 Haibin Wei1,2,3†
Haibin Wei1,2,3† Yujie Yin
Yujie Yin Wenwen Yang
Wenwen Yang Jinyan Zhu
Jinyan Zhu Lin Chen
Lin Chen Rui Guo
Rui Guo Songtao Li
Songtao Li- 1School of Public Health, Zhejiang Chinese Medical University, Hangzhou, China
- 2Department of Biobank, Zhejiang Cancer Hospital, Hangzhou, Zhejiang, China
- 3School of Life Science, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China
- 4Department of Clinical Nutrition, Affiliated Zhejiang Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China
Pro-inflammatory factor-associated vascular cell adhesion molecule 1 (VCAM1) activation initiates cardiovascular events. This study aimed to explore the protective role of nuciferine on TNFα-induced VCAM1 activation. Nuciferine was administrated to both high-fat diet (HFD)-fed mice and the TNFα-exposed human vascular endothelial cell line. VCAM1 expression and further potential mechanism(s) were explored. Our data revealed that nuciferine intervention alleviated VCAM1 activation in response to both high-fat diet and TNFα exposure, and this protective effect was closely associated with autophagy activation since inhibiting autophagy by either genetic or pharmaceutical approaches blocked the beneficial role of nuciferine. Mechanistical studies revealed that Akt/mTOR inhibition, rather than AMPK, SIRT1, and p38 signal pathways, contributed to nuciferine-activated autophagy, which further ameliorated TNFα-induced VCAM1 via repressing AP1 activation, independent of transcriptional regulation by IRF1, p65, SP1, and GATA6. Collectively, our data uncovered a novel biological function for nuciferine in protecting VCAM1 activation, implying its potential application in improving cardiovascular events.
Introduction
Vascular cell adhesion molecule 1 (VCAM1) is a transmembrane protein and consists of an intracellular (C-terminal) and extracellular (N-terminal) portion. The extracellular portion contains several immunoglobulin (Ig)-like domains, which are responsible for tight adhesion with immune cells through the interaction with α4β1, α4β7, α9β1, and αDβ2 integrins (Elices et al., 1990; Ulyanova et al., 2005). VCAM1 rarely expresses in endothelial cells under physiological conditions. However, it could be quickly induced/activated directly or indirectly by various stimuli, such as pro-inflammatory factors [e.g., tumor necrosis factor alpha (TNFα), interleukin-1 beta (IL-1β), and interferon-gamma (IFN-γ)] (Carlos et al., 1990; Neish et al., 1992; Paleolog et al., 1992), oxidative stresses [e.g., reactive oxygen species (ROS) and oxidative modification of low-density lipoprotein (ox-LDL)] (Yoshida et al., 2000; Lee et al., 2007; Zhu et al., 2008), and infections (e.g., coronavirus disease 2019, human immunodeficiency virus, and human cytomegalovirus) (Liu et al., 2005; Zhao et al., 2018; Robles et al., 2022). Endothelial dysfunction and ongoing cardiovascular inflammation are caused by immune cells penetrating the arterial wall as a result of activated VCAM1 on endothelial cells, which attracts leukocytes, monocytes, and neutrophils (Goswami et al., 2021). Unsettled cardiovascular inflammation further aggravates VCAM1 activation, which leads to the initiation of cardiovascular events. Accumulated clinical and animal studies have shown that VCAM1 is activated in different cardiovascular events, such as atherosclerosis, stroke, and heart failure (Troncoso et al., 2021). Cardiovascular events are the leading causes of death globally (Khoury et al., 2021), but effective preventions or interventions are still limited (Lu and Thum, 2019). There is an urgent need for studies that try to identify novel therapeutic approaches. As a reversible process, VCAM1 activation is recognized as an ideal therapeutic target for the prevention/intervention of cardiovascular events at an early stage or other endothelial dysfunction-related diseases (Carter et al., 2002; Park et al., 2013; Furuta et al., 2021).
Nelumbo nucifera Gaertn (also known as lotus) is an edible medicinal plant recorded by both Ayurveda and Chinese traditional medicine (Wang et al., 2015). Lotus is constituted mainly by lotus leaves, flower, rhizomes, and seeds, among which lotus leaf is commonly consumed as tea and medicine. Nuciferine (C19H21NO2) is the main bioactive component obtained from lotus leaves. As an aporphine alkaloid, nuciferine has been reported with extensive pharmacological functions, including anti-inflammatory, anti-oxidant, anti-obesity, anti-hepatic steatosis, and anti-tumor properties (Wu et al., 2017; Bai et al., 2022; Du et al., 2022). Since inflammation, oxidative stress, and obesity are risk factors for VCAM1 activation and further cardiovascular events (Ross, 1999; Davis et al., 2011; Yannakoulia and Panagiotakos, 2021), we proposed that nuciferine protects pro-inflammatory factor-induced activation of VCAM1. To the best of our knowledge, limited study has been conducted to investigate the beneficial role of nuciferine against VCAM1 activation. In the present study, we reported for the first time that nuciferine intervention improved both high-fat diet (HFD) and TNFα-induced VCAM1 activation. Further mechanistic study revealed that nuciferine-stimulated autophagy via suppressing Akt/mTOR contributed to its preventive role against VCAM1 activation.
Materials and methods
Chemicals and reagents
Nuciferine (purity by HPLC ≥98%) was obtained from Chengdu Must Bio-Technology Co., Ltd. (Chengdu, China), and dissolved in dimethyl sulfoxide (DMSO) in a 10 mmol/L stock. The chemical structure of nuciferine was drawn using ChemDraw 21.0.0 software (PerkinElmer, Waltham, MA). Rapamycin, SB202190, chloroquine (CQ), asiatic acid, and MK2206 were purchased from Selleck (Shanghai, China). CQ was dissolved in sterilized water, while rapamycin, SB202190, asiatic acid, and MK2206 were dissolved in DMSO. Recombinant human TNFα was bought from R&D Systems (Minneapolis, MN) and dissolved in sterile phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA) before use. Information about all antibodies is shown in Table 1.
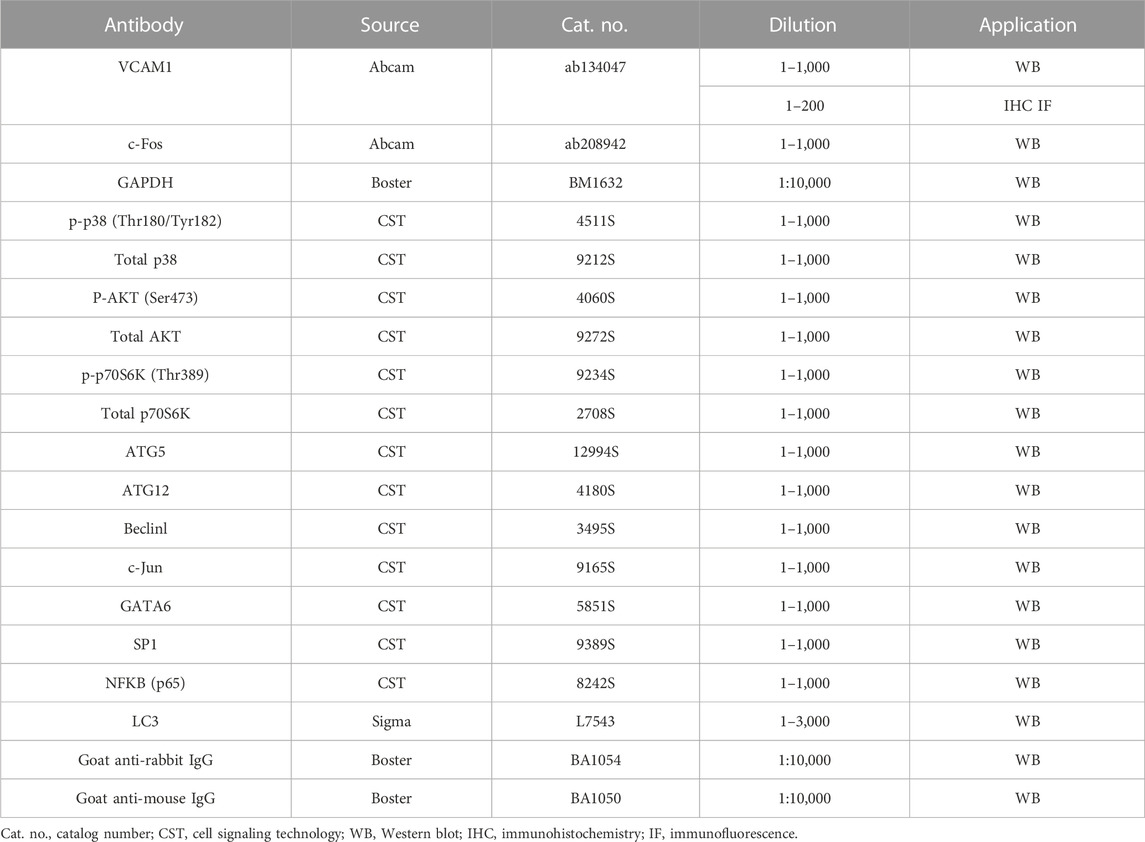
TABLE 1. Antibodies information.
Animal handling
C57BL/6J male mice (6 weeks old) were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China), and kept in a specific pathogen-free (SPF) laboratory with free access to food and water. All animal experiments were approved by the Laboratory Animal Management and Ethics Committee of Zhejiang Chinese Medical University (Approval No. IACUC-20210423-08). After 1 week of acclimatization, all mice were randomly assigned into a control group (n = 5), model group (n = 5), and nuciferine group (n = 5). The mice in the control group were fed a normal-fat diet (NFD; TP26352, TROPHIC Animal Feed High- Tech Co., Ltd., Nantong, China). The mice in the model group were fed an HFD (TP26304, TROPHIC Animal Feed High- Tech Co., Ltd.). Mice in the NUC group were fed an HFD containing 0.06% (w/w of the HFD) nuciferine supplement. The dosage of nuciferine referred to the low dose that Zhang et al. reported (Zhang et al., 2018). Caloric compositions of each diet are listed in Table 2. After 16 weeks of feeding, all mice were anesthetized and euthanized, and the plasma and aortic arch tissues were collected and stored in −80°C for further experiments.
