 Kelsey J. Jesser
Kelsey J. Jesser Willy Valdivia-Granda
Willy Valdivia-Granda Jessica L. Jones
Jessica L. Jones Rachel T. Noble
Rachel T. Noble- 1Institute of Marine Sciences, University of North Carolina at Chapel Hill, Morehead City, NC, United States
- 2Orion Integrated Biosciences, New Rochelle, NY, United States
- 3Gulf Coast Seafood Laboratory, Division of Seafood Science and Technology, U.S. Food and Drug Administration, Dauphin Island, AL, United States
Vibrio parahaemolyticus is a ubiquitous and abundant member of native microbial assemblages in coastal waters and shellfish. Though V. parahaemolyticus is predominantly environmental, some strains have infected human hosts and caused outbreaks of seafood-related gastroenteritis. In order to understand differences among clinical and environmental V. parahaemolyticus strains, we used high quality DNA sequencing data to compare the genomes of V. parahaemolyticus isolates (n = 43) from a variety of geographic locations and clinical and environmental sample matrices. We used phylogenetic trees inferred from multilocus sequence typing (MLST) and whole-genome (WG) alignments, as well as a novel classification and genome clustering approach that relies on protein motif fingerprints (MFs), to assess relationships between V. parahaemolyticus strains and identify novel molecular targets associated with virulence. Differences in strain clustering at more than one position were observed between the MLST and WG phylogenetic trees. The WG phylogeny had higher support values and strain resolution since isolates of the same sequence type could be differentiated. The MF analysis revealed groups of protein motifs that were associated with the pathogenic MLST type ST36 and a large group of clinical strains isolated from human stool. A subset of the stool and ST36-associated protein motifs were selected for further analysis and the motif sequences were found in genes with a variety of functions, including transposases, secretion system components and effectors, and hypothetical proteins. DNA sequences associated with these protein motifs are candidate targets for future molecular assays in order to improve surveys of pathogenic V. parahaemolyticus in the environment and seafood.
Introduction
Vibrio parahaemolyticus is a native member of bacterial flora in coastal ecosystems worldwide (1) and is a leading cause of illness associated with seafood (2, 3). V. parahaemolyticus bioaccumulates in oysters and other filter feeders during warm months and has been shown to proliferate rapidly in waters >15°C (4). When conditions are optimal for growth, virtually 100% of oysters have detectable concentrations of V. parahaemolyticus or other potentially pathogenic Vibrio species (5). Consumption of uncooked or mishandled seafood, often raw oysters, is a major mode of infection for V. parahaemolyticus, which causes an estimated 45,000 cases of gastrointestinal illness each year in the United States (6) and accounts for almost 50% of food poisoning outbreaks in Taiwan, Japan, and Southeast Asia (7, 8). Increasing rates of vibriosis have been reported around the world, especially at high latitudes where the increase has been correlated to rising sea surface temperatures (9–11). The prevalence of V. parahaemolyticus in densely populated coastal areas, as well as the economic value of the tourism and seafood industries, underscores the importance of accurate detection, quantification, and monitoring measures for this pathogen. However, measuring the abundance of disease-causing V. parahaemolyticus is difficult because most strains isolated from environmental sources are considered nearly exclusively environmental and nonpathogenic. Ecological and genomic similarities between known virulent strains and strictly environmental strains of V. parahaemolyticus make differentiating the organisms that actually cause disease challenging, but is vital when considering the risk V. parahaemolyticus populations may pose to human health (12).
Because not all strains of V. parahaemolyticus are considered truly pathogenic, putative virulence genes that are epidemiologically correlated with disease-causing strains are used to predict public health risks (13, 14). The tdh (thermostable direct hemolysin) and trh (thermostable-related hemolysin) genes are considered major virulence factors for V. parahaemolyticus (14), and many molecular detection methods, including several real-time quantitative PCR (qPCR) assays, have been developed based on these genes [e.g., (14–16)]. However, there have been multiple reports of clinical strains that do not have tdh and/or trh [e.g., (17–19)], contributing to concerns that these genes may not be reliable markers for virulence. The occurrence of tdh and trh in environmental isolates is typically 1–10% but can be much higher depending on sample location, source, and detection method, indicating that these genes may have environmental functions not related to human virulence (20). Other genetic markers that are thought to be important for pathogenicity include type-III secretion systems (T3SS) and effector proteins, urease genes, and genes involved in bacterial adherence and biofilm formation (21). Wagley et al. (22) showed that tdh-/trh- (nontoxigenic) strains were phylogenetically similar to other virulent V. parahaemolyticus strains despite not carrying virulence genes typically associated with disease-causing strains. This study also demonstrated that both nontoxigenic and toxigenic (tdh+ and/or trh+) strains can cause disease in the Galleria mellonella moth model, suggesting that there are unknown genes that are important for virulence in nontoxigenic strains.
Because of the challenges associated with detecting and quantifying pathogenic V. parahaemolyticus in the environment, understanding the relatedness of pathogenic and presumptive nonpathogenic environmental strains is key to gaining insights into the genomic differences between pathogenic and nonpathogenic V. parahaemolyticus strains. Molecular studies exploring the relatedness between V. parahaemolyticus isolates have employed a range of phylogenetic approaches, including single gene analyses (23, 24) and multilocus sequence typing (MLST) (25, 26) to investigate how clinical and environmental strains segregate into phylogenetic groups based on genetic similarity. MLST-based phylogenetic methods in particular have been used to illuminate the evolutionary patterns associated with the emergence of virulent phenotypes. However, despite being widely-used to study the molecular epidemiology of V. parahaemolyticus, MLST-based approaches, which rely on sequencing internal housekeeping gene loci, have limited phylogenetic resolution due to the relatively small amount of sequence data that is used. This is especially problematic for very similar strains or clonal populations which have caused disease outbreaks. For example, MLST phylogenetic methods are unable to differentiate ST36 strains, which have been tied to disease outbreaks associated with raw oyster consumption and improperly handled cooked shellfish in the United States and Europe (26–28).
Increasingly, phylogenetic studies are focused on genome-wide approaches, which have strain-level resolution and have become popular as whole-genome (WG) sequencing technologies have become less expensive and more widely available. A number of phylogenetic approaches which incorporate WG sequencing data have been utilized for V. parahaemolyticus and other bacterial pathogens. WG approaches for V. parahaemolyticus have relied on the alignment of core-genome genes, single nucleotide polymorphisms (SNPs), or draft or complete genome sequences (29–31). A study by Turner et al. (31) which relied on the alignment of WG sequences found enhanced phylogenetic resolution for pathogenic V. parahaemolyticus strains, which enabled the analysis of subclade diversity for pathogenic ST36 and ST3 strains. Whistler et al. (32) also used a WG method to achieve enhanced phylogenetic resolution for ST36 strains. Similar conclusions regarding the usefulness of WG-based phylogenetic analyses have been reached for a range of bacterial groups, including enterotoxigenic E. coli (33) and other bacterial species which are relevant to public health (30).
