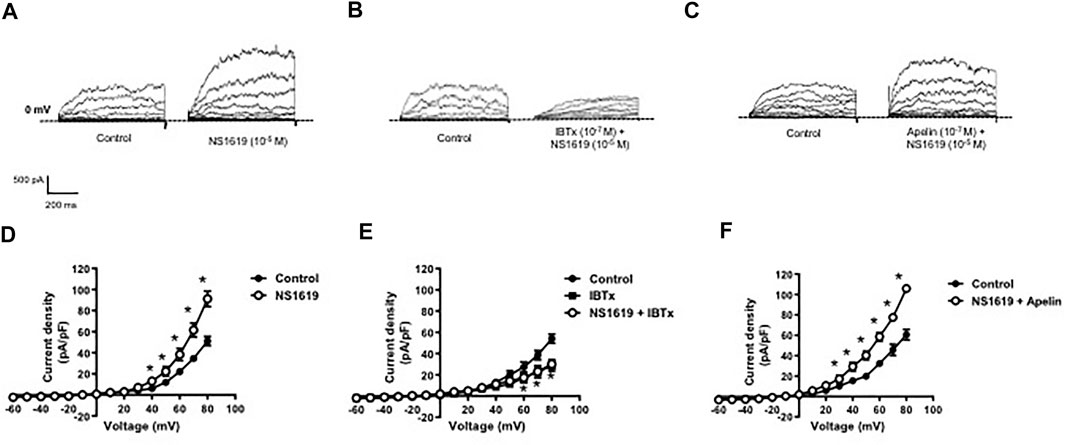
FIGURE 2. Effect of apelin on NS1619-induced BKCa currents in coronary arterial myocytes. Whole cell BKCa currents induced in response to NS1619 (10-5 M) were recorded in freshly isolated coronary arterial myocytes. Representative traces showing the current recordings from smooth muscle cells after treatment with (A) NS1619 (10−5 M, 2 min) alone or (B) in the presence of IBTx (10−7 M, 5 min) or (C) apelin (10−7 M, 5 min). (D–F) Summary I–V curves of BKCa currents at baseline and after treatment with (D) NS1619 (10−5 M) alone or in the presence of (E) IBTx (10−7 M) or (F) apelin (10−7 M). Dotted line represents zero current level (at 0 mV). Values are presented as the mean ± S.E.M. (n = 6–7). *p < 0.05 vs. control current density.
Vascular Relaxation Studies (NO/cGMP-Induced Relaxation)
In coronary arterial segments contracted with five-HT (10−7 M), the NO donor, DEA NONOate (DEA, 10−9 − 3 × 10−5 M), caused concentration-dependent relaxation (Figure 3). Iberiotoxin (10−7 M) caused a significant rightward shift in the DEA concentration-response curve (Figure 3A); however, DEA-induced relaxations were unaffected in the presence of apelin (10−7 M) [pD2 = 6.38 ± 0.20 vs. 6.15 ± 0.28 in the absence and presence of apelin, respectively; p > 0.05 (n = 5)] (Figure 3B). Similarly, relaxations induced by acetylcholine (10−9 − 3 × 10−6 M), an endothelium-dependent vasodilator that acts via NO release from endothelial cells, were significantly inhibited by iberiotoxin (Figure 3C), but not by apelin [pD2 = 7.16 ± 0.17 vs. 7.38 ± 0.30 in the absence and presence of apelin, respectively; p > 0.05 (n = 6)] (Figure 3D). The stable cGMP analog, 8-Bromo-cGMP [8-Br-cGMP (10−9 – 10−4 M)], also caused concentration-dependent relaxation of isolated coronary arteries. The response to 8-Br-cGMP was significantly attenuated in the presence of iberiotoxin (Figure 3E), but not apelin [pD2 = 5.25 ± 0.16 vs. 5.41 ± 0.44 in the absence and presence of apelin, respectively; p > 0.05 (n = 4)] (Figure 3F).
