 Haitham Elbir
Haitham Elbir Laurent Abi-Rached
Laurent Abi-Rached Pierre Pontarotti
Pierre Pontarotti Niyaz Yoosuf
Niyaz Yoosuf Michel Drancourt
Michel Drancourt- 1URMITE, UMR63, CNRS 7278, IRD 198, INSERM 1095, Aix Marseille Université, Marseille, France
- 2Equipe ATIP, Centre National de la Recherche Scientifique, Laboratoire d’Analyse, Topologie, Probabilités – Unité Mixte de Recherche 7353, Aix Marseille Université, Marseille, France
- 3Equipe EBM, Centre National de la Recherche Scientifique, Laboratoire d’Analyse, Topologie, Probabilités – Unité Mixte de Recherche 7353, Aix-Marseille Université, Marseille, France
Background: Relapsing fever borreliae are vector-borne bacteria responsible for febrile infection in humans in North America, Africa, Asia, and in the Iberian Peninsula in Europe. Relapsing fever borreliae are phylogenetically closely related, yet they differ in pathogenicity and vectors. Their long-term taxonomy, based on geography and vector grouping, needs to be re-apprised in a genomic context. We therefore embarked into genomic analyses of relapsing fever borreliae, focusing on species found in Africa.
Results: Genome-wide phylogenetic analyses group Old World Borrelia crocidurae, Borrelia hispanica, B. duttonii, and B. recurrentis in one clade, and New World Borrelia turicatae and Borrelia hermsii in a second clade. Accordingly, average nucleotide identity is 99% among B. duttonii, B. recurrentis, and B. crocidurae and 96% between latter borreliae and B. hispanica while the similarity is 86% between Old World and New World borreliae. Comparative genomics indicates that the Old World relapsing fever B. duttonii, B. recurrentis, B. crocidurae, and B. hispanica have a 2,514-gene pan genome and a 933-gene core genome that includes 788 chromosomal and 145 plasmidic genes. Analyzing the role that natural selection has played in the evolution of Old World borreliae species revealed that 55 loci were under positive diversifying selection, including loci coding for membrane, flagellar, and chemotaxis proteins, three categories associated with adaption to specific niches.
Conclusion: Genomic analyses led to a reappraisal of the taxonomy of relapsing fever borreliae in Africa. These analyses suggest that B. crocidurae, B. duttonii, and B. recurrentis are ecotypes of a unique genomospecies, while B. hispanica is a distinct species.
Introduction
Among spirochetes, the genus Borrelia includes the Lyme disease group of borreliae in North America and Europe and the relapsing fever group of borreliae in North America, Africa, Asia, and in the Iberian Peninsula in Europe (1). Six relapsing fever species have representatives that have been cultured, whereas three uncultured species are detected on the basis of original sequences (2). Eight of the species defined by sequence analysis are found in Africa, including the four definite human pathogens Borrelia hispanica, B. crocidurae, B. duttonii, and B. recurrentis. In Africa, these relapsing fever borreliae are transmitted by Ornithodoros ticks and the Pediculus humanus body louse (3–7).
Although relapsing fever borreliae are genetically very similar, they differ in vector, host range, and disease spectrum. B. recurrentis is a host-restricted species that is documented in human and human lice, most commonly producing deadly infections if left untreated (7). B. crocidurae is found in humans and in several rodent species, causing a febrile tick-borne infection less severe than the louse-borne infection in humans (8). Finally, B. duttonii has a narrow host range and is associated with mortality and pregnancy loss (9).
Relapsing fever borreliae were initially classified on the basis of vector and geography (1). However, the coexistence of several species in the same geographic area such as B. hispanica and B. crocidurae in North Africa (10, 11) and B. duttonii and an unnamed Borrelia sp. (AB105117) (12) in Tanzania questioned this conventional classification. In addition, 16S rRNA gene sequence similarity between such defined species was above the borderline that typically distinguishes other bacterial species (13).
Therefore, revisiting the long-term taxonomy of relapsing fever borreliae in Africa is needed. The recent availability of genome sequences for B. crocidurae, B. duttonii, B. recurrentis, B. hispanica (14–16), B. turicatae, and B. hermsii (unpublished) provides such an opportunity.
Here, we performed comparative genomics and genome-wide phylogenetic analyses to address the taxonomy of relapsing fever borreliae in a phylogenetic context, focusing on borreliae circulating in Africa.
Materials and Methods
Identification of Orthologs
All chromosomal and plasmidic sequences were retrieved from GenBank for genomes: B. crocidurae (GenBank accession number CP003426), B. duttonii (CP000976), B. recurrentis (CP000993), B. hispanica (AYOU00000000), Borrelia miyamotoi (CP006647), Borrelia persica (AYOT00000000), Borrelia hermsii (CP000048), Borrelia turicatae (CP000049), Borrelia bissettii (CP002746), Borrelia garinii (CP003151), Borrelia alzelii (CP002933), and Borrelia burgdorferi (CP00222801). A bidirectional best hit (BBH) approach with an expect value cut-off of 10–3 was used to identify orthologous chromosomal genes; only genes having a similarity >30% and length coverage >70% were kept.
Phylogenetic Analysis
To compare the genomes of the relapsing fever borreliae, each set of orthologous chromosomal gene sequences was first aligned using MAFFT (17). Bayesian phylogenetic analyses were then performed using MRBAYES3.2.1 (18) with a GTR+ model of nucleotide substitution; sampling was performed through three independent runs (each having one cold chain and three heated chains), which were run for 1,000,000 generations or until the average standard deviation of split frequencies for the three runs was <0.01. Trees were sampled every 200 generations and the first one-fourth of the trees was discarded before a consensus tree was generated. Phylogenetic analysis of the telomere resolvase gene was conducted using a maximum likelihood approach with MEGA5 (19).
