 Ridwan Bin Rashid
Ridwan Bin Rashid Jannatul Ferdous
Jannatul Ferdous Suhella Tulsiani
Suhella Tulsiani Peter Kjaer Mackie Jensen
Peter Kjaer Mackie Jensen Anowara Begum
Anowara Begum- 1Department of Microbiology, University of Dhaka, Dhaka, Bangladesh
- 2Section for Global Health, Institute of Public Health, University of Copenhagen, Copenhagen, Denmark
- 3Copenhagen Centre for Disaster Research, Copenhagen, Denmark
Vibrio cholerae O1 and O139 has been known for its ability to cause epidemics. These strains produce cholera toxin which is the main cause of secretory diarrhea. V. cholerae non-O1 and non-O139 strains are also capable of causing gastroenteritis as well as septicemia and peritonitis. It has been proven that virulence factors such as T6SS, hapA, rtxA, and hlyA are present in almost all V. cholerae strains. It is imperative that viable but non-culturable cells of V. cholerae are also detected since they are also known to cause diarrhea. Thus, the aim of this study was to develop an assay that detects all V. cholerae regardless of their serotype, culturable state, and virulence genes present, by targeting the species specific conserved ompW sequence. The developed assay meets these goals with 100% specificity and is capable of detecting as low as 5.46 copy number of V. cholerae. Detection is rapid since neither lengthy incubation period nor electrophoresis is required. The assay had excellent repeatability (CV%: 0.24–1.32) and remarkable reproducibility (CV%: 1.08–3.7). Amplification efficiencies in the 89–100% range were observed. The assay is more economical than Taqman-based multiplex real-time PCR assays. Compared to other real-time assays, the ompW assay is specific and sensitive, has better repeatability and reproducibility, and is more economical.
Introduction
Vibrio cholerae is a Gram-negative, comma shaped facultative pathogen responsible for causing cholera. The global incidence of cholera was about 2.8 million cases per year, with 91,000 deaths (1). V. cholerae O1 has been the etiological agent for several cholera epidemics. The serogroup O139 was responsible for cholera outbreaks in India and other countries in Asia during 1992 (2) and was also isolated during the outbreak in November 2000 in India (3) and March–April 2002 in Bangladesh (4).
Vibrio cholerae O1 and O139 serogroups express toxin coregulated pilus which confers the bacteria the ability to colonize the intestine while the cholera toxin is associated with secretory diarrhea (5). Depending on severity, the infectious dose for V. cholerae varies from 106 to 1011 cells (6).
Toxigenic and non-toxigenic non O1, non-O139 have been documented as incriminating in several outbreaks in developing countries (7–10). In non-CT-producing vibrios, virulence factors such as type 3 secretion systems, hemolysin (HlyA), repeat in toxin (RTX), and heat-stable enterotoxin have major roles in causing infections (11). Hasan et al. (10) reported 98% V. cholerae strains carried hemagglutinin protease hap (98%) irrespective of their source, i.e., clinical or environmental. Other virulence factors present are T6SS (94–99%), rtxA (96%), toxR (87%), and hlyA (83%), and all these virulence factors might be responsible for diarrhea caused by non-toxigenic non-O1/non-O139 variants.
Vibrio cholerae in the viable but non-culturable (VBNC) state can express virulence factors required to produce infection (12). The VBNC cells have the capacity to revert to the culturable state and colonize the intestine (13) the mechanism of which is largely unknown (14). These organisms may go undetected if conventional culture based methods are used (15). Conventional identification of V. cholera, which may be done by biochemical tests, is time consuming and laborious. Available commercial biochemical identification systems, such as dipstick test used for the detection of O1 and O139 strains, are not always accurate (16). V. cholerae has been shown to possess similar biochemical properties with other species in the Genus Vibrio and Aeromonas, hence complicating an accurate identification (17).
Compared to conventional PCR, real-time PCR is less labor intensive, more safe, and rapid due to the elimination of gel electrophoresis. It has greater sensitivity and can detect minute amounts of target amplicons that might be missed by the conventional PCR. Real-time PCR can directly target genomic DNA and thus eliminate extensive incubation periods (18). Furthermore, VBNC cells can be detected which might be missed by culture-based methods. The ompW sequence is highly conserved among V. cholerae species belonging to different biotypes and/or serogroups (17). Hence, the ompW gene could be used as a target for species-specific detection, identification, and quantification.
A number of assays exist for the detection of V. cholerae (19–24) but many of these assays lack empirical data for reproducibility and repeatability. Some of these assays have not been validated in terms of detecting non-specific products that might accompany the amplification reaction. Furthermore, a number of assays are based on virulence factors that might not be present in certain strains and might yield false negative results.
The aim of this study was to develop an assay that detects and quantifies both O1/O139 and non O1/O139 disease causing strains of Vibrio spp. In addition, the assay would be able to quantify VBNC cells that cannot be detected or quantified by conventional methods.
Materials and Methods
Assay Controls and Growth Conditions
A total of 28 bacterial strains were used as assay controls. V. cholerae strains were grown in alkaline peptone water for enrichment, and all other strains were grown in nutrient broth for 24 h in 37°C. Genomic DNA from overnight cultures controls were extracted and purified according to the manufacturer’s instructions by QIAamp DNA mini kit (Qiagen, Hilden, Germany).
Sample Preparation and Spiking
Four different types of samples were taken for experiment: (i) drinking water, (iii) pond water, (ii) boiled rice, and (iii) shrimp. Rice sample were prepared by homogenization of 25 g of boiled rice with 225 μL of phosphate-buffered saline (1 L distilled H2O, 10 g L−1 NaCl, 0.25 g L−1 KCl g L−1, 1.8 g L−1 Na2HPO4, 0.3 g L−1 KH2PO4; pH 7.4) in a Stomacher Lab Blender (Seward Stomacher® 80, Lab Biomaster, UK). Shrimp sample was also prepared by following the same procedure for rice. All of the samples were spiked with different concentrations of V. cholerae CT+ O139, V. cholerae CT+ O1, and V. cholerae CT− non-O1/non-O139. Prior to the evaluation of this assay for these environmental samples, absence of V. cholerae was confirmed by qPCR. DNA extraction was conducted using QiaAmp® DNA mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instruction.
PCR Primer Design
The Outer Membrane Protein W-OMPW Sequence of eight reference strains (Table 1) was downloaded from the NCBI database. The primer design was accomplished by FastPCR 6.05 (PrimerDigital, Helsinki, Finland). Primers that conformed to the criteria and summarized in Table 3 were analyzed for their complementarity with the reference sequence by Basic Local Alignment Search Tool (NCBI, Bethesda, MD, USA). The forward and reverse primer sequences were checked, and the pair that had the highest identity with the Query Sequences (reference sequences) was selected for further analysis (Table 2).

Table 1. Vibrio cholerae ompW sequences with their GenBANK accession numbers used for primer designing.
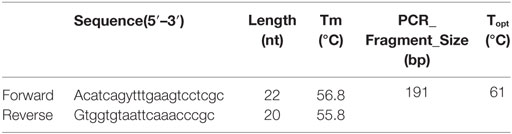
Table 2. ompW gene primers used for real-time PCR along with their properties.
