 Qiang Wang1,2Yuemei Xi1,2Binyang Chen1,2Hairong Zhao1,2Wei Yu1,2
Qiang Wang1,2Yuemei Xi1,2Binyang Chen1,2Hairong Zhao1,2Wei Yu1,2 De Xie1,2Weidong Liu1,2Furong He1,2Chenxi Xu1,2
De Xie1,2Weidong Liu1,2Furong He1,2Chenxi Xu1,2 Jidong Cheng1,2*
Jidong Cheng1,2*- 1Department of Internal Medicine, Xiang’an Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, China
- 2Xiamen Key Laboratory of Translational Medicine for Nucleic Acid Metabolism and Regulation, Xiamen, China
Cisplatin is a widely used and potent anti-neoplastic agent, but severe and inescapable side effects in multiple normal tissues and organs limit its application, especially nephrotoxicity. Molecular mechanisms of cisplatin nephrotoxicity involve mitochondrial damage, oxidative stress, endoplasmic reticulum stress, inflammation, apoptosis, necroptosis, etc. Receptor of advanced glycation end products (RAGE) is a multiligand pattern recognition receptor, engaged in inflammatory signaling and mitochondrial homeostasis. Whether inhibition of RAGE alleviates cisplatin-induced nephropathy has not been investigated. Here, we revealed that RAGE deficiency attenuates cisplatin-induced acute nephrotoxicity, as evidenced by reduced apoptosis, inflammation, lipid accumulation, restored mitochondrial homeostasis and fatty acid oxidation in renal tubular epithelial cells (TECs). In vitro studies showed that, the RAGE-specific inhibitor FPS-ZM1 attenuated the cisplatin-induced decrease of cell viability and fatty acid oxidation in the normal rat renal TEC line NRK-52E cells. Taken together, RAGE knockout mitigated cisplatin-induced acute nephrotoxicity by inhibiting apoptosis, inflammation, and restoring fatty acid oxidation in TECs, suggesting that RAGE inhibition could be a therapeutic option for cisplatin-induced acute nephrotoxicity.
Introduction
Acute kidney injury (AKI) is a prevalent complication in hospitalized patients with an incidence of 10–15% (Ronco et al., 2019). Cisplatin is a frequently utilized and effective chemotherapeutic agent in clinical settings, which renders it a common cause of AKI. The exploration of the molecular mechanisms of cisplatin nephrotoxicity has never halted; however, the mechanisms have yet been fully defined, so therapies that can abolish cisplatin-induced AKI always remain lacking (McSweeney et al., 2021). Studies have demonstrated that cisplatin nephrotoxicity links to mitochondrial damage, oxidative stress, endoplasmic reticulum stress, inflammation, apoptosis, necroptosis (Xu et al., 2015; Zahedi et al., 2017; Lu et al., 2020; Mapuskar et al., 2021), etc. According to recent reports, impaired fatty acid oxidation (FAO) plays a key role in the process of cisplatin nephrotoxicity (Chiba et al., 2019; Jang et al., 2020). What’s more, compromised FAO in renal TECs is considered an essential pathogenesis of renal interstitial fibrosis (Kang et al., 2015).
RAGE is a multiligand pattern recognition receptor implicated in inflammatory signaling (Coughlan et al., 2009; Adeshara et al., 2018). Meanwhile, RAGE modulates glucose and lipid metabolism, e.g., senescence-induced RAGE promotes hepatic steatosis by suppressing FAO (Song et al., 2014; Wan et al., 2020). It was found that RAGE blockade could relieve tubular and glomerular damage resulting from diabetes as well as glomerulosclerosis induced by adriamycin (Guo et al., 2008; Matsui et al., 2017; Sanajou et al., 2019). However, whether RAGE inhibition can alleviate cisplatin-induced AKI, mainly characterized by TEC injury, and its underlying mechanisms remain unexplored. Thus, we used RAGE global knockout (RAGE-/-) mice to pursue the role of RAGE in cisplatin-induced AKI in vivo; moreover, we revealed the role of RAGE-specific inhibitor FPS-ZM1 in cisplatin-induced NRK-52E cellular insult in vitro and aimed to further dissect the potential molecular mechanisms.
Materials and Methods
Reagents
Cisplatin (#T1564) is from Topsicence (United States); FPS-ZM1 (#HY-19370) is from MedChemExpress (United States); rabbit anti-RAGE (#ab3611) is from Abcam (United Kingdom); rabbit anti-Bax (#CPA1092), and Bcl-2 (#CPA1095) antibodies are from Cohesion Bioscience (United Kingdom); rabbit anti-Phospho-NF-κB p65 (Ser536) (#3033) and NF-κB p65 (#3034) antibodies are from Cell Signaling Technology (United States); rat anti-F4/80 antibody (#14-4801-82) is from Invitrogen United States; mouse anti-GAPDH (#AC033), rabbit anti-β-actin (#AC026), Cpt1a (#A5307) and PGC-1α (#A12348) and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (#AS014) antibodies are from ABclonal (China). One Step Terminal transferase dUTP nick-end labelling (TUNEL) Apoptosis Assay Kit (#C1090) is from Beyotime (China). SuperKine™ West Femto Maximum Sensitivity Substrate (#BMU102-CN) is from Abbkine (China).
Cell Culture and Treatment
Normal rat renal TEC line NRK-52E is from Center for Excellence in Molecular and Cellular Sciences, Chinese Academy of Sciences (China), routinely cultured with DMEM (#PM150210, Procell, China) containing 5% fetal bovine serum in incubator of 37°C, 5% carbon dioxide. To evaluate the role of FPS-ZM1 in cisplatin-induced cellular insult, NRK-52E cells were pretreated with FPS-ZM1 alone for 6 h, followed by co-treatment with cisplatin for 24 h.
Mice
RAGE-/- mice of C57BL/6J background were kindly shared by Prof. Ben Lu (Central South University, China) (Deng et al., 2018), and the genotypes of the mice was confirmed with real-time PCR and western blot (Supplementary Figure S1). All mice were kept at Xiamen University Laboratory Animal Center (XMULAC) under a 12-h light/dark cycle, provided with standard chow diet and water ad libitum.
Mice were randomized into four groups: 1) wild type (WT) mice receiving vehicle (0.5% sodium carboxymethylcellulose), 2) WT mice receiving cisplatin (dissolved in 0.5% sodium carboxymethylcellulose to form a homogeneous suspension, 20 mg/kg body weight, single intraperitoneal injection), 3) RAGE-/- mice receiving vehicle, and 4) RAGE-/- mice receiving cisplatin. Mice were sacrificed 72 h following cisplatin administration. All mice for experiments were male, SPF grade, 6–8 weeks weighting 18∼22 g. All procedures were approved by the Animal Care and Use Committee of Xiamen University.
Renal Function Assessment
Renal function is indicated by serum creatinine (CREA) and blood urea nitrogen (BUN), both of which are measured by fully automated biochemistry analyzer (#BS-240VET, Mindray, China).
Oil Red O Staining
Frozen sections of renal tissue were maintained in Oil Red O working solution at room temperature for 1 h, washed 3 times with double distilled water, and re-stained with hematoxylin. After being rinsed with tap water, the sections were mounted with glycerol gelatin, observed and photographed under a light microscope (#DM2700 P, Leica, Germany).
Cell Counting Kit-8 Assay
Cells were seeded in 96-well plates (8,000 cells, 100 μl medium per well). After allowed to adhere overnight, cells were pretreated with FPS-ZM1 for 6 h followed by co-treatment with cisplatin for 24 h. Finally, 10 μl CCK-8 solution was added to each well, and the absorbance at 450 nm was detected after 2-h incubation.