Bayesian Concordance Analysis
The Bayesian concordance analysis was carried out with BUCKy 1.4.0 (20) using the phylogenetic trees generated in the Bayesian analysis with MRBAYES. For the discordance parameter α, we investigated three values representing the range of possible values: “0.01,” “1” (default value), and “2.” These values approximately translate into prior probabilities that two randomly sampled genes share the same tree of 0.99, 0.50, and 0.33, respectively. The final analysis used three runs of one million generations with eight chains (one cold chain and seven hot chains) and a discordance parameter α set to “1.”
Selection Analyses
To generate codon alignments, we first aligned each set of orthologous protein sequences with MAFFT (17). The amino acids were then replaced by the corresponding codons using PAL2NAL (21). Estimation of the non-synonymous (dN)/synonymous (dS) substitution rate ratio (ω) was performed using the maximum likelihood method implemented in the CODEML program from the PAML package (22) with the F3X4 model of codon frequencies. The ω ratio measures the selective pressure on coding sequences, with <1, =1, and >1 indicating negative purifying selection, neutral evolution, and positive diversifying selection, respectively. For each gene, the tree topology with the highest joint probably in the concordance analysis was used for these analyses and three sets of likelihood ratio tests were conducted to compare null models that do not allow ω > 1 (M1a, M7, and M8a) with models that do (M2a and M8). Significance was assessed by comparing twice the difference in likelihood between the models (2ΔL) to a χ2 distribution with one (M8a/M8) or two (M1a/M2 and M7/M8) degrees of freedom (22).
Results
Relapsing Fever Borreliae Core and Pan Genome
The genomes of relapsing fever and Lyme disease borreliae include a linear chromosome, 1–15 linear plasmids, and 1–9 circular plasmids. This genome structure was compared to that of the 3,974 bacterial genomes described in the GOLD database. This analysis identified 755 fragmented genomes (19%), with 371 bacterial genomes consisting of one chromosome and one plasmid and 364 bacterial genomes including 3–11 DNA fragments (chromosome and plasmid). The remaining bacterial genomes were Borrelia, which are fragmented into 2–21 DNA fragments. In addition to Borrelia, linear chromosomes were only observed in Streptomyces davawensis and Agrobacterium tumefaciens (23, 24). The unusual linear DNA fragments of Borrelia have covalently closed telomeres (25). Phylogenetic analysis of the telomere resolvase gene grouped the Borrelia species into Lyme and relapsing fever groups. Furthermore, the Borrelia and the A. tumefaciens telomere resolvase genes clustered together and away from homologous gene in virus and eukaryotes (Figure 1).
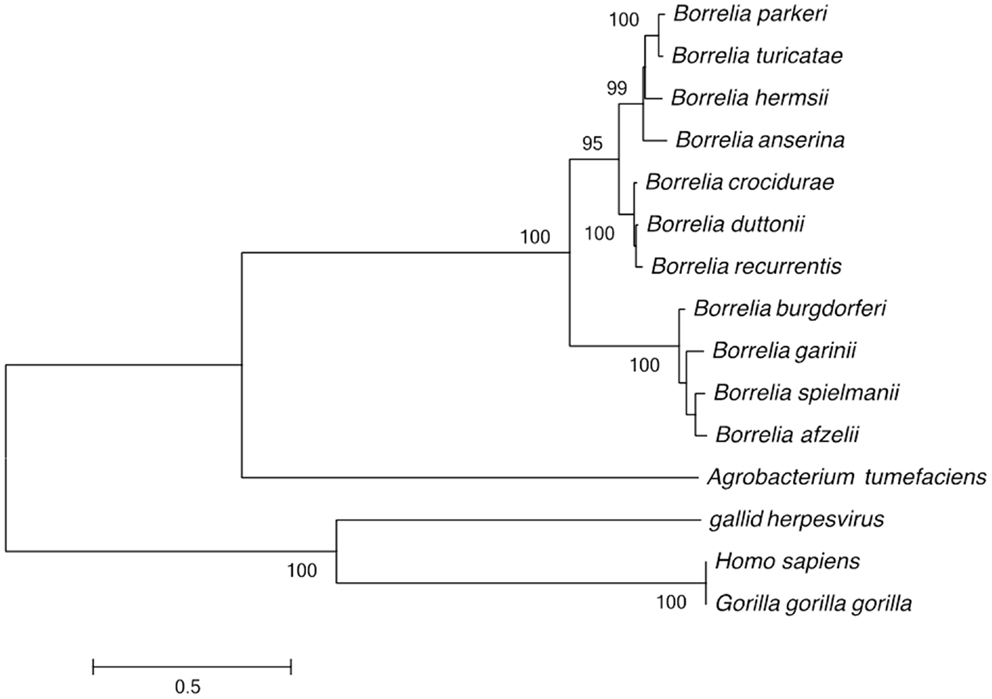
Figure 1. Phylogenetic tree based on the Borrelia telomere resolvase gene sequence, separating the Lyme disease group from the recurrent fever group; and separating the Old World from the New World recurrent fever Borrelia. Only bootstrap values >80% are indicated at nodes. The bar indicates a 0.5% divergence in nucleotidic sequences.
Borrelia hermsii and B. turicatae chromosomes are collinear, whereas Old World borreliae genomes collinearity is disrupted by 1,134-bp insertion sequences (IS) IS605 OrfB (B. duttonii, B. recurrentis, and B. crocidurae) or 414-bp transposase IS200 (only in B. crocidurae). These IS are also carried by the plasmids of the three species and have high sequence identity [97–98%; Ref. (16)]. Chromosome collinearity is further disrupted by a 5-kbp duplication in B. duttonii (16). Also, genes rpsU, ftsK, and bacA that encode putative membrane proteins are duplicated in B. recurrentis (16). The main chromosomal differences between B. duttonii, B. recurrentis, and B. crocidurae exist in the 5′ regions of each chromosome; whereas in New World B. hermsii and B. turicatae, the 5′ regions are more conserved.