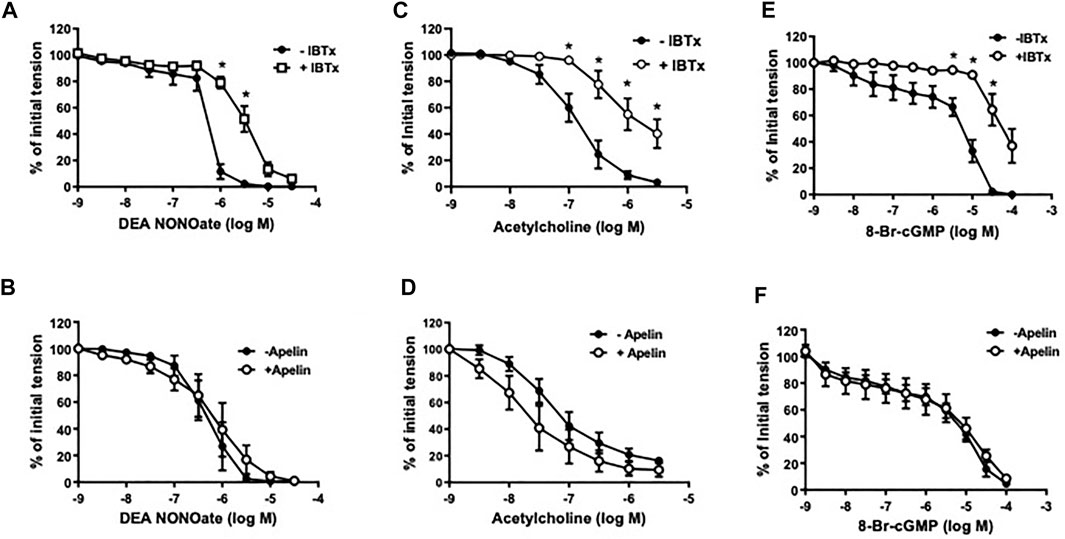
FIGURE 3. Effect of apelin and iberiotoxin on NO-cGMP dependent vasorelaxation of coronary arteries. Log concentration-response curves in producing relaxation of coronary arteries for DEA NONOate (A,B), acetylcholine (C,D) or 8-Bromo-cGMP (E,F) in the absence and presence of IBTx (10−7 M) (A, C, E) or apelin (10−7 M) (B, D, F). Values are presented as the mean ± S.E.M. (n = 4–6). *p < 0.05 vs. control (in the absence of inhibitor).
Intracellular Signaling Studies (Cai2+ and PI3 Kinase Activity)
The functional status of coronary smooth muscle APJ receptors was assessed using intracellular calcium imaging. Apelin (10-7 M) caused a robust increase in intracellular Ca2+ levels in freshly isolated smooth muscle cells (Figures 4A–C). The apelin-induced rise in intracellular Ca2+ was comparable to that observed by depolarization with 60 mM K+, which served as a positive control (Figure 4C). The APJ receptor antagonist, F13A (10-7 M, 5 min), abolished the apelin-induced response, thus confirming functional activation of smooth muscle APJ receptors in response to apelin in coronary arteries (Figure 4C).