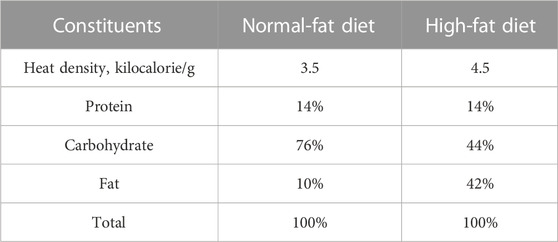
TABLE 2. Caloric compositions of each diet.
Enzyme-linked immunosorbent assay
VCAM1 content in mice plasma was determined using a commercial enzyme-linked immunosorbent assay (ELISA) kit obtained from Cusabio (CSB-E04754m, Wuhan, China). The assay was carried out according to the manufacturer’s instructions. In brief, plasma and standard were first diluted with sample dilution and then added to the coated assay plate, followed by incubation at 37°C. Two hours later, after the liquid was removed, the assay plate was incubated with a biotin antibody at 37°C for 1 h and washed three times. HRP-avidin was then added, and the plate was incubated again at 37°C. One hour later, after washing five times, a TMB substrate was added and incubated at 37°C for 20 min. At last, a stop solution was added to stop the reaction, and the optical density (OD) at 450 nm and 570 nm was determined using a FLUOstar Omega microplate reader (BMG LABTECH, Ortenberg, Germany). The final data were calculated by subtracting the OD at 570 nm from the OD at 450 nm.
Immunohistochemistry
Aortic arch tissues were fixed in 5% paraformaldehyde, embedded in an optimal cutting temperature (OCT) compound (4583, SAKURA, Torrance, CA), and frozen in liquid nitrogen. Frozen sections of 8 μm were cut using a frozen section machine (HM525, Thermo Scientific, MA) and then incubated with 10% goat serum. After that, the sections were incubated with a primary antibody against VCAM1 (1:50) at 4°C overnight, washed with PBS, and then, incubated with the Alexa Fluor® 488-conjugated Goat Anti-Rabbit IgG (H + L) secondary antibody (1:150, DW-GAR4881, Dawen Biotec, Hangzhou, China). The primary and secondary antibodies were diluted in 10% goat serum. Eventually, nuclei were stained with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) staining solution (C1005, Beyotime, Shanghai, China) for 10 min. Images were captured using a digital pathology scanner (Olympus, VS120-S6-W, Tokyo, Japan).
Cell culture
The human vascular endothelial cell line, EA.hy926, was obtained from the American Type Culture Collection (ATCC) (Manassas, VA) and cultured with Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (CellMax, Beijing, China). The cell line was routinely maintained on the 100-mm tissue culture dish in the 37°C incubator with humidified 95% air and 5% CO2.
Cell viability assay
4 × 103 cells/well were seeded onto a 96-well tissue culture plate; 16 h later, the culture medium was replaced with a fresh medium with nuciferine (0, 1, 2.5, 5, 10, and 20 μmol/L) for another 24 h. Cell viability was determined using a cell-counting kit 8 (CCK8) (Bimake, Houston, TX) as we previously described (Zhou et al., 2020).
RNA interfering
Genes targeting small interfering RNA (siRNA) and scrambled siRNA (as the negative control) were synthesized by GenePharma (Shanghai, China). 10 nmol/L siRNA was delivered to cells with RNAiMAX as we previously described (Zhou et al., 2020). Human ATG5, c-Jun, and c-Fos siRNA-targeting sequences (5′-3′) are CCTTTGGCCTAAGAAGAAA (ATG5), GUCAUGAACCACGUUAACA (c-Jun), and GCAAUAGUGUGUUCUGAUU (c-Fos).
Western blot assay
Cells were harvested by scratching and resuspended in pre-cold PBS. RIPA buffer (Boster, Wuhan, China) or a nuclear and cytoplasmic protein extraction kit (CWBIO, Beijing, China) was utilized to extract the total or nuclear protein, respectively. Western blot assay was carried out following a previous protocol (Li et al., 2013). Final data were visualized under a ChemiScope Series machine from Clinx Science Instruments Co., Ltd. (3300 Mini) (Shanghai, China). The internal control for total protein (GAPDH) or nuclear protein (Histone H3) was employed. Relative protein expressions in all blots were quantized using ImageJ (NIH Image, Bethesda, MD). Representative blots from at least three repeats were shown.
RNA extraction and quantitative polymerase chain reaction
Total RNA was extracted using a total RNA extractor (Sangon Biotech, Shanghai, China). Reverse transcription was then carried out to generate cDNA from RNA using HiScript III All-in-one RT SuperMix (Vazyme, Nanjing, China). SYBR green was utilized to determine the relative gene expression level with the 2−ΔΔCT method. ACTB was set as the reference gene. Sequences for gene-specific primers are listed in Table 3.
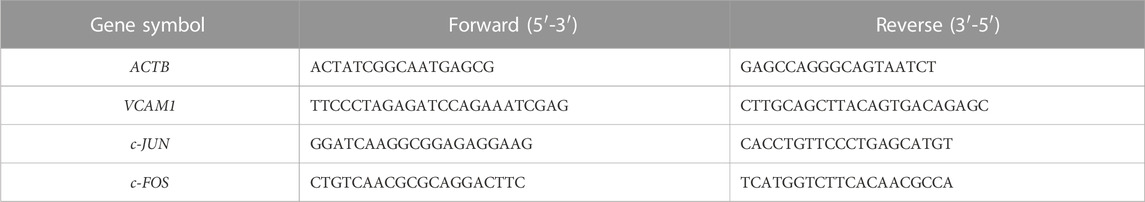
TABLE 3. Sequences of gene-specific primers.
Analysis of the autophagic flux
Lentivirus encoding GFP-LC3 (GeneChem, Shanghai, China) was delivered to cells to visualize autophagic flux. The autophagic flux was determined using a laser scanning confocal microscope (LSM 880, ZEISS, Jena, Germany). GFP-positive autophagic puncta were counted and quantified in each group (at least 20 cells were included per group) according to a previous study.
Statistical analysis
All data were expressed as the mean ± SD. Statistical analysis was performed using unpaired Student’s t-test with GraphPad Prism 8.02 software (GraphPad Software, San Diego, CA). When p < 0.05, differences between the compared groups were considered to be significant.
Results
Nuciferine protects TNFα-induced VCAM1 in the vascular endothelium
The chemical structure of nuciferine is shown in Figure 1A. As an aporphine alkaloid, nuciferine might be toxic when consumed beyond a certain concentration; therefore, cell viability was first assessed in the presence of nuciferine (0, 1, 2.5, 5, 10, 20, and 40 μmol/L) in endothelial cells. No significant cytotoxicity was observed when the dose of nuciferine was lower than 5 μmol/L (Figure 1B). Then, a commonly employed cellular model for VCAM1 activation was established by TNFα (10 ng/mL, 12 h) treatment in the EA.hy926 human vascular endothelial cell line. As shown in Figures 1C, D, TNFα treatment robustly stimulated VCAM1 expression at both transcriptional and protein levels. To investigate the protective role of nuciferine against VCAM1 activation, cells were pretreated with incremental doses of nuciferine (0, 1, 2.5, and 5 μmol/L) for 1 h before TNFα exposure. Our data clearly showed that nuciferine (the optimal dose is 5 μmol/L) significantly reversed TNFα-induced VCAM1 activation (Figures 1C, D). To consolidate our findings, we established an obesity-associated VCAM1 elevation model via feeding mice with an HFD, and changes in plasma TC, plasma TG, plasma HDL-C, plasma LDL-C, and mouse body weight confirmed the success of the animal model (Supplementary Figures S4A–E). As shown in Figures 1E, F, the HFD induced a significant increase in VCAM1 in both the arterial endothelium and blood while nuciferine administration rescued HFD-increased VCAM1 (Figures 1E, F). These data clearly indicated the protective ability of nuciferine against VCAM1 activation.
