 Ilias Karaiskos
Ilias Karaiskos Styliani Lagou
Styliani Lagou Konstantinos Pontikis
Konstantinos Pontikis Vasiliki Rapti
Vasiliki Rapti Garyphallia Poulakou
Garyphallia Poulakou- 1First Department of Internal Medicine-Infectious Diseases, Hygeia General Hospital, Athens, Greece
- 2Third Department of Medicine, School of Medicine, Sotiria General Hospital, National and Kapodistrian University of Athens, Athens, Greece
- 3ICU First Department of Respiratory Medicine, School of Medicine, Sotiria General Hospital, National and Kapodistrian University of Athens, Athens, Greece
The recent expansion of multidrug resistant and pan-drug-resistant pathogens poses significant challenges in the treatment of healthcare associated infections. An important advancement, is a handful of recently launched new antibiotics targeting some of the current most problematic Gram-negative pathogens, namely carbapenem-producing Enterobacteriaceae (CRE) and carbapenem-resistant P. aeruginosa (CRPA). Less options are available against carbapenem-resistant Acinetobacter baumannii (CRAB) and strains producing metallo-beta lactamases (MBL). Ceftazidime-avibactam signaled a turning point in the treatment of KPC and partly OXA- type carbapenemases, whereas meropenem-vaborbactam was added as a potent combination against KPC-producers. Ceftolozane-tazobactam could be seen as an ideal beta-lactam backbone for the treatment of CRPA. Plazomicin, an aminoglycoside with better pharmacokinetics and less toxicity compared to other class members, will cover important proportions of multi-drug resistant pathogens. Eravacycline holds promise in the treatment of infections by CRAB, with a broad spectrum of activity similar to tigecycline, and improved pharmacokinetics. Novel drugs and combinations are not to be considered “panacea” for the ongoing crisis in the therapy of XDR Gram-negative bacteria and colistin will continue to be considered as a fundamental companion drug for the treatment of carbapenem-resistant Enterobacteriaceae (particularly in areas where MBL predominate), for the treatment of CRPA (in many cases being the only in vitro active drug) as well as CRAB. Aminoglycosides are still important companion antibiotics. Finally, fosfomycin as part of combination treatment for CRE infections and P. aeruginosa, deserves a greater attention. Optimal conditions for monotherapy and the “when and how” of combination treatments integrating the novel agents will be discussed.
Introduction
Treatment and outcomes of healthcare-acquired infections have been threatened by the worldwide increasing incidence of antimicrobial resistance among Gram-negative bacteria (1). New terminology was launched to describe extensively drug-resistant (XDR) and pan drug-resistant (PDR) Gram-negative microorganisms (2), with very limited treatment options and major representatives Klebsiella pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa (1, 3). Several official reports including those of the World Health Organization (WHO), the Infectious Diseases Society of America (IDSA) and the UK Government in 2015 have designated antimicrobial resistance as one of the major problems affecting human health and health economy (4–6). As a result of the pressure exerted from regulatory bodies and social waves, we have a handful of already launched new antibiotics targeting some of the current most problematic Gram-negative pathogens, namely Klebsiella pneumoniae carbapenemase (KPC)- producing Enterobacteriaceae and multi-drug-resistant (MDR) P. aeruginosa. There are more new antimicrobials in the final stage of development, some of them targeting pathogens expressing metallo-beta lactamases, MDR A. baumannii and other problematic pathogens, whereas novel non-antibiotic approaches are in development, in order to confront pathogens' ability to develop resistance to new antibiotic classes (7–9). Currently, ceftazidime-avibactam, ceftolozane-tazobactam, meropenem-vaborbactam, and eravacycline have been added to our armamentarium in US and Europe, whereas plazomicin has already got Food and Drug Adminstration (FDA) approval. For all recently launched antimicrobials, evidence about the optimal use outside registration trials is accumulating, though not clear yet. Unmet clinical needs may be geographically diverse and different from regulatory-approved indications (10). Furthermore, lessons learned from the previous antibiotic dawn, call for vigilance for emergence of resistance in every new antibiotic. Prudent use of these precious additions in our antimicrobial armamentarium is important to ensure their longevity (1). In this narrative review, first we will appraise old- revived (colistin, fosfomycin) or established (tigecycline-aminoglycosides-carbapenems) antibiotics that are being used in the treatment of XDR pathogens, with focus on their drawbacks as optimal treatment options and their potential as treatment components along with new antibiotics. Then we will present a summary of new antibiotics centered in real-life use and try to define the most appropriate candidate-patient to whom they must be prescribed. Finally, the “when and how” of combination treatments will be appraised.