Comparing the gene content of B. hermsii against that of other Borrelia revealed five genes that were lost in some or all the non-B. hermsii borreliae: three hypothetical proteins (AAX17236, AAX17110, and AAX16935) that are unique to B. hermsii, one hypothetical protein (AAX16910) missing from relapsing fever borreliae but present in the Lyme group borreliae, and the glutathione peroxidase-like BsaA gene (AAX17028) that is absent in non-B. hermsii borreliae but that has an ortholog in Geobacillus thermoglucosidasius and Bacillus species. A similar analysis using B. turicatae as a reference revealed one hypothetical protein (AAX17575) absent in non-B. turicatae borreliae, a second hypothetical protein (AAX18156) absent from relapsing fever borreliae but present in Lyme group borreliae, and the crossover junction endodeoxyribonuclease RuvC (AAX17369) gene that is absent in non-B. turicatae borreliae but has an ortholog in Clostridium acidurici. Comparison of New World and Old World relapsing borreliae genomes identified two genes missing from the New World relapsing fever borreliae: RNA polymerase sigma-54 factor (ACH93394) and antigen P35-like protein (ACH92961). Conversely, one gene encoding the DNA repair protein RadA (AAX17754) is missing from the Old World relapsing fever borreliae genome.
In summary, the relapsing fever Borrelia pan genome includes 2,514 genes, and the number of protein coding genes (CDS) ranges from 990 in B. recurrentis to 2,140 in B. crocidurae. Of these, 933 (37.1%) represent the core genome and encode 788 core chromosome proteins and 145 core plasmid proteins. Interestingly, while the core genes represent <50% of the pan genome, they represent more than 81% of the genes classified into the cluster of orthologous group (COG) categories (Figure 2), consistent with the core genes representing conserved groups not only in Borrelia but also across other bacterial genera.
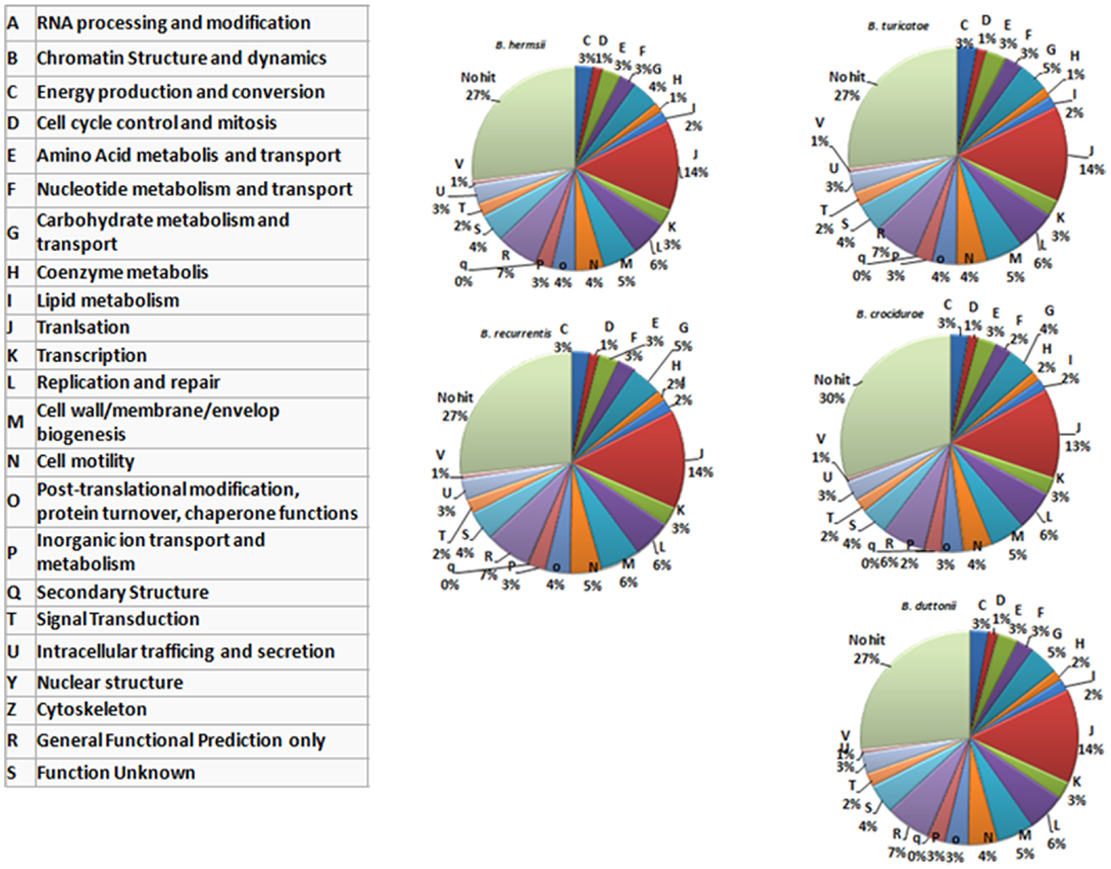
Figure 2. Functional classification of the genes encoded by five recurrent fever Borrelia (chromosomal genes). The protein-coding sequences are classified according to categories determined in the cluster of orthologous group (COG) database, as indicated in the left-hand table.