In the current study, we utilized high-quality WG shotgun sequencing data for 43 V. parahaemolyticus strains isolated from both clinical and environmental sample matrices between 2006 and 2010 from geographic locations in the United States and Prince Edward Island (PEI), Canada. We compared the relationships between strains using MLST and WG phylogenetic methods, as well as a novel classification approach based on protein motifs that are identified in raw sequencing data using DNA scanning algorithms (34). The protein motif method uses short protein fragments specific to a given taxonomic group or functional category to reveal relationships between bacterial pathogens and molecular markers associated with virulence. Together, a group of protein motifs constitutes a motif fingerprint (MF) for an isolate that can be used to identify genomic regions associated with pathogenicity. The MF method is based on the knowledge that each bacterial species and strain has distinctive short protein-coding sequences that can be used to distinguish between and classify microorganisms (35), and has recently been used alongside phylogenetics to investigate the molecular evolution of epizootic hemorrhagic disease viruses (36, 37). Importantly, MFs are not limited to functional or virulence genes, and MF clustering is not limited to vertically transferred phylogenetic relationships. Instead, each MF is specific to a pathogen family, genus, species, or strain, and may incorporate multiple individual protein motifs, thus avoiding biases or assumptions commonly associated with the identification of gene targets associated with bacterial pathogens. Additionally, this MF approach is advantageous since it can be used to screen, scan, and directly compare raw sequencing datasets without the need for a genome assembly step. The objective of this research was to compare the results of MLST and WG phylogenetic clustering with MF clustering for both clinical and environmental V. parahaemolyticus isolates. We demonstrate that the phylogenetic and MF clustering methods are complementary and describe how MF clustering can be used to identify molecular targets associated with virulent V. parahaemolyticus strains.
Materials and Methods
Strain Information and Typing
A total of 43 V. parahaemolyticus strains were used in this study. Metadata, including strain serovar, MLST type, location and year isolated, sample matrix, and tdh/trh typing, for all strains is listed in Table 1. Raw WG shotgun sequencing data were downloaded from the NCBI SRR database for 4 V. parahaemolyticus strains (NCBI BioSamples SAMN01923894, SAMN01940374, SAMN02741394, and SAMN02741402). Raw WG shotgun sequencing data and associated metadata for an additional 39 strains were provided by the Food and Drug Administration (FDA) Gulf Coast Seafood Laboratory (GCSL). Data for the FDA strains, which were sequenced as part of the University of California at Davis 100K Pathogen Genome Project, are also available in NCBI (see NCBI BioSample IDs in Table 1). V. parahaemolyticus tdh/trh gene presence or absence was determined for the FDA strains at the FDA GCSL using the protocol described in Nordstrom et al. (14) for multiplex qPCR with an internal amplification control. Serotyping of the FDA isolates was as described in (17). For strains where raw sequence data were downloaded directly from NCBI, tdh/trh gene presence or absence data was collected from previously published studies (39, 40), with the exception of SAMN01923894. To our knowledge, SAMN01923894 has not been PCR-typed for tdh or trh, though we did find a tdh homolog in a RASTtk (41) annotation of the draft genome. However, because the genome is not closed, we cannot be sure whether trh is truly absent. For this reason, SAMN01923894 is listed in Table 1 and in all figures as “not typed.”
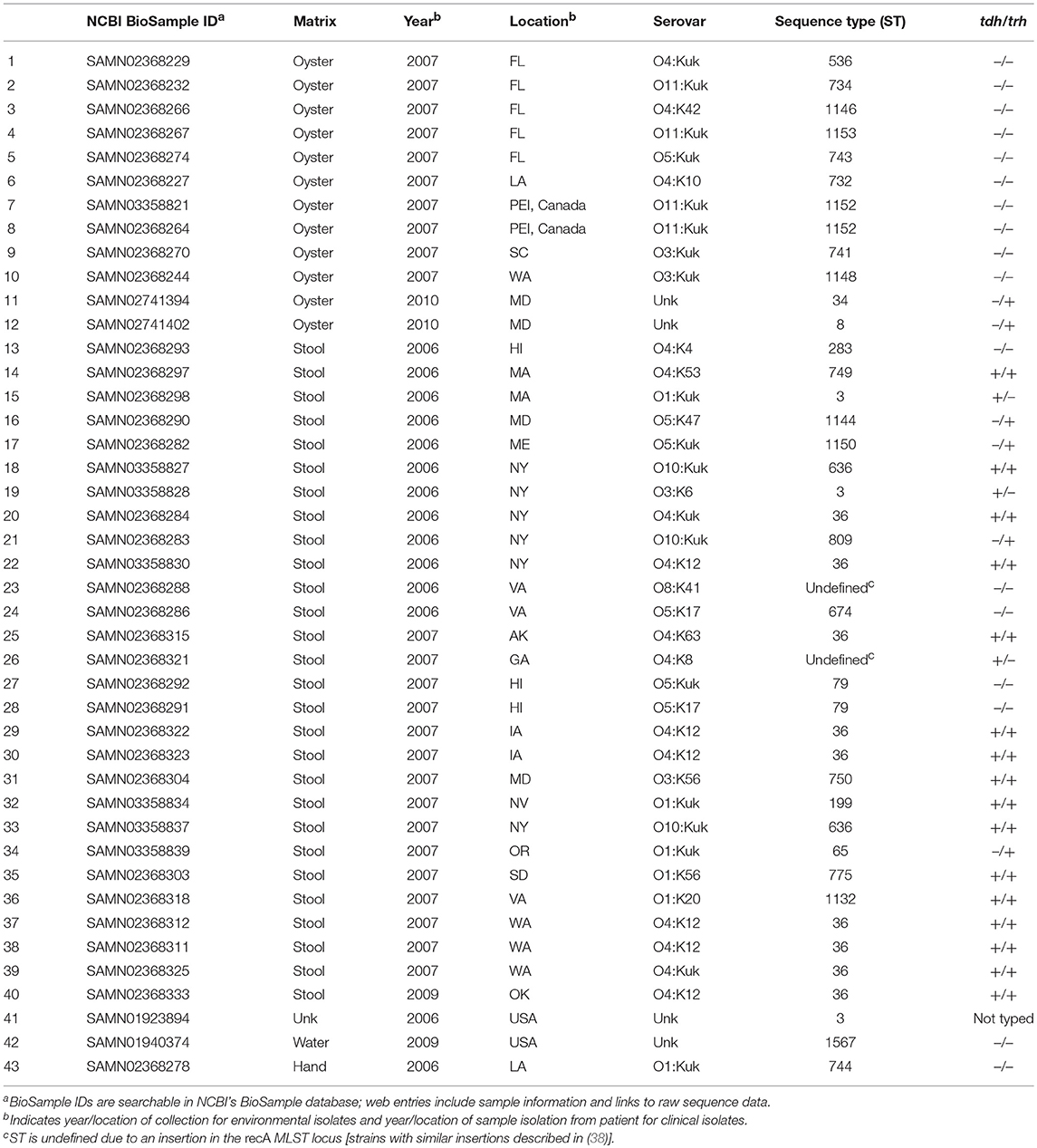
Table 1. Vibrio parahaemolyticus isolates.
Genome Assembly
Raw sequencing reads were trimmed prior to genome assembly using the JGI bbduk tool (k = 27, ktrim = l, hdist = 1, minlength = 50). High-quality draft genomes were assembled using SPAdes v. 3.10.0 (42) or Velvet v. 1.2.10 (43) prokaryotic genome assemblers as implemented in Geneious v. 11.0.4 (44). K-mer sizes for the SPAdes assemblies were selected automatically by the software and the careful option was selected to reduce mismatches and short indels. Velvet assemblies were run using the manual option with k-mer size = 33. The best assembly was evaluated based on N50 values and the number and length of assembled contigs. Based on these criteria, either the Velvet or SPAdes assembly was selected for each isolate. Contigs < 200 bp in length were filtered from the assemblies. Genome scaffolding was done using the Medusa web server (45) with all closed V. parahaemolyticus genomes in NCBI (n = 19; accessed January 2018) used as comparison genomes. Genome assembly statistics are listed in Table S1.