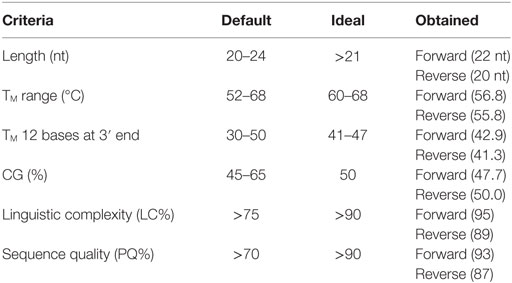
Table 3. Primer parameters obtained for the designed primers together with the default and ideal range as stated by Kalendar et al. (25).
Calculation of the Physical Parameters of Primers
Primer quality was calculated by the consecutive summation of the points according to the parameters: total sequence and purine–pyrimidine sequence complexity, the melting temperatures of the whole primer, and of the 12 bases from each of the terminal 3′ and 5′. The melting temperature of the 12 bases at the 3′ terminus is calculated by nearest neighbor thermodynamic parameters (26). Linguistic complexity measurements (Eqs 1–3) were performed using the alphabet-capacity L-gram method (27, 28). The Tm was calculated by the nearest neighbor thermodynamic parameters (26, 29). The optimal annealing temperature (Ta) was calculated by the Eq. 4 (30).
Real-time PCR Conditions
A Mastermix consisted of 12.5 μL 2× Power SYBR green® PCR master mix containing passive reference of ROX dye (Applied Biosystems, Life Technologies, Warrington, UK), 2.5 μL of 100 nM each sense and antisense primer, 2.5 μL of DEPC treated H2O, and 5 μL of template DNA. The thermal conditions were maintained under the following conditions: polymerase activation at 95°C for 5 min, followed by 40 cycles of 30 s at 95°C for and 1 min at 60°C. The real-time PCR was performed using the machine Applied Biosystems StepOne™ (48-well).
Specificity of the qPCR Assay
In order to investigate the capability of the assay to distinguish between target and non-target, DNA from 10 isolates of E. coli, 5 isolates of Enterococcus spp., 6 isolates of Salmonella spp., 3 isolates from Vibrio spp., and 7 isolates of V. cholerae were used as templates. The concentration of all DNA samples from the isolates was kept almost same (approximately 10 ng/μL) by diluting with DEPC-treated water or concentrating by DNA concentrator (Eppendorf Concentrator 5301).
Melt Curve Analysis and Detection of Non-Specific Products
Four dilutions of two V. cholerae strains were subjected to qPCR as stated above, and the reaction mixtures containing the SYBR Green PCR products were gradually warmed to 95°C at a ramp rate of 0.3°C/s with continuous fluorescence acquisition. The melting curves were created by plotting the derivative reporter vs the temperature. The melting curve analysis was performed with duplicates of four serial dilutions of template DNA ranging from 106 to 103 gene copies per reaction using the ABI software version 2.2.2. The SYBR green PCR products were also resolved for identity in 1.5% agarose gel by electrophoresis.
Sensitivity and Limits of Detection (LOD)
The DNA sample of V. cholerae was then serially diluted (10-fold) upto 7-log10 (5.46 × 105 copy numbers down to 5.46 × 10−1) in DEPC-treated water. Five microliters from each dilution were used as template for detection. Distilled water was used as no template control.
Calibration Standards for Standard Curves
To estimate the number of cells in a reaction, the mass of a single bacterial genomic DNA was calculated. The genome size of one V. cholerae was 4,033,460 bp (NCBI Genbank10952301). The molecular mass of the genome was found by multiplying the genome size with the mass of base pair. The molecular mass of V. cholerae was found to be 4.52 fg. The starting concentration of each stock DNA was measured by ColibriMicrovolume Spectrometer (Titertek-Berthold, Berthold Detection Systems GmbH, Bleichstrasse, Pforzheim, Germany) at absorbance 260 nm. To establish the number of cells in final reaction mixture, the stock concentration was divided by the molecular mass of the specific bacteria. The 7-log serial dilution (1:10) of the stock DNA was prepared in triplicate and the corresponding cell numbers were calculated in the final PCR reaction mixture.
Repeatability and Reproducibility
The precision of the PCR assays was evaluated for dilutions ranging from 5.46 × 105 gene copies per reaction down to 5.46 × 10−1 copy numbers. The dilutions were tested in four replicates in two separate PCR runs. The SD of the CT values of each concentration was then calculated by using Eqs 1 and 2.
where is the mean CT value and n is the number of observations. The value obtained was used to calculate the coefficient of variation, CV, with Eq. 2.
The intra-assay precision (repeatability) was assessed by calculating the coefficient of variation (CV%) for individual runs. The inter-assay precision (reproducibility) was calculated by determining the coefficient of variation (CV%) of both runs combined.
Ethical Clearance
The study did not involve any human or animal related issues. Therefore, we did not seek any ethical clearance in this study. Besides, the lab is facilitated with biosafety level II functions. The test and control strains of this study fall under the BSL II category.
Results
Physical Parameters of Primers
The physical parameters of the primers obtained are summarized in Table 3. Sequence quality and TM 12 bases at 3′ end of both forward and reverse primers, LC and length of forward primer, and CG% of reverse primer were all in the ideal range (see Table 3). All the others parameters were within the default range.
Repeatability and Reproducibility
The intra- and inter-run precision obtained has been summarized in Table 4. The coefficient of variation for the first replicate varied from 0.24 to 1.32 and for the second replicate the CV% ranged from 0.48 to 1.1. The CV% for the inter-run reproducibility varied from 1.08 to 3.79. The amplification plot and standard curve have been shown (Figures 1–4).
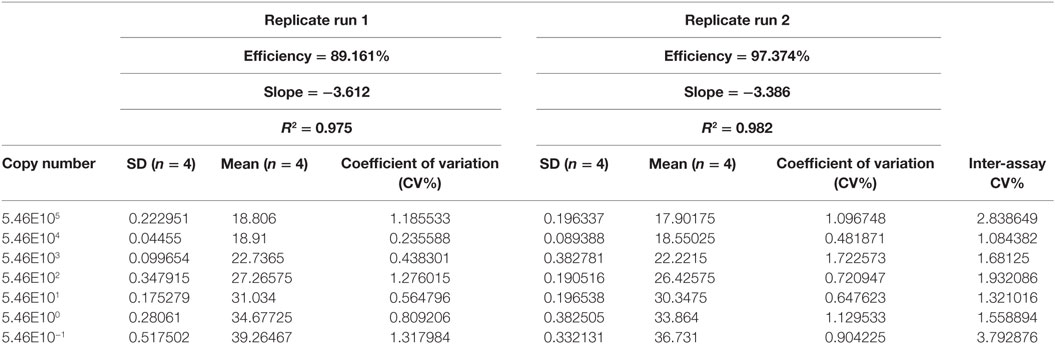
Table 4. Comparison of sensitivity of detection and precision of two replicate runs.