Prevalent Mechanisms of Resistance Among Difficult-to-treat Pathogens
New drugs are called to encounter pathogens with accumulating mechanisms of resistance. Carbapenem resistance is a pivotal event in the generation of XDR pathogens, because a potent antibiotic class is inactivated, along with all beta-lactams, when the underlying mechanism is the production of a carbapenemase, which acts as a broad beta-lactamase (11). Clinical implications are profound, due to the exhaustion of therapeutic options. Enterobacteriaceae, with Klebsiella pneumoniae the most representative, P. aeruginosa and A. baumannii are the most common pathogens in clinical practice that harbor carbapenemases. Table 1 shows the most relevant mechanisms of resistance encountered among XDR and PDR isolates (11–14). The majority of acquired carbapenemases belong to either Ambler class A, or class B (metallo-beta-lactamases–MBLs), or class D (oxacillinases–OXAs) (13). Extended-spectrum beta-lactamases (ESBLs), although not conferring resistance to carbapenems, are important contributors of resistance traits because ESBL-, and carbapenemsae- encoding plasmids are frequently vectors of resistance determinants for other antimicrobial classes, such as aminoglycosides (aminoglycoside-modifying enzymes or 16S rRNA methylases) and fluoroquinolones (Qnr, or efflux pumps) (11–13).
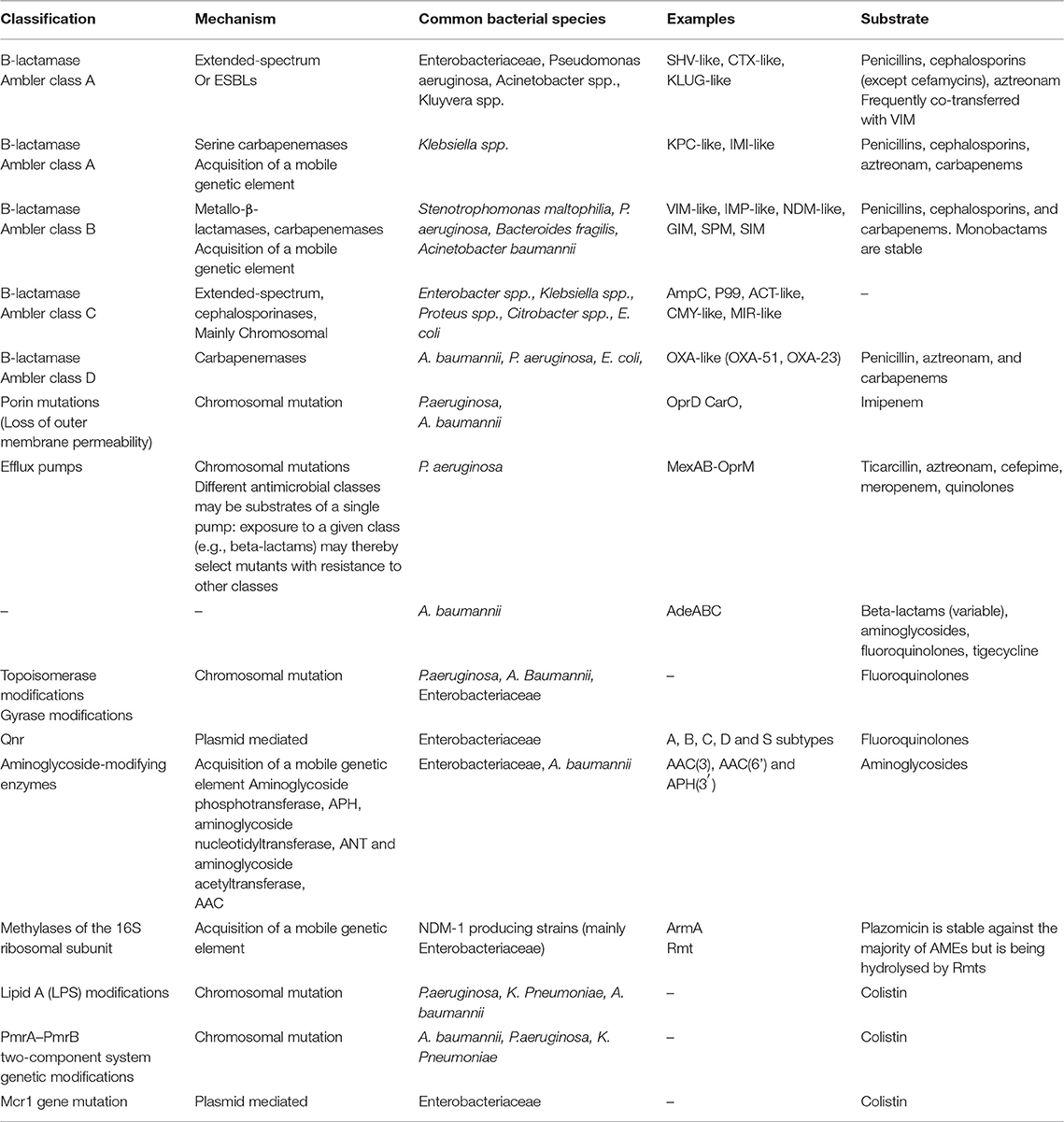
Table 1. Prevalent mechanisms of resistance among pathogens with extended-drug resistance (XDR) (11–14).
Outer membrane modifications and particularly mutations in membrane porins may confer resistance to carbapenems without resistance to other beta-lactams. Efflux pumps confer resistance to carbapenems without carbapenemase production; they may affect multiple antibiotics. Aminoglycoside-modifying enzymes confer resistance to aminoglycoside class; usually they are located in mobile genetic elements along with other mechanisms of resistance (11–13). Finally resistance to colistin was chromosomally mediated for years; a plasmid-mediated mechanism through mutation in mcr1 gene is a new threat, first identified from Enterobacteriaceae in China (14).