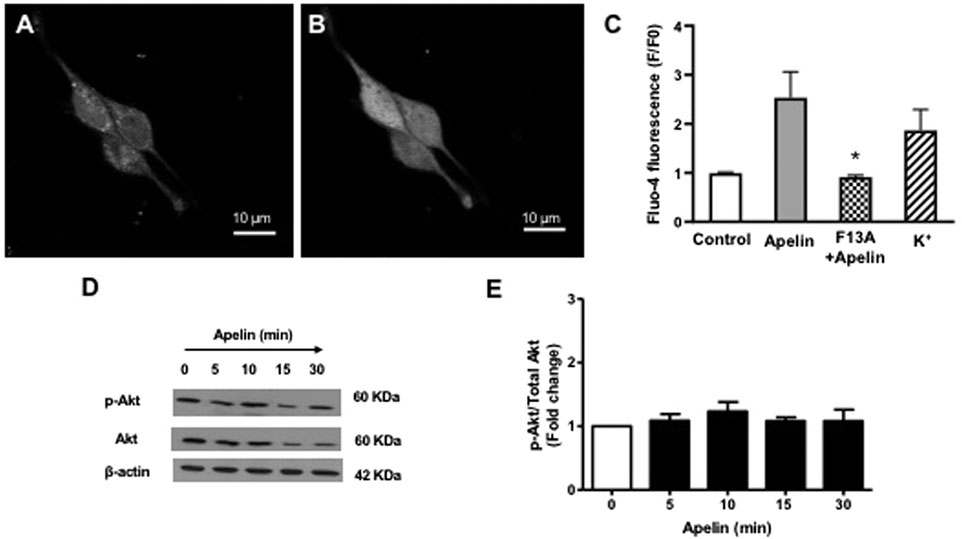
FIGURE 4. Effect of apelin-APJ signaling on coronary intracellular Ca2+ levels and PI3-kinase activity. Representative images of freshly isolated coronary arterial smooth muscle cells loaded with Fluo-four AM (5 µM) (A) before and (B) after exposure to apelin (10−7 M). (C) Bar graph summarizing changes in fluorescence (F/F0) in response in apelin alone (10−7 M) or in the presence of F13A (10−7 M); 60 mM K+ was used as a positive control for the experiments (n = 3). (D) Representative blots showing changes in phosphorylation of Akt (p-Akt) or Akt (total) protein expression in a time-dependent manner following treatment with apelin (10−7 M). (E) Summary data showing effect of apelin on PI3-kinase activity expressed as a ratio of p-Akt/Akt protein levels (n = 3). Values are presented as the mean ± S.E.M. *p < 0.05 vs. apelin alone; **p < 0.05 vs. control.
Since apelin is known to increase PI3 kinase activity in cerebral arteries (Modgil et al., 2013), experiments were performed to evaluate the effect of apelin on PI3 kinase activity in coronary arteries. PI3-kinase activity was detected by measuring the ratio of phosphorylated Akt to total Akt, using immunoblot analysis. Apelin (10-7 M) treatment caused no significant change in PI3-kinase activity in coronary arteries, as indicated by unchanged P-Akt/total Akt ratio (Figures 4D,E).
Discussion
The present study was designed to evaluate the effects of apelin on BKCa channels in coronary arterial smooth muscle cells. The key findings are: 1) apelin has no inhibitory effect on BKCa currents in coronary arterial smooth muscle cells; and 2) apelin does not inhibit NO/cGMP-induced relaxations, which are dependent on BKCa channels in coronary arteries (Darkow et al., 1997; Tunstall et al., 2011). These results are in striking contrast to our previous findings in cerebral arteries (Mughal et al., 2018b; Mughal et al., 2020), and clearly demonstrate that distinct molecular mechanisms underly the vasomotor effects of apelin in coronary and cerebral arteries.
BKCa channels are expressed in vascular smooth muscle cells from rat cerebral and coronary arteries (Modgil et al., 2013; Mughal et al., 2018a; Mughal et al., 2018b). In cerebral arteries, apelin inhibits smooth muscle BKCa channels and thereby impairs endothelium-dependent relaxations mediated by NO and an EDH pathway (Mughal et al., 2018b; Mughal et al., 2020). The results of the present study demonstrate that in coronary arterial myocytes apelin failed to inhibit increases in BKCa current density induced by membrane depolarization or by pharmacological activation with NS1619. Consistent with these electrophysiological findings, apelin was without effect on coronary arterial relaxation caused by endothelium-derived NO (i.e. acetylcholine), exogenously administered NO (i.e., DEA NONOate), and the stable, cell permeable cGMP analogue, 8-Br-cGMP. The absence of an inhibitory effect of apelin cannot be attributed to the lack of involvement of BKCa channels in NO/cGMP pathway-induced relaxation of rat coronary arteries since iberiotoxin, a potent and selective BKCa channel blocker (Kaczorowski and Garcia., 2016; Su et al., 2016), markedly impaired the responses to ACh, DEA, and 8-Br-cGMP. That apelin had no effect on these responses also cannot be explained by an absence of APJ receptors, which are indeed expressed in rat coronary arterial smooth muscle cells (Mughal et al., 2018a). Taken together, these data indicate that APJ receptors are not effectively coupled to BKCa channels in coronary arterial smooth muscle cells.