Quantitative Real-Time PCR
Total RNA was extracted and purified using RNA extraction kit with mini spin columns (#AG21022, Accurate Biology, China), 2 mg of which was then subject to reverse transcription with Evo M-MLV Mix Kit (#AG11728, Accurate Biology, China). Relative quantification of mRNA was performed on a Real-Time PCR Detection System (#CFX96, Bio-Rad, United States) using SYBR Green Mix (#AH0104-B, SparkJade Biotechnology, China), and then calculated by 2−ΔΔCT method. Primer sequences are listed in Supplementary Table S1.
Western Blot
Western blot assay was performed as previously described (Wang et al., 2021). Briefly, protein lysates from cells or mouse renal cortex were harvested with radioimmunoprecipitation assay buffer (#EA0002, SparkJade Biotechnology, China). Aliquots were electrophoretically separated by 10∼13% SDS-PAGE and transferred to PVDF membranes (#BSP0161, PALL, United States), which were incubated at 4°C overnight with proper primary antibodies followed by HRP-labeled secondary antibody at room temperature for 1 h. Finally, images were captured by Azure C280 system (United States) and analyzed with ImageJ software (National Institutes of Health, United States).
Transmission Electron Microscopy
Briefly, 1 mm3 upper pole renal cortex was sequentially harvested and immobilized in 2.5% neutral glutaraldehyde fixative and 1% osmium acid at 4°C. Tissues were dehydrated in gradient acetone and then embedded, followed by cut into 50 nm ultrathin sections using ultramicrotome. Finally, sections were double stained with uranyl acetate and lead nitrate. Transmission electron microscope (#HT-7800, Hitachi, Japan) was used for mitochondrial observation and image acquisition.
Assessment of Tubular Damage
Fresh murine kidneys were paraffin-embedded after immobilization in 4% paraformaldehyde, and then cut into 5-μm-thick sections for subsequent hematoxylin-eosin (HE) staining and Periodic Acid Schiff (PAS) staining. Tubular injury in PAS-stained sections was rated as per the proportion of damage area: 0 = normal, 1 = 1∼25, 2 = 26∼50, 3 = 51∼75, and 4 = 76∼100%. Finally, TUNEL fluorescence staining was carried out for renal tubular apoptotic cell evaluation according to the manufacturer’s instructions.
Immunohistochemical Staining of F4/80
Immunohistochemistry was performed as previously described (Zou et al., 2016). Briefly, fresh kidney tissues were embedded in paraffin and sectioned into 5 μm following immobilization in paraformaldehyde solution. Then, the sections were deparaffinized, hydrated, antigen-repaired, and endogenous peroxidase activity was abrogated by 3% hydrogen peroxide. Rat anti-F4/80 antibody (1:200) and goat HRP-conjugated anti-rat secondary antibody (1:200) were used in sequence for section incubation. Finally, antigen of F4/80 was localized by chromogenic substrate of DAB working solution.
Statistical Analysis
Date are expressed as mean ± standard error of the mean (SEM) and calculated with One-way or Two-way analysis of variance (ANOVA) when comparing means between groups in the GraphPad Prism software (version 9.0.0, United States). p < 0.05 was considered statistically significant.
Results
RAGE Products Knockout Attenuated Cisplatin-Induced Nephropathy
We first assessed if RAGE blockade could attenuate cisplatin-induced kidney injury using RAGE knockout mice. Mice were subject to intraperitoneal injection of a single dose of cisplatin (20 mg/kg). After 72 h, blood and kidneys were harvested for subsequent examinations. Western blot showed that cisplatin significantly upregulated the protein level of renal RAGE, which was eliminated by RAGE knockout (Figures 1A,B). The macroscopic kidneys of WT mice receiving cisplatin appeared grayish white, indicating the notable nephrotoxicity of cisplatin. The macroscopic kidneys of RAGE-/- mice receiving cisplatin showed significant attenuation of renal graying, suggesting that RAGE knockout may ameliorate the cisplatin-induced nephrotoxicity (Figure 1C). Consistently, HE and PAS staining showed dramatic tubular necrosis triggered by cisplatin, whereas deletion of RAGE significantly diminished the lesion (Figures 1D,E), which was semi-quantified in Figure 1F. Meanwhile, we detected canonical markers of renal injury and discovered that cisplatin significantly upregulated the mRNA level of KIM-1 in kidney, serum concentrations of creatinine (CREA) and blood urea nitrogen (BUN), while RAGE knockout partially reversed these markers (Figures 1G–I). In summary, RAGE knockout attenuated cisplatin-induced renal injury in mice.
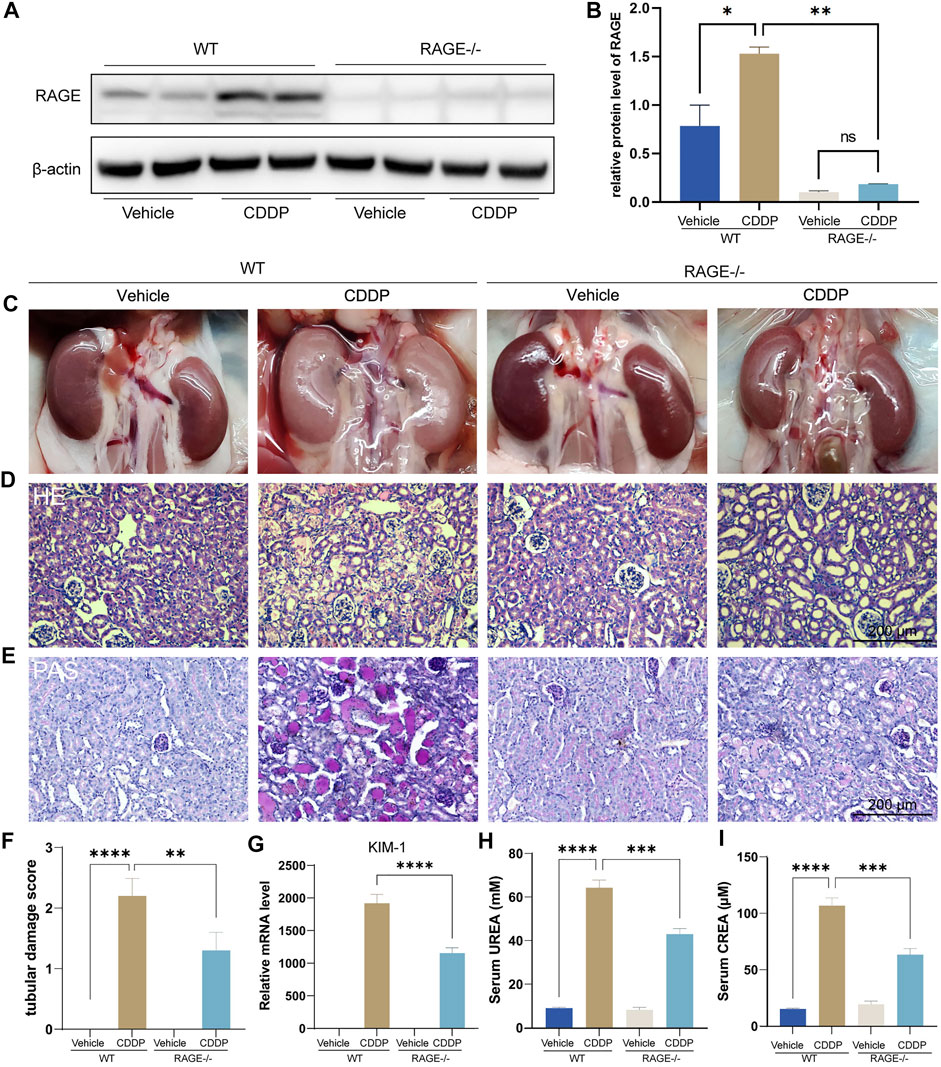
FIGURE 1. RAGE knockout attenuated cisplatin-induced nephropathy. The protein level of RAGE was detected by western blot (A) and semi-quantified in a histogram (B). (C) Representative macroscopic murine kidneys. (D) HE staining of paraffin sections of mouse kidneys. (E) PAS staining and (F) corresponding tubular injury scores of paraffin sections of mouse kidney. (G) Detection of mRNA level of KIM-1, a marker of kidney injury by qPCR. (H,I) Determination of UREA and CREA levels in mice. One-way ANOVA was used to compare means between groups. Data are expressed as mean ± SEM. Scare bar: 200 μm. **p < 0.01, ***p < 001, ****p < 0.0001. WT, wild type; CDDP, cisplatin.