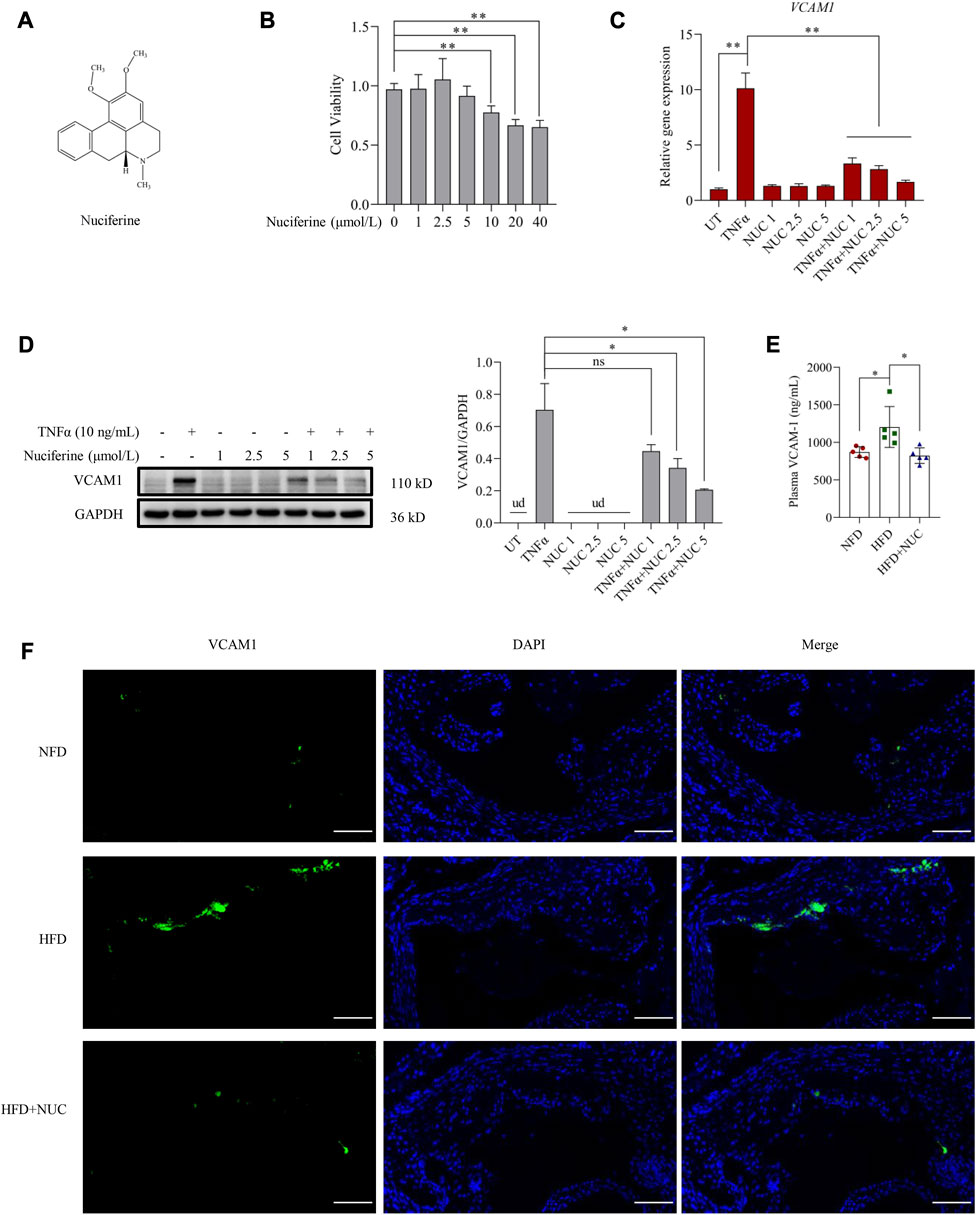
FIGURE 1. Nuciferine represses VCAM1 activation both in vitro and in vivo. (A) Chemical structure of nuciferine. (B) Cell viability test in EA.hy926 cells under 24 h treatment of nuciferine (0, 1, 2.5, 5, 10, 20, and 40 μmol/L). Relative mRNA level of VCAM1 verified by qPCR (C), protein level of VCAM1 verified by Western blot assay (D) (left panel), and the relative quantification data (D) (right panel), while EA.hy926 cells were pretreated with nuciferine (0, 1, 2.5, and 5 μmol/L) for 2 h, followed by 12 h incubation with TNFα (10 ng/mL) or vehicle. (E) Plasma VCAM1 level examined by the ELISA method from the indicated mice. (F) VCAM1 immunofluorescence staining of arterial endothelia tissues from the indicated mice; nuclei were visualized by DAPI staining (blue), and VCAM1 was visualized by an FITC-conjugated secondary antibody (green); the final data were captured under a fluorescence digital pathology scanner (magnification ×200; scale bar, 100 μm). All values are denoted as the means ± SD from five animal samples or at least three independent batches of cells. Each group contains the same amount of solvent. ud, undetectable. ∗p < 0.05 and ∗∗p < 0.01 indicate statistically significant differences.
Autophagy regulates VCAM1 activation in challenge to TNFα
Autophagy induction provided an effective strategy to improve VCAM1 activation (Wei et al., 2020). In this study, we observed that pre-incubation of human vascular endothelial cells with rapamycin, a commonly used autophagy agonist, significantly reversed TNFα-induced VCAM1 activation at both mRNA (Figure 2A) and protein levels (Figure 2B). Moreover, we established an instantaneous autophagy suppression model by transfecting endothelial cells with specific interfering RNA targeting on ATG5 (siATG5). The data revealed that ATG5 was effectively silenced by siRNA (Supplementary Figure S1), while autophagy suppression aggravated TNFα-induced VCAM1 activation at both mRNA and protein levels (Figures 2C, D). These results collectively indicated that autophagy negatively regulates VCAM1 expression in human vascular endothelial cells when challenged to TNFα exposure.
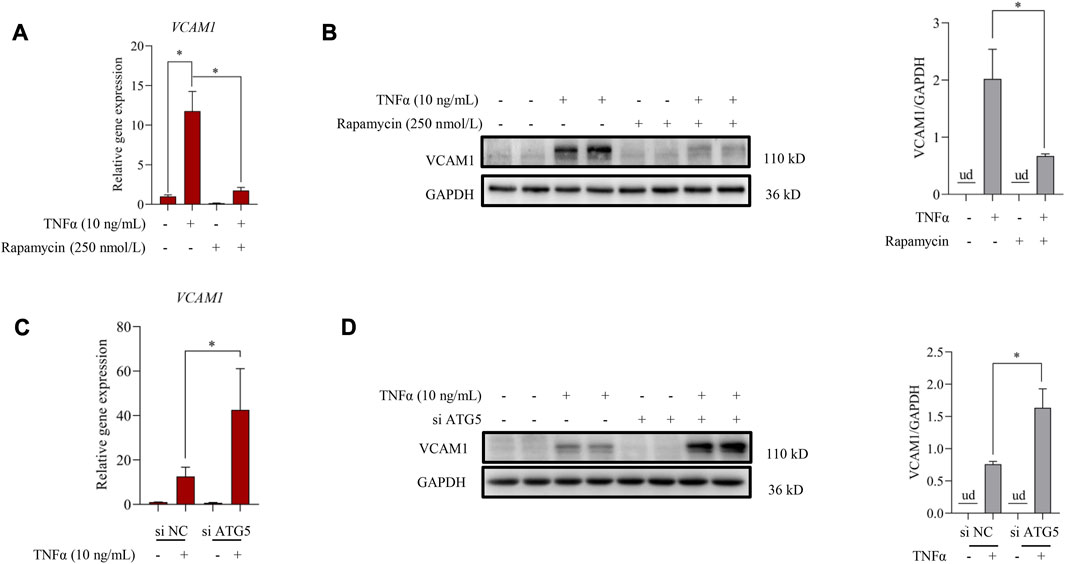
FIGURE 2. Autophagy contributes to TNFα-induced VCAM1 activation. (A) Relative mRNA level of VCAM1. (B) VCAM1 protein expression and the quantification data; EA.hy926 cells were pretreated with rapamycin for 2 h, followed by 12 h TNFα stimulation. (C) Relative VCAM1 gene expression level, (D) VCAM1 protein expression level and the quantification data; EA.hy926 cells were treated with si ATG5 or scrambled siRNA (si NC) for 24 h and then incubated with TNFα or vehicle for another 12 h. All values are denoted as the means ± SD from at least three independent tests. Each group contains the same amount of solvent. ud, undetectable. ∗p < 0.05 and ∗∗p < 0.01 indicate statistically significant differences.
Autophagy stimulation contributes to nuciferine-alleviated VCAM1 activation
We further investigated whether autophagy stimulation contributes to nuciferine-improved VCAM1 activation. According to our knowledge, the regulation of nuciferine on autophagy in vascular endothelial cells remains unknown. Here, we observed for the first time that nuciferine enhanced autophagy in human vascular endothelial cells based on the following findings: 1) nuciferine incubation increased microtubule-associated protein 1 lightchain 3 (MAP1LC3/LC3) autophagic puncta in the GFP-LC3-lentivirus-infected vascular endothelial cells (Figure 3A); 2) nuciferine treatment enhanced the expression of LC3 Ⅱ (lipidated form of LC3, a widely used marker for autophagic activation (Figure 3B); 3) nuciferine intervention promoted autophagic flux (Figures 3A, B) since nuciferine stimulated both LC3 puncta and LC3 Ⅱ expression in the presence of CQ, a well-employed autophagy antagonist through inhibiting autophagosome degradation in lysosome; and 4) nuciferine promoted the expression of autophagy initiation-related proteins, including Beclin1, ATG5, and ATG12 (Figure 3C). These data implied that nuciferine is a potential phytogenic autophagy inducer in vascular endothelial cells. More importantly, we explored whether autophagy stimulation was involved in nuciferine-alleviated VCAM1 activation. Autophagy was chemically or genetically suppressed by CQ or siRNA knocking down ATG5, respectively. Our data showed that nuciferine failed to improve TNFα-induced VCAM1 activation after autophagy repression (Figures 3D, E), implying that nuciferine-stimulated autophagy contributed to its preventive benefit against VCAM1 activation.