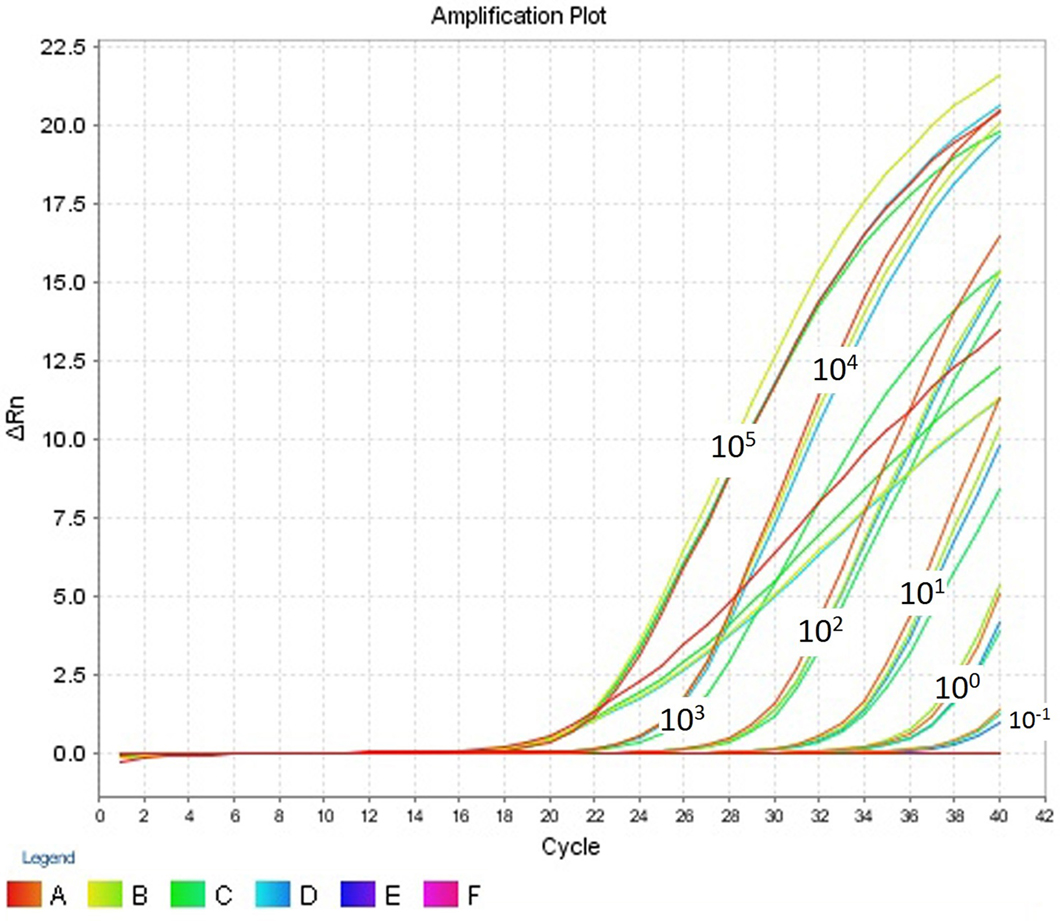
Figure 1. Amplification plot (ΔRn vs Cycle) for testing the sensitivity and precision of the first replicate run.
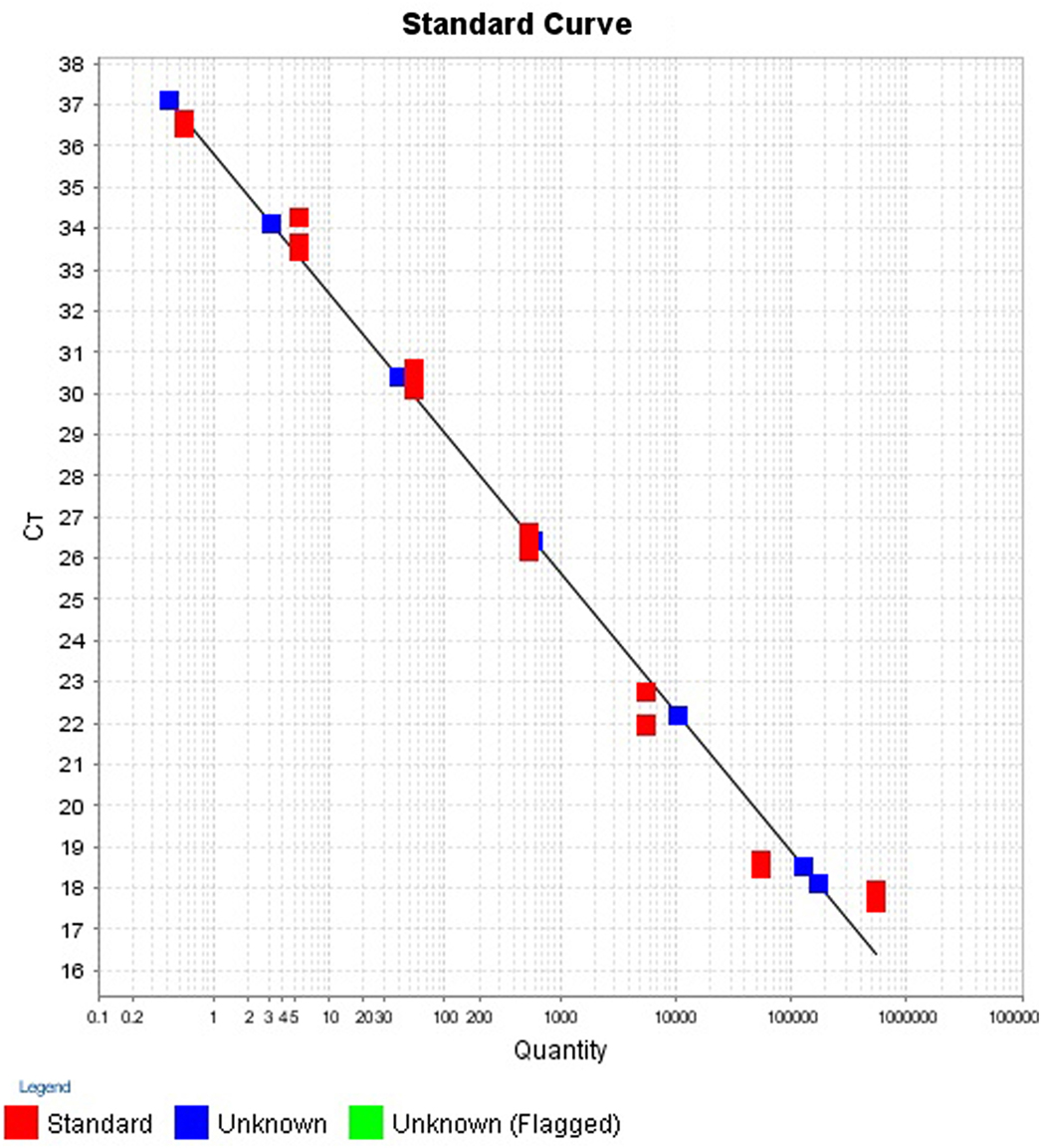
Figure 2. Standard curve for the quantification of samples in first replicate run.