Old and Established Antibiotics
Colistin
Colistin, systemically administered in the form of the prodrug colistin methanesulfonate (CMS), is a revived antibiotic that has been addressed as the last resort antibiotic of the twenty-first century due to the steep increase of resistance rates and the lack of new effective antimicrobial agents (15). The antimicrobial spectrum of colistin includes MDR and XDR Gram-negatives regardless of mechanism of resistance, mainly K. pneumoniae, A. baumannii, and P. aeruginosa, whereas Proteae are inherently resistant (15). In vitro activities of colistin and novel combination ceftazidime/avibactam (CAZ-AVI) against isolates of Enterobacteriaceae collected in Europe as part of the International Network For Optimal Resistance Monitoring (INFORM) global surveillance program from 2012 to 2015 revealed CAZ-AVI as the most potent active agent with a susceptibility rate of 99.4%, whereas susceptibility to colistin was 82.8% (16). Regarding P. aeruginosa, colistin was the most active drug followed by CAZ-AVI with a susceptibility rate of 99.6 and 92.4%, respectively (17).
During the last decade, a better characterization of the colistin pharmacokinetics (PKs) has been made feasible due to advanced technology, resulting in minimization of hydrolysis of CMS to colistin during workup, introducing colistin into a revolutionary period (15). The necessity of a loading dose and longer dosing intervals (18, 19) as well as a dosing scheme based on creatinine clearance (CrCL) have been proposed (20) and assimilated in clinical practice (19–21). The updated colistin daily maintenance doses have been shown to achieve target attainment of clinically relevant plasma colistin concentrations (around 2 mg/L) for approximately 90% for patients with CrCl < 80 ml/min (22). However, the recent observation that a high CrCL of >80 mL/min decreases the ability to achieving the appropriate steady-state colistin levels attributed to a higher amount of colistin cleared by the kidneys, probably mandates either the necessity of combinations or higher dosing (5, 8, 19, 22).