Functional Classification of the Plasmidome
The B. recurrentis, B. duttonii, and B. crocidurae pan-plasmidome includes 706 genes, 162 of which are core plasmid genes, 166 genes being conserved in B. crocidurae and B. recurrentis, and 180 genes being conserved between B. duttonii and B. recurrentis. Interestingly, only 8–12% of the pan-plasmidome genes are assigned a functional category: this contrasts with the results obtained for the chromosomal genome, where 70–73% of the genes are assigned to a COG category (Figure 2), and is consistent with the plasmid genes evolving more rapidly than their chromosomal counterparts. Among the plasmidic genes, the most represented categories of genes are those involved in cell cycle control, cell division, chromosome partitioning translation while the least represented category is energy production and conversion. In B. crocidurae and B. duttonii, plasmidic genes could be assigned to 11 different categories and to 7 categories in B. recurrentis plasmidome. In particular, coenzyme transport and metabolism is missing from B. recurrentis (Figure 3). The B. crocidurae plasmidome encodes 607 proteins, 90% of which have homologs in the non-redundant database and an additional 9% are assigned putative function according to COG database. Some short genes are also present as remnants and thus represent potentially degraded genes. In the other species, plasmid sequence collinearity is more frequently interrupted by non-coding sequences and species-specific genes: accordingly, a low 42% density of CDs is due to large intergenic spacers. The five genomes encode glycoside hydrolase genes (CH23 and CH73) and glycosyl transferase family GT28 and GT51 (26) previously shown to be required for peptidoglycan synthesis (27), suggesting susceptibility to β-lactamines. Further comparisons against the Antibiotic Resistance Genes Database (28) indicate a complete absence of antibiotic resistance, in line with field observations that in Africa, antibiotics are regularly effective against relapsing fevers (29). Among studied species, only B. crocidurae contains one putative copy of clustered regularly interspaced short palindromic repeats (CRISPRs).
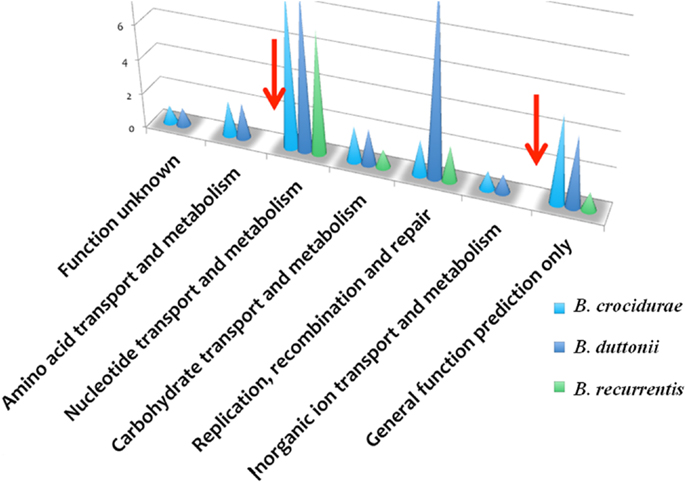
Figure 3. Functional classification of plasmidic genes according to COG categories showing gene categories, which are missing in B. duttonii (dark blue symbol) and B. crocidurae (light blue symbol), compared to B. recurrentis (green symbol). Red arrows indicate known functions missing in B. recurrentis.
Genome-Wide Phylogenetic Analyses Reveal a Major Evolutionary Split
In order to further investigate the relationships between the B. duttonii, B. recurrentis, and B. crocidurae genomes, we used the 788 core chromosome genes and compared the phylogenetic signal obtained for each individual gene using a concordance analysis. This analysis reveals that the five genomes form two major clades: one containing B. duttonii, B. recurrentis, and B. crocidurae and second containing B. hermsii and B. turicatae; all 788 genes investigated support this clustering (concordance factor = 1).
The concordance analysis also reveals a second well-supported split in the B. duttonii, B. recurrentis, and B. crocidurae clade, with the first two forming a group and the third one being an outgroup (concordance factor = 0.94). Consistent with this, only 16 of the 788 genes supported an alternative grouping for these three genomes (joint probability in the concordance analysis ≥0.5): 10 supported a B. recurrentis–B. crocidurae group and 6 a B. duttonii–B. crocidurae group. The tree topology was also conserved after including other relapsing and Lyme species (Figure 4). Further comparison of nucleotide sequences across all Borrelia genomes for the 788 core chromosome genes indicates an average nucleotide identity >99% between B. duttonii, B. recurrentis, and B. crocidurae (Table 1). Consistent with such a high identity at the nucleotide level, 173 proteins display identical amino acid sequences between B. duttonii and B. recurrentis and 108 between B. crocidurae and B. duttonii. Across these three species, 58 proteins display no sequence variation: those sequences notably include proteins involved in translation, ribosomal structure, and biogenesis (33%) as well as proteins involved in cell motility (10%); the remaining conserved proteins represent a large part (80%) of the COG functions.
