 Carolina Iglesias-Lopez
Carolina Iglesias-Lopez Antònia Agustí
Antònia Agustí Antonio Vallano
Antonio Vallano Merce Obach
Merce Obach- 1Department of Pharmacology, Therapeutics and Toxicology, Universitat Autònoma de Barcelona, Barcelona, Spain
- 2Clinical Pharmacology Service, Vall d’Hebron University Hospital, Barcelona, Spain
- 3Medicines Department, Catalan Healthcare Service, Barcelona, Spain
Several advanced therapy medicinal products (ATMPs) have been approved in the European Union (EU). The aim of this study is to analyse the methodological features of the clinical trials (CT) that supported the marketing authorization (MA) of the approved ATMPs in the EU. A systematic review of the characteristics of pivotal CT of ATMPs approved in the EU until January 31st, 2021 was carried out. A total of 17 ATMPs were approved and 23 CT were conducted to support the MA (median, 1, range, 1–3). Of those studies, 8 (34.78%) were non-controlled and 7 (30.43%) used historical controls. Only 7 (30.4%) were placebo or active-controlled studies. Among all CT, 21 (91.3%) were open-label and 13 (56.52%) had a single-arm design. To evaluate the primary endpoint, 18 (78.26%) studies used an intermediate and single variable. The median (IQR) number of patients enrolled in the studies was 75 (22–118). To date, ATMPs’ approval in the EU is mainly supported by uncontrolled, single-arm pivotal CT. Although there is a trend toward an adaptive or a life cycle approach, a switch to more robust clinical trial designs is expected to better define the benefit and the therapeutic added value of ATMPs.
Introduction
Advanced therapy medicinal products (ATMPs) are a medicinal class that includes gene, cell and tissue therapies. The success of ATMP development and the approval of these therapies in the European Union (EU) has been crucial to the growth of clinical research during the last few years in this field, particularly for gene therapy.
Multiple indications are being targeted, most of them being refractory and recurrent stages of a disease that lacks effective therapeutic alternatives, and a significant proportion of them affecting the paediatric population (Alamo et al., 2019). With the introduction of ATMPs that can cover unmet needs and have the potential to cure life-threatening diseases, biological therapies initiated a shift from traditional clinical development pathway to an accelerated and highly product-specific one. The adaptive pathway concept and Priority Medicines scheme (PRIME) were launched in the EU specifically to speed the access of products targeting a significant unmet medical need. Several approved ATMPs were granted a PRIME designation and accelerated marketing authorisation application assessment during their development, allowing early access to these medicines (Iglesias-Lopez et al., 2021a).
Due to the type of target diseases, the inherent complexity of these products, and their accelerated developments, less comprehensive clinical data might be generated. These characteristics may lead to uncertainties in the benefit/risk profile for the product at the time of marketing authorization (MA). The aim of this study is to further analyse the clinical development of the current approved ATMPs’. Here, we describe the methodological features of the clinical trials that have driven ATMPs to their European approval and we compare the gene therapy trials versus the cell and tissue engineered trials.
Methods
A systematic review of the pivotal trials’ features that supported the MA of the ATMPs approved in the EU was carried out using the following approach:
1) Search strategy: Data collection was primarily extracted from European Public Assessment Reports on the European Medicines Agency (EMA) website (www.ema.europa.eu). The search was carried out until January 31st, 2021. In addition, a search for the main clinical trials of the approved ATMPs was conducted using ClinicalTrials.gov database and the related publications.
2) Eligibility criteria: Only products classified as ATMPs according to the EMA criteria (European Medicines Agency, 2010; Iglesias-Lopez et al., 2019) and authorised under centralised procedure in the EU have been considered. Combined ATMPs class, i.e., ATMP combined with a medical device, have been grouped according to the main ATMP category: gene therapy medicinal product, somatic cell therapy medicinal products or tissue engineered products. Only those trials identified or referenced as pivotal, and therefore, decisive for the MAA were analysed.
3) Data extraction and collected variables: The authors designed specific data extraction forms using Excel 2019 (Microsoft Corporation, Redmond, WA, United States) to collect information. For each ATMP the following variables were collected: type of ATMP, pharmacotherapeutic group, ATC code, therapeutic area (according to MeSH terms), diseases and other circumstances for its use (according to chapter’s title from the international version of the ICD-10), number of assessed clinical indications and pivotal clinical trials conducted. For each pivotal clinical trial, the following variables were selected: phase, design, type of randomization, type of control, type of study blinding, number of arms, participating centres, type of hypothesis and primary endpoint, presence and type of health-related quality of life (HRQoL) endpoints, presence of pre-specified analysis, duration of the main phase of the study, pivotal trial ongoing at the time of MAA, overall number of patients that participated in the study (enrolled, on intervention arm or control arm and safety set), age and sex of population, existence or absence of previous treatments, and geographic location of the pivotal trial. To determine if the study was ongoing at the time of the submission, the MAA submission date and the final data collection date for the primary outcome measure of the pivotal clinical trial were reviewed. Standard definitions of analysis set were used to classify among intended to treat (ITT), modified ITT (mITT) and per protocol set (PP) following ICH (E9) and EMA guidelines (ICH, 1998b; European Medicnes Agency, 2007). To assign the type of hypothesis in the case of two variables being used to evaluate the primary endpoint, the most robust variable was selected, i.e., final versus surrogate variables.
4) Statistical analysis: Statistical analysis for categorical and continuous variables was made using means of proportions, mean, standard deviation (SD), median, quartiles 25 and 75 (Q25, Q75), and range (minimum and maximum). The statistical analysis was performed using SAS® 9.4 (SAS Institute Inc., Cary, NC, United States).
Results
A total of 17 ATMPs have been approved in the EU (Table 1) and 23 main trials were conducted to support the MA for these products (median, 1, range, 1–3). The ATMPs trials by disease area, according to ICD-10 classification, included: neoplasms (7), endocrine, nutritional and metabolic diseases (2), diseases of the blood and blood-forming organs and certain disorders involving the immune mechanism (2), diseases of the eye and adnexa (2), diseases of the nervous system (1), diseases of the musculoskeletal system and connective tissue (3), diseases of the digestive system (1). In addition, there were 6 ATMPs for rare inherited disorders and 6 for neoplasms in which 4 were indicated for haematological malignancies and 2 for solid tumours. The detailed results of this study are presented in Table 2 by type of ATMP, in Table 3 for gene therapy studies and in Table 4 for cell and tissue therapy studies.