The current clinical efficacy of colistin is extrapolated from reported studies focusing mainly on bacteremias and VAP caused by MDR and XDR Gram-negative pathogens. However, the major drawbacks of publications are their retrospective, non-randomized design, with a variety of dosages, and the absence of a loading dose, whereas the simultaneous administration of other active in vitro antibiotics renders inconclusive the efficacy of monotherapy with colistin (15). The issue of combination therapy with colistin has received considerable critical attention and is a debated and controversial topic (23, 24). Regarding carbapenem resistant Enterobacteriaceae (CRE), the combination of colistin with another active in vitro antibiotic has been reported to be beneficial in terms of survival, as depicted in a big number of patients and despite their retrospective source of the studies (23–25). The combination of meropenem (if MIC is ≤8 mg/L) with gentamicin or tigecycline or colistin have resulted significantly in reduction of mortality particularly in patients with septic shock, high mortality score, or rapidly fatal underlying diseases (25, 26). However, in low-risk bloodstream infections (BSIs) and non-bacteremic urinary and abdominal infections, monotherapy seems as an adequate therapeutic choice (25). There are currently minimal and of low-quality clinical evidence suggesting superiority of monotherapy over combination therapy for treatment of carbapenem resistant P. aeruginosa (CRPA). However, many clinicians, taking into consideration PK/PD limitations of colistin and the development of resistances favor combination treatment (23, 24). Similarly, colistin has been frequently used in combination for the treatment of carbapenem resistant A. baumannii (CRAB), with the same concerns regarding the suboptimal PK of polymyxins (23, 24). This concept has recently been challenged by randomized control trials focusing on infections caused by CRAB demonstrating superior efficacy of monotherapy of colistin vs. combination therapy (27–29). The most recent study enrolled 151 patients with CRAB infections treated with colistin alone vs. colistin plus meropenem and rates of clinical failure (83 vs. 81%, p = 0.64) and mortality (46 vs. 52%, p = 0.40) were found similar in both groups. However, the unexpected high rates of clinical failure and mortality, highlights the necessity of novel agents to effectively treat CRAB infections (29).
Fosfomycin
Having been discovered in the ‘60s, fosfomycin in its intravenous form, fosfomycin disodium (FD), was soon almost abandoned due to the availability of other agents and the peculiarities of susceptibility testing (30). It was not until about a decade ago that it was reintroduced into clinical practice, with a new role in the ongoing fight against XDR and PDR Gram-negative bacteria in critically ill patients (30, 31).
Fosfomycin exerts bactericidal action against susceptible organisms and possesses a unique mechanism of inhibiting the first step of peptidoglycan synthesis, making cross-resistance with other agents unlikely (32). FD has straightforward pharmacokinetics, though less well studied in the critically ill. High concentrations are achieved in serum and urine, well-above minimum inhibitory concentrations (MICs) of susceptible organisms, while penetration to compartments relevant to the ICU setting (lung, cerebrospinal fluid, abscess fluid) is satisfactory (33–35). FD has very few toxicity concerns (36, 37). Hypokalemia and sodium overload stand out as the most important; however, they are usually controlled in the ICU setting.
In contemporary in vitro studies fosfomycin was active against more than 80% of Staphylococcus aureus, Enterococcus faecium, ESBL-producing Escherichia coli, and K. pneumoniae and slightly less against carbapenem-resistant (CR) K. pneumoniae (38). Less information was available for P. aeruginosa, but at least in one study, 80.6% of strains were inhibited by 128 mg/L of fosfomycin (39). Against a recent collection of 396 K. pneumoniae isolates originating from Greek hospitals, fosfomycin was the third most active agent, being active against 58% of strains by using the strict EUCAST criteria for E. coli (where susceptibility break point is 32 mg/L as opposed to 64 mg/L for CLSI) (40). These results were confirmed in a recent collection of US bacterial isolates (41). Of great interest to intensivists is the absence of activity against A. baumannii strains (30). The in vivo development of resistance during treatment is the most feared complication of FD use (41) driving repeated recommendations for use only within combination regimens (8, 30). Recent reports however even with monotherapy did not verify this fear (37).
A great heterogeneity is represented in intravenous fosfomycin studies, further plagued by the absence of control groups. Grabein et al were able to identify only 10 controlled studies with solely one having been published the current decade; fosfomycin had similar clinical efficacy when compared with various agents (37). Recently, the results of a multicenter, randomized, double-blind phase 2/3 trial were announced, showing non-inferiority of intravenous fosfomycin to piperacillin/tazobactam, in patients with complicated urinary tract infections (cUTIs), or acute pyelonephritis (42). These results however should be extrapolated with caution within the ICU environment, since only one in three pathogens exhibited a resistance phenotype.