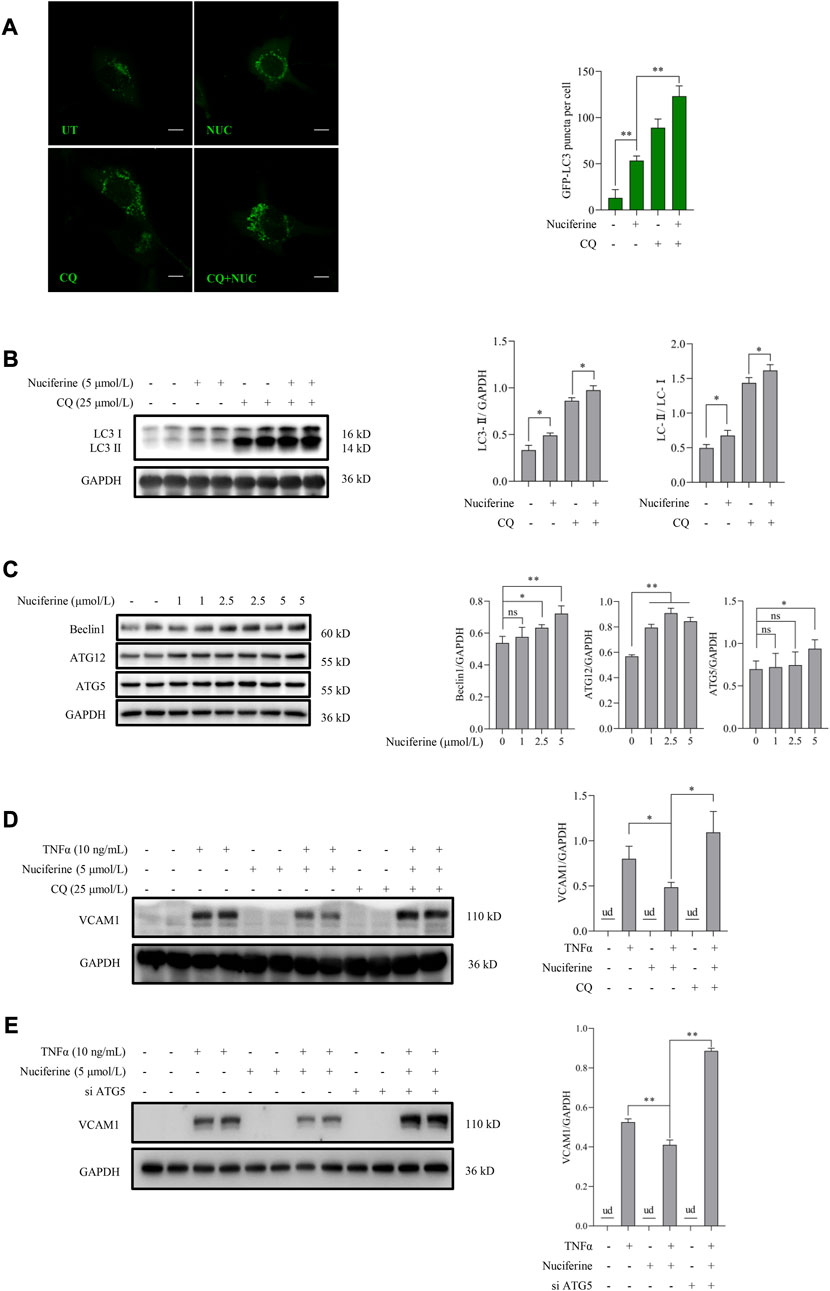
FIGURE 3. Nuciferine stimulates autophagy to protect against TNFα-induced VCAM1 activation. (A) GFP-LC3 puncta detected by confocal and bar graphs of the quantification data. The quantification data were generated from at least 20 different cells in each group. Scale bar, 20 μm. (B) LC3 Ⅰ/Ⅱ protein level and the quantification data. Cells in (A,B) were treated with CQ for 2 h ahead of nuciferine treatment. (C) Immunoblots of autophagy initiation-related proteins (Beclin1, ATG12, and ATG5) and the quantification data in EA.hy926 cells treated with nuciferine (0, 1, 2.5, and 5 μmol/L) for 12 h. (D,E) Protein level of VCAM1, as well as the quantification data in EA.hy926 cells, pretreated with CQ for 2 h (D) or siRNAs for 24 h (E) and then nuciferine (5 μmol/L) for 2 h, followed by 12 h TNFα incubation. All values are denoted as the means ± SD from at least three independent tests. Each group contains the same amount of solvent. ud, undetectable. ∗p < 0.05 and ∗∗p < 0.01 indicate statistically significant differences. ns, no significant differences.
p38 MAPK is irrelevant to nuciferine-reduced VCAM1 via autophagy activation
We aimed to explore the upstream signaling pathways through which nuciferine stimulated autophagy to prevent VCAM1 activation. The involvement of several autophagy regulatory signaling pathways, including AMP-activated protein kinase (AMPK) (Kim et al., 2011; Zhou et al., 2019), silent information regulator sirtuin 1 (SIRT1) (Yuan et al., 2020; Lohanathan et al., 2022), and p38 mitogen-activated protein kinases (p38 MAPK) (Webber, 2010; Song et al., 2020), which have been reported to be modulated by nuciferine or its analog, was investigated in this study. Our data indicated that neither phosphorylated AMPK nor SIRT1 was stimulated by nuciferine incubation (Figure 4A), which preliminarily ruled out their participation in nuciferine-promoted autophagy. However, phosphorylated p38 (p-p38) was significantly reduced by nuciferine treatment when compared with that in the control group (Figure 4A). Correspondingly, p38 MAPK pathway inhibition by its special chemical antagonist SB202190 markedly promoted autophagic flux in vascular endothelial cells (Figure 4B). SB202190 treatment also partially protected the endothelial cells from TNFα-induced VCAM1 activation (Figure 4C). However, p38 MAPK induction by its pharmacological agonist asiatic acid could not block the protective role of nuciferine against TNFα-induced VCAM1 activation (Figure 4D). These data collaboratively indicated that p38 inhibition stimulated autophagy and improved TNFα-induced VCAM1 activation; however, such regulation was not the main mechanism of the beneficial effect of nuciferine on VCAM1 reduction.
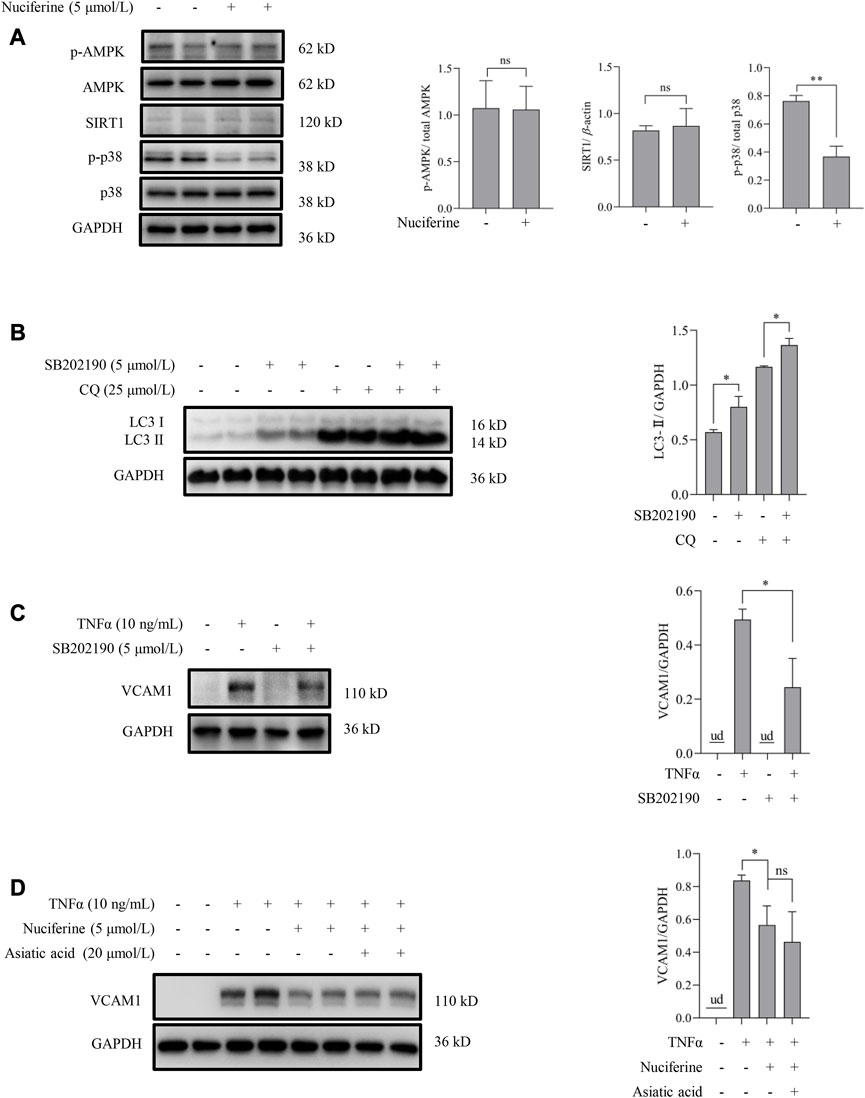
FIGURE 4. p38 MAPK is irrelevant to autophagy activation and VCAM1 reduction in response to nuciferine. (A) Upstream molecules for autophagy induction (p-AMPK, SIRT1, and p-p38) in the presence of nuciferine (6 h) detected by Western blot assay and the quantification data. (B) LC3 Ⅰ/Ⅱ protein level and the quantification data in EA.hy926 cells pretreated with CQ for 2 h, followed by SB202190 (5 μmol/L) treatment for 12 h. (C) Immunoblots of VCAM1 and the quantification data in cells pretreated with SB202190 (5 μmol/L) for 2 h, followed TNFα treatment for 12 h (D) VCAM1 protein expression and the quantification data in cells pretreated with nuciferine (5 μmol/L) and asiatic acid for 2 h, followed by TNFα treatment for 12 h. All values are denoted as the means ± SD from at least three independent tests. Each group contains the same amount of solvent. ud, undetectable. ∗p < 0.05 indicate statistically significant differences. ns, no significant differences.
Akt pathway contributes to nuciferine-stimulated autophagy and -protected VCAM1 activation
Akt inhibition has been reported to be associated with autophagy activation via regulating the mammalian target of rapamycin (mTOR) pathway (Takeuchi et al., 2005). In this study, we first tested the regulatory role of the Akt pathway on autophagy in human vascular endothelial cells. Our data showed that inhibiting Akt using a pan Akt antagonist MK-2206 significantly increased the autophagic flux (Figure 5A). Subsequently, the involvement of the Akt/mTOR pathway in nuciferine-stimulated autophagy and -protected VCAM1 activation was evaluated. Our data revealed that nuciferine treatment reduced the basal level of phosphorylated Akt (p-Akt on Ser473) and phosphorylated p70 S6 kinase (p-p70S6K on Thr389), a well-recognized target to reflect mTOR activity (Figure 5B). Similar with nuciferine treatment, Akt inhibition by MK-2206 also protected the endothelial cells from TNFα-induced VCAM1 activation (Figure 5C). More importantly, Akt activation, using its special physiological agonist insulin, strongly abolished nuciferine-induced autophagy activation (Figure 5D) and blocked the preventive role of nuciferine on VCAM1 activation (Figure 5E). These results indicated that Akt suppression contributed to nuciferine-stimulated autophagy and further protection on VCAM1 activation.