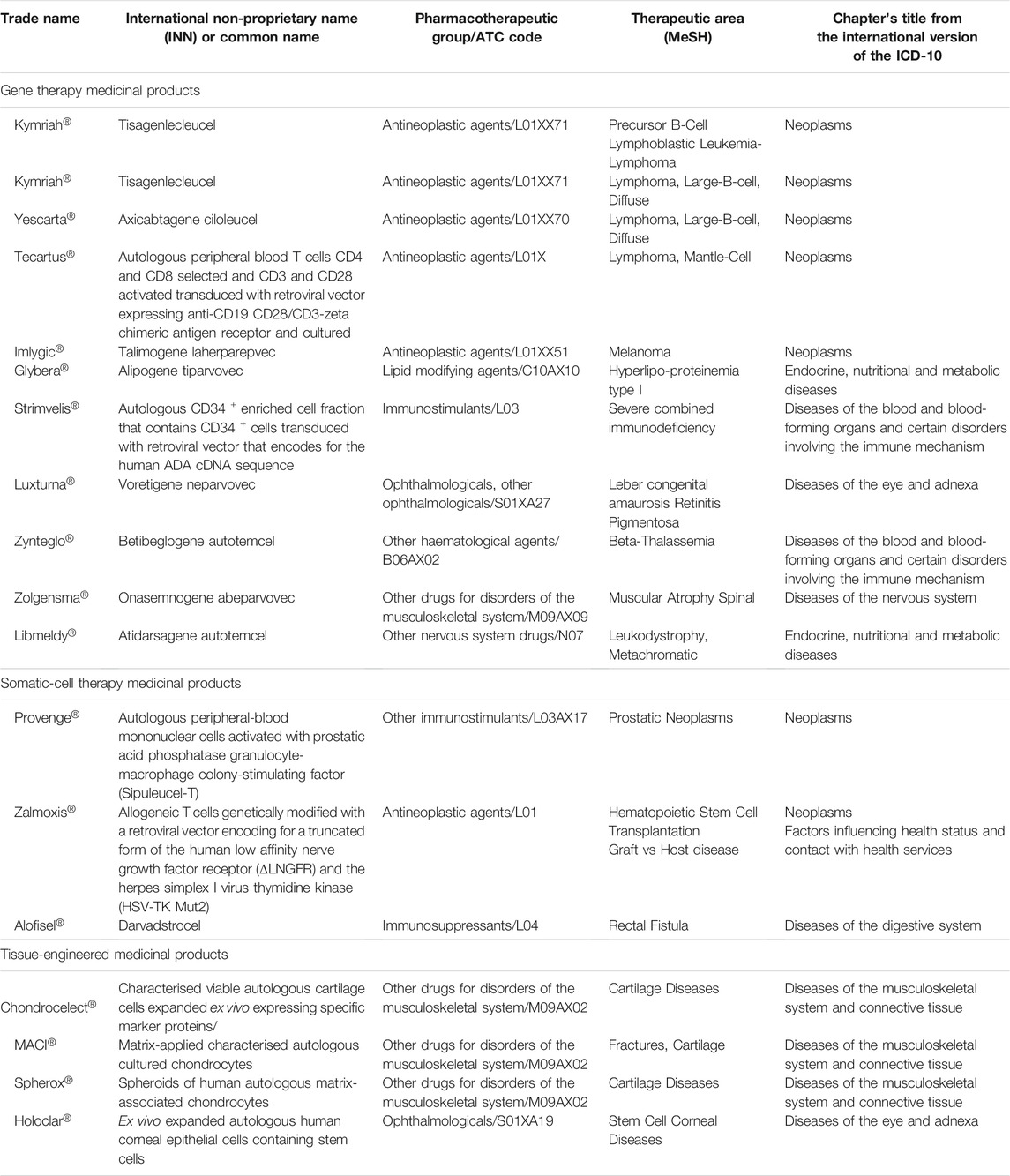
TABLE 1. Approved ATMPs in the European Union and therapeutic indication.
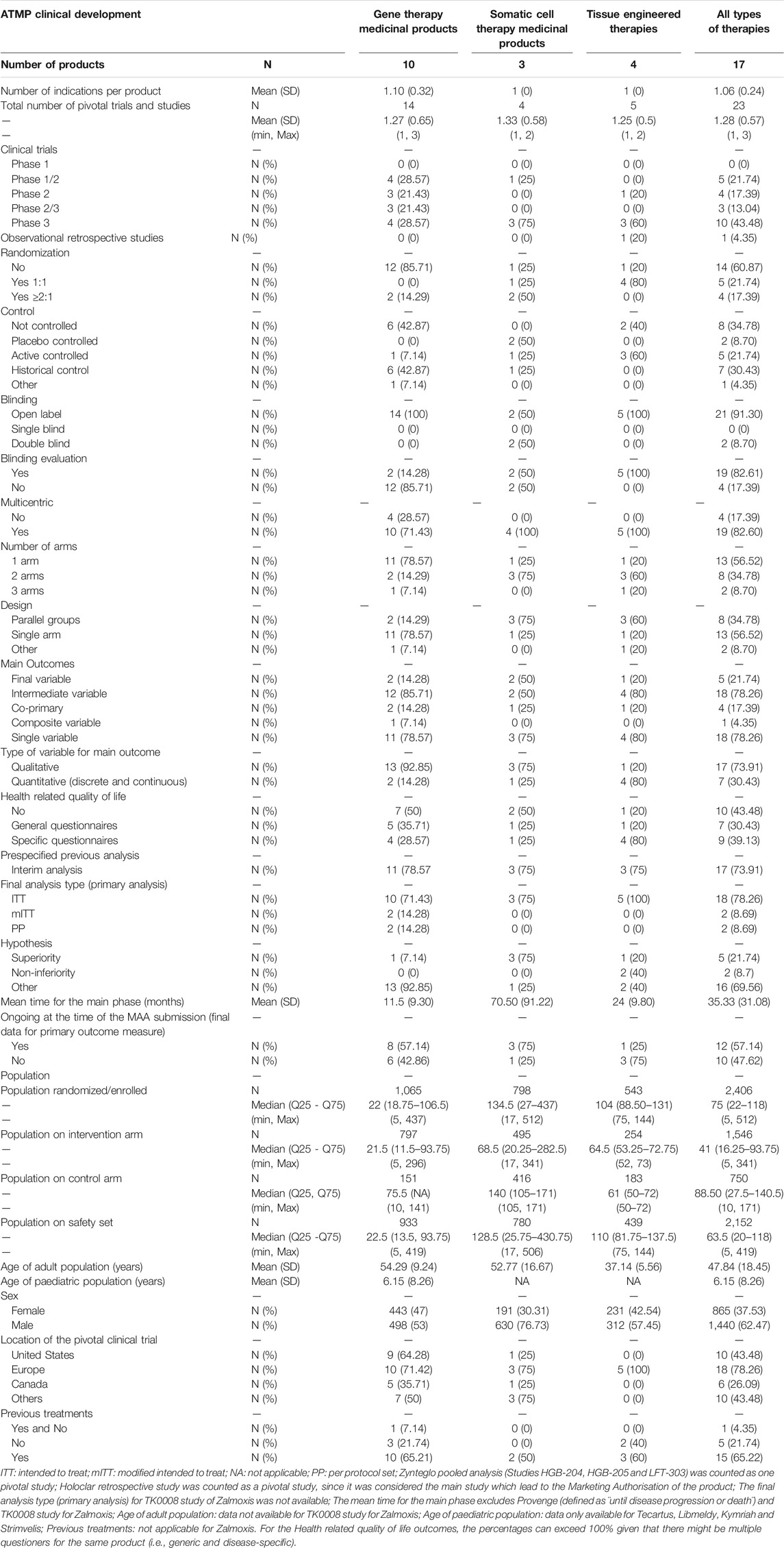
TABLE 2. Design features of pivotal clinical trials for the approved advanced therapy medicinal products in the European Union.