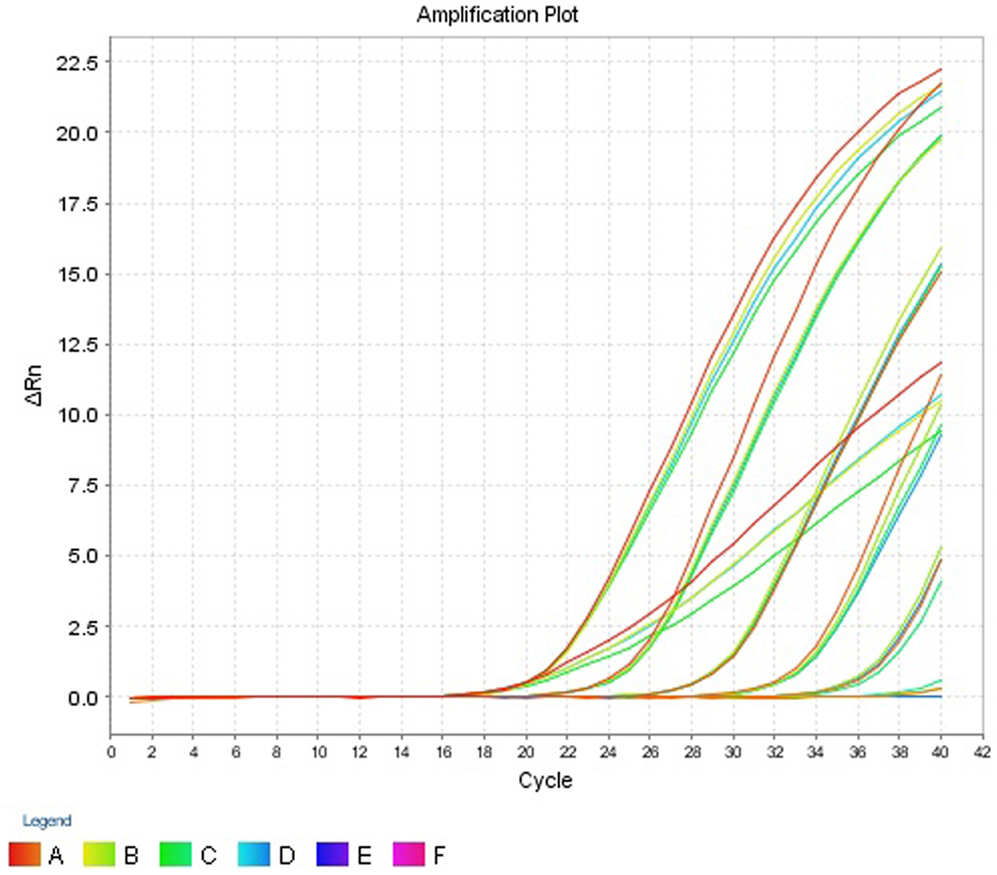
Figure 3. Amplification plot (ΔRn vs Cycle) for testing the sensitivity and precision of the second replicate run.
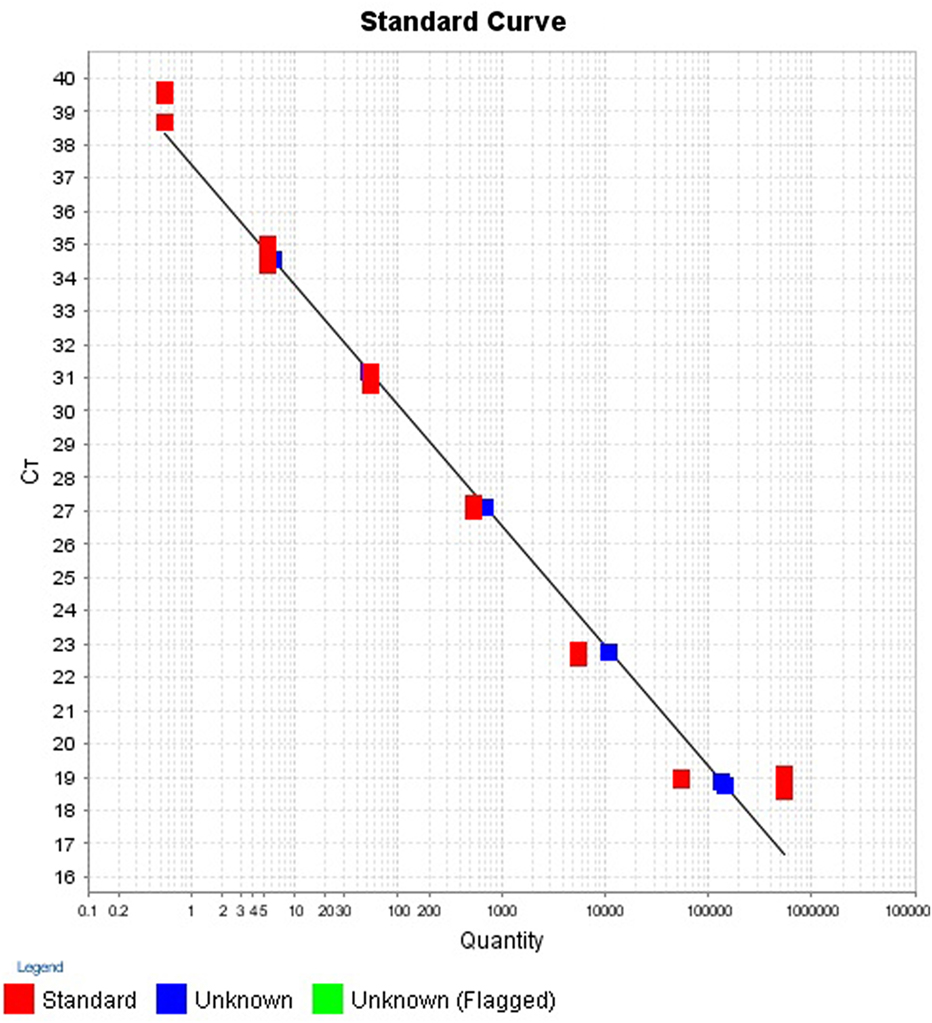
Figure 4. Standard curve for the quantification of samples in second replicate run.
Sensitivity and LOD
The LOD or analytical sensitivity was found to be 5.46 copies since among 8 replicates. The 5.46 was the lowest gene copies that were consistently detected. For higher dilution, i.e., 0.546 copy number, the assay failed to register a CT value in 2 of the 8 replicates.
Specificity
The assay registered CT values which ranged from 18.778 to 19.697 for the 4 V. cholerae strains and was detectable in the amplification plot (Figure 5). Two E. coli strains, EHEC and EIEC, had CT values of 35.073 and 38.439, respectively. The CT values for all other strains were undetermined. Strains which had CT values of less than 35 were considered as ompW positive. Hence, the assay was able to correctly detect V. cholerae and gave a negative result for all other strains, thus proving the assay was V. cholerae specific. The results have been summarized in Table 5.