Fosfomycin is not extensively used against MDR infections, with only 10 among 342 patients with bloodstream infections by CRE in the INCREMENT cohort being treated with FD (43). These data imply that FD is still regarded as salvage treatment for CR infections or treatment for breakthrough infections in patients already receiving anti-XDR treatment. Indeed, a few years ago our group was able to show that almost 2/3 of 48 critically ill patients suffering from CR infections, were already receiving colistin for a median of 13.5 days (31). Apart from the fear of resistance development during treatment, several reasons might account for FD under-prescription. Absence of high-quality evidence of efficacy could be implicated, along with difficulties of unobstructed access to FD with only few countries excepted (44). Lately FD has been approved for use in at least 14 European member states (45), while in the US ZTI-01 (Contepo®) has been granted Qualified Infectious Disease Product and Fast Track designations by the US Food and Drug Administration in several indications (44). In addition, several laboratory issues may stand in the way of FD use, such as the poor performance of diffusion gradient and broth microdilution techniques in comparison with gold standard agar dilution (39, 46–48), the particularity of disc diffusion susceptibility determination (49), the absence of susceptibility thresholds adapted to intravenous use (49, 50) and the difference in proposed susceptibility thresholds between CLSI and EUCAST (50, 51). Finally, labeling information in most European countries encourages salvage use (52). A few years ago our group showed that less than half of 48 fosfomycin-treated critically ill patients, received the first dose of the drug within the first 24 h from infection onset, and these patients tended to have better outcomes than their delayed initiation counterparts (31). As antimicrobial treatment exerts its maximum effect when applied early (53), surveillance cultures (54), or rapidly detection of carbapenemase production in microbiological samples (9), may early identify patients that might benefit from FD treatment.
It is undisputable that there is a place for FD within the ICU, against CR infections (8, 23). Probably, the greatest unmet need that FD is qualified to cover are XDR and PDR CRE infections, especially in the presence of colistin resistance or production of metallo-β-lactamases. Against CR pseudomonal infections, more agents are available and fosfomycin MICs are higher (44), however FD might be occasionally useful. It is questionable whether recent results implying modest in vivo synergistic effects with colistin against A. baumannii infections (28) will expand its use in this indication. Regarding specific sites of infection, it is anticipated that FD will have its best performance in the urinary tract. Nevertheless, currently available data imply comparable activity in other conditions, as well, such as ventilator-associated pneumonia (VAP) or bloodstream infections (BSIs) (31, 31). Doses up to 16–24 g/daily have been used in MDR infections, which seem justified based on pathogens MICs and fosfomycin pharmacokinetic properties (55). However, according to a recent population pharmacokinetic (PK) study, significant variance in exposure exists in critically ill patients under modern dosage regimens. This variance is not thoroughly explained by differences in renal function and identifies an area where future research is warranted (56). It is evident that an unexplored potential exists for the intravenous form of fosfomycin.
Combination treatment is the rule for CRE infections in the severely ill host, while for pseudomonal infection it is frequently recommended, even in the absence of carbapenem resistance (7, 8, 26, 57, 58). In the case of FD, combination treatment is being proposed with greater intensity, as a means to overcome selection of resistant mutants. The dogma was recently challenged by the results of the previously mentioned ZEUS study (42), showing that there might be certain patient subgroups that may inconsequently deviate from the norm of combined treatment. Regarding the companion agent choice, no specific recommendation can be made on the companion antimicrobial, since consistent synergistic patterns have not been observed (32, 59–63) and only rarely has antagonism between fosfomycin and another agent been noted (61). Recent in vitro evidence implies that co-administration with aminoglycosides might be beneficial in terms of efficacy, while according to older reports fosfomycin might lessen aminoglycoside toxicity (64, 65). However, the addition of nebulized amikacin-fosfomycin at a 5:2 ratio to standard treatment did not lead to improved VAP outcomes in a recent randomized-controlled trial (RCT) (66). No synergy with colistin has been demonstrated, but here is in vitro and in vivo evidence of synergy with carbapenems (61, 67, 68) and it is extremely interesting to see whether these effects are replicated (or potentiated) with modern, more active against CRE beta lactams, such as novel beta-lactam beta-lactamase inhibitor combinations.