One possible explanation for the differential effect of apelin in cerebral vs coronary arterial smooth muscle cells could be related to the intracellular signaling pathways stimulated in response to apelin-APJ receptor activation. Binding of apelin to APJ receptors in cerebral arterial smooth muscle cells stimulates the PI3 kinase/Akt signaling pathway, which is required for apelin-induced inhibition of BKCa channel function (Modgil et al., 2013). That apelin did not activate the PI3K/Akt-signaling pathway in coronary arterial smooth muscle cells provides a plausible mechanistic explanation as to why APJ receptors may not be efficiently linked to BKCa channels in coronary myocytes; however, the possibility that another intracellular signaling pathway plays a role in the lack of inhibitory effect of apelin on BKCa channel function cannot be ruled out. The absence of effect of apelin on BKCa channels and PI3 kinase/Akt signaling is not due to an inability to activate APJ receptor signaling in coronary arterial smooth muscle cells, since apelin caused a rapid and robust increase in intracellular Ca2+ levels. The apelin-induced rise in intracellular Ca2+ levels was attenuated by the APJ receptor antagonist, F13A, thus confirming that smooth muscle APJ receptors are indeed responsive to apelin in coronary arteries. It is also worth noting that although apelin caused an increase in intracellular Ca2+ levels in coronary smooth muscle cells, it does not cause contraction of isolated coronary arteries (Mughal et al., 2018a). This opens the possibility that apelin/APJ receptor signaling in coronary arterial smooth muscle cells may be involved in other cellular functions beyond the regulation of vascular tone.
The coronary circulation is controlled by multiple regulatory systems, including, for example, metabolic autoregulation, the sympathetic nervous system, circulating vasoactive molecules, and mediators produced by endothelial cells located within the blood vessel wall (Goodwill et al., 2017). The extent to which such systems play a role in regulating vasomotor tone varies between epicardial arteries (conductance vessels) and small arteries and arterioles (resistance vessels and microcirculation). For example, NO is a principal regulator of epicardial coronary vasomotor tone, whereas metabolic autoregulation and EDH pathways play a more prominent role in smaller resistance vessels and the microcirculation (Deussen et al., 2012; Ellinsworth et al., 2016; Goodwill et al., 2017). The present findings show a lack of inhibitory effect of apelin/APJ receptor signaling on the NO/cGMP/BKCa channel pathway in epicardial arteries, but whether or not this is true for other regions of the coronary circulation remains to be determined.
As with all biological studies, there are some limitations that should be considered. Although pre-constricted coronary arterial rings were used to investigate the effects of apelin on the vasodilators used in this study, it is possible that the vascular effects of apelin may differ in unstimulated arterial rings with intrinsic myogenic tone. In addition, our present findings are limited to arteries obtained from healthy adult male rats. It is well known that responses to vasoactive mediators may vary depending on the age and sex of the animals, as well as in pathological conditions. Thus, future studies with female rats, various age groups, and appropriate models of cardiovascular disease will be needed to further enhance our understanding of the vasomotor effects of apelin.
The results of the present study build on our knowledge of the actions of apelin in coronary arteries. The ability of apelin to cause endothelium-dependent relaxation (Mughal et al., 2018a), coupled with a lack of inhibitory effect on BKCa channels makes it unlikely that activation of APJ receptors in coronary arteries would adversely affect epicardial coronary flow by creating a vasoconstrictive environment. It could be expected that apelin or other APJ receptor agonists in development will not interfere with the vasodilator effects of endogenous BKCa channel openers, nor would these agents be likely to elicit coronary vasospasm, which has been hypothesized to be related to reduced K conductance in coronary artery smooth muscle cells (Dick and Tune, 2010).