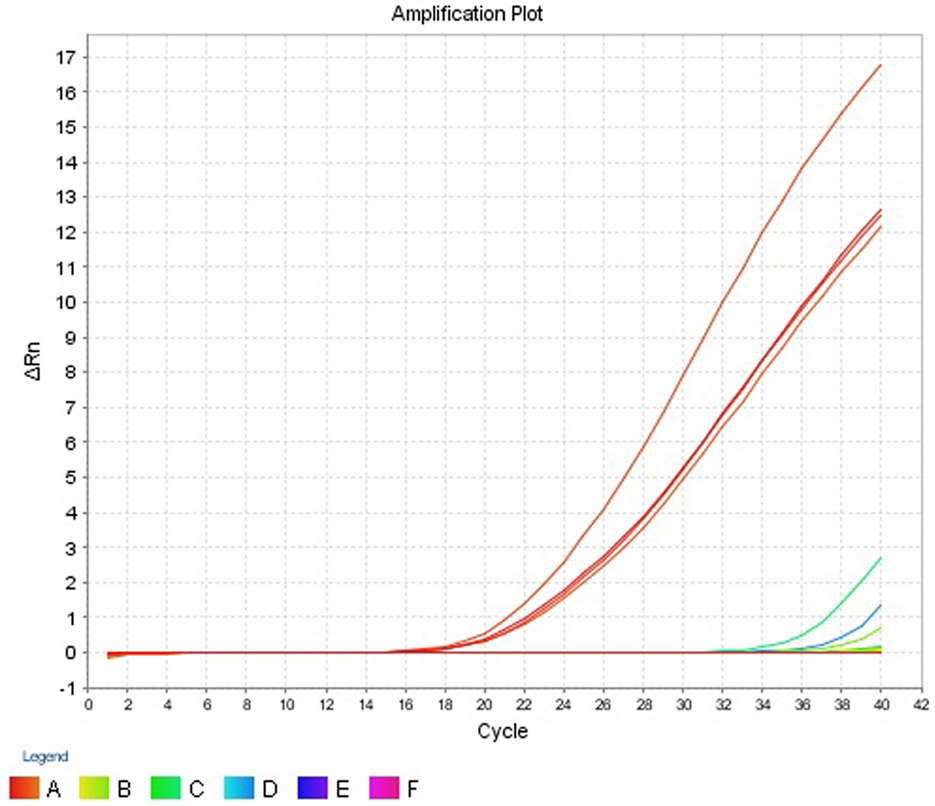
Figure 5. Amplification plot (ΔRn vs Cycle) obtained for specificity test.
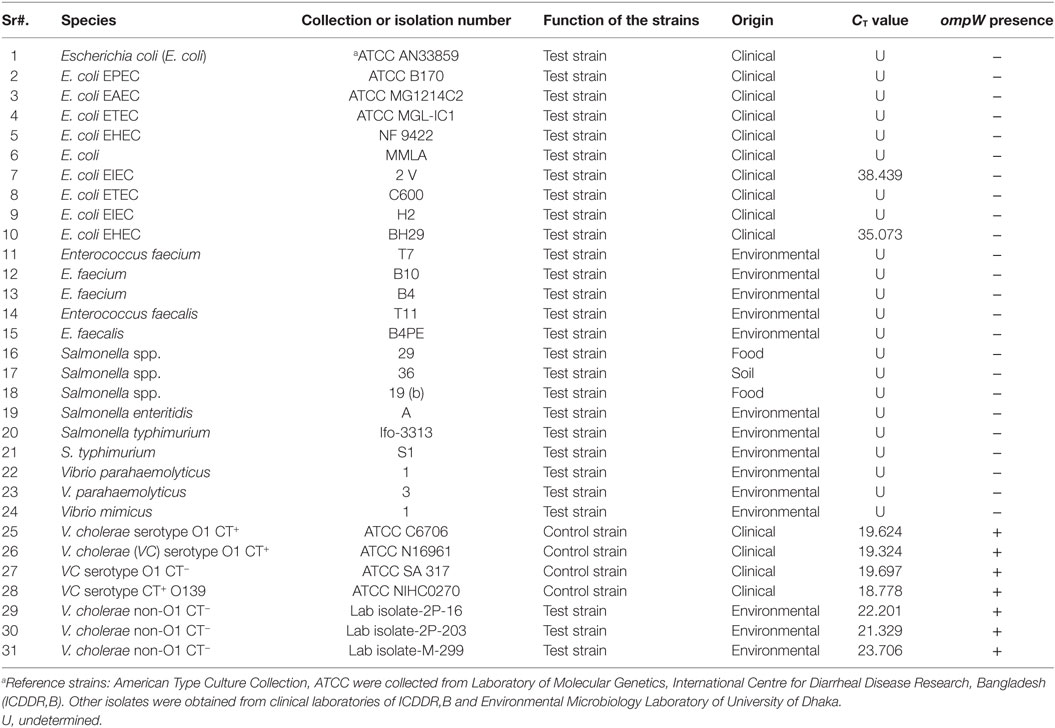
Table 5. Detection of ompW gene for specificity test.
Melt Curve Analysis and Detection of Non-Specific Products
In the melt curve (Figure 6), a single distinct peak was seen, indicating that all the PCR products had similar Tm values which was approximately 78.46°C. Agarose gel electrophoresis of SYBR green PCR products gave a single distinct band of about 191 bp (Figure 7). It could be concluded that neither secondary non-specific products nor primer dimers were formed.
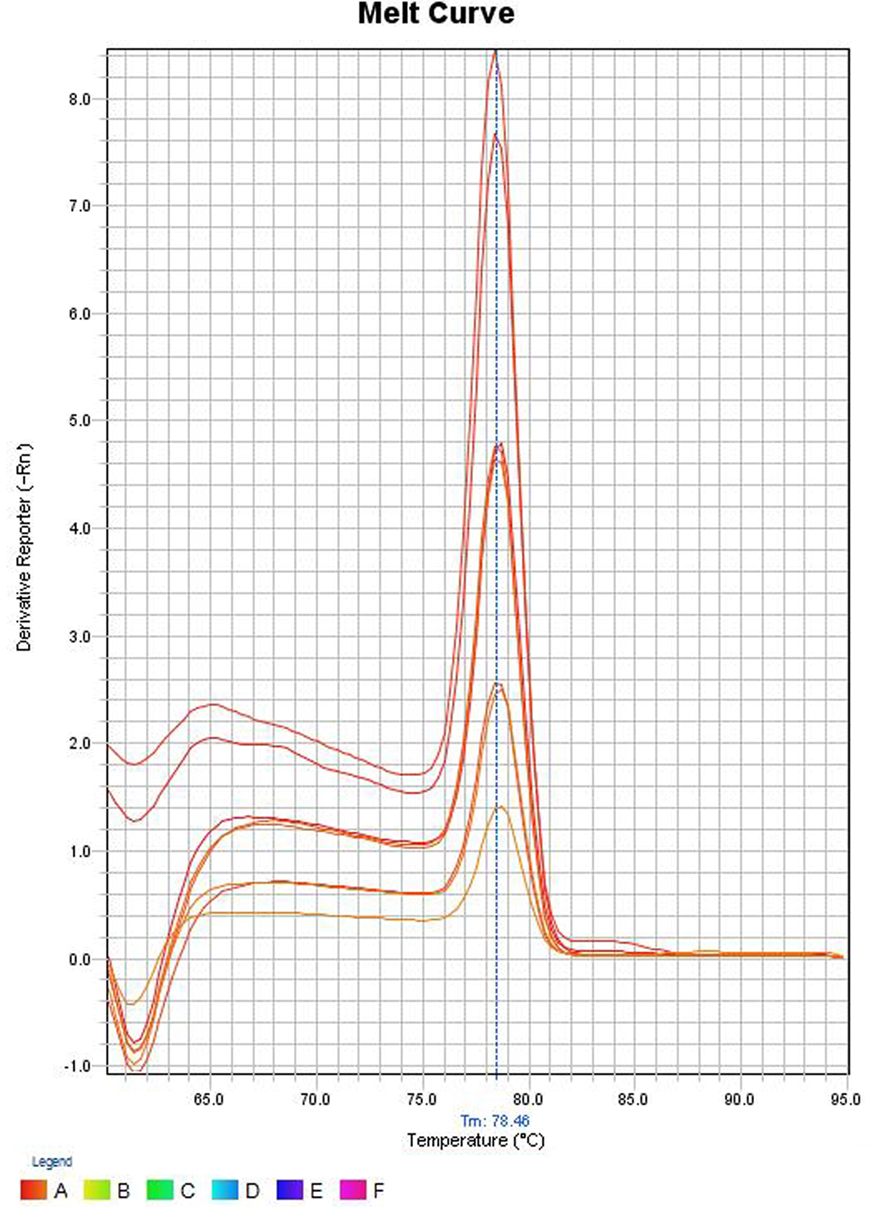
Figure 6. Melt curve of SYBR green PCR products. The Y-axis represents the derivative reporter (−Rn) while x-axis represents the temperature (°C). The figure shows a melting temperature (31) of human ompW PCR products as 78.46°C.