RAGE Products Knockout Reduced Cisplatin-Induced Apoptosis of Nephrocytes
Apoptosis of proximal renal tubules is an essential mechanism of cisplatin-induced nephrotoxicity (Kaushal et al., 2008; Ozkok and Edelstein, 2014; Zhang et al., 2017). To assess if RAGE knockout could exert an effect on cisplatin-induced tubular apoptosis, we performed TUNEL staining and examined mRNA and protein levels of apoptosis-related genes. The number of TUNEL-positive cells (red fluorescence) was significantly increased in WT mice subjected to cisplatin, which was attenuated by RAGE knockout (Figure 2A). Cisplatin decreased the mRNA level of anti-apoptotic gene Bcl-2, increased the mRNA level of pro-apoptotic gene BAX in WT mouse kidneys, and as a result, the Bcl-2/BAX ratio was downregulated, while RAGE knockdown reverted these modifications (Figure 2B). Consistently, RAGE knockout curtailed the overexpression of BAX protein triggered by cisplatin (Figure 2C). Although RAGE knockout failed to rescue the expression of downregulated anti-apoptotic protein Bcl-2, the decreased ratio of Bcl-2/BAX was still restored (Figure 2C). All these were semi-quantified in the Figure 2D. In conclusion, genetic blockade of RAGE reduced cisplatin-induced renal tubular apoptosis.
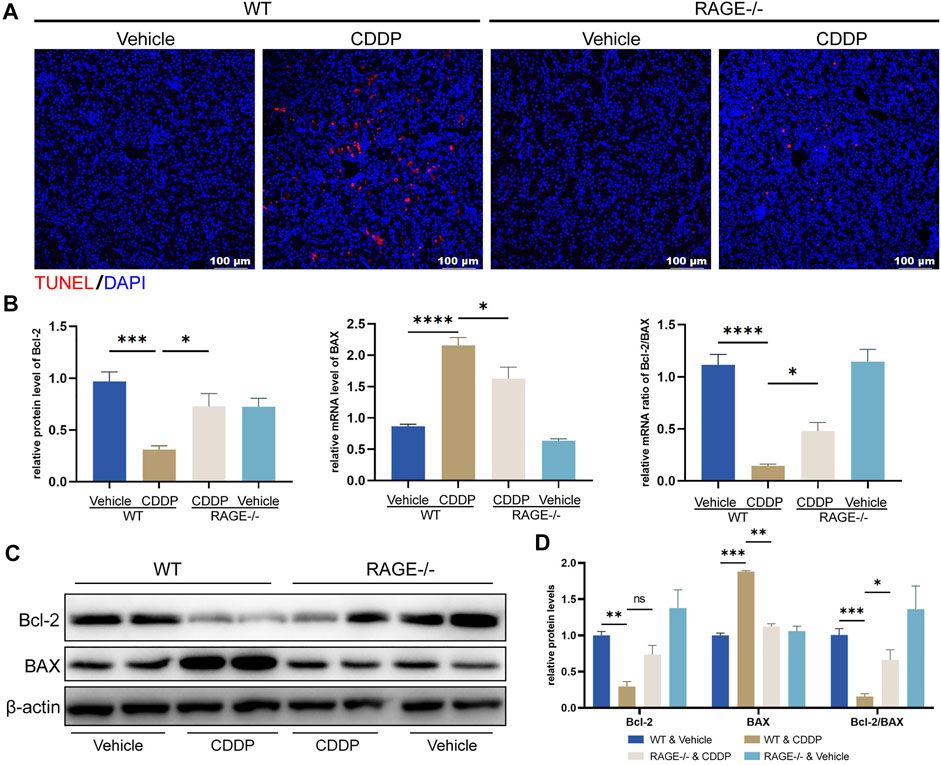
FIGURE 2. RAGE knockout reduced cisplatin-induced apoptosis of nephrocytes. (A) TUNEL staining of paraffin sections of mouse kidneys. (B) The mRNA levels of Bcl-2 and BAX were detected by qPCR, and the ratio of Bcl-2/BAX was calculated. One-way ANOVA was used to compare means between groups. (C) The protein levels of Bcl-2 and BAX were detected by western blot and then semi-quantified in a histogram with Two-way ANOVA (D). Data are expressed as mean ± SEM. Scale bar: 100 μm. *p < 0.05, **p < 0.01, ***p < 001, ****p < 0.0001. ns, not significant.
RAGE Products Knockout Mitigated Cisplatin-Induced Renal Inflammation
Renal inflammation response, especially immune cell infiltration, is a typical mechanism of cisplatin nephrotoxicity (Pabla and Dong, 2008; Manohar and Leung, 2018). To evaluate renal inflammation, we determined the relative transcript levels of pro-inflammatory factors and used immunohistochemistry to assess renal immune infiltration. The results showed that cisplatin markedly upregulated the transcript levels of tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), and cyclooxygenase-2 (COX-2) in the renal cortex tissue and dramatically increased macrophage infiltration (F4/80-positive), while RAGE knockout significantly alleviated these alterations (Figures 3A,B). Given that NF-κB is a central element mediating cisplatin-induced renal inflammation (Ozkok et al., 2016; Yu et al., 2018), We further investigated the expression level of NF-κB p65, and the results showed that both p-p65 and total p65 were significantly upregulated in mice allocated cisplatin. However, overexpression of p-p65 was not fully due to the increase of total p65, because the ratio of p-p65 to total p65 still surged when cisplatin was administrated, which was partly but significantly quenched in RAGE knockout mice (Figures 3C,D). Taken together, RAGE suppression attenuated cisplatin-induced murine renal inflammation.