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the North Dakota State University Animal Care and Use Committee.
Author Contributions
Participated in research design: AM, CS, SO’R. Conducted experiments: AM. Performed data analysis: AM, CS, SO’R. Wrote or contributed to the writing of the manuscript: AM, SO’R.
Funding
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute (Grant HL124338).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bolotina, V. M., Najibi, S., Palacino, J. J., Pagano, P. J., and Cohen, R. A. (1994). Nitric Oxide Directly Activates Calcium-dependent Potassium Channels in Vascular Smooth Muscle. Nature 368, 850–853. doi:10.1038/368850a0
Boucher, J., Masri, B., Daviaud, D., Gesta, S., Guigné, C., Mazzucotelli, A., et al. (2005). Apelin, a Newly Identified Adipokine Up-Regulated by Insulin and Obesity. Endocrinology 146, 1764–1771. doi:10.1210/en.2004-1427
Brame, A. L., Maguire, J. J., Yang, P., Dyson, A., Torella, R., Cheriyan, J., et al. (2015). Design, Characterization, and First-In-Human Study of the Vascular Actions of a Novel Biased Apelin Receptor Agonist. Hypertension 65, 834–840. doi:10.1161/hypertensionaha.114.05099
Brayden, J., and Nelson, M. (1992). Regulation of Arterial Tone by Activation of Calcium-dependent Potassium Channels. Science 256, 532–535. doi:10.1126/science.1373909
Campbell, W. B., Gebremedhin, D., Pratt, P. F., and Harder, D. R. (1996). Identification of Epoxyeicosatrienoic Acids as Endothelium-Derived Hyperpolarizing Factors. Circ. Res. 78, 415–423. doi:10.1161/01.res.78.3.415
Darkow, D. J., Lu, L., and White, R. E. (1997). Estrogen Relaxation of Coronary Artery Smooth Muscle Is Mediated by Nitric Oxide and cGMP. Am. J. Physiology-Heart Circulatory Physiol. 272, H2765–H2773. doi:10.1152/ajpheart.1997.272.6.h2765
Deussen, A., Ohanyan, V., Jannasch, A., Yin, L., and Chilian, W. (2012). Mechanisms of Metabolic Coronary Flow Regulation. J. Mol. Cell Cardiol. 52, 794–801. doi:10.1016/j.yjmcc.2011.10.001
Dick, G. M., and Tune, J. D. (2010). Role of Potassium Channels in Coronary Vasodilation. Exp. Biol. Med. (Maywood) 235, 10–22. doi:10.1258/ebm.2009.009201
Dogan, M. F., Yildiz, O., Arslan, S. O., and Ulusoy, K. G. (2019). Potassium Channels in Vascular Smooth Muscle: a Pathophysiological and Pharmacological Perspective. Fundam. Clin. Pharmacol. 33, 504–523. doi:10.1111/fcp.12493
Dopico, A. M., Bukiya, A. N., and Jaggar, J. H. (2018). Calcium- and Voltage-Gated BKchannels in Vascular Smooth Muscle. Pflugers Arch. 470, 1271–1289. doi:10.1007/s00424-018-2151-y
Earley, S., Heppner, T. J., Nelson, M. T., and Brayden, J. E. (2005). TRPV4 Forms a Novel Ca 2+ Signaling Complex with Ryanodine Receptors and BK Ca Channels. Circ. Res. 97, 1270–1279. doi:10.1161/01.res.0000194321.60300.d6
Ellinsworth, D. C., Sandow, S. L., Shukla, N., Liu, Y., Jeremy, J. Y., and Gutterman, D. D. (2016). Endothelium-derived Hyperpolarization and Coronary Vasodilation: Diverse and Integrated Roles of Epoxyeicosatrienoic Acids, Hydrogen Peroxide, and gap Junctions. Microcirculation 23, 15–32. doi:10.1111/micc.12255