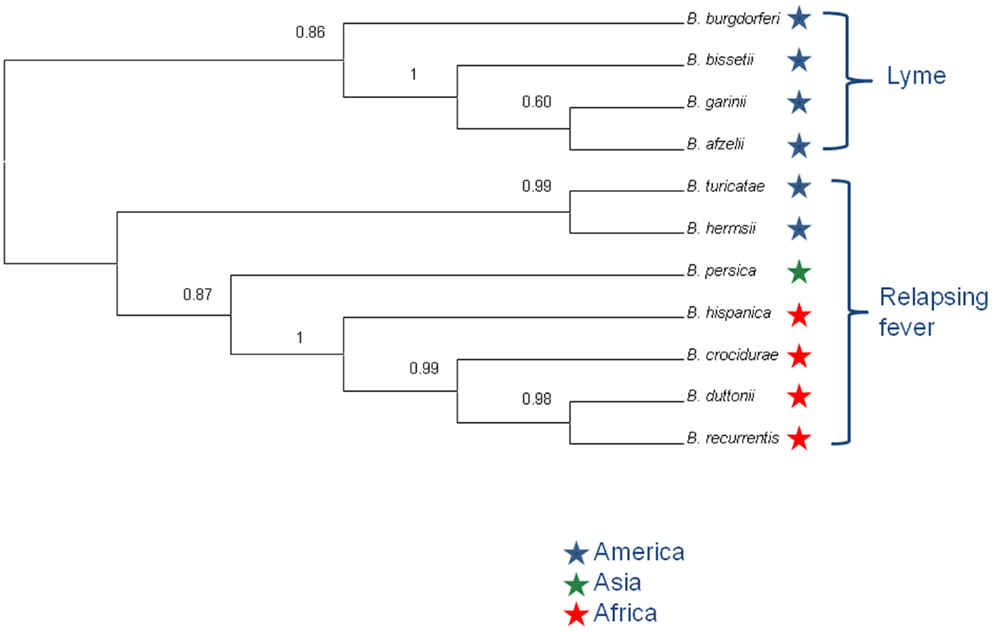
Figure 4. Primary concordance analysis. The tree shows the topology of Borrelia sp., grouped into the Lyme disease and relapsing fever group. Concordance factor values are indicated at nodes.
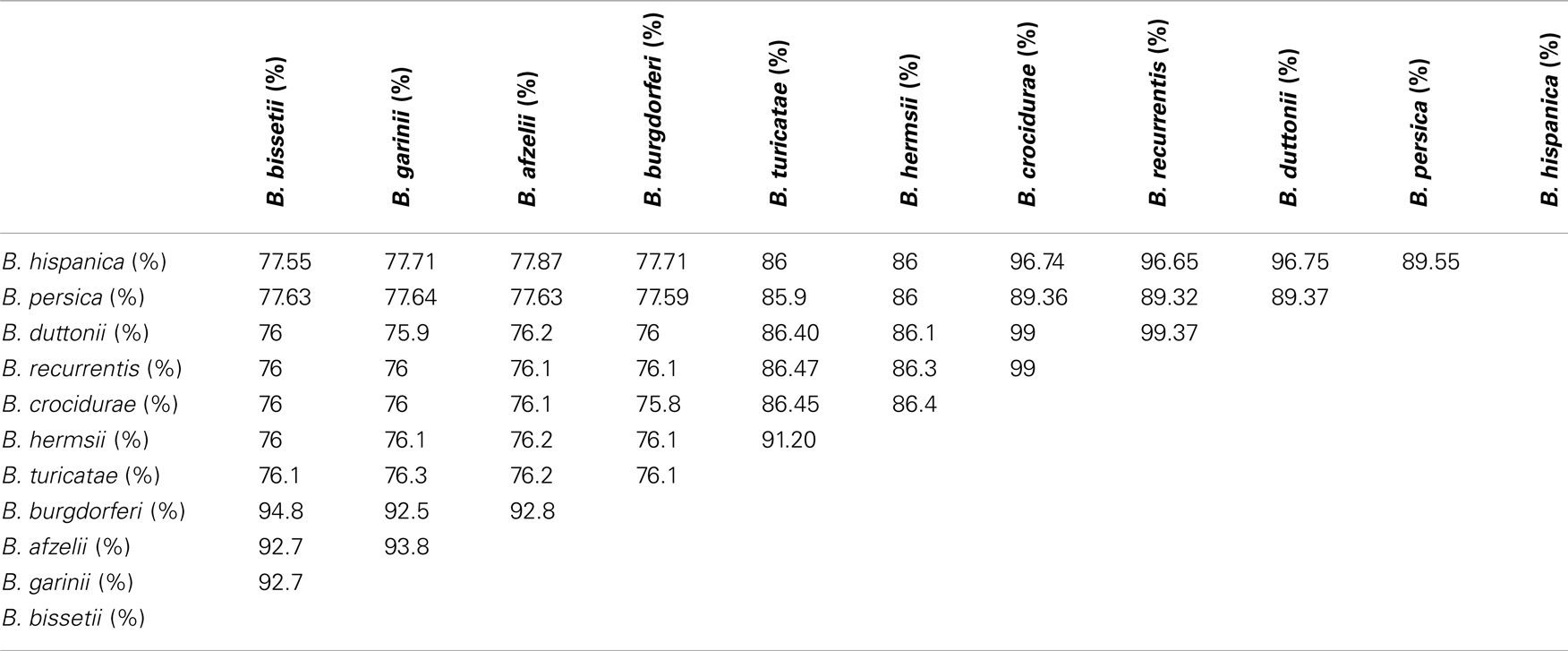
Table 1. Average nucleotide identity among Borrelia species.
Consistent with their high nucleotide identity (86–99%), there was no significant difference in codon usage between the five relapsing fever borreliae, with the most frequently used codons in the chromosome (as measured by the relative synonymous codon usage or RSCU) being AGA, which encodes arginine (RSCU, 3.8), and TTA, which encodes leucine (RSCU, 2.6). Similarly, the least used codons are CGG and CGC that encode arginine and CTG, which encodes leucine.
Positive Diversifying Selection Contributed to the Evolution of the Borrelia Genomes
To investigate how natural selection contributed to the evolution of the five relapsing fever borreliae genomes, we performed a genome-wide analysis of positive selection: significant evidence of positive diversifying selection was observed for 55 of the 788 core chromosome genes (α ≤ 0 with at least two of the three methods used; Table 2). Of these 55 genes, 8 (14.5%) are involved in translation, ribosomal structure, and biogenesis, 5 (9%) in replication, recombination, and repair, and 4 (7.2%) in cell wall biogenesis, cell motility, and signal transduction mechanisms.