Tigecycline
The glycyclcycline tigecycline has been used in the last decade as a salvage treatment for infections caused by CRE and CRAB (69). Treatment outcomes have been hampered by the low serum concentrations of the drug in the approved dosing regimen and the low penetration in the epithelial lining fluid (ELF) of mechanically ventilated patients (70). A higher dose of tigecycline (HDT) (100 mg twice daily after 200 mg of loading dose) has been suggested, particularly for VAP/HAP, A. baumannii infections and bacteraemic infections, although all the above represent off-label use of the drug (71, 72). Decreased fibrinogen levels have been reported with augmented dose regimens of tigecycline (73). A systematic review encompassing 263 patients from 11 studies (including only one RCT), reported significantly higher rates of gastrointestinal adverse events (nausea, diarrhea and vomiting) with HDT compared to standard dose (74).
Combination treatments of tigecycline seem mandatory, given the above-mentioned characteristics of the drug and the early warnings about increased mortality, particularly in critically ill patients and when given as monotherapy (75). Before the advent of the new antimicrobials listed in the next section, possible companions were limited to colistin and meropenem (25, 69, 76). Tigecycline-colistin or -polymyxin B combinations have shown in vitro and in vivo synergy in animal model studies with KPC-2 (77–81). Real life data on colistin-tigecycline combination were captured in the INCREMENT retrospective cohort, in which high-risk patients with CRE-BSI on tigecycline-containing combinations (79 patients) had lower mortality compared with those who received colistin monotherapy (27).
Small size and heterogeneity are the rule of almost all reports of HDT in the treatment of MDR pathogens. In a study of 30 postoperative patients admitted in the ICU with two or more positive blood cultures with KPC producing K. pneumoniae, HDT was compared with standard dose combined with colistin in both arms. Significantly lower mortality was demonstrated in the HDT group (82). De Pascale et al. studied HDT in a cohort of patients with VAP by CRE (27 patients) or CRAB (28 patients), in which HDT was the only predictor of clinical cure (83). A retrospective study of 40 patients with BSI caused by CPKP (23 on HDT and 17 on standard-dose tigecycline combinations) found no difference in hospital mortality (84). A systematic review of 2016 compiled 25 studies (21 controlled and 5 single -arm studies) of tigecycline in the treatment of CRE infections (85). No difference in overall mortality, clinical response and microbiological response was found between tigecycline-containing arms and comparator arms. Subgroup analyses elucidated a significantly lower 30-day mortality for patients on tigecycline combination regimens than those who received monotherapy (OR = 1.83 [95% CI = 1.07–3.12; P = 0.03]) and other antibiotic regimens (OR = 0.59 [95% CI = 0.39–0.88; P = 0.01]), respectively. In addition, ICU mortality was significantly lower in HDT compared to standard dose (OR, 12.48; 95% CI, 2.06–75.43; P = 0.006) (85). A great heterogeneity of the studies included in this meta-analysis was reported (85, 86).
Tigecycline will continue to be a valuable antibiotic, as part of combination regimens in the new era of antibiotics (75). HDT is almost mandatory when treatment of difficult to -treat organisms is considered, probably excluding intraabdominal non-bacteremic infections. As part of empiric regimens, it can offer coverage against A. baumannii, depending on local epidemiological data (82). Emerging data on possible synergy with new antibiotics such as plazomicin, need further evaluation in order to define other possible combinations (87).
Aminoglycosides
The worldwide expansion of XDR pathogens and particularly that of CRE has brought into light aminoglycosides, which may retain activity even in XDR isolates (88). In vitro susceptibility rates may vary significantly, depending on the dissemination of aminoglycoside modifying enzymes, which are frequently co-transferred along with other resistance genes on mobile genetic elements (89–91). It is of concern that expression of 16S rRNA methyltranferases, enzymes which confer resistance to all aminoglycosides, is especially common in NDM-producing CRE (40). In a recent multicenter study from Greece, 396 consecutive CRKP isolates were tested for susceptibility to antibiotics and mechanisms of carbapenem resistance. Gentamicin exhibited 61.9% susceptibility against this collection of strains, being higher for KPC-producers and dual KPC-VIM producers (69.1 and 90.1%, respectively); in vitro susceptibility against NDM-1, VIM, and OXA-48 producers was 42.6, 38.2, and 28.6%, respectively (92). Taking into account that colistin resistance was 40.6%, with the highest rates (>90%) observed among isolates exhibiting dual VIM-KPC production, it easily understood how important have become aminoglycosides in the contemporary armamentarium.