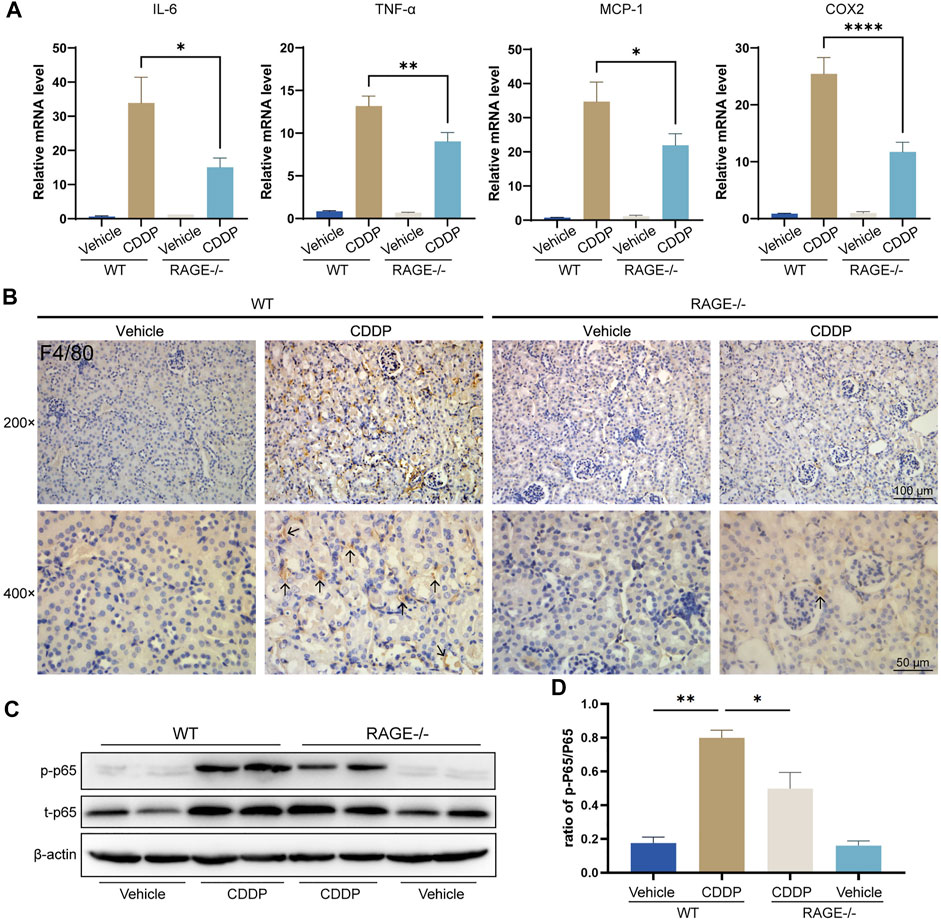
FIGURE 3. RAGE knockout mitigated cisplatin-induced renal inflammation. (A) qPCR detection of mRNA levels of inflammation-related indicators in mouse kidney, including pro-inflammatory factors IL-6 and TNF-α, chemokine MCP-1, and COX-2. (B) Immunohistochemical staining of mouse kidney paraffin sections. Brown marked by arrows means F4/80-positive, which indicates infiltration of macrophages. (C) The protein levels of phosphorylated p65 and total p65 were detected by western blot and then semi-quantified in a histogram (D). One-way ANOVA was used to compare means between groups. Data are expressed as mean ± SEM. Scale bar: 100 or 50 μm as indicated in the figure. *p < 0.05, **p < 01, ****p < 0.0001. p, phosphorylated; t, total.
RAGE Products Knockout Restored Cisplatin-Induced Mitochondrial Homeostasis
Mitochondrial homeostasis is essential for the survival of renal TECs (Szeto, 2017; Wang et al., 2020). Cisplatin significantly disturbs the mitochondrial homeostasis of renal TECs, resulting in nephrotoxicity (Wang et al., 2018; Lu et al., 2020). To determine if RAGE deficiency restores mitochondrial homeostasis in the kidney of mice subject to cisplatin, we performed TEM. Mitochondrial morphology of renal TECs in RAGE-/- mice was comparable to that of WT mice. After cisplatin administration for 3 days, WT mice showed significant swelling and reduced number of mitochondria as well as disappearance of mitochondrial cristae (Figures 4A,B). Similar alterations were indicated by mitochondrial copy number according to the qPCR result (Figure 4C). Considering that PGC-1α is a central regulator of mitochondrial biosynthesis (Qian et al., 2019; Fontecha-Barriuso et al., 2020; Popov, 2020), we further examined its mRNA and protein levels and found that RAGE knockout rescued the cisplatin-induced PGC-1α suppression (Figures 4D–F). Collectively, RAGE knockout restored cisplatin-induced mitochondrial homeostasis.
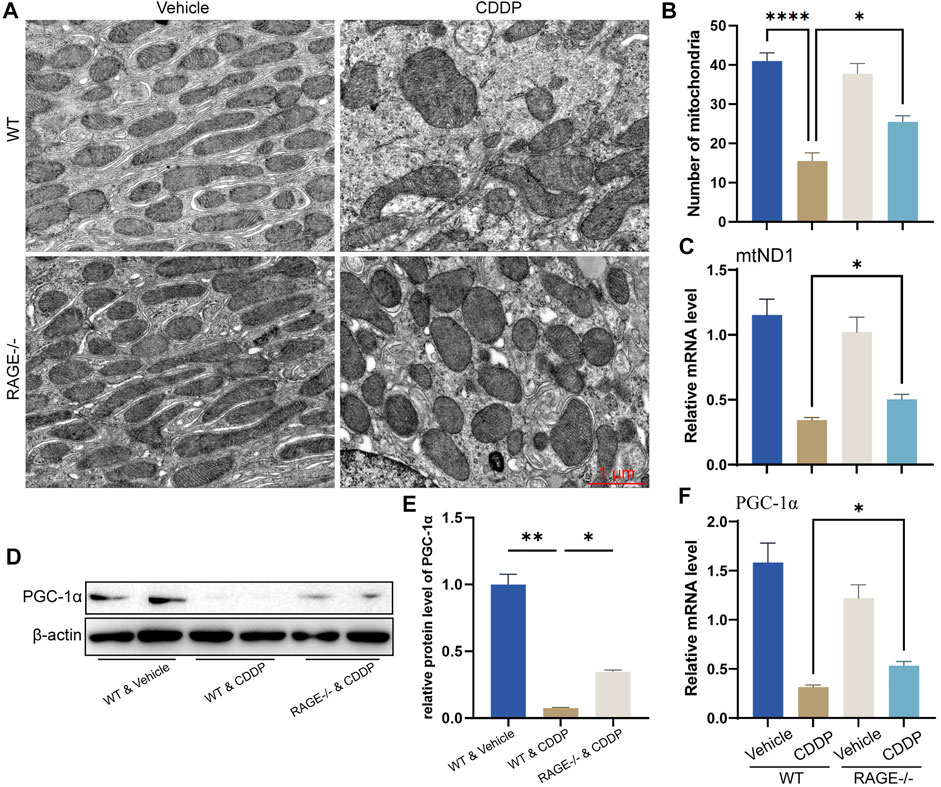
FIGURE 4. RAGE knockout restored cisplatin-induced mitochondrial homeostasis. (A) Mitochondrial morphology of mouse renal tubular epithelial cells was observed using transmission electron microscopy (TEM). (B) The number of mitochondria was counted according to TEM. (C) The mRNA level of mtND1, indicating the relative copy number of cellular mitochondria. (D) The protein level of PGC-1α was detected by western blot and then semi-quantified in a histogram (E). (F) The mRNA level of PGC-1α determined by qPCR. One-way ANOVA was used to compare means between groups. Data are expressed as mean ± SEM. Scale bar: 1 μm. *p < 0.05, **p < 01, ****p < 0.0001.
RAGE Products Knockout Diminished Cisplatin-Induced Renal Lipid Accumulation and FAO Impairment
FAO is the primary energy source for renal TECs (Kang et al., 2015). Cisplatin blocks this process, thereby preventing cellular access to energy and ultimately causing lipid accumulation and cell injury (Li et al., 2020). Oil Red O staining showed that TECs of WT mice receiving cisplatin exhibited significant lipid accumulation, while RAGE knockout significantly attenuated this alteration (Figure 5A). Further qPCR results revealed a significant reduction in transcription levels of FAO-related genes Ehhadh and Cpt1a in WT mice receiving cisplatin and a marked improvement in the RAGE-/- mice receiving cisplatin (Figure 5B). Cpt1a was taken for further validation at the protein level (Figures 5C,D). In summary, RAGE knockout diminished cisplatin-induced renal lipid accumulation and FAO impairment.