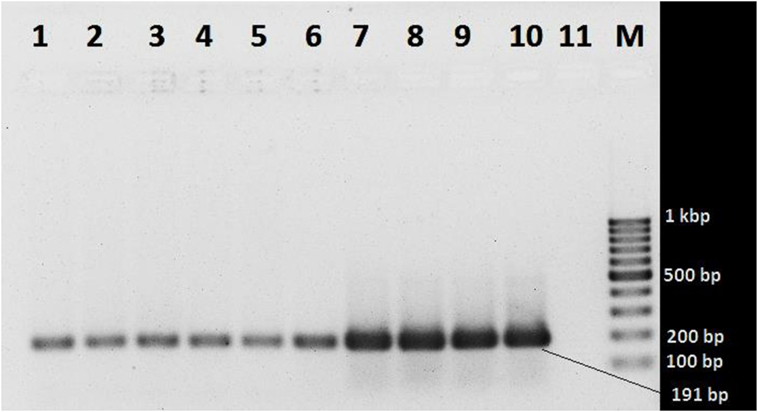
Figure 7. Agarose gel electrophoresis of SYBR green PCR products. Lanes 1 and 2 (103 gene copy): Vibrio cholerae O1 ATCC N16961 and V. cholerae O139 ATCC NIHC0270, respectively; lanes 3 and 4 (104 copies): V. cholerae O1 ATCC N16961 and V. cholerae O139 NIHC0270 ATCC, respectively; lanes 5 and 6 (105 copies): V. cholerae O1 ATCC N16961 and V. cholerae O139NIHC0270ATCC, respectively; lanes 7 and 8 (106 copies): V. cholerae O1 ATCC N16961 and V. cholerae O139NIHC0270ATCC, respectively; lanes 9 and 10 (107 copies): V. cholerae O1 ATCC N16961 and V. cholerae O139NIHC0270ATCC, respectively; Lane 10 (M): molecular weight marker (100 bp DNA Ladder, Karl Roth, Germany), 11 no template control.
Discussion
We have developed a real-time assay with designed primers for the detection and quantification of V. cholerae. The assay was based on SYBR Green PCR Mastermix and targeted the ompW gene, which is present in all species of V. cholerae. Initially, the physical properties of primers were assessed, followed by validation of sensitivity, precision, specificity, and melt curve analysis.
The LC describes nucleotide arrangement and composition of a sequence and the likelihood of PCR success of each primer. LC values of 80 and higher serve as excellent candidate primers. The primers developed had LC values of 89 and 95 for reverse and forward primers, respectively. Low-complexity regions such as Simple Sequence Repeats, imperfect direct or inverted repeats, triple-stranded DNA structures, and G/C quadruplexes (32) were unlikely to be formed if primers with high LC values are used. The parameter “Primer Quality” determines the possibility of primer dimer formation since dimers reduces the PQ value. The designed primers had PQ values of 87 and 93 for reverse and forward primers, respectively. Thus, these high values suggest that self-complementarity was not apparent. Two terminal C/G bases, recommended for increased PCR efficiency (33) were present in the designed primers.
The efficiency of a PCR assay is the amount of DNA that is amplified in each cycle. An efficiency of 100% indicates the target DNA has been doubled. The efficiencies obtained for the replicates 1 and 2 were 89.16 and 97.37%, respectively. Generally, efficiencies ranging from 90 to 100% are considered to be satisfactory. Inadequate primer design, production of non-specific amplicons and primer dimers may be responsible for reduced efficiencies (34). This is, however, only an estimate of the PCR efficiency and a real test sample, such as food, may contain inhibitory substances that decrease the PCR efficiency (35).
The precision of the assay was assessed by calculating both repeatability (intra-assay precision) and reproducibility (inter-assay precision). The coefficient of variation (CV%) for the repeatability ranged from 0.24 to 1.32 for both replicates. The CV% for the reproducibility varied from 1.08 to 3.79. The reproducibility is an important parameter since changed conditions such as different equipment and operators might affect the outcome. Pipetting and other human errors might account for poor precision. The precision usually increases with decreasing gene copy concentration (34) but this pattern was not observed for the developed assay. Retesting is required if the % CV of the PCR replicates exceeded 30% (36). All the CV% values for the assay were acceptable.
Specificity is ability to detecting chosen gene in the presence of non-specific DNA (34). The specificity is an important parameter since, in clinical and food samples, DNA from a wide range of organisms might be present. The developed assay was able to correctly detect the 7 V. cholerae and gave CT values that ranged from 18.778 to 23.706. Though the assay did not give any CT values for the 22 non-V. cholerae strains (Table 5), two E. coli strains—E. coli EIEC 2V, E. coli EHEC BH29 showed CT values of 38.439 and 35.073 respectively. Since the cut point CT value for ABI StepOne real-time machine is between >8 and <35, these CT values of E. coli strains can be considered as negative results.
The LOD is the lowest gene copy number that the assay is able to consistently detect (37). A satisfactory LOD is 10 gene copies per reaction, and the assay was able to meet this requirement by consistently detecting 5.46 copies of the gene. The LOD sheds light on how sensitive the assay is.
The assay was evaluated for its ability to detect V. cholerae O1/O139 and non-O1/non-O139 in food and environmental samples over different dilutions. It was observed that drinking water, pond water, shrimp, and boiled rice spiked with these strains registered CT values that ranged from 16.33 to 26.78 (Table 6).