MLST Phylogenetic Tree Building
MLST loci were extracted in silico from assembled genomes using the online tool at the Center for Genomic Epidemiology [CGE, https://cge.cbs.dtu.dk/services/MLST/; (46)], which obtains MLST allele sequence and profile data from PubMLST.org (47). The MLST scheme used for V. parahaemolyticus was first published by Gonzalez-Escalona et al. (48), and relies on internal sequences of 7 housekeeping gene loci which span both of the V. parahaemolyticus chromosomes (recA, dnaE, gyrB, dtdS, pntA, pyrC, and tnaA). MLST sequences were downloaded from CGE's webservice and imported into Geneious. In Geneious, MLST sequences were concatenated and a 3,682 bp sequence alignment was built using MUSCLE v. 3.8.425 (49). A maximum-likelihood phylogenetic tree was inferred using RAxML v. 8.2.11 (50) as implemented in Geneious with the general time-reversible gamma substitution model. RAxML tree-building started with a complete random tree and the best scoring maximum likelihood tree was selected after 1,000 replicates of rapid bootstrapping. Rapid bootstrapping was also used to calculate branch support values for the MLST tree. Two strains, SAMN02368288 and SAMN02368321, had undefined STs due to insertions in the recA MLST locus [see (38) for a description of similar strains]. These strains were not included in the MLST alignment or tree.
WG Phylogenetic Tree Building
Scaffolds for V. parahaemolyticus isolates were aligned using Mugsy, an aligner for closely-related genomes which identifies collinear regions using a segment-based progressive multiple alignment system (51). The Mugsy WG alignment was converted from maf to fasta format with one entry per genome using the Galaxy web-platform's converter tool, which joins and converts conserved alignment blocks shared by all genomes in the alignment (52). TrimAL (53) with the strictplus algorithm was used to remove spurious and poorly aligned positions and divergent regions in the WG alignment, with a resulting core sequence alignment of 4,629,130 bp. An approximate maximum-likelihood phylogenetic tree was inferred using FastTree v. 2.1.5 (54) with the general time-reversible model. The reliability of each split in the phylogenetic tree was assessed using both FastTree support values, which are based on the Shomodaira-Hasegawa test of three alternate topologies around each split, and 1,000 bootstrap replicates, which were generated using PHYLIP SEQBOOT software (55). Both MLST and WG phylogenetic trees were midpoint rooted and annotated using the ggtree package (56) in R v. 3.5.1 (57).
Protein Motif Fingerprint Discovery
Motif discovery analyses were performed on Orion Integrated Biosciences servers using MF generation (MF-gen) and CHAST algorithms to identify protein fragments associated with precise taxonomies via an exhaustive search of GenBank protein databases as described in Corpas et al. (58) and Wilson et al. (36). Briefly, all protein entries in GenBank were divided into 12-amino acid subsequences (motifs) that were position-independent and did not contain overlaps. Every known proteome across >6.7 million taxonomies assigning organism strain, serotype, species, family, and superfamily were searched against this motif library and each motif was assigned a detailed and specific taxonomic label. In addition, we used a library of MFs covering >600,000 plasmids. We classified three types of MFs: (i) MF-type I are segments specific to a given taxonomic group (e.g., Vibrio species or V. parahaemolyticus strains), (ii) MF-type II are shared by the host and pathogen only, and may have been co-opted by the pathogen to influence immune signaling or regulatory/metabolic pathways, and (iii) MF-type III are non-specific segments shared in more than two species. Only MF-types I and II were used to scan the raw, unassembled sequence reads of the 43 V. parahaemolyticus isolates via perfect matching after a 6-frame translation process.
MF Clustering
Vibrio-associated motifs were selected and manually filtered for those that were highly variable across the 43 V. parahaemolyticus genomes. Motif abundance counts were normalized across isolates into a matrix using Genesis v. 1.7.7 (59), where the presence of an MF in each bacterial genome was presented in an MF event matrix (MFEM) where each row (g) represented a normalized MF occurrence count and each column represented an MF event (n) in a given strain. The distance between strains was determined in Genesis as described in (36), where the MFEM = g × n array was clustered using an average linkage hierarchical clustering algorithm using the Pearson correlation coefficient (r) between g and n. Select protein motif sequences associated with genome clusters of interest were assigned putative gene functions using blastp (60).
Results
A total of 43 high-quality draft genomes of V. parahaemolyticus strains isolated in the United States and PEI, Canada between 2006 and 2010 were compared using both MLST and WG phylogenetics and a novel protein motif clustering analysis. Of these 43 genomes, 29 (67.4%) were isolated from clinical matrices (human samples), 13 (30.2%) were isolated from water or oysters, and 1 was from an unknown sample matrix. All but one isolate was assayed for the presence of tdh and trh genes using PCR. Of the 42 isolates for which tdh/trh typing was done, 17 (40.4%) were tdh+/trh+, 3 (7.1%) were tdh+/trh–, 5 (11.9%) were tdh–/trh+, and 17 (40.4%) were tdh–/trh–. Of the 29 clinical isolates, 16 (55.1%) were tdh+/trh+, 3 (10.3%) were tdh+/trh–, 4 (13.8%) were tdh–/trh+, and 6 (20.7 %) were tdh–/trh–. Serotyping of the 39 FDA isolates revealed 20 unique serotypes. Isolate metadata is summarized in Table 1.
MLST
In silico MLST analyses revealed 30 unique MLST types, indicating a high degree of genetic diversity amongst strains. There were 2 strains that had recA insertions that resulted in undefined MLST types. The number of MLST types covered in this study is comparable to others which have focused on isolates from North America, though the number of MLST types reported varies depending on the number of isolates analyzed and the geographic and temporal range of the study [see (26, 61, 62) for relevant examples]. The most common ST types associated with the 29 clinical isolates included in the present study were ST36 (31.0%), and ST3 (10.3%), both of which have been associated with outbreaks of gastrointestinal illness (63, 64) and have been reported as common clinical MLST types (26). MLST-tdh/trh typing results in the present study are aligned with previous findings. For example, ST36 isolates included in this study were found to be tdh+/trh+, as has been previously described for ST36 strains (28, 61). MLST types for all isolates are summarized in Table 1.
Phylogenetic Trees
The WG phylogenetic tree (Figure 1), which was inferred using a 4,629,130 bp multiple sequence alignment, had improved taxonomic resolution in comparison to the MLST phylogenetic tree (Figure 2), which was inferred using a 3,682 bp multiple sequence alignment. The largest clusters in the phylogenetic trees were labeled (Figures 1, 2) and membership of the V. parahaemolyticus strains in these clusters is summarized in Table 2. The WG tree was fully bifurcating, but the MLST tree contained relationships that were not fully resolved because MLST sequences for isolates with the same MLST type were identical. The WG phylogeny had improved branch support values at most nodes. Based on visual inspection, no clear trends tying tdh/trh typing to clustering results were observed in the MLST phylogenetic tree. In the WG phylogenetic tree, clinical nontoxigenic strains were clustered together and with tdh–/trh+ strains. The tdh+/trh+ clinical strains also largely clustered together in the WG phylogeny, though some tdh+/trh+ clinical strains grouped more closely with environmental, nontoxigenic strains. WG trees annotated with serovar, MLST type, and the location/year strains were isolated are available in the supplementary material (Figures S1–S3).