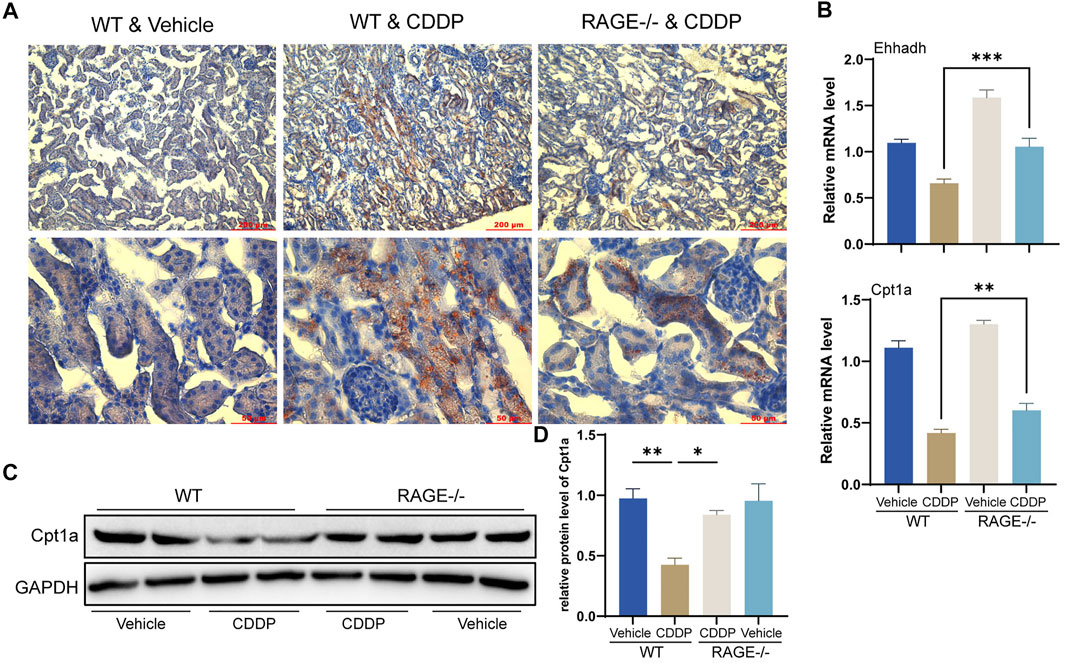
FIGURE 5. RAGE knockout diminished cisplatin-induced renal lipid accumulation and FAO impairment. (A) Oil Red O staining was performed to assess lipid deposition in the kidney tissues. (B) The histogram indicated mRNA levels of FAO-related genes Ehhadh and Cpt1a. (C) Cpt1a was selected for validation of protein expression with subsequent semi-quantification (D). One-way ANOVA was used to compare means between groups. Data are expressed as mean ± SEM. Scale bar: 200 or 50 μm as indicated in the figure. *p < 0.05, **p < 0.01, ***p < 0.001.
The RAGE Products‐Specific Inhibitor FPS-ZM1 Restored Cisplatin-Suppressed NRK-52E Cell Viability
Nephroprotection of RAGE deficiency was verified on NRK-52E cells with FPS-ZM1, a specific inhibitor of RAGE (Deane et al., 2012). First, we confirmed upregulation of RAGE induced by cisplatin and inhibitory effect of FPS-ZM1 on RAGE at mRNA and protein levels in NRK-52E cells (Figures 6A–C). Then, we showed that FPS-ZM1 at no more than 20 μM was not toxic to NRK-52E cells (Supplementary Figure S2); cisplatin reduced cell viability, which was visibly and statistically reversed by FPS-ZM1 (Figures 6D,E). The inhibitory effect of FPS-ZM1 on apoptosis was further illustrated by the modification in the expression level of cleaved Caspase-3 protein (Figures 6F,G). These results are in agreement with the aforementioned findings of animal experiments, further suggesting that inhibition of RAGE directly attenuates the toxicity of cisplatin on renal TECs.

FIGURE 6. The RAGE-specific inhibitor FPS-ZM1 restored cisplatin-suppressed NRK-52E cell viability. The protein level of RAGE in NRK-52E cells was accessed by western blot (A) and semi-quantified (B). (C) The mRNA level of RAGE in NRK-52E cells determined by qPCR. (D) The viability status of NRK-52E cells observed under a microscope. (E) Quantification of cell viability by CCK-8. The protein level of RAGE cleaved caspase 3 (F) was assessed by western blot, followed by semi-quantification (G). One-way ANOVA was used to compare means between groups. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; FPS, FPS-ZM1.
FPS-ZM1 Diminished Cisplatin-Induced Lipid Accumulation and FAO Impairment of NRK-52E Cells
To evaluate and validate the effect of FPS-ZM1 on lipid metabolism in NRK-52E cells, we performed Oil red O staining and observed that cisplatin prominently elicited cellular lipid deposition, which was significantly restrained by FPS-ZM1 (Figure 7A). This is consistent with the in vivo findings. Further qPCR results showed that cisplatin significantly inhibited FAO in NRK-52E cells, while FPS-ZM1 blocked this process, thereby inhibiting lipid deposition (Figure 7B). In addition, we also evaluated the synthesis rate of free fatty acid. Unexpectedly, cisplatin significantly halted the fatty acid synthesis capacity of NRK-53E cells indicated by reduced mRNA levels of ACC, SCD1 and FASN genes, while FPS-ZM1 dramatically restored their mRNA levels (Figure 7C). Collectively, FPS-ZM1 diminished cisplatin-induced lipid accumulation and FAO impairment of NRK-52E cells.
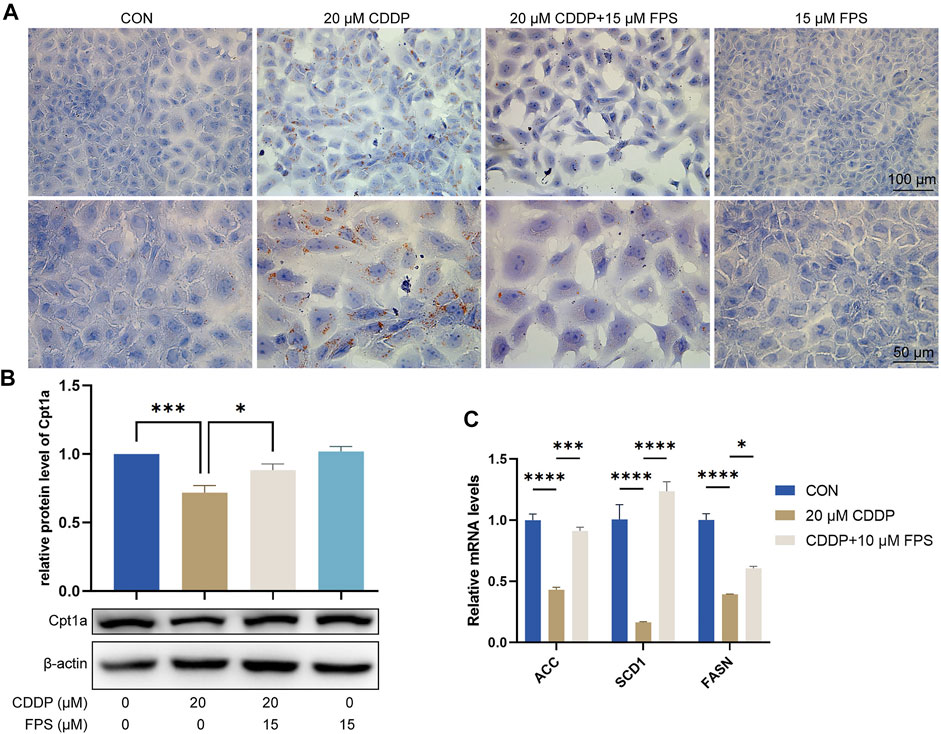
FIGURE 7. FPS-ZM1 diminished cisplatin-induced renal lipid accumulation and FAO impairment. (A) Photomicrographs illustrated lipid deposition in NRK-52E cells by using Oil red O staining. (B) Protein expression profiling of Cpt1a by western blot. One-way ANOVA was used to compare means between groups. (C) The mRNA levels of fatty acid synthesis-related genes ACC, SCD1, and FASN determined by qPCR. Two-way ANOVA was used to compare means between groups. Data are expressed as mean ± SEM. Scale bar: 100 or 50 μm as indicated in the figure. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Discussion
Cisplatin is a widely used anticancer agent, yet frequently accompanied by nephrotoxicity with elusive mechanisms that may involve mitochondrial damage, impaired FAO, oxidative stress, endoplasmic reticulum stress, inflammation, apoptosis, necroptosis, etc. RAGE is a multiligand pattern recognition receptor, engaged in the regulation of inflammation, apoptosis, and FAO, and expressed in multiple cells, including renal TECs (Morcos et al., 2002). Previous studies have showed that RAGE involves epithelial-mesenchymal transition of renal TECs and adriamycin-induced glomerulosclerosis (Oldfield et al., 2001; Guo et al., 2008), which led us to decipher the role of RAGE in cisplatin nephrotoxicity.