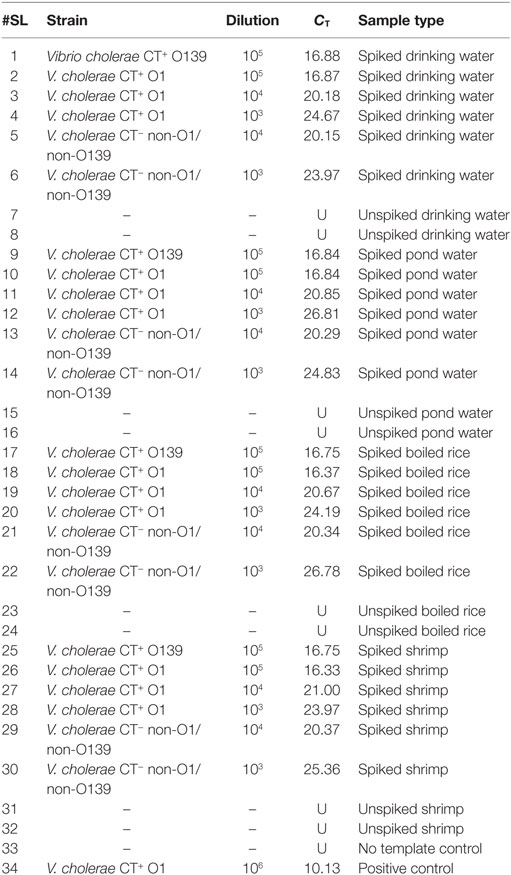
Table 6. Evaluation of the assay using direct environmental samples.
To assess if the assay is affected by interference from non-target DNA, unspiked drinking water, pond water, shrimp, and boiled rice were examined by qPCR. Before this assessment, absence of V. cholerae was confirmed. Results showed that no CT values were obtained for these unspiked food and water samples. Thus, this assay is suitable for detecting both V. cholerae O1/O139 and non-O1/non-O139 in food and environmental samples since non-specific amplification was not seen in negative controls.
Melt curve analysis was done to assess whether secondary products such as primer dimers or non-specific products were formed. The melt curve gave a single peak with a Tm value of about 78.46°C. Agarose gel electrophoresis of SYBR Green PCR products gave a single band at 191 bp. These results suggest that the amplification was specific and only one type of amplicon was produced. Non-specific products hamper the efficiency of the assay and affect precision. Non-specific products were absent suggests that the primer design was adequate. The primers were specific and intended amplicons were produced. We can conclude the primers were not complementary to one another since primer dimers were not produced.
Many assays have been developed for detection and quantification of V. cholerae (19–24). Though impressive none of these presented any statistical figures (such as coefficient of variation) which would inform us about the reproducibility and repeatability. Many of these assays did not undergo melt curve analysis or the PCR products were not subjected to agarose gel electrophoresis and hence we do not know their status regard the formation of non-specific products. Since they are multiplex in nature, they add to the cost and hence are not suitable for purposes. For instance, during quality control testing of seafood where only quantification is required to see if the levels in food is acceptable to the standards set by the governing bodies.
An extremely impressive multiplex real-time assay has been developed by Bliem and colleagues (38). The assay is multiplex in nature, and hence the use of multiple primers might add to the cost. The assay developed by Bliem and colleagues had inter-assay variance of 2–28% for ompW. But our assay, which utilizes a primer for ompW gene with different sequence, was more precise with inter-assay variance of 1.08–3.79.
Future objectives of our study might include the optimization of this assay to detect and quantify V. cholerae in food, water, and clinical samples. Some samples might contain inhibitory substances that decrease PCR efficiency (35) and hence optimization of the methods involving sample processing, DNA extraction, and assay itself might be required.
Author Contributions
PJ and AB are the principal investigators of the project and contributed to the manuscript revision and final version approval to be published. RR conducted the study in the laboratory, performed statistical analysis, and wrote the first draft of the manuscript. ST contributed to revising the manuscript critically for important intellectual content. JF contributed to the study designing, implementation, manuscript reviewing, and revising it critically. The authors have agreed to be accountable for answering questions related to the accuracy and integrity of the work appropriately done.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was funded by the project entitled “Combating Cholera Caused by Climate Change in Bangladesh, C5” (Grant no. 12-040KU) from Danish International Development Agency (DANIDA). DFC is responsible for the administration of all DANIDA funded development research projects.
References
1. Ali M, Lopez AL, You Y, Kim YE, Sah B, Maskery B, et al. The global burden of cholera. Bull World Health Organ (2012) 90(3):209–18. doi: 10.2471/BLT.11.093427
2. Ramamurthy T, Yamasaki S, Takeda Y, Nair GB. Vibrio cholerae O139 Bengal: odyssey of a fortuitous variant. Microbes Infect (2003) 5(4):329–44. doi:10.1016/S1286-4579(03)00035-2
3. Khuntia HK, Pal BB, Meher PK, Chhotray GP. Environmental Vibrio cholerae O139 may be the progenitor of outbreak of cholera in coastal area of Orissa, Eastern India, 2000: molecular evidence. Am J Trop Med Hyg (2008) 78(5):819–22.
4. Faruque SM, Chowdhury N, Kamruzzaman M, Ahmad QS, Faruque A, Salam MA, et al. Reemergence of epidemic Vibrio cholerae O139, Bangladesh. Emerg Infect Dis (2003) 9(9):1116–22. doi:10.3201/eid0909.020443
5. Dutta D, Chowdhury G, Pazhani GP, Guin S, Dutta S, Ghosh S, et al. Vibrio cholerae non-O1, non-O139 serogroups and cholera-like diarrhea, Kolkata, India. Emerg Infect Dis (2013) 19(3):464–7. doi:10.3201/eid1903.121156
6. Schmid-Hempel P, Frank SA. Pathogenesis, virulence, and infective dose. PLoS Pathog (2007) 3(10):e147. doi:10.1371/journal.ppat.0030147
7. Rudra S, Mahajan R, Mathur M, Kathuria K, Talwar V. Cluster of cases of clinical cholera due to Vibrio cholerae 010 in east Delhi. Indian J Med Res (1996) 103:71–3.
8. Dalsgaard A, Forslund A, Bodhidatta L, Serichantalergs O, Pitarangsi C, Pang L, et al. A high proportion of Vibrio cholerae strains isolated from children with diarrhoea in Bangkok, Thailand are multiple antibiotic resistant and belong to heterogenous non-O1, non-O139 O-serotypes. Epidemiol Infect (1999) 122(02):217–26. doi:10.1017/S0950268899002137
9. Onifade T, Hutchinson R, Van Zile K, Bodager D, Baker R, Blackmore C. Toxin producing Vibrio cholerae O75 outbreak, United States, March to April 2011. Euro Surveill (2011) 16(20):19870.