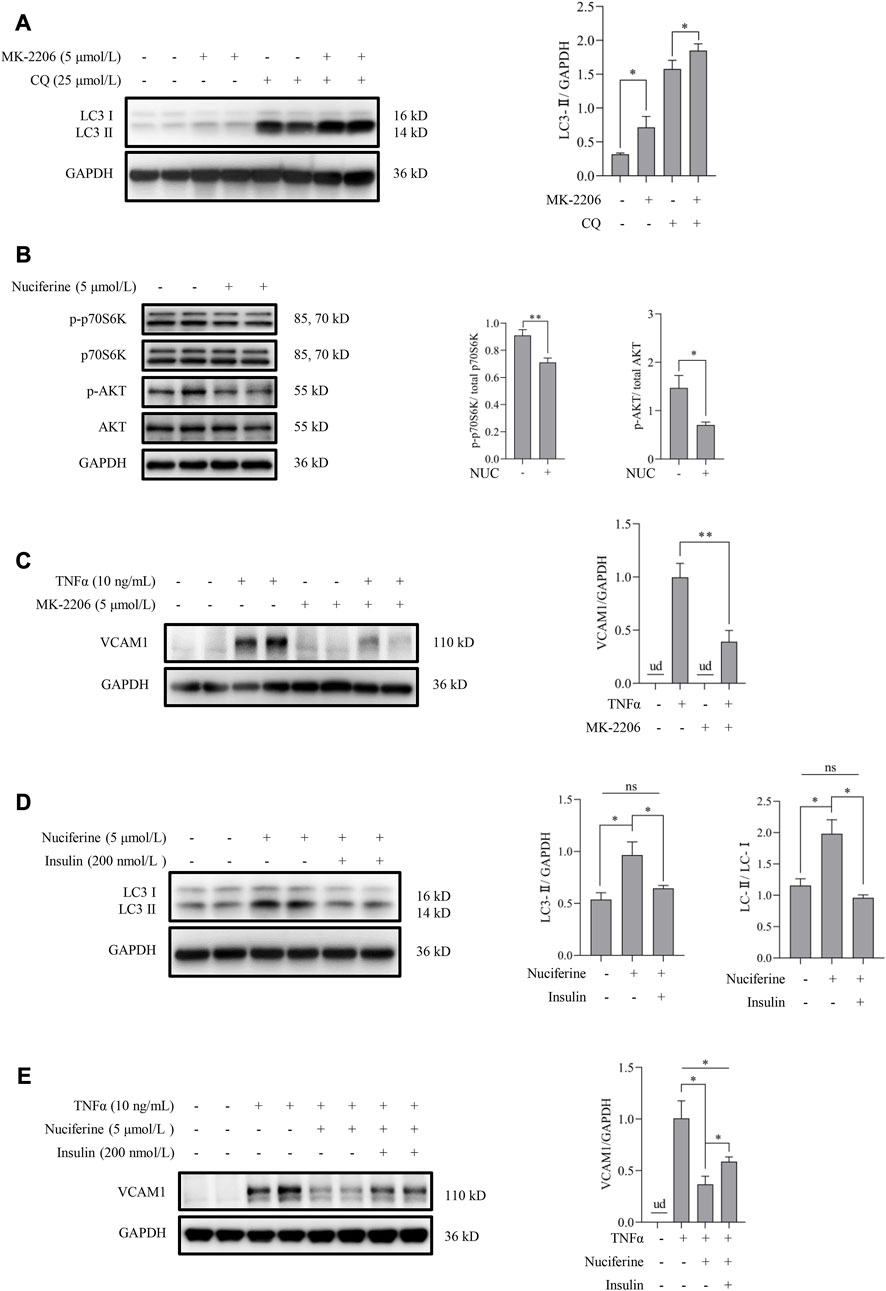
FIGURE 5. Nuciferine activates autophagy through Akt/mTOR inhibition to repress VCAM1 activation. (A) LC3 Ⅰ/Ⅱ protein level and the quantification data in cells pretreated with CQ for 2 h, followed by MK-2206 (5 μmol/L) treatment for 12 h. (B) Status of molecules in Akt/mTOR signaling (p-Akt, Akt, p-p70S6K, and p70S6K) under nuciferine treatment for 6 h and the quantification data. (C) VCAM1 protein expression and the quantification data in cells pretreated with MK-2206 (5 μmol/L) for 2 h, followed by TNFα treatment for 12 h. (D) LC3 Ⅰ/Ⅱ protein level and the quantification data in cells pretreated with insulin (200 nmol/L) for 2 h, followed by nuciferine treatment for 12 h. (E) VCAM1 protein expression and the quantification data in cells pretreated with nuciferine (5 μmol/L) and insulin (200 nmol/L) for 2 h, followed by TNFα treatment for 12 h. All values are denoted as the means ± SD from at least three independent tests. Each group contains the same amount of solvent. ud: undetectable. ∗p < 0.05 and ∗∗p < 0.01 indicate statistically significant differences. ns, no significant differences.
Nuciferine alleviates TNFα-induced AP1 activation
Transcriptional activation by nuclear factors, such as activating protein 1 (AP1, the heterodimeric form of c-Fos and c-Jun), nuclear factor-κB (NFκB/p65), GATA-binding protein 6 (GATA6), interferon regulatory factor 1 (IRF1), and specificity protein 1 (SP1), is the main regulation manner for VCAM1 generation (Iademarco et al., 1992; Neish et al., 1992; Neish et al., 1995a; Neish et al., 1995b). As shown in Figure 1C, nuciferine treatment robustly prevented the TNFα-induced transcriptional activity of VCAM1, which spurred us to explore the potential transcriptional regulation mechanism(s) behind nuciferine-reduced VCAM1. We first analyzed the nuclear levels of the aforementioned nuclear factors, and our data showed that TNFα exposure triggered the nuclear expressions of AP1 (c-Fos and c-Jun), IRF1, and GATA6 but not those of NFκB/p65 and SP1, while nuciferine intervention significantly reduced the nuclear levels of c-Fos and c-Jun (components of the heterodimeric AP1) without affecting IRF1 and GATA6 (Figure 6A). Then, to confirm the decisive role of AP1 (c-Fos and c-Jun) in TNFα-induced VCAM1 activation in the present cell setting, specific siRNAs were introduced to knock down c-Fos or c-Jun, respectively. The data revealed that c-Fos or c-Jun was effectively silenced by siRNA (Supplementary Figures S2, S3). Upon genetic knockdown of c-Fos or c-Jun, the TNFα-induced VCAM1 protein was largely abolished (Figures 6B, C). These findings implied that nuciferine alleviated TNFα-induced AP1 activation to prevent VCAM1 activation.
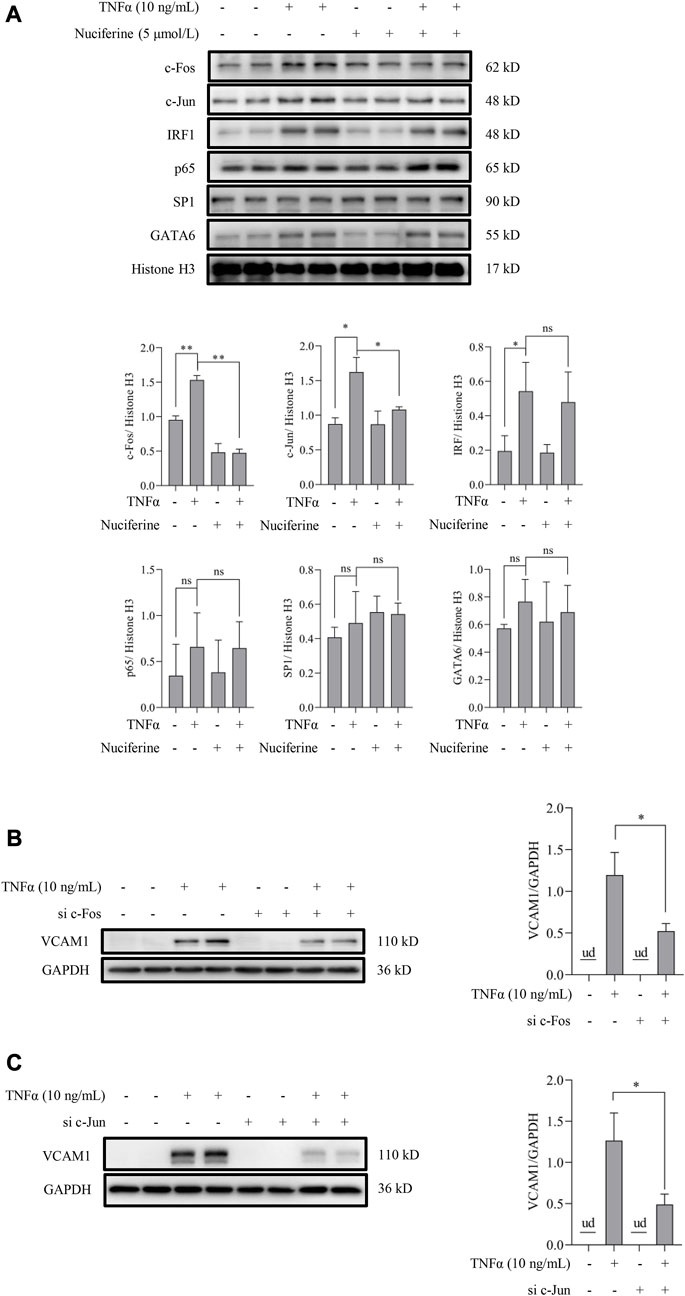
FIGURE 6. Nuciferine represses TNFα-induced nuclear translocation of AP1. (A) Protein abundance for transcriptional factors of VCAM1 (c-Fos, c-Jun, IRF1, p65, SP1, and GATA6) in the nuclear extracts and the relative quantification data. EA.hy926 cells were pretreated with nuciferine for 2 h, followed by 1 h TNFα incubation. Histone H3 was utilized as the loading control for nuclear proteins. Immunoblots of VCAM1 under 12 h TNFα stimulation in the presence of c-Fos- (B) and c-Jun-specific (C) siRNAs (si c-Fos and si c-Jun) or scrambled siRNA (positive control). All values are denoted as the means ± SD from at least three independent tests. Each group contains the same amount of solvent. ud: undetectable. ∗p < 0.05 and ∗∗p < 0.01 indicate statistically significant differences. ns, no significant differences.