The present study revealed for the first time that RAGE knockout significantly attenuated cisplatin-induced nephrotoxicity, as evidenced by reduced renal apoptosis, inflammation, lipid accumulation, as well as restored mitochondrial homeostasis and FAO. In vitro experiments showed that the RAGE-specific inhibitor FPS-ZM1 counteracted the inhibitory effect of cisplatin on cell viability and FAO of the rat renal TEC line NRK-52E. These findings elaborate the essential role of RAGE in cisplatin nephrotoxicity, suggesting that RAGE inhibition holds promise as a new therapeutic strategy to mitigate cisplatin nephropathy.
Renal TECs are highly susceptible to apoptosis, which renders apoptosis an important mechanism of cisplatin nephrotoxicity (Havasi and Borkan, 2011). Pharmacological inhibition or gene knockout of RAGE could alleviate apoptosis in renal cells in a diverse range of settings (Zhou et al., 2012; Hagiwara et al., 2018; Mao et al., 2018). We therefore investigated if RAGE deletion could attenuate cisplatin-induced apoptosis in renal TECs. Both in vivo and in vitro experiments suggested that RAGE suppression restored cisplatin-induced apoptosis in renal TECs.
Cisplatin leads to renal inflammation (Hsing et al., 2021; Imig et al., 2021). NF-κB is a canonical signal that mediates renal inflammation (Sanz et al., 2010; Ozkok et al., 2016; Liu et al., 2017). RAGE can mediate inflammatory signaling by regulating NF-κB (Volz et al., 2012; Ali et al., 2015; Dwir et al., 2020). Our results showed that RAGE deficiency reduced cisplatin-activated NF-κB and decreased the transcription of NF-κB downstream pro-inflammatory factors TNF-α and IL-6, chemokine MCP-1, and COX-2, as well as macrophage infiltration in the kidney. These results suggest that RAGE/NF-κB signaling may mediate cisplatin-induced renal inflammation.
Mitochondrial homeostasis is essential for the survival of renal TECs (Yu et al., 2018; Wang et al., 2020), whereas prone to impairment by cisplatin (Szeto, 2017; Lu et al., 2020). Studies indicate that modulation of RAGE improves mitochondrial injury (Yu et al., 2017; Mao et al., 2018; Syed et al., 2020). We revealed that RAGE knockout reversed the decrease in mitochondrial number of renal TECs caused by cisplatin. These data further suggest a link between RAGE and mitochondrial homeostasis.
Apart from being a source of energy, fatty acids are also engaged in the formation of mitochondrial membrane phospholipids. Calcium-independent Phospholipase A2γ can repair damaged mitochondrial membrane phospholipids by hydrolyzing damaged acyl chains to make them re-esterify with fatty acids and thus maintain mitochondrial survival and function, including FAO (Elimam et al., 2013).
FAO is the primary source of energy for renal TECs (Kang et al., 2015), but susceptible to inhibition by cisplatin, which leads to failed access to sufficient energy for cells and ultimately causes lipid accumulation and cellular insult (Jang et al., 2020). Previous studies suggest that RAGE is implicated in the regulation of FAO (Wan et al., 2020). We revealed that knockout of RAGE halted cisplatin-induced lipid accumulation and FAO impairment in murine kidney, which was further confirmed by in vitro experiments. Given that FAO mainly occurs in mitochondria, it is consistent with the aforementioned results and further elaborates a tight link between RAGE and mitochondria. Finally, we unexpectedly identified a restorative effect of FPS-ZM1 on fatty acid synthesis capacity. Considering that the predominant energy source of renal TECs is lipids rather than glucose (Kang et al., 2015), NRK-52E cells may need to convert glucose to lipids before oxidizing it for energy supply. This may explain why FPS-ZM1 attenuates lipid accumulation in cells when restores fatty acid synthesis, i.e., it facilitates both the supply and utilization of the energy substance, lipids. Of course, further studies are warranted to confirm this.
In conclusion, RAGE deficiency ameliorates cisplatin nephrotoxicity by reducing apoptosis, inflammation, and restoring FAO in TECs. It suggests that RAGE may serve as a promising therapeutic target for the treatment of cisplatin nephrotoxicity.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Animal Care and Use Committee of Xiamen University.