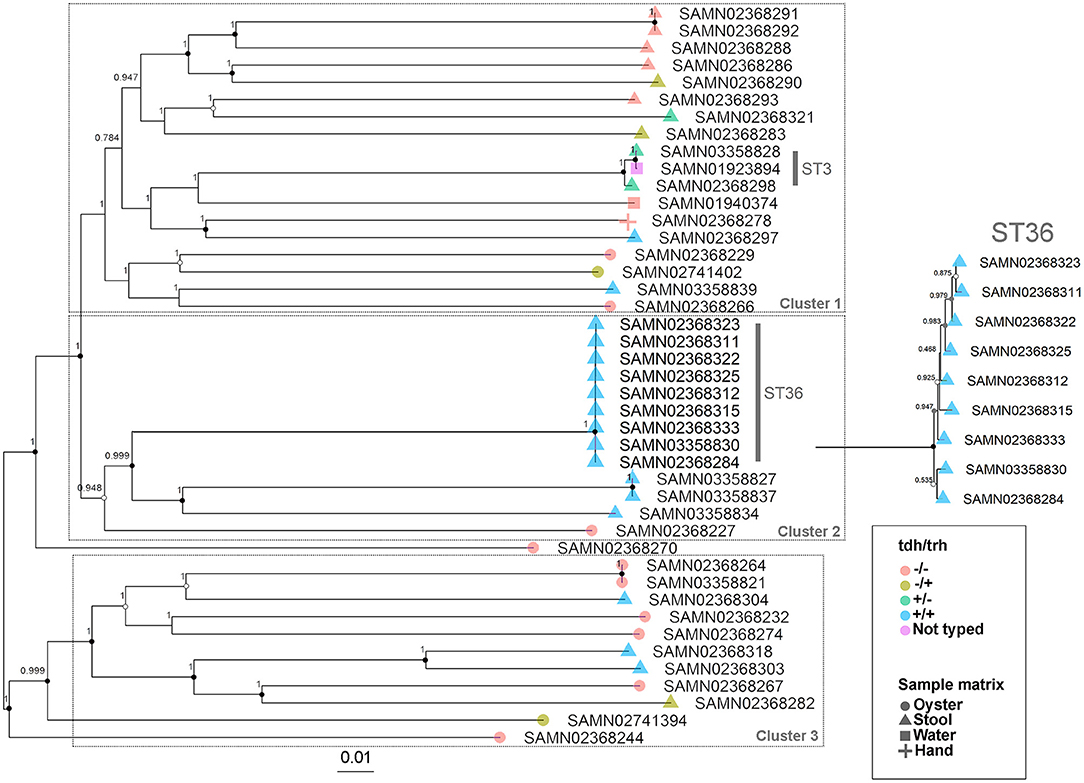
Figure 1. Approximate maximum-likelihood phylogenetic tree based on a whole genome (WG) sequence alignment of 43 V. parahaemolyticus genomes. Colors indicate hemolysin gene (tdh and trh) presence or absence and shape indicates sample isolation source. Inset is an enlargement of the ST36 clade to illustrate subclade diversity and resolution. Nodes are labeled with FastTree support values. Circles at nodes indicate bootstrap support values (1000 replicates) of >0.9 (black), >0.7 (gray), and >0.5 (white). Numbered clusters correspond with clusters listed in Table 2.
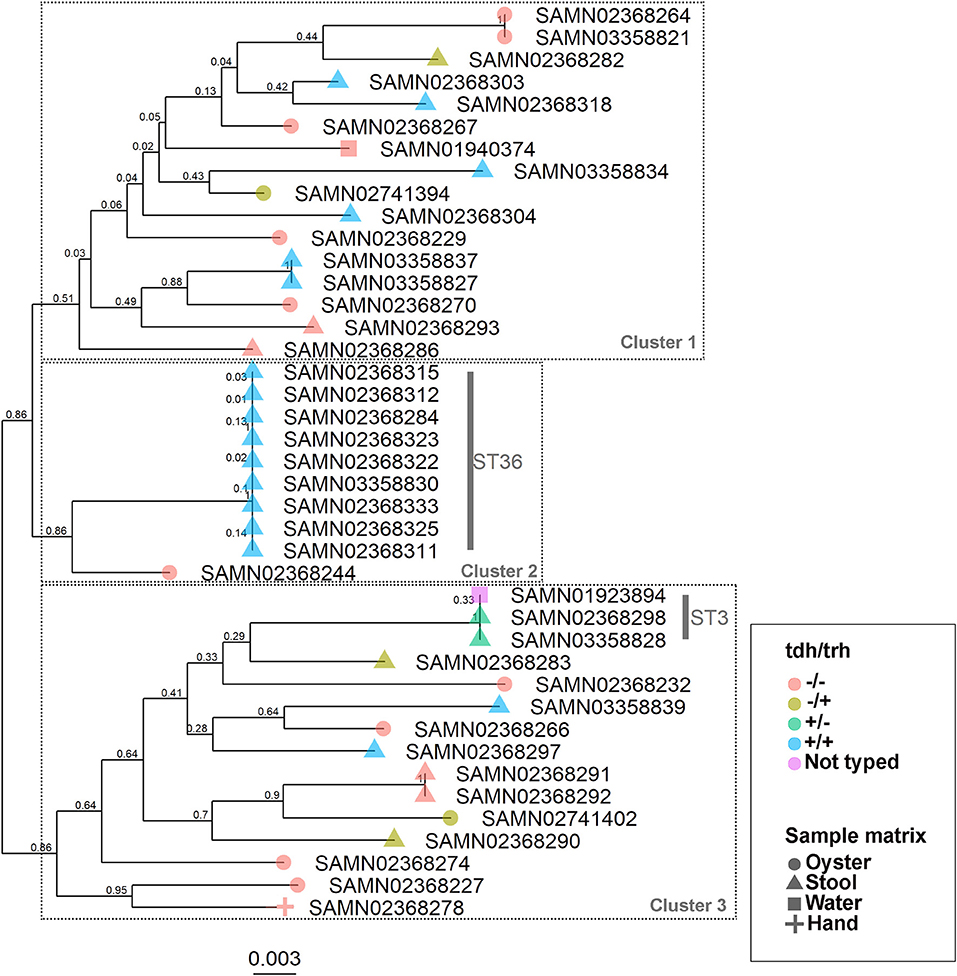
Figure 2. Maximum-likelihood phylogenetic tree based on an alignment of multilocus sequence typing (MLST) loci for 43 V. parahaemolyticus genomes. Colors indicate hemolysin gene (tdh and trh) presence or absence and shape indicates sample isolation source. Nodes are labeled with bootstrap support values (1000 replicates). Numbered clusters correspond with clusters listed in Table 2.

Table 2. Cluster membership of Vibrio parahaemolyticus isolates in the WG and MLST phylogenies and the MF clustering analysis.