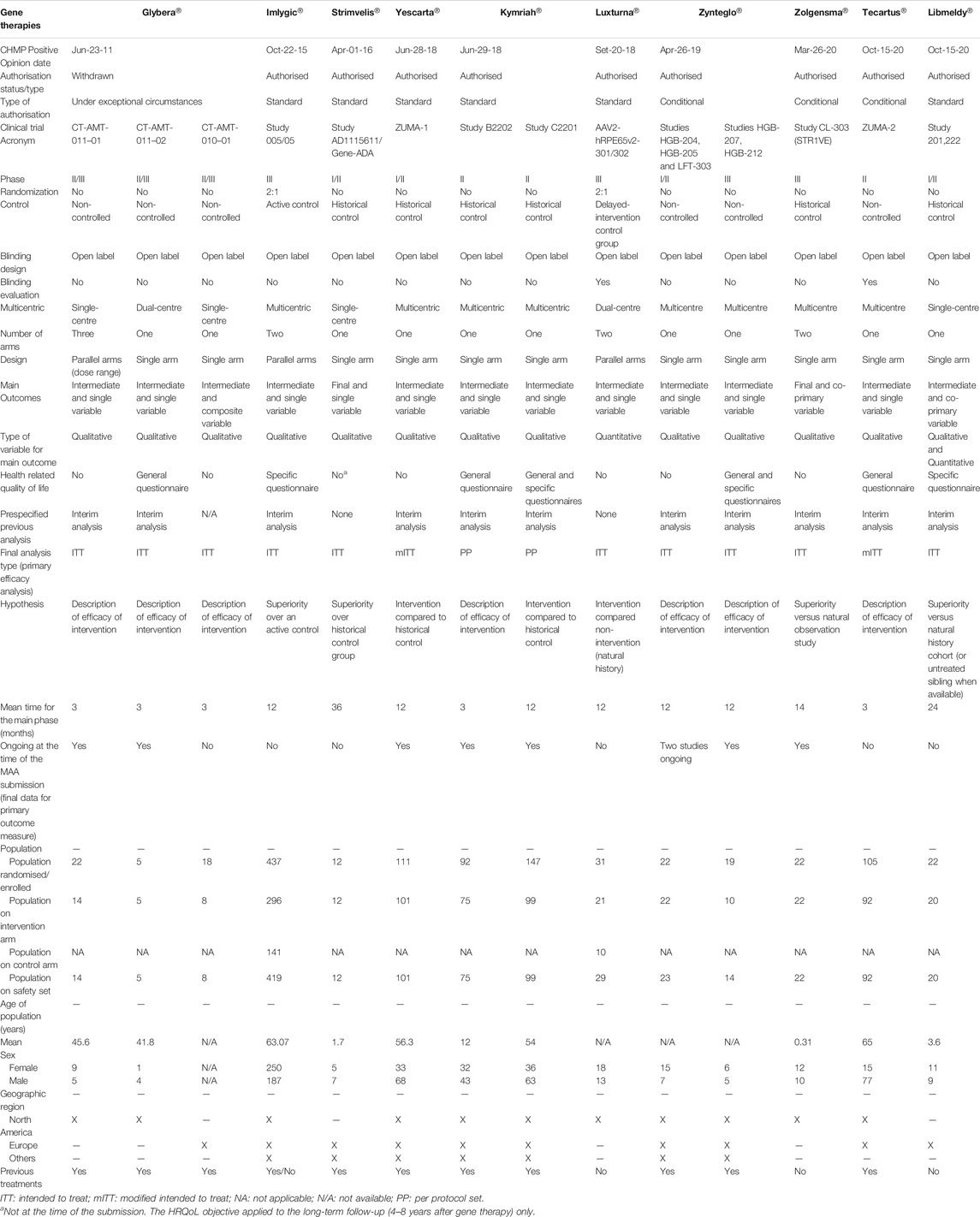
TABLE 3. Design features of pivotal clinical trials for the approved gene therapy medicinal products in the European Union.
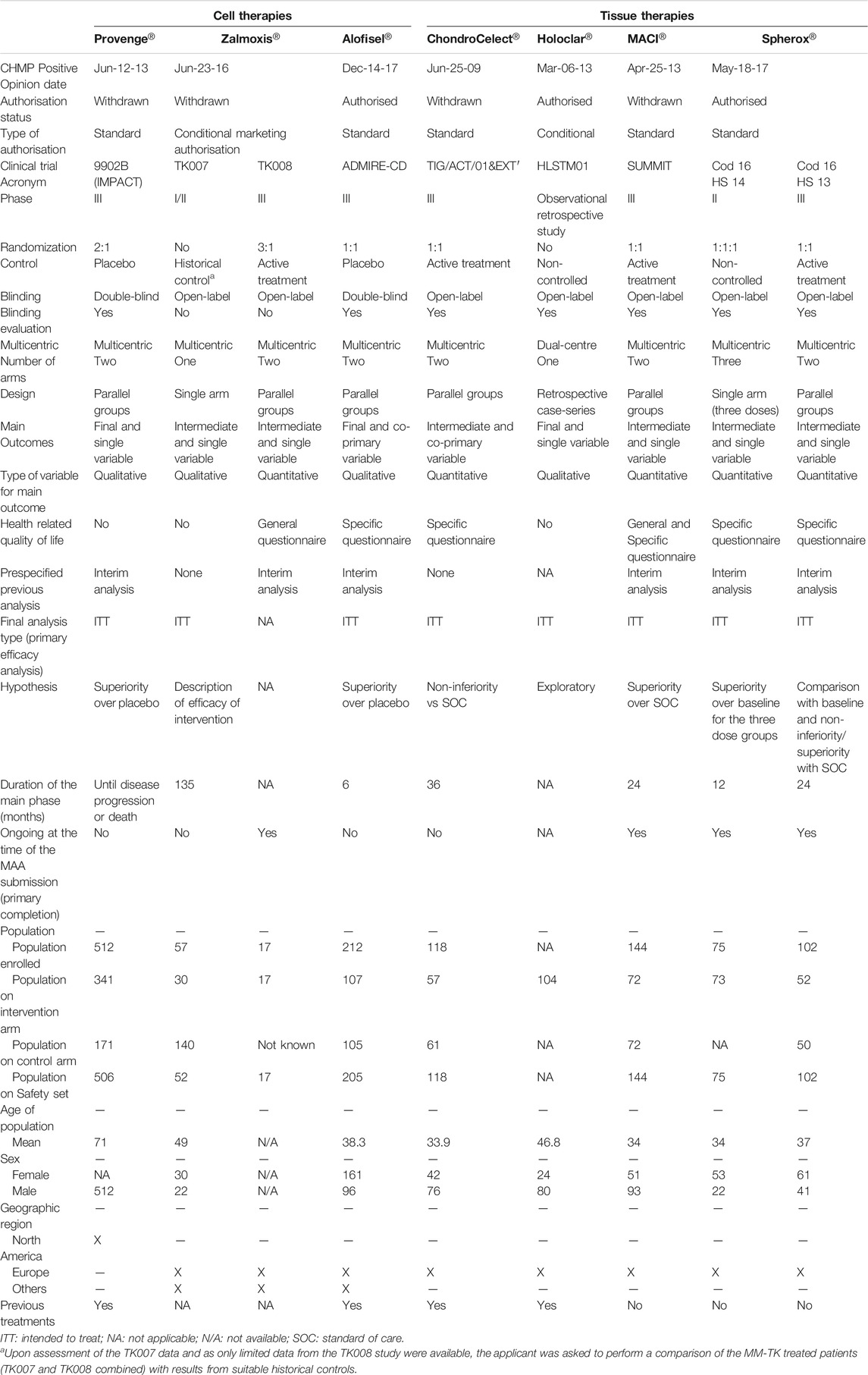
TABLE 4. Design features of pivotal clinical trials for the approved cell and tissue engineered therapy medicinal products in the European Union.