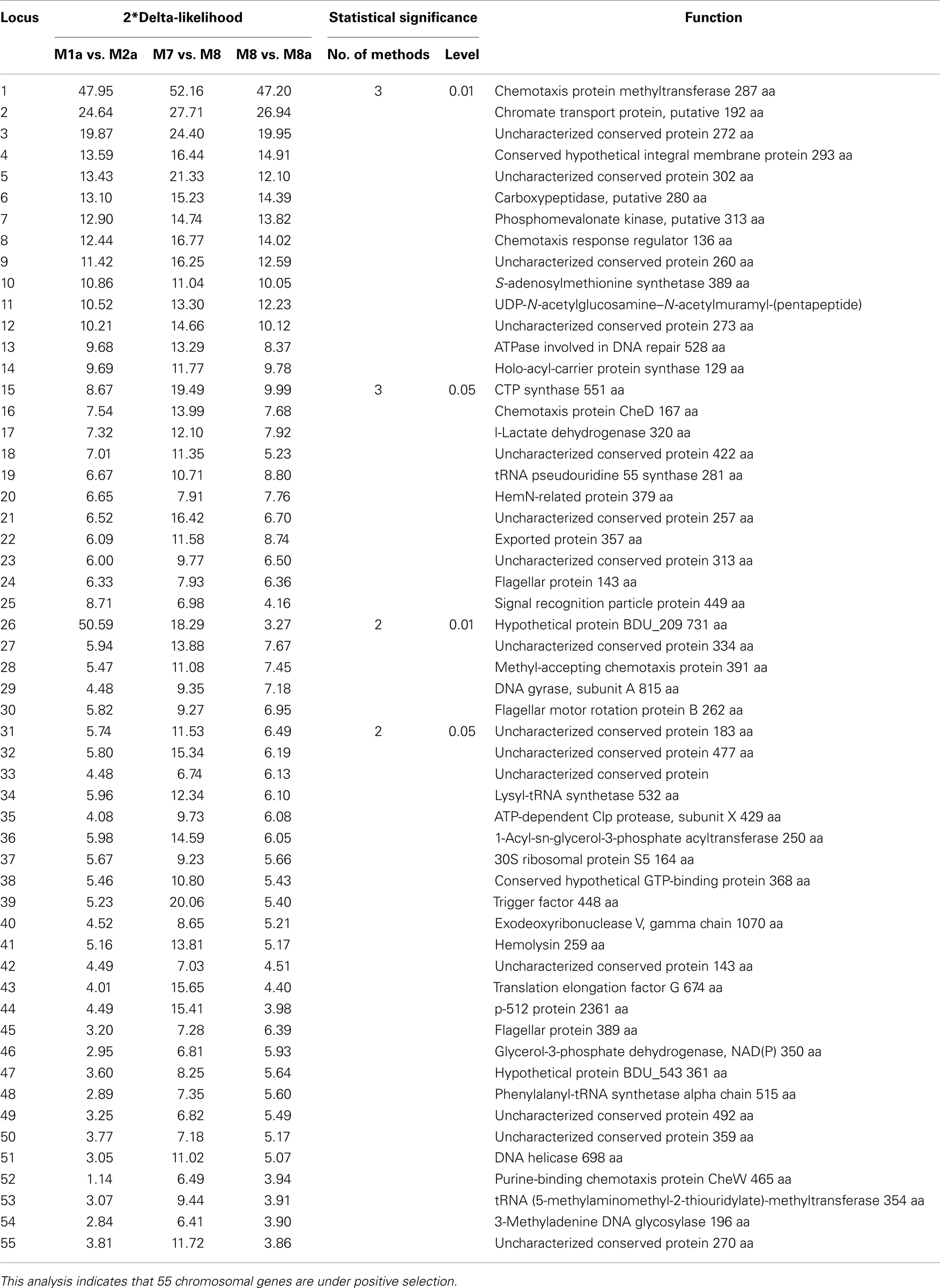
Table 2. Analysis of positive selection in five Borrelia species.
Discussion
Thanks to the sequencing of several African relapsing fever borreliae (14–16), it is now possible to assess relationships between African and American relapsing fever borreliae. Interestingly, all recurrent fever Borrelia shares an overall genome architecture with Lyme disease group borreliae that is unique, in that they exhibit a massively linear genome. This unique topology is maintained by one plasmidic copy of telomere resolvase, an enzyme rarely found in bacteria. Borrelia telomere resolvase is related to that of A. tumefaciens, a plant pathogen unrelated to Borrelia.
Whole chromosome comparisons across American and African relapsing fever borreliae show extensive conservation of gene content and gene order, only disrupted by missing genes in one group. As for genes shared between B. duttonii, B. recurrentis, and B. crocidurae, B. turicatae, and B. hermsii, the average nucleotide identity is ~86%. The high number of impaired genes in B. recurrentis might be indicative of a decaying genome (16). In bacteria, RecA and RadA proteins play a critical role in DNA repair processes (30): both genes are present in the New World relapsing fever borreliae that also have impaired genes. In contrast, B. crocidurae and B. duttonii encode RecA but not RadA and have a lower number of impaired genes, while B. recurrentis lacks both proteins and contains many impaired genes. The accurate pan-genome size is probably bigger than the one we estimated.
Close relatedness between Borrelia species is further reflected in their gene content as they share >90% of their genes. Similarly, a high level of functional conservation was observed, as many orthologous proteins share high sequence identity. While the chromosomes have the same codon usage, different codon usage distinguishes the chromosome and the plasmids in the same species. Indeed, the majority of housekeeping genes are chromosomal whereas the plasmids mostly encode surface proteins responsible for antigenic variability and host-interactions. It became apparent that some plasmid genes code for unique functions not represented in the chromosome, in addition to some function that are shared with chromosomal genes. Plasmids are frequently subject to genomic rearrangement and, not surprisingly, borreliae plasmids are not totally collinear and conserved in terms of gene content and orientation but they still share syntenic sequences representing the core plasmid. As for the African relapsing fever borreliae chromosome, 90% of its gene content is included in the core genome and is thus common to the three sequenced genomes; the remaining 10% represent species-specific genes that lie in the 5′ extremities. Interestingly, the variable sequences in these 5′ extremities are related to plasmid sequences, a phenomenon already observed for the B. burgdorferi chromosome (31).