MF Clustering
Hierarchical clustering was used to group genomes and protein motifs (Figure 3). A large group of stool isolates was identified in the protein motif analysis (Figure 3, cluster indicated in green) that included 22 of 29 (75.9%) clinical strains. Of these stool cluster isolates, 15 (68.2%) were tdh+, 19 (86.4%) were trh+, and 3 (13.6%) were nontoxigenic. The stool genome cluster was defined by two protein motifs. One of these, assigned the specific taxonomic label “O29774: V. parahaemolyticus (TH3996),” was present in all but one of the stool-associated genomes in the group. Motifs with this taxonomic label were found in the coding sequences of several hypothetical proteins, transposases, a hemolysin, and putative proteins associated with the cell membrane, conjugation, and the T3SS apparatus and effectors (see Table S2 for the list of top blast hits).
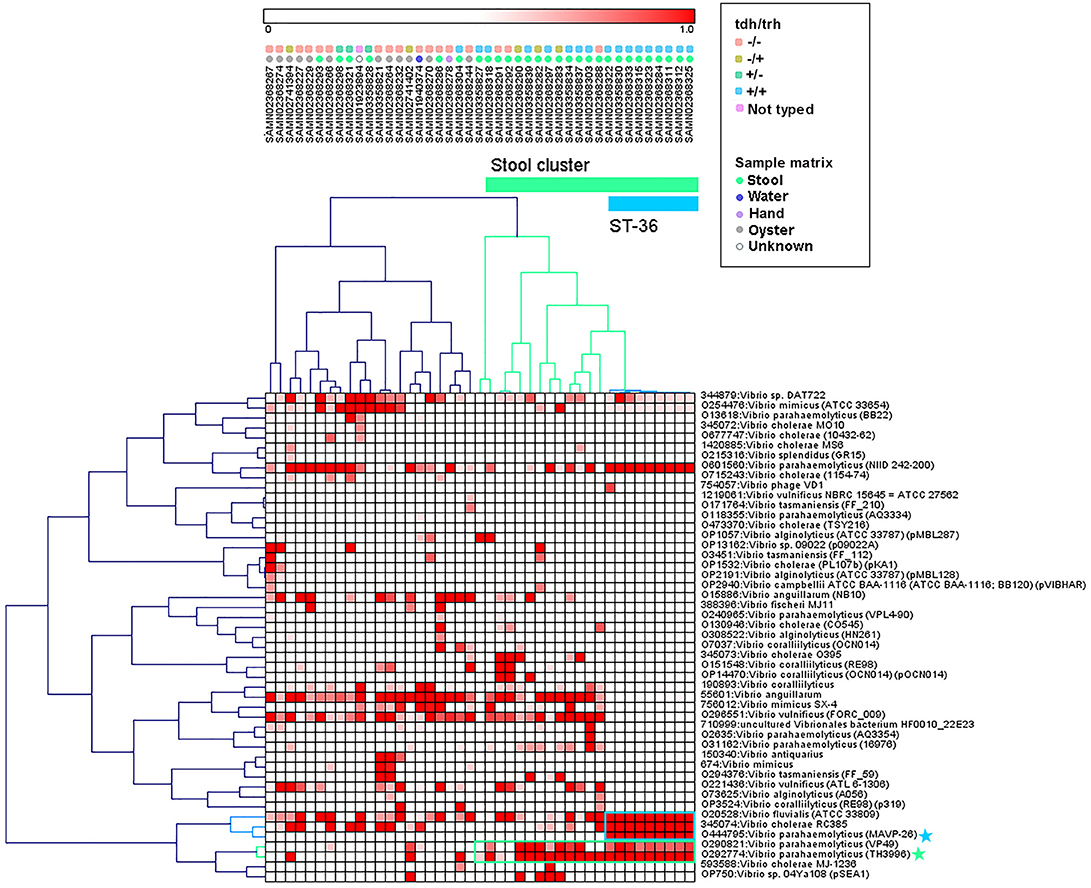
Figure 3. Pearson correlation average linkage hierarchical clustering of Vibrio motif fingerprints (MFs) across 43 V. parahaemolyticus genomes revealed a large cluster of stool isolates (green) and clustered ST36 isolates together (blue). Protein motifs associated with these clusters are designated by colored boxes, and specific motif taxonomies that are proposed as targets for quantitative molecular assay design are labeled with colored stars.
Within the MF stool cluster, we observed a group of 9 strains with very similar MFs that corresponded to ST36 (Figure 3, cluster indicated in blue), a clonal sequence type that is tdh+/trh+ and has caused widespread human illness (65). This ST36 genome cluster was defined by a small group of protein motifs, one of which, labeled “O444795: V. parahaemolyticus (MAVP _26),” was specific to the ST36 genomes. Motifs associated with this taxonomic label were identified in the coding sequences of a wide array of functional genes, including hypothetical proteins, transcriptional regulators, transporter proteins, an RTX toxin, and a capsular biosynthesis protein (Table S2). Membership of individual V. parahaemolyticus isolates in either the stool-associated or the ST36 MF clusters is summarized in Table 2.
Discussion
Comparison of WG and MLST Phylogenetic Trees
We observed differences in strain clustering at more than one position between the phylogenetic trees inferred using the WG and MLST sequence alignments (Figures 1, 2). This has been previously observed for V. parahaemolyticus (31) and multiple other bacterial species (30). Phylogeny inferred using MLST loci, while reproducible, discriminatory, and potentially useful for epidemiological surveillance and disease outbreak investigations, cannot fully represent genome phylogeny because it utilizes relatively little sequence data and because MLST gene loci do not exemplify the entire genome. For these reasons, MLST trees should be interpreted with caution. In the present study, the amount of sequence data represented by the MLST alignment (3,682 bp) was just 0.08% of that utilized in the WG (4,629,130 bp) alignment. The lower branch support values computed for the MLST tree compared to the WG tree are likely due to the relatively small number of informative sites in the MLST alignment. However, because MLST-based analyses have been so widely used in epidemiological studies of V. parahaemolyticus, MLST typing information is still essential to interpret WG sequence data in the context of previous research and disease outbreaks.
The importance of the improved taxonomic resolution of the sequence-based WG phylogeny is exemplified by the fully bifurcating trees for strains of the same sequence type (see Table 1 for MLST sequence types and Figure 1 for the WG phylogeny). Though little differentiation was observed for the 9 ST36 strains in the WG tree, the WG phylogenetic approach did allow us to observe relationships between strains that we were unable to discern in the MLST phylogeny. These strain-level differences for very closely-related strains are key to understanding the evolution of pathogenic types as they move through the environment and human populations. A previous WG study of V. parahaemolyticus (28) found that ST36 strains from a disease outbreak in Maryland could be differentiated from historical strains isolated on the west coast of the United States using a genome-wide SNP phylogenetic analysis. These results, as well as the results of the current study, underscore the value of fully-resolved WG phylogenetic trees for V. parahaemolyticus as a means to define the diversity and evolution of disease-causing types.