Author Contributions
JC designed the experiments. QW, YX, BC, HZ, WY, DX, WL, FH, and CX performed the experiments. QW, YX, and BC analyzed the data. JC and QW wrote the article.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81570772 and 81703742), the Natural Science Foundation of Fujian Province (2020J01018) and the Gout Research Foundation (Japan, 2019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.907133/full#supplementary-material
References
Adeshara, K. A., Agrawal, S. B., Gaikwad, S. M., and Tupe, R. S. (2018). Pioglitazone Inhibits Advanced Glycation Induced Protein Modifications and Down-Regulates Expression of RAGE and NF-Κb in Renal Cells. Int. J. Biol. Macromol. 119, 1154–1163. doi:10.1016/j.ijbiomac.2018.08.026
Ali, T., Badshah, H., Kim, T. H., and Kim, M. O. (2015). Melatonin Attenuates D-Galactose-Induced Memory Impairment, Neuroinflammation and Neurodegeneration via RAGE/NF-K B/JNK Signaling Pathway in Aging Mouse Model. J. Pineal Res. 58 (1), 71–85. doi:10.1111/jpi.12194
Chiba, T., Peasley, K. D., Cargill, K. R., Maringer, K. V., Bharathi, S. S., Mukherjee, E., et al. (2019). Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect against AKI. J. Am. Soc. Nephrol. 30 (12), 2384–2398. doi:10.1681/ASN.2019020163
Coughlan, M. T., Thorburn, D. R., Penfold, S. A., Laskowski, A., Harcourt, B. E., Sourris, K. C., et al. (2009). RAGE-induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. J. Am. Soc. Nephrol. 20 (4), 742–752. doi:10.1681/asn.2008050514
Deane, R., Singh, I., Sagare, A. P., Bell, R. D., Ross, N. T., LaRue, B., et al. (2012). A Multimodal RAGE-specific Inhibitor Reduces Amyloid β-mediated Brain Disorder in a Mouse Model of Alzheimer Disease. J. Clin. Invest 122 (4), 1377–1392. doi:10.1172/jci58642
Deng, M., Tang, Y., Li, W., Wang, X., Zhang, R., Zhang, X., et al. (2018). The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-dependent Lethality in Sepsis. Immunity 49 (4), 740–e7. e747. doi:10.1016/j.immuni.2018.08.016
Dwir, D., Giangreco, B., Xin, L., Tenenbaum, L., Cabungcal, J. H., Steullet, P., et al. (2020). Correction: MMP9/RAGE Pathway Overactivation Mediates Redox Dysregulation and Neuroinflammation, Leading to Inhibitory/excitatory Imbalance: a Reverse Translation Study in Schizophrenia Patients. Mol. Psychiatry 25 (11), 3105–2904. doi:10.1038/s41380-019-0393-510.1038/s41380-020-0716-6
Elimam, H., Papillon, J., Takano, T., and Cybulsky, A.V. (2013). Complement‐mediated Activation of Calcium-independent Phospholipase A2γ: Role of Protein Kinases and Phosphorylation. J. Biol. Chem. 288 (6), 3871–3885. doi:10.1074/jbc.M112.396614
Fontecha-Barriuso, M., Martin-Sanchez, D., Martinez-Moreno, J. M., Monsalve, M., Ramos, A. M., Sanchez-Niño, M. D., et al. (2020). The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 10 (2). doi:10.3390/biom10020347
Guo, J., Ananthakrishnan, R., Qu, W., Lu, Y., Reiniger, N., Zeng, S., et al. (2008). RAGE Mediates Podocyte Injury in Adriamycin-Induced Glomerulosclerosis. J. Am. Soc. Nephrol. 19 (5), 961–972. doi:10.1681/ASN.2007101109
Hagiwara, S., Sourris, K., Ziemann, M., Tieqiao, W., Mohan, M., McClelland, A. D., et al. (2018). RAGE Deletion Confers Renoprotection by Reducing Responsiveness to Transforming Growth Factor-β and Increasing Resistance to Apoptosis. Diabetes 67 (5), 960–973. doi:10.2337/db17-0538
Havasi, A., and Borkan, S. C. (2011). Apoptosis and Acute Kidney Injury. Kidney Int. 80 (1), 29–40. doi:10.1038/ki.2011.120
Hsing, C.-H., Tsai, C.-C., Chen, C.-L., Lin, Y.-H., Tseng, P.-C., Satria, R. D., et al. (2021). Pharmacologically Inhibiting Glycogen Synthase Kinase-3β Ameliorates Renal Inflammation and Nephrotoxicity in an Animal Model of Cisplatin-Induced Acute Kidney Injury. Biomedicines 9 (8), 887. doi:10.3390/biomedicines9080887
Imig, J. D., Hye Khan, M. A., Burkhan, A., Chen, G., Adebesin, A. M., and Falck, J. R. (2021). Kidney-Targeted Epoxyeicosatrienoic Acid Analog, EET-F01, Reduces Inflammation, Oxidative Stress, and Cisplatin-Induced Nephrotoxicity. Int. J. Mol. Sci. 22 (6). doi:10.3390/ijms22062793
Jang, H. S., Noh, M. R., Jung, E. M., Kim, W. Y., Southekal, S., Guda, C., et al. (2020). Proximal Tubule Cyclophilin D Regulates Fatty Acid Oxidation in Cisplatin-Induced Acute Kidney Injury. Kidney Int. 97 (2), 327–339. doi:10.1016/j.kint.2019.08.019
Kang, H. M., Ahn, S. H., Choi, P., Ko, Y. A., Han, S. H., Chinga, F., et al. (2015). Defective Fatty Acid Oxidation in Renal Tubular Epithelial Cells Has a Key Role in Kidney Fibrosis Development. Nat. Med. 21 (1), 37–46. doi:10.1038/nm.3762
Kaushal, G. P., Kaushal, V., Herzog, C., and Yang, C. (2008). Autophagy Delays Apoptosis in Renal Tubular Epithelial Cells in Cisplatin Cytotoxicity. Autophagy 4 (5), 710–712. doi:10.4161/auto.6309
Li, M., Li, C. M., Ye, Z. C., Huang, J., Li, Y., Lai, W., et al. (2020). Sirt3 Modulates Fatty Acid Oxidation and Attenuates Cisplatin-Induced AKI in Mice. J. Cell Mol. Med. 24 (9), 5109–5121. doi:10.1111/jcmm.15148
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017). NF-κB Signaling in Inflammation. Signal Transduct. Target Ther. 2. doi:10.1038/sigtrans.2017.23
Lu, Q., Wang, M., Gui, Y., Hou, Q., Gu, M., Liang, Y., et al. (2020). Rheb1 Protects against Cisplatin-Induced Tubular Cell Death and Acute Kidney Injury via Maintaining Mitochondrial Homeostasis. Cell Death Dis. 11 (5), 364. doi:10.1038/s41419-020-2539-4
Manohar, S., and Leung, N. (2018). Cisplatin Nephrotoxicity: a Review of the Literature. J. Nephrol. 31 (1), 15–25. doi:10.1007/s40620-017-0392-z
Mao, Y. X., Cai, W. J., Sun, X. Y., Dai, P. P., Li, X. M., Wang, Q., et al. (2018). RAGE-dependent Mitochondria Pathway: a Novel Target of Silibinin against Apoptosis of Osteoblastic Cells Induced by Advanced Glycation End Products. Cell Death Dis. 9 (6), 674. doi:10.1038/s41419-018-0718-3
Mapuskar, K. A., Steinbach, E. J., Zaher, A., Riley, D. P., Beardsley, R. A., Keene, J. L., et al. (2021). Mitochondrial Superoxide Dismutase in Cisplatin-Induced Kidney Injury. Antioxidants (Basel) 10 (9). doi:10.3390/antiox10091329
Matsui, T., Higashimoto, Y., Nishino, Y., Nakamura, N., Fukami, K., and Yamagishi, S. I. (2017). RAGE-aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 66 (6), 1683–1695. doi:10.2337/db16-1281
McSweeney, K. R., Gadanec, L. K., Qaradakhi, T., Ali, B. A., Zulli, A., and Apostolopoulos, V. (2021). Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers (Basel) 13 (7). doi:10.3390/cancers13071572