Together with variation in plasmid content, positive selection represents one of the main molecular mechanisms contributing to vector–host adaptation. Our study revealed that 55 genes were the targets of positive diversifying selection during the evolution of the recurrent fever borreliae. Although these genes are randomly distributed in the chromosome, they code for proteins involved in translation, ribosomal structure and biogenesis, replication, recombination and repair, cell membrane biogenesis, cell motility, and signal transduction: these mechanisms are related to host interaction and might thus be important factors to restrict the habitats of Borrelia species to specific niche (i.e., host and vector adaptation).
Conclusion
Current taxonomy of the relapsing fever borreliae derives from geographical and tick-specific relationships comforted by analyses of single genes or of several concatenated genes (32). The coexistence of different species in the same geographic region questioned the accuracy of this conventional classification and here we aimed to gain a more accurate picture by using a genome-wide phylogenetic approach, in line with recent recommendations to incorporate genome sequence data into classification of bacteria (33). This analysis grouped the relapsing fever borreliae into a clade A that includes Old World B. hispanica, B. recurrentis, B. crocidurae, and B. duttonii and a clade B that includes New World B. turicatae and B. hermsii. This division is supported by the 788 genes used. The concordance analysis also strongly suggests that B. duttonii and B. recurrentis are closely related and form a clade that has B. crocidurae as an outgroup, strengthening a previous hypothesis that B. recurrentis is a subset of B. duttonii (16). Accordingly, the average nucleotide identity is 99% between B. crocidurae, B. duttonii, and B. recurrentis, 96% between B. hispanica and (B. crocidurae, B. duttonii, and B. recurrentis), and 91% between B. turicatae and B. hermsii, while the ANI between the two groups is at 86%. It was suggested that average nucleotide identity values of ~94% correspond to the cut-off for bacterial species definition (34), so that the average nucleotide identity for B. duttonii, B. crocidurae, and B. recurrentis would be well above that level and the three Old World species would thus be ecotypes of the same genomospecies. These data suggest updating the long-term taxonomy of these borreliae, in delineating a new species Borrelia africana, comprising of three subspecies crocidurae, duttonii, and recurrentis. Also, data here presented allow refining molecular tests for the accurate detection and identification of relapsing fever borreliae, in a point-of-care format (35).
Author Contributions
Pierre Pontarotti and Michel Drancourt designed the study. Haitham Elbir, Laurent Abi-Rached, and Niyaz Yoosuf performed analyses. Haitham Elbir, Laurent Abi-Rached, Pierre Pontarotti, and Michel Drancourt analyzed the data and prepared the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cutler SJ, Abdissa A, Trape JF. New concepts for the old challenge of African relapsing fever borreliosis. Clin Microbiol Infect (2009) 15:400–6. doi: 10.1111/j.1469-0691.2009.02819.x
2. Elbir H, Raoult D, Drancourt M. Relapsing fever borreliae in Africa. Am J Trop Med Hyg (2013) 89:288–92. doi:10.4269/ajtmh.12-0691
3. Parola P, Diatta G, Socolovschi C, Mediannikov O, Tall A, Bassene H, et al. Tick-borne relapsing fever borreliosis, rural senegal. Emerg Infect Dis (2011) 17:883–5. doi:10.3201/eid1705.100573
4. Fukunaga M, Ushijima Y, Aoki LY, Talbert A. Detection of Borrelia duttonii, a tick-borne relapsing fever agent in central Tanzania, within ticks by flagellin gene-based nested polymerase chain reaction. Vector Borne Zoonotic Dis (2001) 1:331–8. doi:10.1089/15303660160025949
5. Sarih M, Garnier M, Boudebouch N, Bouattour A, Rihani A, Hassar M, et al. Borrelia hispanica relapsing fever, Morocco. Emerg Infect Dis (2009) 15:1626–9. doi:10.3201/eid1510.090403
6. Schwan TG, Anderson JM, Lopez JE, Fischer RJ, Raffel SJ, McCoy BN, et al. Endemic foci of the tick-borne relapsing fever spirochete Borrelia crocidurae in Mali, West Africa, and the potential for human infection. PLoS Negl Trop Dis (2012) 6:e1924. doi:10.1371/journal.pntd.0001924
7. Ramos J, Malmierca E, Reyes F, Wolde W, Galata A, Tesfamariam A, et al. Characteristics of louse-borne relapsing fever in Ethiopian children and adults. Ann Trop Med Parasitol (2004) 98:191–6. doi:10.1179/000349804225003136
8. Vial L, Diatta G, Tall A, Bael H, Bouganali H, Durand P, et al. Incidence of tick-borne relapsing fever in West Africa: longitudinal study. Lancet (2006) 368:37–43. doi:10.1016/S0140-6736(06)68968-X
9. Jongen VH, van Roosmalen J, Tiems J, van Holten J, Wetsteyn JC. Tick-borne relapsing fever and pregnancy outcome in rural Tanzania. Acta Obstet Gynecol Scand (1997) 76:834–8. doi:10.3109/00016349709024361
10. Diatta G, Souidi Y, Granjon L, Arnathau C, Durand P, Chauvancy G, et al. Epidemiology of tick-borne borreliosis in Morocco. PLoS Negl Trop Dis (2012) 6:e1810. doi:10.1371/journal.pntd.0001810