The present study corroborates previous findings on the usefulness of WG phylogenetic approaches for V. parahaemolyticus (31, 32). However, although WG sequencing methods are becoming increasingly affordable and accessible, it is important to note that the computational capacity required to utilize WG sequence alignments to infer phylogenetic relationships of closely related bacterial strains may be prohibitive. For example, constructing the alignment used to infer the WG phylogenetic tree for the 43 V. parahaemolyticus isolates presented in the current study took approximately 10 hours and 9 GB memory on a high-performance computing cluster. In contrast, the MLST alignment took <1 min and was computed locally on a desktop computer. Tree building and bootstrapping also required significantly more time and memory for the WG vs. the MLST phylogenetic analysis. As computational capacity and knowledge are both increasing in the field, WG alignments will likely become more feasible in the future. In the meantime, other phylogenetic methods which rely on WG sequencing data can be used. For example, phylogenies inferred using SNPs have been suggested as an alternative to WG sequence alignments because they cover the entire genome and are less time and resource-intensive. Though we did not construct a SNP-based phylogenetic tree in the present study, previous research has shown similar tree topologies and branch support values for SNP and WG phylogenies for several bacterial pathogens (30). A core genome MLST (cgMLST) method has also been reported for V. parahaemolyticus (29). We did not construct a phylogeny using cgMLST loci in the present study, but this method has been shown to produce fast typing results and meaningful, high resolution phylogenies using coding sequences identified in WG datasets.
MF Clustering
The protein MF clustering results provided fresh insights into the genomic characteristics of V. parahaemolyticus strains as they relate to virulence gene presence/absence and isolation source which were unbiased by previous assumptions about strain pathogenicity. Hierarchical clustering of MFs associated with select Vibrio taxa (Figure 3) produced two genome clusters of particular interest. The first was a large cluster of clinical V. parahaemolyticus strains isolated from human stool (Figure 3, cluster indicated in green). The second was a cluster of genomes that corresponded to ST36 (Figure 3, cluster indicated in blue), that had very similar MF profiles to one another and were observed within the larger stool-associated cluster.
The large stool-associated cluster included ~75% of clinical V. parahaemolyticus isolates in our dataset. Though this large cluster of clinical isolates was mostly composed of toxigenic strains, the tdh/trh profiles did not align perfectly with the MF clustering or clinical isolation sources. Approximately 25% of the clinical isolates were not associated with the stool-related MF cluster, and the clinical strains that did not group with the stool-related cluster had MF profiles more similar to environmental strains. Most of the clinical isolates that were not in the stool-related cluster were nontoxigenic, but 4 were tdh+ and/or trh+. Conversely, three of the isolates that were in the stool-associated cluster were nontoxigenic. Together, these results support the idea that tdh and trh gene presence may not be sufficient for defining pathogenic strains because there are unknown genomic factors associated with virulence. On the other hand, broadly speaking, most isolates in the stool-associated cluster did have either tdh (~70%) and/or trh (~85%) and, based on visual inspection, the MF analysis more clearly clustered strains based on tdh/trh virulence gene typing than was observed in either the MLST or WG phylogenetic trees. Despite some caveats, the presence of tdh/trh hemolysin genes did seem to be associated with genome-wide differences linked to virulence in the MF analysis.
The second MF cluster of interest was a group of tdh+/trh+ ST36 genomes, which were clustered together within the larger stool-associated cluster. Given the genomic similarity and phylogenetic relatedness of ST36 strains (Figure 1) it was unsurprising that these strains also clustered in the MF analysis. We consider this an excellent example of the complementarity of the MF analysis with the phylogenetic and MLST methods we used, because we may not have recognized this cluster as ST36 without the MLST results or immediately realized how closely related these strains were if we had not done the phylogenetic analyses. The fact that the ST36 MF cluster was found within the larger stool-associated cluster was interesting, since this pattern was not observed in the WG phylogeny (Figure 1). Based on the MF patterns we observed, it seems that ST36 strains may have genomic signatures of virulence which are also present in other, non-ST36 pathogenic strains.
Future Directions: MFs to Quantitative Molecular Assays
In order to utilize the MF data for the protection of shellfish consumers, the next step in this line of research is to use the sequences of protein motifs associated with the specific genome clusters we identified in the MF analysis to define genomic indicators of virulent V. parahaemolyticus strains. The genome regions containing protein motifs, and indeed the motif sequences themselves, can then be targeted for the development of novel qPCR assays. The hemolysin genes tdh and trh are currently the most commonly used markers for virulent V. parahaemolyticus in seafood and the environment. As previously discussed, these markers are useful for predicting the abundance of virulent V. parahaemolyticus, but the emergence of nontoxigenic clinical strains that do not carry either hemolysin gene is cause for concern. Using the MF approach, we believe we have the capacity to develop qPCR primers and probes that can be used to improve predictions of V. parahaemolyticus virulence potential by capturing some of these nontoxigenic but pathogenic strains. Assays designed using protein motif targets could be used in conjunction with current qPCR methodologies to improve predictions of virulence potential. Though protein motif fingerprinting of bacterial isolates is a novel and powerful bioinformatics tool, the process of developing quantitative assays from protein motif sequences is nontrivial because each protein motif taxonomic label is associated with hundreds to thousands of unique motif sequences. In order to design a functional gene assay using motif sequences, motifs must first be searched against public databases for gene functions and cross-reactivity. The next step is to ensure that the DNA coding regions which include the protein motif sequences are suitable for PCR assay designs by filtering them based on their thermodynamic characteristics. Once suitable motifs have been selected, they can be used to design qPCR primers and probes.
Conclusions
As next-generation sequencing technologies continue to advance, it is possible that sequence-based methods could one day become fully quantitative and rapid enough to completely replace PCR-based quantitative methods for environmental and-seafood related surveys of virulent V. parahaemolyticus. In the meantime, our results suggest that MF clustering shows great promise for identifying new genomic indicators of virulent strains that are not constrained by previous ideas about V. parahaemolyticus virulence traits. Future assays developed using specific motif sequences could be used to improve predictions of potentially pathogenic V. parahaemolyticus in the environment and shellfish and contribute to improved public health outcomes.
Author Contributions
The original idea for this project was developed by KJ, RN, and WV-G. KJ analyzed the data and drafted the manuscript with input and final approval from WV-G, JJ, and RN. WV-G conducted the initial protein motif fingerprinting motif analyses and provided input on the evaluation of the resultant data. JJ and the FDA GCSL contributed genomic sequencing data and associated metadata used in the analyses.
Funding
Research reported in this publication was supported by grants from the UNC Research Opportunities Initiative and NSF AIR (award number 162023) to RN. Additional seed funding was provided for this work by the Office of the UNC Vice Chancellor for Research.
Conflict of Interest Statement
The motif fingerprinting software was developed by WV-G from Orion Integrated Biosciences.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2019.00066/full#supplementary-material
References
1. Wu Y, Wen J, Ma Y, Ma X, Chen Y. Epidemiology of foodborne disease outbreaks caused by Vibrio parahaemolyticus, China, 2003-2008. Food Control. (2014) 46:197–2002. doi: 10.1016/j.foodcont.2014.05.023
2. Su YC, Liu C. Vibrio parahaemolyticus: a concern of seafood safety. Food Microbiol. (2007) 24:549–58. doi: 10.1016/j.fm.2007.01.005
3. Chen AJ, Hasan NA, Haley BJ, Taviani E, Tarnowski M, Brohawn K, et al. Characterization of pathogenic Vibrio parahaemolyticus from the Chesapeake Bay, Maryland. Front Microbiol. (2017) 8:2460. doi: 10.3389/fmicb.2017.02460
4. Daniels NA, MacKinnon L, Bishop R, Altekruse S, Ray B, Hammond RM, et al. Vibrio parahaemolyticus infections in the United States, 1973-1998. J Infect Dis. (2000) 181:1661–6. doi: 10.1086/315459
5. Pruzzo C, Gallo G, Canesi L. Persistence of vibrios in marine bivalves: the role of interactions with haemolymph components. Environ Microbiol. (2005) 7:761–72. doi: 10.1111/j.1462-2920.2005.00792.x,
6. Centers for Disease Control and Prevention. Vibrio Species Causing Vibriosis. (2017). Retrieved from https://www.cdc.gov/vibrio/index.html (Accessed August 29, 2018).