Regarding the design of the studies, 13 (56.52%) were Phase 2/3 and Phase 3 trials, 9 (39.13%) were Phase 1/2 or Phase 2 trials, and 1 (4.35%) was a retrospective study. For all types of therapies, 8 (34.78%) trials were non-controlled, 7 (30.44%) where active- or placebo-controlled, and 7 (30.43%) used an historical control as comparator. Differences were observed between gene and non-gene therapies (Supplementary Table S1 in Supplementary material). Six (42.87%) gene therapy studies were non-controlled and 6 (42.87%) used a historical control, whilst cell and tissue therapies studies were mainly controlled (66.66%). A total of 14 (60.87%) studies were not randomized. Similarly, differences in the existence of randomization between gene and non-gene therapies studies were also observed. Most of the studies for gene products lacked randomisation (85.71%), whereas this was present in 75% of the cell therapies studies and 80% of the tissue therapies studies. A total of 21 (91.30%) were open-label studies; all gene and tissue therapy studies were open-label, and this was also the approach for 50% of cell products trials. However, there is a difference in the blinding evaluation of the relevant endpoints between gene and non-gene therapy studies, as such evaluation is mostly absent in the case of gene therapies (85.71%) but is present in the case of cell and tissue engineered therapies (50% for cell therapy studies and 100% tissue engineered therapy studies). A total of 13 (56.52%) studies were single-arm trials and 10 (43.48%) had two or more arms. A difference in the number of arms between gene and non-gene therapy studies was also observed, where single-arm studies comprised 78.57% of total trials for gene therapy products versus the two- or three-arm designs present in 75% of cell therapy studies and 80% of tissue therapy studies. Accordingly, there are some differences in the design between gene and non-gene therapies studies, mainly in the parallel designs for cell and tissue engineered therapy studies versus single-arm designs for gene therapy studies. Of all studies analysed, 19 (82.60%) were multicentric.
Regarding the methodology used in these pivotal studies, 16 (69.56%) of the studies did not use a superiority or non-inferiority hypothesis but an alternative premise, e.g., comparison with historical controls. There is a difference between gene and non-gene therapies studies, where this type of alternative premises was mainly used for gene therapies trials (92.85%), while standard superiority or non-inferiority tests were used more frequently for cell and tissue engineered therapies trials (75 and 60%, respectively). To evaluate the primary objective, 18 (78.26%) of the trials used an intermediate and single main variable, which was mainly qualitative (73.91%). Final and quantitative variables were used in 5 (21.74%) and 7 (30.43%), respectively, which represents a smaller proportion (Table 5). Of these confirmatory studies, 18 (78.26%) used the intention-to-treat (ITT) principle in assessing the primary efficacy, 2 (14.28%) gene therapy trials used modified intention-to-treat (mITT) and 2 (14.28%) used per protocol set (PP). A total of 16 (69.56%) analysed studies included HRQoL questionnaires, 9 (39.13%) of those being disease-specific. No differences were observed in the type of HRQoL questionnaires between gene and non-gene products studies, i.e., generic versus disease-specific variables.
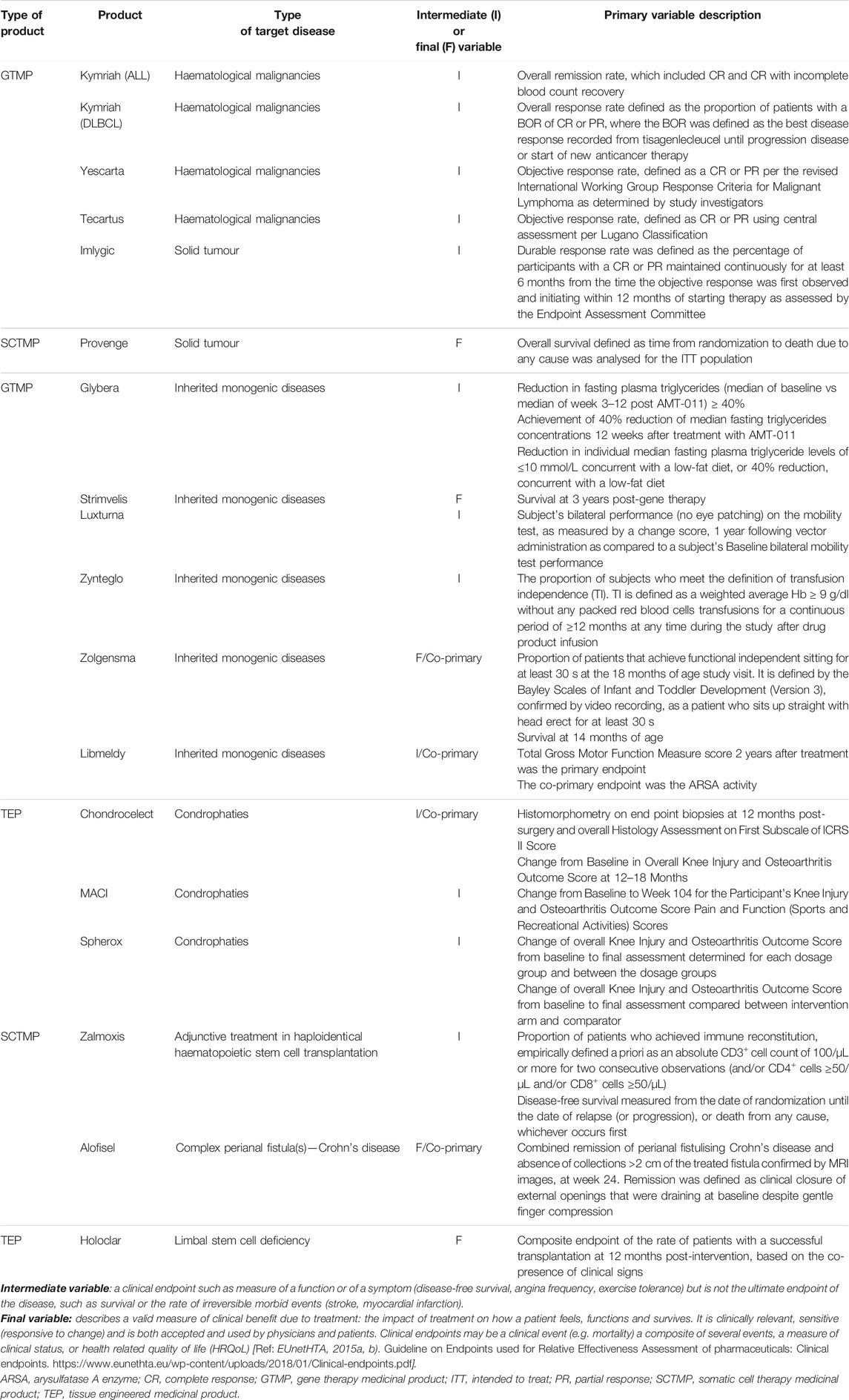
TABLE 5. Primary clinical variables of pivotal clinical trials for the approved ATMPs in the European Union.