Földes, G., Horkay, F., Szokodi, I., Vuolteenaho, O., Ilves, M., Lindstedt, K. A., et al. (2003). Circulating and Cardiac Levels of Apelin, the Novel Ligand of the Orphan Receptor APJ, in Patients with Heart Failure. Biochem. Biophysical Res. Commun. 308, 480–485. doi:10.1016/s0006-291x(03)01424-4
Folino, A., Montarolo, P. G., Samaja, M., and Rastaldo, R. (2015). Effects of Apelin on the Cardiovascular System. Heart Fail. Rev. 20, 505–518. doi:10.1007/s10741-015-9475-x
Gerbier, R., Alvear-Perez, R., Margathe, J. F., Flahault, A., Couvineau, P., Gao, J., et al. (2017). Development of Original Metabolically Stable Apelin-17 Analogs with Diuretic and Cardiovascular Effects. FASEB J. 31, 687–700. doi:10.1096/fj.201600784R
Ghatta, S., Nimmagadda, D., Xu, X., and O'Rourke, S. T. (2006). Large-conductance, Calcium-Activated Potassium Channels: Structural and Functional Implications. Pharmacol. Ther. 110, 103–116. doi:10.1016/j.pharmthera.2005.10.007
Goodwill, A. G., Dick, G. M., Kiel, A. M., and Tune, J. D. (2017). Regulation of Coronary Blood Flow. Compr. Physiol. 7, 321–382. doi:10.1002/cphy.c160016
Gurzu, B., Cristian Petrescu, B., Costuleanu, M., and Petrescu, G. (2006). Interactions between Apelin and Angiotensin II on Rat portal Vein. J. Renin Angiotensin Aldosterone Syst. 7, 212–216. doi:10.3317/jraas.2006.040
Jackson, W. F. (2017). Potassium Channels in Regulation of Vascular Smooth Musclecontraction and Growth. Adv. Pharmacol. 78, 89–144. doi:10.1016/bs.apha.2016.07.001
Jia, Y. X., Lu, Z. F., Zhang, J., Pan, C. S., Yang, J. H., Zhao, J., et al. (2007). Apelin Activates L-Arginine/nitric Oxide Synthase/nitric Oxide Pathway in Rat Aortas. Peptides 28, 2023–2029. doi:10.1016/j.peptides.2007.07.016
Kaczorowski, G. J., and Garcia, M. L. (2016). Developing Molecular Pharmacology of BK Channels for Therapeutic Benefit. Int. Rev. Neurobiol. 128, 439–475. doi:10.1016/bs.irn.2016.02.013
Katugampola, S. D., Maguire, J. J., Matthewson, S. R., and Davenport, A. P. (2001). [125 I]-(Pyr1 )Apelin-13 Is a Novel Radioligand for Localizing the APJ Orphan Receptor in Human and Rat Tissues with Evidence for a Vasoconstrictor Role in Man. Br. J. Pharmacol. 132, 1255–1260. doi:10.1038/sj.bjp.0703939
Kleinz, M. J., and Davenport, A. P. (2004). Immunocytochemical Localization of the Endogenous Vasoactive Peptide Apelin to Human Vascular and Endocardial Endothelial Cells. Regul. Peptides 118, 119–125. doi:10.1016/j.regpep.2003.11.002
Kleinz, M. J., Skepper, J. N., and Davenport, A. P. (2005). Immunocytochemical Localisation of the Apelin Receptor, APJ, to Human Cardiomyocytes, Vascular Smooth Muscle and Endothelial Cells. Regul. Peptides 126, 233–240. doi:10.1016/j.regpep.2004.10.019
Lee, D. K., Saldivia, V. R., Nguyen, T., Cheng, R., George, S. R., and O’Dowd, B. F. (2005). Modification of the Terminal Residue of Apelin-13 Antagonizes its Hypotensive Action. Endocrinology 146, 231–236. doi:10.1210/en.2004-0359