7. Alam M, Chowdhury WB, Bhuiyan NA, Islam A, Hasan NA, Nair GB, et al. Serogroup, virulence, and genetic traits of Vibrio parahaemolyticus in the estuarine ecosystem of Bangladesh. Appl Environ Microbiol. (2009) 75:6268–74. doi: 10.1128/AEM.00266-09
8. Martinez-Urtaza J, Lozano-Leon A, DePaola A, Ishibashi M, Shimada K, Nishibuchi M, et al. Characterization of pathogenic Vibrio parahaemolyticus isolates from clinical sources in Spain and comparison with Asian and North American pandemic isolates. J Clin Microbiol. (2004) 42:4672–8. doi: 10.1128/JCM.42.10.4672-4678.2004
9. Martinez-Urtaza J, Bowers JC, Trnanes J, DePaola A. Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res Int. (2010) 43:1780–90. doi: 10.1016/j.foodres.2010.04.001
10. Broberg CA, Calder TJ, Orth K. Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microb Infect. (2011) 13:992–1001. doi: 10.1016/j.micinf.2011.06.013
11. Baker-Austin C, Trinanes JA, Taylor NG, Hartnell R, Siitonen A, Martinez-Urtaza J. Emerging Vibrio risk at high latitudes in response to ocean warming. Nat Clim Chang. (2013) 3:73–7. doi: 10.1038/nclimate1628
12. Bisha B, Simonson J, Janes M, Bauman K, Goodridge LD. A review of the current status of cultural and rapid detection of Vibrio parahaemolyticus. Int J Food Sci Technol. (2012) 47:885–99. doi: 10.1111/j.1365-2621.2012.02950.x
13. Nishibuchi M, Kaper JB. Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect Immun. (1995) 63:2093–99.
14. Nordstrom JL, Vickery MC, Blackstone GM, Murray SL, DePaola A. Development of a multiplex real-time PCR assay with an internal amplification control for the detection of total and pathogenic Vibrio parahaemolyticus bacteria in oysters. Appl Environ Microbiol. (2007) 73:5840–7. doi: 10.1128/AEM.00460-07
15. Blackstone GM, Nordstrom JL, Vickery MC, Bowen MD, Meyer RF, DePaola A. Detection of pathogenic Vibrio parahaemolyticus in oyster enrichments by real time PCR. J Microbiol Methods. (2003) 53:149–55. doi: 10.1016/S0167-7012(03)00020-4
16. Ward LN, Bej AK. Detection of Vibrio parahaemolyticus in shellfish by use of multiplexed real-time PCR with TaqMan fluorescent probes. Appl Environ Microbiol. (2006) 72:2031–42. doi: 10.1128/AEM.72.3.2031-2042.2006
17. Jones JL, Ludeke CH, Bowers JC, Garrett N, Fischer M, Parsons MB, et al. Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J Clin Microbiol. (2012) 50:2343–52. doi: 10.1128/JCM.00196-12
18. Li Y, Xie X, Shi X, Lin Y. Vibrio parahaemolyticus, southern coastal region of China, 2007-2012. Emerg Infect Dis. (2014) 20:685–88. doi: 10.3201/eid2004.130744
19. Pazhana GP, Bhowmik SK, Ghosh S, Guin S, Dutta S, Rajendran K, et al. Trends in the epidemiology of pandemic and non-pandemic strains of Vibrio parahaemolyticus isolated from diarrheal patients in Kolkata, India. PLoS Negl Trop Dis. (2014) 8:e2815. doi: 10.1371/journal.pntd.0002815
20. Raghunath P. Roles of thermostable direct hemolysin (TDH) and TDH-related hemolysin (TRH) in Vibrio parahaemolyticus. Front Microbiol. (2015) 5:805. doi: 10.3389/fmicb.2014.00805
21. Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, et al. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet. (2003) 361:743–9. doi: 10.1016/S0140-6736(03)12659-1
22. Wagley S, Borne R, Harrison J, Baker-Austin C, Ottaviani D, Leoni F, et al. Galleria mellonella as an infection model to investigate virulence of Vibrio parahaemolyticus. Virulence. (2018) 9:197–207. doi: 10.1080/21505594.2017.1384895
23. Hou XL, Cao QY, Pan JC, Chen Z. Classification and identification of Vibrio cholerae and Vibrio parahaemolyticus isolates based on gyrB gene phylogenetic analysis. Acta Microbiol Sinica. (2006) 46:884–9.
24. Montieri S, Suffredini E, Ciccozzi M, Croci L. Phylogenetic and evolutionary analysis of Vibrio parahaemolyticus and Vibrio alginolyticus isolates based on the toxR gene sequence. New Microbiol. (2010) 33:359–72. Available online at: http://www.newmicrobiologica.org/pub/allegati/2010_4/micro4_11_montieri.pdf
25. Chowdhury NR, Stine OC, Morris JG, Nair GB. Assessment of evolution of pandemic Vibrio parahaemolyticus by multilocus sequence typing. J Clin Microbiol. (2004) 42:1280–2. doi: 10.1128/JCM.42.3.1280-1282.2004
26. Turner JW, Paranjpye RN, Landis ED, Biryukov SV, Gonzalez-Escalona N, Nilsson WB, et al. Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest of the United States. PLoS ONE. (2013) 8:e55726. doi: 10.1371/journal.pone.0055726
27. Martinez-Urtaza J, Baker-Austin C, Jones JL, Newton AE, Gonzalez-Aviles GD, DePaola A. Spread of Pacific Northwest Vibrio parahaemolyticus strain. N Eng J Med. (2013) 369:1573–4. doi: 10.1056/NEJMc1305535
28. Haendiges J, Timme R, Allard MW, Myers RA, Brown EW, Gonzalez-Escalona N. Characterization of Vibrio parahaemolyticus clinical strains from Maryland (2012-2013) and comparisons to locally and globally diverse V. parahaemolyticus strains by whole-genome sequence analysis. Front Microbiol. (2015) 6:125. doi: 10.3389/fmicb.2015.00125
29. Gonzlez-escalona N, Jolley KA, Reed E, Martinez-Urtaza J. Defining a core genome multilocus sequence typing scheme for the global epidemiology of Vibrio parahaemolyticus. J Clin Microbiol. (2017) 55:1682–97. doi: 10.1128/JCM.00227-17
30. Tsang AK, Lee HH, Yiu SM, Lau SK, Woo PC. Failure of phylogeny inferred from multilocus sequence typing to represent bacterial phylogeny. Sci Rep. (2017) 7:4536. doi: 10.1038/s41598-017-04707-4
31. Turner JW, Berthiaume CT, Morales R, Armbrust EV, Strom MS. Genomic evidence of adaptive evolution in emergent Vibrio parahaemolyticus ecotypes. Elem Sci Anth. (2016) 4:000117. doi: 10.12952/journal.elementa.000117
32. Whistler CA, Hall JA, Xu F, Ilyas S, Sirwakoti P, Cooper VS, et al. Use of whole genome phylogeny and comparisons in the development of multiplex-PCR assay to identify sequence type 36 Vibrio parahaemolyticus. J Clin Microbiol. (2015) 53:1864–72. doi: 10.1128/JCM.00034-15
33. Sahl JW, Steinsland H, Redman JC, Anguioli SV, Nataro JP, Sommerfelt H, et al. A comparative genomic analysis of diverse clonal types of entertoxigenic Escherichia coli reveals pathovar-specific conservation. Infect Immun. (2011) 79:950–60. doi: 10.1128/IAI.00932-10
34. Valdivia-Granda W. The next meta-challenge for bioinformatics. Bioinformation. (2008) 2:358. doi: 10.6026/97320630002358
35. Valdivia-Granda WA. Biodefense oriented genomic-based pathogen classification systems: challenges and opportunities. J Bioterror Biodef. (2012) 3:1000113. doi: 10.4172/2157-2526.1000113
36. Wilson WC, Ruder MG, Jasperson D, Smith TPL, Naraghi-Arani P, Lenhoff R, et al. Molecular evolution of epizootic hemorrhagic disease viruses in North America based on historical isolates using motif fingerprints. Virus Genes. (2016) 52:495–508. doi: 10.1007/s11262-016-1332-z
37. Gonzalez JP, Souris M, Valdivia-Granda W. Global spread of hemorrhagic fever viruses: predicting pandemics. In: MS Salvaoto editor. Hemorrhagic Fever Viruses. New York, NY: Humana Press (2018). p. 3–31.