The mean (SD) time for the main phase of the trial was 35.33 (31.08) months, approximately 1 year for the gene therapies and more than 2 years for cell and tissue engineered therapies. A total of 12 (57.14%) studies were ongoing at the time of submission, meaning that the final data collection for primary outcome measuring was not completed. Globally, 17 (73.91%) of the studies had a prespecified interim analysis, with similar proportion among the three types of ATMPs (75–78.57%).
Regarding the overall population size and location of these studies, the median (IQR 25–75) number of patients enrolled in the analysed ATMPs pivotal clinical trials was 75 (22–118). The mean ± SD age of the adult population included in these confirmatory trials was 48 ± 18.45 years old. There is no sufficient data to establish a mean ± SD age for paediatric populations. The sex distribution is higher for males (62.47%) than for women (37.53%). The analysed clinical trials were equally performed in both women and men, but the overall sex distribution was higher for males due to Provenge®’s indication, i.e., treatment of metastatic castration-resistant prostate cancer. The median (IQR 25–75) sample size in the intervention arm was 41 (16–94) patients and 63 (20–118) for the safety set. More than half of participants in these clinical trials had received previous treatments (65.22%). From the 23 pivotal studies analysed, 18 included sites located in the EU (78.26%), and 10 (43.48%) in the United States of America (US) or in other regions, such as Israel, Japan or Australia.
Discussion
Clinical research on ATMPs has increased during the last few years (Alamo et al., 2019). The introduction of ATMPs and the long-term expectancy of their benefit adds a new challenge for the regulatory agencies. In the present study, we aimed to describe the most relevant methodological features of the clinical trials that have driven ATMPs to their approval. The major findings reveal that the pivotal studies of currently approved advanced therapies typically share the following characteristics: 1) they are small, open-label, non-randomised, single-arm studies without control or using historical ones, and 2) intermediate and single variables are used to evaluate the primary efficacy outcome. In addition, this type of designs is more common for gene therapies than for cell and tissue therapies.
Hanna et al previously reported the methodological characteristics of clinical trials assessing ATMPs in an early development phase based on clinical trials registries (Hanna et al., 2016). The results showed very similar characteristics to those found in this study such as small sample size, non-randomised trials, single-arm trials, surrogate endpoints, and adaptive designs. Coppens et al., also reported that the level of scientific evidence required for the approval might differ among different regulatory agencies (Coppens et al., 2018). Elsallab et al. showed that clinical trials of ATMPs did not meet the same strict standards for clinical evidence that were applied to other biologicals submissions (Elsallab et al., 2020). This previously reported data, together with the results of the present study, highlight the limited clinical evidence upon which the authorisation of most ATMPs is based. Of these approved ATMPs, it was considered that eleven (64.70%) had sufficient data for a full MA, while for the remaining six products, five (29.41%) obtained a conditional approval and one (5.88%) was granted with a MA under exceptional circumstances.
The low disease prevalence, the disease severity and burden, the lack or scarce availability of disease-modifying treatments, the patient population’s heterogeneity and the strong presence of paediatric patient populations comprise some of the factors that could contribute to this type of designs.
The type of target diseases has been one of the key factors that might have given more flexibility in terms of level of evidence required for the MA. Our analysis shows that these designs are more commonly used for the development of gene therapy products, which target orphan diseases such as hematologic cancers or rare inherited monogenic disorders (40 and 60% of approved gene therapies, respectively), usually with unmet medical needs. Gene therapies were mainly authorised after conducting a single open-label study, usually non-randomised and non-controlled or using historical controls, and only few of them being Phase III studies. By contrast, tissue therapies trials consisted of Phase III studies controlled with the standard of care, and two out of three cell therapy trials conducted placebo-controlled studies. The approved tissue therapies primarily cover products for articular cartilage damage or prostate cancer, which are relatively common among the overall population and with several treatments available. Moreover, the target population might have also contributed to these alternative designs for gene therapies products, given that 60% of approved gene therapies target paediatric population, while all of the tissue and cell therapies target adults. The targeted paediatric diseases are life-threatening or with a huge impact on patients’ and caregivers’ quality-of-life, and randomised, controlled trials could have posed ethical concerns, as well as recruitment issues.
It is noteworthy to mention that different types of historical controls were used to compare the efficacy of the intervention: historical references from retrospective studies and retrospective databases, prospective natural history cohorts’ studies, untreated sibling data and within-subject comparison between pre- and post-treatment assessments (Hassan et al., 2012; European Medicines Agency, 2021; Maude et al., 2018; Crump et al., 2017). The current EMA guideline states that orphan products are assessed according to the same standards as those for other products but considering their limitations due to low patient recruitment (European Medicnes Agency, 2006). While the same guideline states that most orphan drugs and paediatric indications submitted for regulatory approval are based on randomised controlled trials and deviation from such standards is uncommon, in the case of the current approved ATMPs, alternative approaches as historical controls were frequently used, i.e., Strimvelis®, Kymriah®, Luxturna®, Zolgensma® and Libmeldy®.