11. Trape JF, Diatta G, Arnathau C, Bitam I, Sarih M, Belghyti D, et al. The epidemiology and geographic distribution of relapsing fever borreliosis in West and North Africa, with a review of the Ornithodoros erraticus Complex (Acari: Ixodida). PLoS One (2013) 8:e78473. doi:10.1371/journal.pone.0078473
12. Kisinza WN, McCall PJ, Mitani H, Talbert A, Fukunaga M. A newly identified tick-borne Borrelia species and relapsing fever in Tanzania. Lancet (2003) 362:1283–4. doi:10.1016/S0140-6736(03)14609-0
13. Ras NM, Lascola B, Postic D, Cutler SJ, Rodhain F, Baranton G, et al. Phylogenesis of relapsing fever Borrelia spp. Int J Syst Bacteriol (1996) 46:859–65. doi:10.1099/00207713-46-4-859
14. Elbir H, Gimenez G, Robert C, Bergström S, Cutler S, Raoult D, et al. Complete genome sequence of Borrelia crocidurae. J Bacteriol (2012) 194:3723–4. doi:10.1128/JB.00118-12
15. Elbir H, Larsson P, Upreti M, Normark J, Bergström S. Genome sequence of the relapsing fever borreliosis species Borrelia hispanica. Genome Announc (2014) 2:e01171–13. doi:10.1128/genomeA.01171-13
16. Lescot M, Audic S, Robert C, Nguyen TT, Blanc G, Cutler SJ, et al. The genome of Borrelia recurrentis, the agent of deadly louse-borne relapsing fever, is a degraded subset of tick-borne Borrelia duttonii. PLoS Genet (2008) 4:e1000185. doi:10.1371/journal.pgen.1000185
17. Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol (2009) 537:39–64. doi:10.1007/978-1-59745-251-9_3
18. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics (2003) 19:1572–4. doi:10.1093/bioinformatics/btg180
19. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol (2011) 28:2731–9. doi:10.1093/molbev/msr121
20. Larget BR, Kotha SK, Dewey CN, Ané C. BUCKy: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics (2010) 26:2910–1. doi:10.1093/bioinformatics/btq539
21. Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res (2006) 34:W609–12. doi:10.1093/nar/gkl315
22. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol (2007) 24:1586–91. doi:10.1093/molbev/msm088
23. Jankowitsch F, Schwarz J, Rückert C, Gust B, Szczepanowski R, Blom J, et al. Genome sequence of the bacterium Streptomyces davawensis JCM 4913 and heterologous production of the unique antibiotic roseoflavin. J Bacteriol (2012) 194:6818–27. doi:10.1128/JB.01592-12
24. Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, Qurollo B, et al. Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science (2001) 294:2323–8. doi:10.1126/science.1066803
25. Barbour AG, Garon CF. Linear plasmids of the bacterium Borrelia burgdorferi have covalently closed ends. Science (1987) 237:409–11. doi:10.1126/science.3603026
26. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res (2009) 37:D233–8. doi:10.1093/nar/gkn663
27. Cayrou C, Henrissat B, Gouret P, Pontarotti P, Drancourt M. Peptidoglycan: a post-genomic analysis. BMC Microbiol (2012) 12:294. doi:10.1186/1471-2180-12-294
28. Liu B, Pop M. ARDB – antibiotic resistance genes database. Nucleic Acids Res (2009) 37:D443–7. doi:10.1093/nar/gkn656
29. Perine PL, Teklu B. Antibiotic treatment of louse-borne relapsing fever in Ethiopia: a report of 377 cases. Am J Trop Med Hyg (1983) 32:1096–100.
30. Roca AI, Cox MM. RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol (1997) 56:129–223. doi:10.1016/S0079-6603(08)61005-3
31. Huang WM, Robertson M, Aron J, Casjens S. Telomere exchange between linear replicons of Borrelia burgdorferi. J Bacteriol (2004) 186:4134–41. doi:10.1128/JB.186.13.4134-4141.2004
32. Cutler SJ, Bonilla EM, Singh RJ. Population structure of East African relapsing fever Borrelia spp. Emerg Infect Dis (2010) 16:1076–80. doi:10.3201/eid1607.091085
33. Ramasamy D, Mishra AK, Lagier J-C, Padhmanabhan R, Rossi M, Sentausa E, et al. A polyphasic strategy incorporating genomic data for the taxonomic description of new bacterial species. Int J Syst Evol Microbiol (2014) 64:384–91. doi:10.1099/ijs.0.057091-0
34. Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. (2005) 102:2567–72. doi:10.1073/pnas.0409727102
Keywords: Borrelia, relapsing fever, genomics, phylogeny, speciation
Citation: Elbir H, Abi-Rached L, Pontarotti P, Yoosuf N and Drancourt M (2014) African relapsing fever borreliae genomospecies revealed by comparative genomics. Front. Public Health 2:43. doi: 10.3389/fpubh.2014.00043
Received: 04 March 2014; Accepted: 29 April 2014;
Published online: 14 May 2014.
Edited by:
Evangelos Giamarellos-Bourboulis, University of Athens Medical School, GreeceReviewed by:
Jason A. Carlyon, Virginia Commonwealth University School of Medicine, USAOliver Goldmann, Helmholtz Centre for Infection Research, Germany
Copyright: © 2014 Elbir, Abi-Rached, Pontarotti, Yoosuf and Drancourt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel Drancourt, URMITE, UMR63, Centre National de la Recherche Scientifique 7278, IRD 198, INSERM 1095, Aix Marseille Université, Marseille 13005, France e-mail:bWljaGVsLmRyYW5jb3VydEB1bml2LWFtdS5mcg==