Luo, X., Liu, J., Zhou, H., and Chen, L. (2018). Apelin/APJ System: a Critical Regulator Ofvascular Smooth Muscle Cell. J. Cel Physiol 233, 5180–5188. doi:10.1002/jcp.26339
Maguire, J. J., Kleinz, M. J., Pitkin, S. L., and Davenport, A. P. (2009). [Pyr1]Apelin-13 Identified as the Predominant Apelin Isoform in the Human Heart. Hypertension 54, 598–604. doi:10.1161/hypertensionaha.109.134619
Mistry, D. K., and Garland, C. J. (1998). Nitric Oxide (NO)-induced Activation of Large Conductance Ca2+ -dependent K+ Channels (BKCa ) in Smooth Muscle Cells Isolated from the Rat Mesenteric Artery. Br. J. Pharmacol. 124, 1131–1140. doi:10.1038/sj.bjp.0701940
Modgil, A., Guo, L., O'Rourke, S. T., and Sun, C. (2013). Apelin-13 Inhibits Large-Conductance Ca2+-Activated K+ Channels in Cerebral Artery Smooth Muscle Cells via a PI3-Kinase Dependent Mechanism. PLoS One 8, e83051. doi:10.1371/journal.pone.0083051
Mughal, A., Anto, S., Sun, C., and O'Rourke, S. T. (2020). Apelin Inhibits an Endothelium-Derived Hyperpolarizing Factor-like Pathway in Rat Cerebral Arteries. Peptides 132, 170350. doi:10.1016/j.peptides.2020.170350
Mughal, A., and O'Rourke, S. T. (2018). Vascular Effects of Apelin: Mechanisms and Therapeutic Potential. Pharmacol. Ther. 190, 139–147. doi:10.1016/j.pharmthera.2018.05.013
Mughal, A., Sun, C., and O'Rourke, S. T. (2018b). Apelin Reduces Nitric Oxide-Induced Relaxation of Cerebral Arteries by Inhibiting Activation of Large-Conductance, Calcium-Activated K Channels. J. Cardiovasc. Pharmacol. 71, 223–232. doi:10.1097/fjc.0000000000000563
Mughal, A., Sun, C., and O’Rourke, S. T. (2018a). Activation of Large Conductance, Calcium-Activated Potassium Channels by Nitric Oxide Mediates Apelin-Induced Relaxation of Isolated Rat Coronary Arteries. J. Pharmacol. Exp. Ther. 366, 265–273. doi:10.1124/jpet.118.248682
Nagano, K., Kwon, C., Ishida, J., Hashimoto, T., Kim, J.-D., Kishikawa, N., et al. (2019). Cooperative Action of APJ and α1A-adrenergic Receptor in Vascular Smooth Muscle Cells Induces Vasoconstriction. J. Biochem. 166, 383–392. doi:10.1093/jb/mvz071
Nelson, M. T., and Quayle, J. M. (1995). Physiological Roles and Properties of Potassium Channels in Arterial Smooth Muscle. Am. J. Physiology-Cell Physiol. 268, C799–C822. doi:10.1152/ajpcell.1995.268.4.c799
Pitkin, S. L., Maguire, J. J., Kuc, R. E., and Davenport, A. P. (2010). Modulation of the Apelin/APJ System in Heart Failure and Atherosclerosis in Man. Br. J. Pharmacol. 160, 1785–1795. doi:10.1111/j.1476-5381.2010.00821.x
Salcedo, A., Garijo, J., Monge, L., Fernández, N., Luis García-Villalón, A., Sánchez Turrión, V., et al. (2007). Apelin Effects in Human Splanchnic Arteries. Role of Nitric Oxide and Prostanoids. Regul. Peptides 144, 50–55. doi:10.1016/j.regpep.2007.06.005
Schinzari, F., Veneziani, A., Mores, N., Barini, A., Di Daniele, N., Cardillo, C., et al. (2017). Beneficial Effects of Apelin on Vascular Function in Patients with central Obesity. Hypertension 69, 942–949. doi:10.1161/hypertensionaha.116.08916