Morcos, M., Sayed, A. A., Bierhaus, A., Yard, B., Waldherr, R., Merz, W., et al. (2002). Activation of Tubular Epithelial Cells in Diabetic Nephropathy. Diabetes 51 (12), 3532–3544. doi:10.2337/diabetes.51.12.3532
Oldfield, M. D., Bach, L. A., Forbes, J. M., Nikolic-Paterson, D., McRobert, A., Thallas, V., et al. (2001). Advanced Glycation End Products Cause Epithelial-Myofibroblast Transdifferentiation via the Receptor for Advanced Glycation End Products (RAGE). J. Clin. Invest 108 (12), 1853–1863. doi:10.1172/jci11951
Ozkok, A., Ravichandran, K., Wang, Q., Ljubanovic, D., and Edelstein, C. L. (2016). NF-κB Transcriptional Inhibition Ameliorates Cisplatin-Induced Acute Kidney Injury (AKI). Toxicol. Lett. 240 (1), 105–113. doi:10.1016/j.toxlet.2015.10.028
Ozkok, A., and Edelstein, C. L. (2014). Pathophysiology of Cisplatin-Induced Acute Kidney Injury. BioMed Res. Int. 2014, 967826. doi:10.1155/2014/967826
Pabla, N., and Dong, Z. (2008). Cisplatin Nephrotoxicity: Mechanisms and Renoprotective Strategies. Kidney Int. 73 (9), 994–1007. doi:10.1038/sj.ki.5002786
Popov, L. D. (2020). Mitochondrial Biogenesis: An Update. J. Cell Mol. Med. 24 (9), 4892–4899. doi:10.1111/jcmm.15194
Qian, X., Li, X., Shi, Z., Bai, X., Xia, Y., Zheng, Y., et al. (2019). KDM3A Senses Oxygen Availability to Regulate PGC-1α-Mediated Mitochondrial Biogenesis. Mol. Cell 76 (6), 885–e7. e887. doi:10.1016/j.molcel.2019.09.019
Ronco, C., Bellomo, R., and Kellum, J. A. (2019). Acute Kidney Injury. Lancet 394 (10212), 1949–1964. doi:10.1016/s0140-6736(19)32563-2
Sanajou, D., Ghorbani Haghjo, A., Argani, H., Roshangar, L., Rashtchizadeh, N., Ahmad, S. N. S., et al. (2019). Reduction of Renal Tubular Injury with a RAGE Inhibitor FPS-ZM1, Valsartan and Their Combination in Streptozotocin-Induced Diabetes in the Rat. Eur. J. Pharmacol. 842, 40–48. doi:10.1016/j.ejphar.2018.10.035
Sanz, A. B., Sanchez-Niño, M. D., Ramos, A. M., Moreno, J. A., Santamaria, B., Ruiz-Ortega, M., et al. (2010). NF-kappaB in Renal Inflammation. J. Am. Soc. Nephrol. 21 (8), 1254–1262. doi:10.1681/asn.2010020218
Song, F., Hurtado del Pozo, C., Rosario, R., Zou, Y. S., Ananthakrishnan, R., Xu, X., et al. (2014). RAGE Regulates the Metabolic and Inflammatory Response to High-Fat Feeding in Mice. Diabetes 63 (6), 1948–1965. doi:10.2337/db13-1636
Syed, A. A., Reza, M. I., Shafiq, M., Kumariya, S., Singh, P., Husain, A., et al. (2020). Naringin Ameliorates Type 2 Diabetes Mellitus-Induced Steatohepatitis by Inhibiting RAGE/NF-κB Mediated Mitochondrial Apoptosis. Life Sci. 257, 118118. doi:10.1016/j.lfs.2020.118118
Szeto, H. H. (2017). Pharmacologic Approaches to Improve Mitochondrial Function in AKI and CKD. J. Am. Soc. Nephrol. 28 (10), 2856–2865. doi:10.1681/ASN.2017030247
Volz, H. C., Laohachewin, D., Seidel, C., Lasitschka, F., Keilbach, K., Wienbrandt, A. R., et al. (2012). S100A8/A9 Aggravates Post-ischemic Heart Failure through Activation of RAGE-dependent NF-Κb Signaling. Basic Res. Cardiol. 107 (2), 250. doi:10.1007/s00395-012-0250-z
Wan, J., Wu, X., Chen, H., Xia, X., Song, X., Chen, S., et al. (2020). Aging-induced Aberrant RAGE/PPARα axis Promotes Hepatic Steatosis via Dysfunctional Mitochondrial β Oxidation. Aging Cell 19 (10), e13238. doi:10.1111/acel.13238
Wang, J., Zhu, P., Li, R., Ren, J., Zhang, Y., and Zhou, H. (2020). Bax Inhibitor 1 Preserves Mitochondrial Homeostasis in Acute Kidney Injury through Promoting Mitochondrial Retention of PHB2. Theranostics 10 (1), 384–397. doi:10.7150/thno.40098
Wang, Q., Zhao, H., Gao, Y., Lu, J., Xie, D., Yu, W., et al. (2021). Uric Acid Inhibits HMGB1-TLR4-NF-Κb Signaling to Alleviate Oxygen-Glucose Deprivation/reoxygenation Injury of Microglia. Biochem. Biophys. Res. Commun. 540, 22–28. doi:10.1016/j.bbrc.2020.12.097
Wang, Y., Tang, C., Cai, J., Chen, G., Zhang, D., Zhang, Z., et al. (2018). PINK1/Parkin-mediated Mitophagy Is Activated in Cisplatin Nephrotoxicity to Protect against Kidney Injury. Cell Death Dis. 9 (11), 1113. doi:10.1038/s41419-018-1152-2
Xu, Y., Ma, H., Shao, J., Wu, J., Zhou, L., Zhang, Z., et al. (2015). A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 26 (11), 2647–2658. doi:10.1681/ASN.2014080741
Yu, X., Meng, X., Xu, M., Zhang, X., Zhang, Y., Ding, G., et al. (2018). Celastrol Ameliorates Cisplatin Nephrotoxicity by Inhibiting NF-Κb and Improving Mitochondrial Function. EBioMedicine 36, 266–280. doi:10.1016/j.ebiom.2018.09.031
Yu, Y., Wang, L., Delguste, F., Durand, A., Guilbaud, A., Rousselin, C., et al. (2017). Advanced Glycation End Products Receptor RAGE Controls Myocardial Dysfunction and Oxidative Stress in High-Fat Fed Mice by Sustaining Mitochondrial Dynamics and Autophagy-Lysosome Pathway. Free Radic. Biol. Med. 112, 397–410. doi:10.1016/j.freeradbiomed.2017.08.012
Zahedi, K., Barone, S., Destefano-Shields, C., Brooks, M., Murray-Stewart, T., Dunworth, M., et al. (2017). Activation of Endoplasmic Reticulum Stress Response by Enhanced Polyamine Catabolism Is Important in the Mediation of Cisplatin-Induced Acute Kidney Injury. PLoS One 12 (9), e0184570. doi:10.1371/journal.pone.0184570
Zhang, D., Pan, J., Xiang, X., Liu, Y., Dong, G., Livingston, M. J., et al. (2017). Protein Kinase Cδ Suppresses Autophagy to Induce Kidney Cell Apoptosis in Cisplatin Nephrotoxicity. J. Am. Soc. Nephrol. 28 (4), 1131–1144. doi:10.1681/asn.2016030337
Zhou, L. L., Cao, W., Xie, C., Tian, J., Zhou, Z., Zhou, Q., et al. (2012). The Receptor of Advanced Glycation End Products Plays a Central Role in Advanced Oxidation Protein Products-Induced Podocyte Apoptosis. Kidney Int. 82 (7), 759–770. doi:10.1038/ki.2012.184
Keywords: rage, cisplatin-induced nephrotoxicity, apoptosis, inflammation, fatty acid oxidation
Citation: Wang Q, Xi Y, Chen B, Zhao H, Yu W, Xie D, Liu W, He F, Xu C and Cheng J (2022) Receptor of Advanced Glycation End Products Deficiency Attenuates Cisplatin-Induced Acute Nephrotoxicity by Inhibiting Apoptosis, Inflammation and Restoring Fatty Acid Oxidation. Front. Pharmacol. 13:907133. doi: 10.3389/fphar.2022.907133
Received: 29 March 2022; Accepted: 12 May 2022;
Published: 30 May 2022.
Edited by:
Edgar Jaimes, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Yunwen Yang, Nanjing Children’s Hospital, ChinaHanan Elimam, University of Sadat City, Egypt
Copyright © 2022 Wang, Xi, Chen, Zhao, Yu, Xie, Liu, He, Xu and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jidong Cheng, amlkb25nY2hlbmczNkBob3RtYWlsLmNvbQ==