Autophagy activation contributes to nuciferine-inhibited AP1
Next, we investigated whether nuciferine-activated autophagy contributed to its preventive role on AP1. To answer this question, the involvement of autophagy in the regulation of TNFα-induced AP1 activation was first investigated. Rapamycin was introduced to activate autophagy as we described previously, and the nuclear contents of AP1 (c-Fos and c-Jun) were detected in endothelial cells. As shown in Figure 7A, TNFα-induced nuclear translocation of both c-Fos and c-Jun was abolished by rapamycin treatment. In addition, genetically repressing autophagy by knocked down ATG5 enhanced the TNFα-promoted AP1 (c-Fos and c-Jun) level in nucleus (Figure 7B). These results implied that autophagy activation was involved in AP1 inactivation. Furthermore, in the presence of CQ, a special autophagy inhibitor, nuciferine failed to reduce TNFα-induced c-Fos and c-Jun in the nuclei (Figure 7C), which was consistent with the protein expression pattern of VCAM1 under the same treatments (Figure 3D). These results collectively suggested that nuciferine stimulated autophagy to prevent TNFα-induced nuclear AP1 increase and further VCAM1 activation.
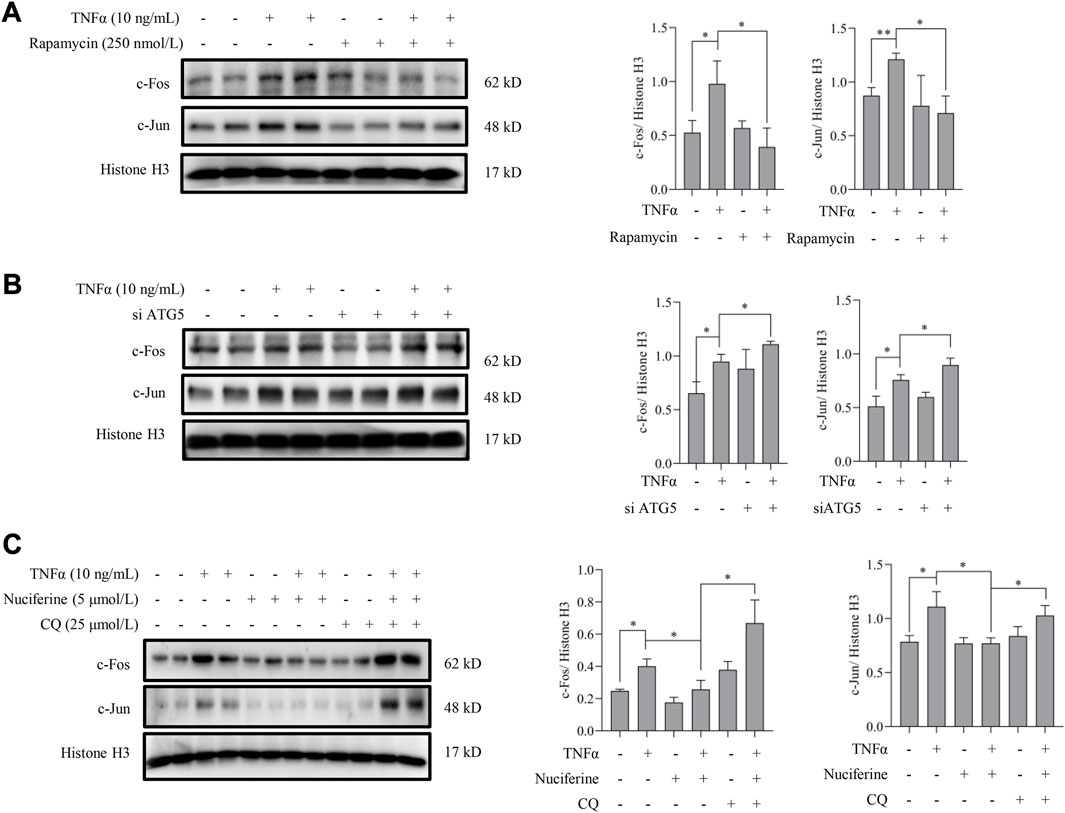
FIGURE 7. Autophagy activation contributes to nuciferine-inhibited AP1. Immunoblots of nuclear AP1 (c-Fos and c-Jun) and the quantification data in EA.hy926 cells (A) pretreated with rapamycin (250 nmol/L) for 2 h or (B) pretreated with si ATG5 (or scrambled siRNA) for 24 h, followed by 1 h TNFα incubation. (C) Protein abundance and the quantification data of AP1 in the nuclei of EA.hy926 cells pretreated with nuciferine and CQ for 2 h and then incubated with TNFα for another 12 h. Histone H3 was utilized as the loading control for nuclear proteins. Each group contains the same amount of solvent. ∗p < 0.05 indicates statistically significant differences.
Discussion
This study reported for the first time the protective role of nuciferine against VCAM1 activation in both obesity-associated mice and human vascular endothelial cells. A mechanism study showed that nuciferine activates autophagy via inhibiting the Akt pathway so that in an autophagy-dependent manner, it reduces the nuclear AP1 (specifically refers to the c-Jun and c-Fos components in this study) level, to repress VCAM1 activation through transcriptional regulation ultimately (Figure 8).
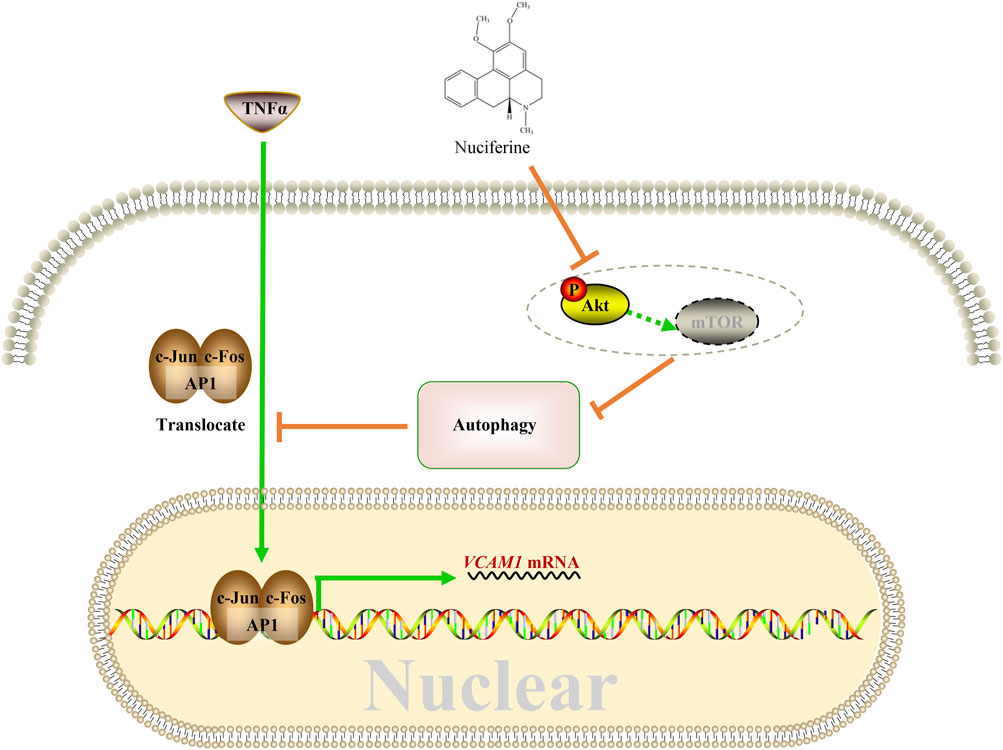
FIGURE 8. Proposed working model.
The level of VCAM1 is increased (i.e., VCAM1 is activated) in different cardiovascular events and is reported to be positively associated with cardiovascular mortality, clinical improvements, and prognosis in patients (Jager et al., 2000; Blum et al., 2006; Castillo et al., 2009). Thus, blockade of VCAM1 activation is recognized as a potential therapeutic approach to improve cardiovascular events. VCAM1 has been reported to be blocked through various approaches, such as neutralizing antibody, genetical modification, or chemical compounds (e.g., AGI-1067), to improve atherosclerosis, rheumatoid arthritis (RA), nonalcoholic steatohepatitis (NASH), and so on (Carter et al., 2002; Park et al., 2013; Furuta et al., 2021). Furthermore, in this study, nuciferine was newly reported as the phytogenic compound to block TNFα-induced VCAM1 activation.
In the following mechanistical studies, we found that nuciferine could increase the protein level of autophagy initiation-related proteins (Beclin1, ATG5, and ATG12) and the number of autophagy puncta, indicating its autophagy-inductive capacity in vascular endothelial cells. This is consistent with the previous study conducted by Shi et al. which showed that nuciferine induces the expression of ATG5 protein, as well as promotes the formation of autophagic puncta and autophagosomes in Caco-2 and HT-29 cell lines (human colonic epithelial tumor cell lines isolated from colorectal adenocarcinoma) (Shi et al., 2021). Since autophagy induction has been considered a therapeutically viable approach for cardiovascular events (Lavandero et al., 2015), compounds that induce autophagy have been investigated for the intervention of these diseases. For example, clonidine and rilmenidine (inducing autophagy through the reduction in cAMP levels) are approved for the clinical treatment of hypertension (Leidal et al., 2018); rapamycin (inducing autophagy through mTOR inhibition) exerted anti-hypertrophic and anti-inflammatory efficiency in the aged heart in an animal study (Flynn et al., 2013). Herein, nuciferine was finally demonstrated to prevent VCAM1 activation through autophagy stimulation in the endothelial cells.