On the other hand, the line of treatment is another factor that might have justified these types of designs so far. As an example of the approved ATMPs, CAR-T therapies are indicated at least as a third-line therapy for relapsed or refractory cancer patients. The four pivotal studies conducted for these products were non-controlled, open-label, Phase II studies where the intervention arm was compared to a historical control. After the approval of the aforementioned therapies, the EMA has published recommendations on clinical considerations on CAR-T-cell product development (European Medicnes Agency, 2020), where it is stated that randomized controlled trial design should be followed even for those cases of late-stage refractory disease. It will be interesting to see how these recommendations are implemented in the near future.
Another important factor observed in the studied designs is the use of surrogate variables instead of a clinically relevant final endpoint. Intermediate endpoints can be used as a primary endpoint for MA, especially when there is a high unmet need, when clinical events are rare/delayed in slowly progressive diseases and a very long follow-up is needed for their assessment, and for rare and/or life-threatening diseases with no therapeutic alternative available (EUnetHTA, 2015b; ICH, 1998a). In the case of all approved gene therapies that target cancer diseases, the proportion of patients with objective overall response rate (ORR) was used as the intermediate primary variable, unlike cell therapy trials that used overall survival (OS) as a final endpoint. OS analysis usually requires a large sample size, a long follow-up and should be evaluated in a randomised, control trial to avoid cofounding factors due to the switch-over of control to intervention or subsequent therapies (Gutman et al., 2013; Pazdur, 2008). However, ORR has been the most commonly used surrogate endpoint in support of accelerated/conditional approvals, but also of standard approvals, since it is directly attributable to a drug’s effect, providing an accurate assessment in single-arm trials conducted in patients with refractory tumours (Food and Drug Administration, 2018a). On the other hand, for gene therapies targeting inherited monogenic diseases, biomarkers were commonly used to predict changes in the desired clinical endpoints, and at least one of the pivotal studies included HRQoL outcomes. Exceptionally for other products, a novel clinical meaningful endpoint, i.e. Luxturna® (Russell et al., 2017), or survival as a final primary outcome were used, i.e. Zolgensma® (Del Rosario et al., 2020; Cech et al., 2012).
These types of non-robust designs for new drugs in areas of high unmet medical need are mainly justified on the basis of ethical reasons, based on the potential life-saving opportunities or quality of life improvement for patients who may not survive or will progress rapidly until robust clinical data is available. On the other hand, the difficulties of conducting standard clinical developments with orphan drugs are well-recognised, and single small trials using alternative approaches have been the basis for numerous MAA in the recent years (Blin et al., 2020; Micallef and Blin, 2020; Picavet et al., 2013; Pontes et al., 2018). This regulatory flexibility sometimes comes at the cost of having a less comprehensive clinical data, and in consequence, greater uncertainty about the product’s benefit-risk balance at the time of MA (Iglesias-Lopez et al., 2021b). In addition, since the introduction of the adaptive pathway concept, the shift towards accelerated clinical developments has also been associated with an intrinsic uncertainty on effectiveness and safety, which can result in promising Phase II results but an unsuccessful Phase III or post-marketing studies (Pharma Intelligence, 2019; Novartis press release, 2021a, b). This highlights the possibility for a patient to receive an early-authorised treatment without meaningful clinical benefits and with exposure to its adverse effects, missing clinical opportunities, and wasting healthcare system resources (Ermisch et al., 2016).
The speeding up access to new drugs is achieved by putting aside traditional Phase III clinical trials in favour of post-marketing evidence generation. This fact is translated into the need to perform long and extensive post-marketing studies, where the costs of evidence generation as well as the costs of therapy are likely to be transferred from the MA holder to healthcare systems (Ermisch et al., 2016; Joppi et al., 2016). It is known, that this post-authorisation commitments can be challenging due to the long-term follow-up, which may lead to delays to complete the studies, and given that patients are more reluctant to participate in a post-marketing trial with all its constraints, if the medicine is already available, above all in those cases where the trial includes randomization (Joint briefing paper, 2015).
Costly treatments with high uncertainties in regard to its benefits, translates to a complex evaluation by the Health Technology Assessment bodies (HTAb), as well as there is industry pressure for corporate pharma and its investors to ensure sustainability in drug development.
Several detailed methodological recommendations for clinical trial designs have been launched to address the shortcomings of carrying out studies in small population (Day et al., 2018; ASTERIX project, 2021; IDEAL project, 2021; Friede et al., 2018) and examples of effective use of a historical control have also been reported (Mulberg et al., 2019). Multi-arm designs and platform designs sharing where a common control is shared have been raised as a potential solution (International Rare Diseases Research Consortium, 2016; Food and Drug Administration, 2018b). Comparator data can also be taken from pragmatic trials, observational studies or registries, but ensuring its quality (EUnetHTA, 2015a). In addition, real world data plays a key role to provide sufficient therapeutic evidence for these type of therapies and efforts are being made for a better use of registries (European Medicines Agency, 2017).
Methodological and clinical guidelines for a specific medical condition is an effective manner of obtaining regulatory guidance and providing a predictable decision-making regulatory framework. Given that ATMPs are innovative and more complex than traditional pharmaceuticals or other biological drugs, some specific requirements related to the study design and methodology, study population, safety, dose selection, as well as preclinical and product controls need to be considered for the development of these therapies. The FDA has launched several guidelines for the development of ATMPs aimed at certain types of conditions based on the acquired experience of the current approved advanced therapies. These guidelines address the point of uncontrolled designs and the need of more robust study designs in order to provide proper evidence of efficacy (Food and Drug Administration, 2020a; Food and Drug Administration, 2020b; Food and Drug Administration, 2021). Although still limited, with the current experience of the approved ATMPs in the EU, the EMA has started to launch new recommendations on the types of study designs and methodologies that can support the MA more robustly (European Medicnes Agency, 2020). This fact might lead to a switch on the current trend used in clinical designs based on uncontrolled pivotal studies or with historical control comparisons to randomised-controlled trials.
The limitations of this study are the small sample size and the fact that further analysis, once more therapies are approved, is required to determine with greater accuracy the most common clinical design and methodology for ATMPs, as well as to elucidate the potential differences between gene therapy trials versus cell and tissue therapy trials. Another limitation is that approved ATMPs have not been compared to other approved medicines. Nevertheless, this is an exhaustive study that evaluates the pivotal trials for approved ATMPs.