38. Gonzalez-Escalona N, Gavilan RG, Brown EW, Martinez-Urtaza J. Transoceanic spreading of pathogenic Vibrio parahaemolyticus with distinctive signatures in the recA gene. PLoS ONE. (2015) 10:e0117485. doi: 10.1371/journal.pone.0117485
39. Haendiges J, Jones J, Myers RA, Mitchell CS, Butler E, Toro M, et al. A nonautochthonous US strain of Vibrio parahaemolyticus isolated from Chesapeake Bay oysters caused the outbreak in Maryland. Appl Environ Microbiol. (2016) 82:3208–16. doi: 10.1128/AEM.00096-16
40. Ronholm J, Petronella N, Leung CC, Pightling AW, Banjeree SK. Genomic features of environmental and clinical Vibrio parahaemolyticus isolates lacking recognized virulence factors are dissimilar. Appl Environ Microbiol. (2016) 82:1102–13. doi: 10.1128/AEM.03465-15
41. Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. (2015) 5:8365. doi: 10.1038/srep08365
42. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
43. Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using Brujin graphs. Genome Res. (2008) 18:821–9. doi: 10.1101/gr.074492.107
44. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. (2012) 28:1647–9. doi: 10.1093/bioinformatics/bts199
45. Bosi E, Donati B, Galardini M, Brunetti S, Sagot MF, Lio P, et al. MeDuSa: a multi-draft based scaffolder. Bioinformatics. (2015) 31:2443–51. doi: 10.1093/bioinformatics/btv171
46. Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, et al. Multilocus sequence typing of total-genome sequenced bacteria. J Clin Microbiol. (2012) 50:1355–61. doi: 10.1128/JCM.06094-11
47. Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. (2010) 11:595. doi: 10.1186/1471-2105-11-595
48. Gonzalez-Escalona N, Martinez-Urtaza J, Romero J, Espejo RT, Jaykus LA, DePaola A. Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by Multilocus sequence typing. J Bacteriol. (2008) 190:2831–40. doi: 10.1128/JB.01808-07
49. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. (2004) 32:1792–7. doi: 10.1093/nar/gkh340
50. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. (2014) 30:1312–3. doi: 10.1093/bioinformatics/btu033
51. Angiuoli SV, Salzberg SL. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics. (2010) 27:334–42. doi: 10.1093/bioinformatics/btq665
52. Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. (2005) 15:1451–5. doi: 10.1101/gr.4086505
53. Capella-Guitierrez S, Silla-Martinez JM, Gabaldon T. trimAL: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. (2008) 25:1972–3. doi: 10.1093/bioinformatics/btp348
54. Price MN, Dehal PS, Arkin AP. FastTree 2- approximately maximum-likelihood trees for large alignments. PLoS ONE. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
55. Felsenstein J. PHYLIP (Phylogeny Inference Package) Version 3.6. Distributed by the author, Department of Genome Sciences, University of Washington, Seattle, WA (2005).
56. Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. (2017) 8:28–36. doi: 10.1111/2041-210X.12628
57. R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2013). Available online at: http://www.R-project.org
58. Corpas M, Valdivia-Granda W, Torres N, Greshake B, Coletta A, Knaus A, et al. Crowdsourced direct-to-consumer genomic analysis of a family quartet. BMC Genomics. (2015) 16:910. doi: 10.1186/s12864-015-1973-7
59. Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. (2002) 18:207–8. doi: 10.1093/bioinformatics/18.1.207
60. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
61. Ludeke CHM, Gonzalez-Escalona N, Fischer M, Jones JL. Examination of clinical and environmental Vibrio parahaemolyticus isolates by multi-locus sequence typing (MLST) and multiple-locus variable-number tandem-repeaat analysis (MLVA). Front Microbiol. (2015) 6:564. doi: 10.3389/fmicb.2015.00564
62. Banerjee SK, Kearney AK, Nadon CA, Peterson C, Tyler K, Bakouche L, et al. Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J Clin Microbiol. (2014) 52:1081–8. doi: 10.1128/JCM.03047-13
63. Han D, Tang H, Lu J, Wang G, Zhou L, Min L, et al. Population structure of clinical Vibrio parahaemolyticus from 17 coastal countries, determined through multilocus sequence analysis. PLoS ONE. (2014) 9:e107371. doi: 10.1371/journal.pone.0107371
64. Martinez-Urtaza J, van Aerle R, Abanto M, Haendiges J, Myers RA, Trinanes J, et al. Genomic variation and evolution of Vibrio parahaemolyticus ST36 over the course of a transcontinental epidemic expansion. mBio. (2017) 8:e01425–e01417. doi: 10.1128/mBio.01425-17
Keywords: Vibrio parahaemolyticus, genomics, MLST, whole-genome sequencing, phylogenetics, protein motif fingerprinting, virulence
Citation: Jesser KJ, Valdivia-Granda W, Jones JL and Noble RT (2019) Clustering of Vibrio parahaemolyticus Isolates Using MLST and Whole-Genome Phylogenetics and Protein Motif Fingerprinting. Front. Public Health 7:66. doi: 10.3389/fpubh.2019.00066
Received: 14 December 2018; Accepted: 06 March 2019;
Published: 08 May 2019.
Edited by:
Fumito Maruyama, Kyoto University, JapanReviewed by:
Chew Chieng Yeo, Sultan Zainal Abidin University, MalaysiaVeranja Chathurani Liyanapathirana, University of Peradeniya, Sri Lanka
Copyright © 2019 Jesser, Valdivia-Granda, Jones and Noble. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelsey J. Jesser, a2Vsc2V5Lmplc3NlckBnbWFpbC5jb20=