Su, Z., Brown, E. C., Wang, W., and MacKinnon, R. (2016). Novel Cell-free High-Throughput Screening Method for Pharmacological Tools Targeting K+ Channels. Proc. Natl. Acad. Sci. USA 113, 5748–5753. doi:10.1073/pnas.1602815113
Tang, X., Qian, L.-L., Wang, R.-X., Yao, Y., Dang, S.-P., Wu, Y., et al. (2017). Regulation of Coronary Arterial Large Conductance Ca2+-Activated K+ Channel Protein Expression and Function by N-3 Polyunsaturated Fatty Acids in Diabetic Rats. J. Vasc. Res. 54, 329–343. doi:10.1159/000479870
Tunstall, R. R., Shukla, P., Grazul-Bilska, A., Sun, C., and O'Rourke, S. T. (2011). MT2 Receptors Mediate the Inhibitory Effects of Melatonin on Nitric Oxide-Induced Relaxation of Porcine Isolated Coronary Arteries. J. Pharmacol. Exp. Ther. 336, 127–133. doi:10.1124/jpet.110.174482
Tykocki, N. R., Boerman, E. M., and Jackson, W. F. (2017). Smooth Muscle Ion Channelsand Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr. Physiol. 7, 485–581. doi:10.1002/cphy.c160011
Wang, W., McKinnie, S. M. K., Farhan, M., Paul, M., McDonald, T., McLean, B., et al. (2016). Angiotensin-Converting Enzyme 2 Metabolizes and Partially Inactivates Pyr-Apelin-13 and Apelin-17. Hypertension 68, 365–377. doi:10.1161/hypertensionaha.115.06892
Wang, W., McKinnie, S. M., Patel, V. B., Haddad, G., Wang, Z., Zhabyeyev, P., et al. (2013). Loss of Apelin Exacerbates Myocardial Infarction Adverse Remodeling and Ischemia-Reperfusion Injury: Therapeutic Potential of Synthetic Apelin Analogues. J. Am. Heart Assoc. 2, e000249. doi:10.1161/jaha.113.000249
Yan, J., Wang, A., Cao, J., and Chen, L. (2020). Apelin/APJ System: an Emerging Therapeutic Target for Respiratory Diseases. Cell. Mol. Life Sci. 77, 2919–2930. doi:10.1007/s00018-020-03461-7
Zhang, D. X., Borbouse, L., Gebremedhin, D., Mendoza, S. A., Zinkevich, N. S., Li, R., et al. (2012). H 2 O 2 -Induced Dilation in Human Coronary Arterioles: Role of Protein Kinase G Dimerization and Large-Conductance Ca 2+ -Activated K + Channel Activation. Circ. Res. 110, 471–480. doi:10.1161/circresaha.111.258871
Keywords: apelin, coronary artery, nitric oxide, vasorelaxation, BKCa channels
Citation: Mughal A, Sun C and O’Rourke ST (2021) Apelin Does Not Impair Coronary Artery Relaxation Mediated by Nitric Oxide-Induced Activation of BKCa Channels. Front. Pharmacol. 12:679005. doi: 10.3389/fphar.2021.679005
Received: 10 March 2021; Accepted: 18 May 2021;
Published: 28 May 2021.
Edited by:
Ana Paula Davel, State University of Campinas, BrazilReviewed by:
Roger Lyrio Santos, Federal University of Espirito Santo, BrazilAndrew P. Braun, University of Calgary, Canada
Copyright © 2021 Mughal, Sun and O’Rourke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen T. O’Rourke, c3RlcGhlbi5vcm91cmtlQG5kc3UuZWR1
†Present address: Amreen Mughal, Department of Pharmacology, University of Vermont, Burlington, United States