Conclusion
The results of our study show that most authorised ATMPs are based on small, open-label, uncontrolled and single-arm pivotal trials using single and intermediate variables to evaluate outcomes. ATMPs are innovative therapies that mainly target orphan diseases and high unmet medical needs. This fact has led to certain methodological. weaknesses in their pivotal clinical trials, which in turn has resulted in limited data to robustly assess the benefit/risk of the product. A gradual shift towards the production of more methodologically sound randomized-controlled trials is expected to better define the benefit and the therapeutic added value of ATMPs.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
Conception and design of the study: CI-L, AV, AA, and MO. Acquisition of data: CI-L. Analysis and interpretation of data: CI-L, AV, AA, and MO. Drafting and revising the manuscript: CI-L. Reviewed and edited the manuscript: CI-L, AA, MO, and AV. All authors have approved the final article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The findings and conclusions in this article should not be construed to represent any agency determination or policy.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.773712/full#supplementary-material
References
Álamo, J. E., Timón, M., Gómez-Platero, C. G., Abad, C. D., González, M. V., et al. (2019). Clinical Trials of Advanced Therapy Investigational Medicinal Products in Spain: Preparing for the European Clinical Trials Regulation. Cell Gene Ther. Insights 5, 1431–1449. doi:10.18609/cgti.2019.147
ASTERIX project (2021). Advances in Small Trials dEsign for Regulatory Innovation and eXcellence. Available at: http://www.asterix-fp7.eu/(Accessed August 28, 2021).
Blin, O., Lefebvre, M. N., Rascol, O., and Micallef, J. (2020). Orphan Drug Clinical Development. Therapie 75, 141–147. doi:10.1016/j.therap.2020.02.004
Cech, D. J., and Martin, S. T. (2012). Chapter 5-Evaluation of Function, Activity, and Participation," in Functional Movement Development Across the Life Span. Elsevier, 88–104. doi:10.1016/b978-1-4160-4978-4.00005-3
Coppens, D. G. M., de Wilde, S., Guchelaar, H. J., De Bruin, M. L., Leufkens, H. G. M., Meij, P., et al. (2018). A Decade of Marketing Approval of Gene and Cell-Based Therapies in the United States, European Union and Japan: An Evaluation of Regulatory Decision-Making. Cytotherapy 20, 769–778. doi:10.1016/j.jcyt.2018.03.038
Crump, M., Neelapu, S. S., Farooq, U., Van Den Neste, E., Kuruvilla, J., Westin, J., et al. (2017). Outcomes in Refractory Diffuse Large B-Cell Lymphoma: Results from the International SCHOLAR-1 Study. Blood 130, 1800–1808. doi:10.1182/blood-2017-03-769620
Day, S., Jonker, A. H., Lau, L. P. L., Hilgers, R. D., Irony, I., Larsson, K., et al. (2018). Recommendations for the Design of Small Population Clinical Trials. Orphanet J. Rare Dis. 13, 195. doi:10.1186/s13023-018-0931-2
Del Rosario, C., Slevin, M., Molloy, E. J., Quigley, J., and Nixon, E. (2020). How to Use the Bayley Scales of Infant and Toddler Development. Arch. Dis. Child. Educ. Pract. Ed. 106, 108–112. doi:10.1136/archdischild-2020-319063
Elsallab, M., Bravery, C. A., Kurtz, A., and Abou-El-Enein, M. (2020). Mitigating Deficiencies in Evidence during Regulatory Assessments of Advanced Therapies: A Comparative Study with Other Biologicals. Mol. Ther. Methods Clin. Dev. 18, 269–279. doi:10.1016/J.OMTM.2020.05.035
Ermisch, M., Bucsics, A., Vella Bonanno, P., Arickx, F., Bybau, A., Bochenek, T., et al. (2016). Payers' Views of the Changes Arising through the Possible Adoption of Adaptive Pathways. Front. Pharmacol. 7, 305. doi:10.3389/fphar.2016.00305
EUnetHTA (2015a). Criteria for the Choice of the Most Appropriate Comparator(s) Summary of Current Policies and Best Practice Recommendations. Available at: https://eunethta.eu/wp-content/uploads/2018/03/Criteria_WP7-SG3-GL-choice_of_comparator_amend2015.pdf (Accessed April 19, 2021).
EUnetHTA (2015b). Guideline on Endpoints Used for Relative Effectiveness Assessment of Pharmaceuticals: Surrogate Endpoints. Available at: https://eunethta.eu/wp-content/uploads/2018/03/surrogate_endpoints.pdf.
European Medicines Agency (2021). Committee for Medicinal Products for Human Use (CHMP) Assessment Report: Libmeldy. Available at: https://www.ema.europa.eu/en/documents/assessment-report/libmeldy-epar-public-assessment-report_en.pdf.
European Medicines Agency (2017). Patient Registry Initiative- Strategy and Mandate of the Cross-Committee Task Force. Available at: www.ema.europa.eu/contact (Accessed July 23, 2021).
European Medicines Agency (2010). Reflection Paper on Classification of Advanced Therapy Medicinal Products. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-classification-advanced-therapy-medicinal-products_en-0.pdf (Accessed March 6, 2020).
European Medicnes Agency (2006). Committe for Medicinal Products for Human Use (CHMP), Guideline on Clinial Trials in Small Populations Populations. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-trials-small-populations_en.pdf (Accessed March 28, 2021).
European Medicnes Agency (2020). Guideline on Quality, Non-clinical and Clinical Aspects of Medicinal Products Containing Genetically Modified Cells. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-medicinal-products-containing-genetically-modified_en-0.pdf (Accessed February 2, 2021).
European Medicnes Agency (2007). Reflection Paper on Methodological Issues in Confirmatory Clinical Trials Planned with an Adaptive Design. London: EMEA Doc. Ref. CHMP/EWP/2459/02 http://www.emea.europa.eu (Accessed May 10, 2021).Available at:
Food and Drug Administration (2018a). Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics Guidance for Industry. Available at: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htmand/orhttps://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (Accessed April 11, 2021).
Food and Drug Administration (2020a). Human Gene Therapy for Hemophilia; Guidance for Industry. Available at: https://www.fda.gov/vaccines-blood-biologics/guidance-compliance- (Accessed February 2, 2021).
Food and Drug Administration (2021). Human Gene Therapy for Neurodegenerative Diseases; Draft Guidance for Industry. Available at: https://www.fda.gov/vaccines-blood-biologics/guidance-compliance- (Accessed February 2, 2021).
Food and Drug Administration (2020b). Human Gene Therapy for Retinal Disorders; Guidance for Industry. Available at: https://www.fda.gov/vaccines-blood-biologics/guidance-compliance- (Accessed February 2, 2021).
Food and Drug Administration (2018b). Master Protocols: Efficient Clinical Trial Design Strategies to Expedite Development of Oncology Drugs and Biologics Guidance for Industry DRAFT GUIDANCE. Available at: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (Accessed July 20, 2021).