10. Hasan NA, Ceccarelli D, Grim CJ, Taviani E, Choi J, Sadique A, et al. Distribution of virulence genes in clinical and environmental Vibrio cholerae strains in Bangladesh. Appl Environ Microbiol (2013) 79(18):5782–5. doi:10.1128/AEM.01113-13
11. Chatterjee S, Ghosh K, Raychoudhuri A, Chowdhury G, Bhattacharya M, Mukhopadhyay A, et al. Incidence, virulence factors, and clonality among clinical strains of non-O1, non-O139 Vibrio cholerae isolates from hospitalized diarrheal patients in Kolkata, India. J Clin Microbiol (2009) 47(4):1087–95. doi:10.1128/JCM.02026-08
12. Mishra A, Taneja N, Sharma M. Viability kinetics, induction, resuscitation and quantitative real-time polymerase chain reaction analyses of viable but nonculturable Vibrio cholerae O1 in freshwater microcosm. J Appl Microbiol (2012) 112(5):945–53. doi:10.1111/j.1365-2672.2012.05255.x
13. Colwell R, Brayton P, Herrington D, Tall B, Huq A, Levine M. Viable but non-culturable Vibrio cholerae O1 revert to a cultivable state in the human intestine. World J Microbiol Biotechnol (1996) 12(1):28–31. doi:10.1007/BF00327795
14. Li L, Mendis N, Trigui H, Oliver JD, Faucher SP. The importance of the viable but non-culturable state in human bacterial pathogens. Front Microbiol (2014) 5:258. doi:10.3389/fmicb.2014.00258
15. Huq A, Colwell RR. A microbiological paradox: viable but nonculturable bacteria with special reference to Vibrio cholerae. J Food Prot (1996) 59(1):96–101. doi:10.4315/0362-028X-59.1.96
16. O’Hara CM, Sowers EG, Bopp CA, Duda SB, Strockbine NA. Accuracy of six commercially available systems for identification of members of the family vibrionaceae. J Clin Microbiol (2003) 41(12):5654–9. doi:10.1128/JCM.41.12.5654-5659.2003
17. Nandi B, Nandy RK, Mukhopadhyay S, Nair GB, Shimada T, Ghose AC. Rapid method for species-specific identification of Vibrio cholerae using primers targeted to the gene of outer membrane protein OmpW. J Clin Microbiol (2000) 38(11):4145–51.
18. Noble R, Weisberg R. A review of technologies for rapid detection of bacteria in recreational waters. J Water Health (2005) 3:381–92. doi:10.2166/wh.2005.051
19. Gubala AJ, Proll DF. Molecular-beacon multiplex real-time PCR assay for detection of Vibrio cholerae. Appl Environ Microbiol (2006) 72(9):6424–8. doi:10.1128/AEM.02597-05
20. Huang J, Zhu Y, Wen H, Zhang J, Huang S, Niu J, et al. Quadruplex real-time PCR assay for detection and identification of Vibrio cholerae O1 and O139 strains and determination of their toxigenic potential. Appl Environ Microbiol (2009) 75(22):6981–5. doi:10.1128/AEM.00517-09
21. Koskela KA, Matero P, Blatny JM, Fykse EM, Olsen JS, Nuotio LO, et al. A multiplatform real-time polymerase chain reaction detection assay for Vibrio cholerae. Diagn Microbiol Infect Dis (2009) 65(3):339–44. doi:10.1016/j.diagmicrobio.2009.07.009
22. Bielawska-Drózd A, Mirski T, Bartoszcze M, Cieślik P, Roszkowiak A, Michalski A. Development of real-time PCR assay for detection of Vibrio cholerae. Pol J Environ Stud (2012) 21(2):279–88.
23. Mehrabadi JF, Morsali P, Nejad HR, Fooladi AAI. Detection of toxigenic Vibrio cholerae with new multiplex PCR. J Infect Public Health (2012) 5(3):263–7. doi:10.1016/j.jiph.2012.02.004
24. Tirapattanun A, Chomvarin C, Wongboot W, Kanoktippornchai B. Application of SYBR green real-time PCR for detection of toxigenic Vibrio cholerae O1 in the aquatic environment. Chiang Mai J Sci (2015) 42(3):588–98.
25. Kalendar R, Lee D, Schulman AH. FastPCR software for PCR primer and probe design and repeat search. Genes Genomes Genomics (2009) 3(1):1–14.
26. SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Nal Acad Sci U S A (1998) 95(4):1460–5. doi:10.1073/pnas.95.4.1460
27. Gabrielian A, Bolshoy A. Sequence complexity and DNA curvature. Comput Chem (1999) 23(3):263–74. doi:10.1016/S0097-8485(99)00007-8
28. Orlov YL, Potapov VN. Complexity: an internet resource for analysis of DNA sequence complexity. Nucleic Acids Res (2004) 32(Suppl 2):W628–33. doi:10.1093/nar/gkh466
29. Allawi HT, SantaLucia J. Thermodynamics and NMR of internal G·T mismatches in DNA. Biochemistry (1997) 36(34):10581–94. doi:10.1021/bi962590c
30. Kalendar R, Lee D, Schulman AH. FastPCR software for PCR, in silico PCR, and oligonucleotide assembly and analysis. Methods Mol Biol (2014) 1116:271–302. doi:10.1007/978-1-62703-764-8_18
31. Huehn S, Eichhorn C, Urmersbach S, Breidenbach J, Bechlars S, Bier N, et al. Pathogenic vibrios in environmental, seafood and clinical sources in Germany. Int J Med Microbiol (2014) 304(7):843–50. doi:10.1016/j.ijmm.2014.07.010
32. Kypr J, Kejnovská I, Renčiuk D, Vorlíčková M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res (2009) 37(6):1713–25. doi:10.1093/nar/gkp026
33. Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J (1997) 72(3):1047. doi:10.1016/S0006-3495(97)78756-3
34. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem (2009) 55(4):611–22. doi:10.1373/clinchem.2008.112797
35. Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonák J, Lind K, et al. The real-time polymerase chain reaction. Mol Aspects Med (2006) 27(2):95–125. doi:10.1016/j.mam.2005.12.007
36. Welzel TM, Miley WJ, Parks TL, Goedert JJ, Whitby D, Ortiz-Conde BA. Real-time PCR assay for detection and quantification of hepatitis B virus genotypes A to G. J Clin Microbiol (2006) 44(9):3325–33. doi:10.1128/JCM.00024-06
37. Burd EM. Validation of laboratory-developed molecular assays for infectious diseases. Clin Microbiol Rev (2010) 23(3):550–76. doi:10.1128/CMR.00074-09
Keywords: Vibrio cholerae, OmpW, CT value, sensitivity and specificity, gene copy number, real-time PCR
Citation: Rashid RB, Ferdous J, Tulsiani S, Jensen PKM and Begum A (2017) Development and Validation of a Novel Real-time Assay for the Detection and Quantification of Vibrio cholerae. Front. Public Health 5:109. doi: 10.3389/fpubh.2017.00109
Received: 09 August 2016; Accepted: 02 May 2017;
Published: 19 May 2017
Edited by:
Khwaja M. Sultanul Aziz, Bangladesh Academy of Sciences, BangladeshReviewed by:
Ashima Kushwaha Bhardwaj, Indian Institute of Advanced Research, IndiaAbu Sayeed Abdullah Al Masud, International University of Business Agriculture and Technology, Bangladesh
Copyright: © 2017 Rashid, Ferdous, Tulsiani, Jensen and Begum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anowara Begum, YW5vd2FyYUBkdS5hYy5iZA==