Then, we attempted to investigate how nuciferine stimulated autophagy. AMPK and SIRT1 pathways are generally accepted upstream signals involved in autophagy stimulation. Referring to previous studies, AMPK activates autophagy in the initiation stage through mTOR complex 1 (mTORC1) inhibition, mammalian autophagy-initiating kinase ULK1 activation through phosphorylation, or Beclin1-VPS34-VPS15 complex activation in the presence of ATG14L (Egan et al., 2011; Kim et al., 2011); SIRT1 mainly activates autophagy via its deacetylase activity to induce the expression of ATGs or fork-head box protein O1 (FOXO1) (Lee et al., 2008; Hariharan et al., 2010). However, nuciferine had no influences on these two molecules under our experimental setting in the human vascular endothelial cells, whereas p38 MAPK signaling was significantly repressed upon nuciferine treatment. p38 MAPK signaling is mainly activated by inflammatory and cellular stress stimuli and involves the negative control of autophagy under different cell settings (Webber and Tooze, 2010). SB202190, an antagonist of the p38 MAPK pathway specifically targeting p38α/β, is demonstrated as an autophagy inducer through p38 inhibition (Comes et al., 2007). Although SB202190 activated autophagy and prevented TNFα-induced VCAM1 activation in our experimental setting as well (Figures 4B, C), activating the p38 MAPK pathway by asiatic acid had no influence on the protective role of nuciferine against TNFα-induced VCAM1 activation (Figure 4D). In fact, it has been proposed that pyridinyl imidazole class inhibitors, such as SB202190, alter autophagy flux and autophagy-promoting gene expression in a cell type-specific, MAPK14/p38α-MAPK11/p38β-independent manner (Menon et al., 2015). Thus, p38 MAPK is irrelevant to autophagy activation and VCAM1 reduction in response to nuciferine.
As nuciferine was found to induce the protein level of Beclin1, ATG12, and ATG5 (Figure 3C), we further verified the involvement of mTOR-related pathways, which governs autophagy and lysosomal biogenesis (Napolitano and Ballabio, 2016). Akt signaling is one of the mTOR-related pathways and could regulate autophagy activity via mTOR. In the current study, nuciferine treatment led to the dephosphorylation of both Akt (Ser473) and p70S6K (Thr389, one of the downstream targets of mTOR), implying a negative regulation of the Akt/mTOR pathway by nuciferine (Figure 5B). As a coincidence to our finding on Akt, nuciferine has been recently reported to block the activation of the PI3K/Akt signaling pathway via calmodulin 4 to suppress the proliferation and migration of the vascular smooth muscle cells as a potential drug against atherosclerosis (Xiao et al., 2023). Thus, we further investigated whether nuciferine stimulated autophagy through the Akt/mTOR pathway. Our data showed that similar to nuciferine, suppressing Akt by the specific antagonist MK-2206 stimulated autophagy and protected human vascular endothelial cells from TNFα-induced VCAM1 activation. In addition, activating Akt with insulin repressed autophagy and diminished protection against TNFα-induced VCAM1 activation induced by nuciferine. Collectively, nuciferine was proved to stimulate autophagy to protect VCAM1 activation through the Akt/mTOR pathway.
Next, the autophagy-dependent protective role of nuciferine against TNFα-induced VCAM1 was elucidated. VCAM1 is rarely expressed under physical conditions but could be quickly activated by a pro-inflammatory factor, including TNFα, which regulates the activation, maturation, and cytokine and chemokine release of leukocytes, thus playing a vital and direct role in endothelial dysfunction (Zhang et al., 2009). Upon stimulation, VCAM1 expression is transcriptionally activated by its transcription factors (Furuta et al., 2021), including AP1, NF-κB (p65), IRF1, GATAs, and SP1(Iademarco et al., 1992; Neish et al., 1992; Neish et al., 1995a; Neish et al., 1995b). In this study, TNFα was found to stimulate the nuclear translocation to activate these transcription factors except for SP1, which might be due to the experiment settings. Among these transcription factors, AP1 is a cluster of heterodimeric transcription factors, which could be activated by various stimuli, including inflammatory cytokines (Wisdom, 1999). c-Jun and c-Fos were picked up to represent AP1 since c-Jun is the most potent subunit among AP1 members and forms a stable heterodimer with c-Fos (Shaulian and Karin, 2002). Furthermore, for the first time, this study proved that nuciferine treatment alleviates TNFα-induced AP1 activation, which contributes to its protection against VCAM1 activation, but the mechanism remains to be elucidated.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by the Laboratory Animal Management and Ethics Committee of Zhejiang Chinese Medical University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
HW: conceptualization, data curation, and writing–original draft. YY: investigation and writing–original draft. WY: validation and writing–original draft. JZ and LC: resources and writing–original draft. RG: software and writing–original draft. ZY: methodology and writing–review and editing. SL: writing–review and editing, project administration, and supervision.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Zhejiang Natural Science Foundation for Distinguished Young Scholars (Grant Number LR20H260001), Natural Science Foundation of China (Grant Numbers 81773422 and 81973041), and Opening Project of Key Laboratory of Integrative Chinese and Western Medicine for the Diagnosis and Treatment of Circulatory Diseases of Zhejiang Province (Grant Numbers 2C32005 and 2C32105).
Acknowledgments
The authors appreciate the technical support from the public platform of the Medical Research Center, Academy of Chinese Medical Science, Zhejiang Chinese Medical University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1264324/full#supplementary-material
References
Bai, X., Liu, X., Li, S., An, H., Kang, X., and Guo, S. (2022). Nuciferine inhibits TMEM16A in dietary adjuvant therapy for lung cancer. J. Agric. Food Chem. 70 (12), 3687–3696. doi:10.1021/acs.jafc.1c08375
Blum, A., Khazim, K., Merei, M., Peleg, A., Blum, N., and Vaispapir, V. (2006). The stroke trial - can we predict clinical outcome of patients with ischemic stroke by measuring soluble cell adhesion molecules (CAM)? Eur. Cytokine Netw. 17 (4), 295–298. doi:10.1684/ecn.2006.0048
Carlos, T. M., Schwartz, B. R., Kovach, N. L., Yee, E., Rosa, M., Osborn, L., et al. (1990). Vascular cell adhesion molecule-1 mediates lymphocyte adherence to cytokine-activated cultured human endothelial cells [published erratum appears in Blood 1990 Dec 1;76(11):2420]. Blood 76 (5), 965–970. doi:10.1182/blood.v76.5.965.965
Carter, R. A., Campbell, I. K., O'Donnel, K. L., and Wicks, I. P. (2002). Vascular cell adhesion molecule-1 (VCAM-1) blockade in collagen-induced arthritis reduces joint involvement and alters B cell trafficking. Clin. Exp. Immunol. 128 (1), 44–51. doi:10.1046/j.1365-2249.2002.01794.x
Castillo, J., Alvarez-Sabin, J., Martinez-Vila, E., Montaner, J., Sobrino, T., Vivancos, J., et al. (2009). Inflammation markers and prediction of post-stroke vascular disease recurrence: The MITICO study. J. Neurol. 256 (2), 217–224. doi:10.1007/s00415-009-0058-4
Comes, F., Matrone, A., Lastella, P., Nico, B., Susca, F. C., Bagnulo, R., et al. (2007). A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 14 (4), 693–702. doi:10.1038/sj.cdd.4402076
Davis, B. K., Wen, H., and Ting, J. P. (2011). The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735. doi:10.1146/annurev-immunol-031210-101405
Du, X., Di Malta, C., Fang, Z., Shen, T., Niu, X., Chen, M., et al. (2022). Nuciferine protects against high-fat diet-induced hepatic steatosis and insulin resistance via activating TFEB-mediated autophagy-lysosomal pathway. Acta Pharm. Sin. B 12 (6), 2869–2886. doi:10.1016/j.apsb.2021.12.012
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331 (6016), 456–461. doi:10.1126/science.1196371
Elices, M. J., Osborn, L., Takada, Y., Crouse, C., Luhowskyj, S., Hemler, M. E., et al. (1990). VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell 60 (4), 577–584. doi:10.1016/0092-8674(90)90661-w
Flynn, J. M., O'Leary, M. N., Zambataro, C. A., Academia, E. C., Presley, M. P., Garrett, B. J., et al. (2013). Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 12 (5), 851–862. doi:10.1111/acel.12109
Furuta, K., Guo, Q., Pavelko, K. D., Lee, J. H., Robertson, K. D., Nakao, Y., et al. (2021). Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J. Clin. Invest. 131 (6), e143690. doi:10.1172/JCI143690
Goswami, S. K., Ranjan, P., Dutta, R. K., and Verma, S. K. (2021). Management of inflammation in cardiovascular diseases. Pharmacol. Res. 173, 105912. doi:10.1016/j.phrs.2021.105912
Hariharan, N., Maejima, Y., Nakae, J., Paik, J., Depinho, R. A., and Sadoshima, J. (2010). Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 107 (12), 1470–1482. doi:10.1161/circresaha.110.227371
Iademarco, M. F., McQuillan, J. J., Rosen, G. D., and Dean, D. C. (1992). Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J. Biol. Chem. 267 (23), 16323–16329. doi:10.1016/s0021-9258(18)42004-2
Jager, A., van Hinsbergh, V. W., Kostense, P. J., Emeis, J. J., Nijpels, G., Dekker, J. M., et al. (2000). Increased levels of soluble vascular cell adhesion molecule 1 are associated with risk of cardiovascular mortality in type 2 diabetes: The hoorn study. Diabetes 49 (3), 485–491. doi:10.2337/diabetes.49.3.485