Friede, T., Posch, M., Zohar, S., Alberti, C., Benda, N., Comets, E., et al. (2018). Recent Advances in Methodology for Clinical Trials in Small Populations: the InSPiRe Project. Orphanet J. Rare Dis. 13, 186. doi:10.1186/s13023-018-0919-y
Gutman, S. I., Piper, M., and Grant, M. D. (2013). Progression-Free Survival: What Does It Mean for Psychological Well-Being or Quality of Life? Rockville, MD: Agency for Healthcare Research and Quality (US). Available at: https://www.ncbi.nlm.nih.gov/books/NBK137763/(Accessed April 11, 2021).
Hanna, E., Rémuzat, C., Auquier, P., and Toumi, M. (2016). Advanced Therapy Medicinal Products: Current and Future Perspectives. J. Mark Access Health Pol. 4, 31036. doi:10.3402/jmahp.v4.31036
Hassan, A., Booth, C., Brightwell, A., Allwood, Z., Veys, P., Rao, K., et al. (2012). Outcome of Hematopoietic Stem Cell Transplantation for Adenosine Deaminase-Deficient Severe Combined Immunodeficiency. Blood 120, 3615–3626. doi:10.1182/blood-2011-12-396879
ICH (1998a). ICH Topic E8 General Considerations for Clinical Trials Step 5 Note for Guidance on General Considerations for Clinical Trials. Available at: http://www.emea.eu.int (Accessed April 11, 2021).
ICH (1998b). ICH Topic E9 Statistical Principles for Clinical Trials Step 5 Note for Guidance on Statistical Principles for Clinical Trials. Available at: http://www.emea.eu.int (Accessed March 27, 2021).
IDEAL project (2021). Integrated DEsign and AnaLysis of Clinical Trials in Small Population Groups. Available at: https://www.ideal.rwth-aachen.de/(Accessed August 28, 2021).
Iglesias-López, C., Agustí, A., Obach, M., and Vallano, A. (2019). Regulatory Framework for Advanced Therapy Medicinal Products in Europe and United States. Erratum in: Front Pharmacol 11, 766. doi:10.3389/fphar.2019.00921
Iglesias-Lopez, C., Obach, M., Vallano, A., and Agustí, A. (2021a). Comparison of Regulatory Pathways for the Approval of Advanced Therapies in the European Union and the United States. Cytotherapy 23, 261–274. doi:10.1016/j.jcyt.2020.11.008
Iglesias-Lopez, C., Agustí, A., Vallano, A., and Obach, M. (2021b). Current landscape of clinical development and approval of advanced therapies. Mol Ther Methods Clin Dev 23, 606–618. doi:10.1016/j.omtm.2021.11.003
International Rare Diseases Research Consortium (2016). Small Population Clinical Trials Task Force Workshop Report and Recommendations. Available at: https://www.irdirc.org/wp-content/uploads/2017/12/SPCT_Report.pdf (Accessed May 16, 2021).
Joint briefing paper (2015). "Adaptive Licensing” or “Adaptive Pathways”: Deregulation under the Guise of Earlier Access Executive. Available at: http://english.prescrire.org/Docu/DOCSEUROPE/(Accessed April 19, 2021).
Joppi, R., Gerardi, C., Bertele', V., and Garattini, S. (2016). Letting post-marketing Bridge the Evidence gap: The Case of Orphan Drugs. BMJ 353, i2978. doi:10.1136/bmj.i2978
Maude, S. L., Laetsch, T. W., Buechner, J., Rives, S., Boyer, M., Bittencourt, H., et al. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 378, 439–448. doi:10.1056/nejmoa1709866
Micallef, J., and Blin, O. (2020). Orphan Drug Designation in Europe: A Booster for the Research and Development of Drugs in Rare Diseases. Therapie 75, 133–139. doi:10.1016/j.therap.2020.02.003
Mulberg, A. E., Bucci-Rechtweg, C., Giuliano, J., Jacoby, D., Johnson, F. K., Liu, Q., et al. (2019). Regulatory Strategies for Rare Diseases under Current Global Regulatory Statutes: A Discussion with Stakeholders. Orphanet J. Rare Dis. 14 (1), 36. doi:10.1186/s13023-019-1017-5
Novartis press release (2021a). Novartis’ Kymriah Fails to Meet Primary Goal in Non-hodgkin Lymphoma Trial. Available at: https://www.clinicaltrialsarena.com/news/novartis-kymriah-lymphoma-trial/(Accessed August 27, 2021).
Novartis press release (2021b). Novartis Kymriah® Demonstrates Consistent Efficacy and Safety Outcomes in US Patients when Used in Real-World Setting | Novartis. Available at: https://www.novartis.com/news/media-releases/novartis-kymriah-demonstrates-consistent-efficacy-and-safety-outcomes-us-patients-when-used-real-world-setting (Accessed April 18, 2021).
Pazdur, R. (2008). Endpoints for Assessing Drug Activity in Clinical Trials. Oncologist 13 (Suppl. 2), 19–21. doi:10.1634/theoncologist.13-S2-19
Pharma Intelligence (2019). Disappointing End for MolMed’s Zalmoxis Cell Therapy in EU. Available at: https://pink.pharmaintelligence.informa.com/PS140998/Disappointing-End-For-MolMeds-Zalmoxis-Cell-Therapy-In-EU (Accessed March 8, 2020).
Picavet, E., Cassiman, D., Hollak, C. E., Maertens, J. A., and Simoens, S. (2013). Clinical Evidence for Orphan Medicinal Products-A Cause for Concern? Orphanet J. Rare Dis. 8, 164. doi:10.1186/1750-1172-8-164
Pontes, C., Fontanet, J. M., Vives, R., Sancho, A., Gómez-Valent, M., Ríos, J., et al. (2018). Evidence Supporting Regulatory-Decision Making on Orphan Medicinal Products Authorisation in Europe: Methodological Uncertainties. Orphanet J. Rare Dis. 13, 206. doi:10.1186/s13023-018-0926-z
Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z. F., Tillman, A., et al. (2017). Efficacy and Safety of Voretigene Neparvovec (AAV2-hRPE65v2) in Patients with RPE65-Mediated Inherited Retinal Dystrophy: a Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 390, 849–860. doi:10.1016/S0140-6736(17)31868-8
Keywords: drug development, drug approval, research design, methods, clinical trials, advanced therapies, cell- and tissue-based therapy, genetic therapy
Citation: Iglesias-Lopez C, Agustí A, Vallano A and Obach M (2021) Methodological Characteristics of Clinical Trials Supporting the Marketing Authorisation of Advanced Therapies in the European Union. Front. Pharmacol. 12:773712. doi: 10.3389/fphar.2021.773712
Received: 10 September 2021; Accepted: 28 October 2021;
Published: 29 November 2021.
Edited by:
Kevin Lu, University of South Carolina, United StatesReviewed by:
Kurt Neumann, Independent researcher, Kerékteleki, HungaryKata Mazalin, Servier, Hungary
Copyright © 2021 Iglesias-Lopez, Agustí, Vallano and Obach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Vallano, YXZhbGxhbm9AY2F0c2FsdXQuY2F0