Khoury, M. K., Yang, H., and Liu, B. (2021). Macrophage biology in cardiovascular diseases. Arterioscler. Thromb. Vasc. Biol. 41 (2), e77–e81. doi:10.1161/atvbaha.120.313584
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13 (2), 132–141. doi:10.1038/ncb2152
Lavandero, S., Chiong, M., Rothermel, B. A., and Hill, J. A. (2015). Autophagy in cardiovascular biology. J. Clin. Invest. 125 (1), 55–64. doi:10.1172/jci73943
Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., et al. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U. S. A. 105 (9), 3374–3379. doi:10.1073/pnas.0712145105
Lee, S., Chung, J., Ha, I. S., Yi, K., Lee, J. E., Kang, H. G., et al. (2007). Hydrogen peroxide increases human leukocyte adhesion to porcine aortic endothelial cells via NFkappaB-dependent up-regulation of VCAM-1. Int. Immunol. 19 (12), 1349–1359. doi:10.1093/intimm/dxm104
Leidal, A. M., Levine, B., and Debnath, J. (2018). Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 20 (12), 1338–1348. doi:10.1038/s41556-018-0235-8
Li, S., Li, Y., Ning, H., Na, L., Niu, Y., Wang, M., et al. (2013). Calcium supplementation increases circulating cholesterol by reducing its catabolism via GPER and TRPC1-dependent pathway in estrogen deficient women. Int. J. Cardiol. 168 (3), 2548–2560. doi:10.1016/j.ijcard.2013.03.057
Liu, K., Chi, D. S., Li, C., Hall, H. K., Milhorn, D. M., and Krishnaswamy, G. (2005). HIV-1 Tat protein-induced VCAM-1 expression in human pulmonary artery endothelial cells and its signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 289 (2), L252–L260. doi:10.1152/ajplung.00200.2004
Lohanathan, B. P., Rathinasamy, B., Huang, C. Y., and Viswanadha, V. P. (2022). Neferine attenuates doxorubicin-induced fibrosis and hypertrophy in H9c2 cells. J. Biochem. Mol. Toxicol. 36 (7), e23054. doi:10.1002/jbt.23054
Lu, D., and Thum, T. (2019). RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat. Rev. Cardiol. 16 (11), 661–674. doi:10.1038/s41569-019-0218-x
Menon, M. B., Dhamija, S., Kotlyarov, A., and Gaestel, M. (2015). The problem of pyridinyl imidazole class inhibitors of MAPK14/p38α and MAPK11/p38β in autophagy research. Autophagy 11 (8), 1425–1427. doi:10.1080/15548627.2015.1059562
Napolitano, G., and Ballabio, A. (2016). TFEB at a glance. J. Cell Sci. 129 (13), 2475–2481. doi:10.1242/jcs.146365
Neish, A. S., Khachigian, L. M., Park, A., Baichwal, V. R., and Collins, T. (1995a). Sp1 is a component of the cytokine-inducible enhancer in the promoter of vascular cell adhesion molecule-1. J. Biol. Chem. 270 (48), 28903–28909. doi:10.1074/jbc.270.48.28903
Neish, A. S., Read, M. A., Thanos, D., Pine, R., Maniatis, T., and Collins, T. (1995b). Endothelial interferon regulatory factor 1 cooperates with NF-kappa B as a transcriptional activator of vascular cell adhesion molecule 1. Mol. Cell Biol. 15 (5), 2558–2569. doi:10.1128/MCB.15.5.2558
Neish, A. S., Williams, A. J., Palmer, H. J., Whitley, M. Z., and Collins, T. (1992). Functional analysis of the human vascular cell adhesion molecule 1 promoter. J. Exp. Med. 176 (6), 1583–1593. doi:10.1084/jem.176.6.1583
Paleolog, E. M., Aluri, G. R., and Feldmann, M. (1992). Contrasting effects of interferon gamma and interleukin 4 on responses of human vascular endothelial cells to tumour necrosis factor alpha. Cytokine 4 (6), 470–478. doi:10.1016/1043-4666(92)90007-e
Park, J. G., Ryu, S. Y., Jung, I. H., Lee, Y. H., Kang, K. J., Lee, M. R., et al. (2013). Evaluation of VCAM-1 antibodies as therapeutic agent for atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 226 (2), 356–363. doi:10.1016/j.atherosclerosis.2012.11.029
Robles, J. P., Zamora, M., Adan-Castro, E., Siqueiros-Marquez, L., Martinez de la Escalera, G., and Clapp, C. (2022). The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin α5β1 and NF-κB signaling. J. Biol. Chem. 298 (3), 101695. doi:10.1016/j.jbc.2022.101695
Ross, R. (1999). Atherosclerosis-an inflammatory disease. N. Engl. J. Med. 340 (2), 115–126. doi:10.1056/nejm199901143400207
Shaulian, E., and Karin, M. (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4 (5), E131–E136. doi:10.1038/ncb0502-e131
Shi, Z., Fang, Z. Y., Gao, X. X., Yu, H., Zhu, Y. W., Ouyang, H. L., et al. (2021). Nuciferine improves high-fat diet-induced obesity via reducing intestinal permeability by increasing autophagy and remodeling the gut microbiota. Food Funct. 12 (13), 5850–5861. doi:10.1039/d1fo00367d
Song, C., Cao, J., Lei, Y., Chi, H., Kong, P., Chen, G., et al. (2020). Nuciferine prevents bone loss by disrupting multinucleated osteoclast formation and promoting type H vessel formation. FASEB J. 34 (3), 4798–4811. doi:10.1096/fj.201902551R
Takeuchi, H., Kondo, Y., Fujiwara, K., Kanzawa, T., Aoki, H., Mills, G. B., et al. (2005). Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 65 (8), 3336–3346. doi:10.1158/0008-5472.CAN-04-3640
Troncoso, M. F., Ortiz-Quintero, J., Garrido-Moreno, V., Sanhueza-Olivares, F., Guerrero-Moncayo, A., Chiong, M., et al. (2021). VCAM-1 as a predictor biomarker in cardiovascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 1867 (9), 166170. doi:10.1016/j.bbadis.2021.166170
Ulyanova, T., Scott, L. M., Priestley, G. V., Jiang, Y., Nakamoto, B., Koni, P. A., et al. (2005). VCAM-1 expression in adult hematopoietic and nonhematopoietic cells is controlled by tissue-inductive signals and reflects their developmental origin. Blood 106 (1), 86–94. doi:10.1182/blood-2004-09-3417
Wang, C., Chen, Y. F., Quan, X. Q., Wang, H., Zhang, R., Xiao, J. H., et al. (2015). Effects of neferine on Kv4.3 channels expressed in HEK293 cells and ex vivo electrophysiology of rabbit hearts. Acta Pharmacol. Sin. 36 (12), 1451–1461. doi:10.1038/aps.2015.83
Webber, J. L. (2010). Regulation of autophagy by p38alpha MAPK. Autophagy 6 (2), 292–293. doi:10.4161/auto.6.2.11128
Webber, J. L., and Tooze, S. A. (2010). Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 29 (1), 27–40. doi:10.1038/emboj.2009.321
Wei, A., Xiao, H., Xu, G., Yu, X., Guo, J., Jing, Z., et al. (2020). Hyperoside protects human umbilical vein endothelial cells against anticardiolipin antibody-induced injury by activating autophagy. Front. Pharmacol. 11, 762. doi:10.3389/fphar.2020.00762
Wisdom, R. (1999). AP-1: one switch for many signals. Exp. Cell Res. 253 (1), 180–185. doi:10.1006/excr.1999.4685
Wu, H., Yang, Y., Guo, S., Yang, J., Jiang, K., Zhao, G., et al. (2017). Nuciferine ameliorates inflammatory responses by inhibiting the TLR4-mediated pathway in lipopolysaccharide-induced acute lung injury. Front. Pharmacol. 8, 939. doi:10.3389/fphar.2017.00939
Xiao, M., Xian, C., Wang, Y., Qi, X., Zhang, R., Liu, Z., et al. (2023). Nuciferine attenuates atherosclerosis by regulating the proliferation and migration of VSMCs through the Calm4/MMP12/AKT pathway in ApoE((-/-)) mice fed with High-Fat-Diet. Phytomedicine 108, 154536. doi:10.1016/j.phymed.2022.154536
Yannakoulia, M., and Panagiotakos, D. (2021). Weight loss through lifestyle changes: impact in the primary prevention of cardiovascular diseases. Heart 107 (17), 1429–1434. doi:10.1136/heartjnl-2019-316376
Yoshida, N., Manabe, H., Terasawa, Y., Nishimura, H., Enjo, F., Nishino, H., et al. (2000). Inhibitory effects of vitamin E on endothelial-dependent adhesive interactions with leukocytes induced by oxidized low density lipoprotein. Biofactors 13 (1-4), 279–288. doi:10.1002/biof.5520130142
Yuan, P., Hu, Q., He, X., Long, Y., Song, X., Wu, F., et al. (2020). Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death Dis. 11 (2), 141. doi:10.1038/s41419-020-2343-1
Zhang, C., Deng, J., Liu, D., Tuo, X., Xiao, L., Lai, B., et al. (2018). Nuciferine ameliorates hepatic steatosis in high-fat diet/streptozocin-induced diabetic mice through a PPARα/PPARγ coactivator-1α pathway. Br. J. Pharmacol. 175 (22), 4218–4228. doi:10.1111/bph.14482
Zhang, H., Park, Y., Wu, J., Chen, X., Lee, S., Yang, J., et al. (2009). Role of TNF-alpha in vascular dysfunction. Clin. Sci. (Lond) 116 (3), 219–230. doi:10.1042/CS20080196
Zhao, J., Zhong, F., Yu, H., Chen, Z., Wang, M., and Chen, J. (2018). Human cytomegalovirus infection-induced autophagy was associated with the biological behavioral changes of human umbilical vein endothelial cell (HUVEC). Biomed. Pharmacother. 102, 938–946. doi:10.1016/j.biopha.2018.03.156
Zhou, L., Ning, H., Wei, H., Xu, T., Zhao, X., Fu, A., et al. (2020). A novel STAT3-mediated GATA6 pathway contributes to tert-butylhydroquinone- (tBHQ-) protected TNFα-activated vascular cell adhesion molecule 1 (VCAM-1) in vascular endothelium. Oxid. Med. Cell Longev. 2020, 6584059. doi:10.1155/2020/6584059
Zhou, L., Wang, Q., Zhang, H., Li, Y., Xie, S., and Xu, M. (2019). YAP inhibition by nuciferine via AMPK-mediated downregulation of HMGCR sensitizes pancreatic cancer cells to gemcitabine. Biomolecules 9 (10), 620. doi:10.3390/biom9100620
Keywords: nuciferine, VCAM1, autophagy, AP1, AKT/mTOR, vascular endothelium
Citation: Wei H, Yin Y, Yang W, Zhu J, Chen L, Guo R, Yang Z and Li S (2023) Nuciferine induces autophagy to relieve vascular cell adhesion molecule 1 activation via repressing the Akt/mTOR/AP1 signal pathway in the vascular endothelium. Front. Pharmacol. 14:1264324. doi: 10.3389/fphar.2023.1264324
Received: 20 July 2023; Accepted: 14 September 2023;
Published: 28 September 2023.
Edited by:
Alejandro Urzua, University of Santiago, ChileCopyright © 2023 Wei, Yin, Yang, Zhu, Chen, Guo, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songtao Li, bGlzb25ndGFvQHpjbXUuZWR1LmNu
†These authors have contributed equally to this work