Aminoglycosides have been traditionally used as part of combination regimens and only in urinary tract infections they have been used as monotherapy. Response rates of 88% have been reported with aminoglycoside monotherapy in UTIs compared to only 64% with polymyxins (93, 94). Treatment of other body compartments is hampered by the PK/PD obstacles of the drug; monotherapies of CRE except UTIs have been associated with unacceptably high mortality rates of 80% (95). Therefore, combinations with other antibiotic(s) active against the targeted pathogen are recommended. Observational studies including large cohorts of patients with KPC-KP infections have shown that the combination of a carbapenem with an aminoglycoside was associated with the lowest mortality rate, provided that the MIC of the pathogen against meropenem was ≤8 mg/L (25, 26, 95). Aerosolised aminoglycosides may represent an important alternative for VAP, due to suboptimal PK/PD targets attained with iv administration (96, 97). Despite promising preliminary studies, RCTs failed to demonstrate improved mortality when an inhaled aminoglycoside was added as definitive treatment of HAP and VAP through an optimized device; lack of target population (i.e., MDR and XDR infections) among persons enrolled in the study may have obscured any added benefit from aerosolized antibiotics (66, 97).
Current optimal use of aminoglycosides has important drawbacks. First, established breakpoints of resistance differ between the two major relevant societies, being set at 16 mg/L (CLSI) and 8 mg/L (EUCAST) for amikacin; and 4 mg/L (CLSI) and 2 mg/L (EUCAST) for gentamicin (51, 98). Many MDR and XDR pathogens have borderline susceptibilities against these breakpoints, a fact further complicated by great the difficulty to predict PK/PDs of aminoglycosides in critically ill patients (88, 99–104). Recent reports from critically ill patients have shown that for isolates with an MIC of 16 mg/L for amikacin and 4 mg/L for gentamicin, the necessary doses to achieve therapeutic concentrations in plasma that achieve the PK/PD target would be up to 30–40 and 8–10 mg/kg for amikacin and gentamicin, respectively. Even with the more conservative EUCAST breakpoints, the PK/PD target of 8 mg/L for amikacin would not be achieved despite administration of higher doses than approved (101–103). When the desired therapeutic concentration is not in the blood but in the lung parenchyma, further PD obstacles arise (104). Given the PK/PD limitations of aminoglycosides, a high dose has been recommended followed by renal replacement therapy as a measure of minimizing associated renal toxicity (105).
Clinical experience with aminoglycosides as monotherapy or as “active monotherapy” when co-administered antibiotics were inactive in vitro against the targeted pathogen. However, small published series verified PK/PD considerations and consistently reported clinical failure with isolates having MICs on the breakpoint of susceptibility, whereas clinical success was demonstrated in infections by isolates with low MICs (106, 107). According to the PK/PD data listed above, monotherapy with aminoglycosides would be very risky for critically ill patients for infections in compartments outside the urinary tract (88, 93, 95, 108, 109).
Short duration courses of aminoglycosides (5–7 days) given as once-daily regimens are associated with less nephrotoxicity compared to multiple daily dosing (110, 111). Even high doses of aminoglycosides were well tolerated in terms of nephrotoxicity (101, 102). Acute kidney injury rates (AKI) between 12 and 17% have been reported in reports of critically ill patients; septic shock and prolonged administration (>10 days) were associated with increased nephrotoxicity risk (110, 112). Consequently, therapeutic drug monitoring should be undertaken, to ensure that therapeutic and non-toxic levels will be delivered to the patient. As shown in Table 2, amikacin and gentamicin are recommended to be given once daily, without a loading dose (111). Higher doses recommended for patients treated for serious CRE infections to attain a maximum serum concentration of 30 and 60 mg/ml, respectively corresponding to a PD target of 8 times the MIC of the pathogen (111, 123). Peak levels of 15–20 mg/ml for gentamicin and tobramycin and 20–40 mg/ml for amikacin are targeted with once-daily regimens. Trough levels indicative of low nephrotoxicity risk are 1 mg/ml for gentamicin or tobramycin, and 1–4 mg/ml for amikacin (111, 123).
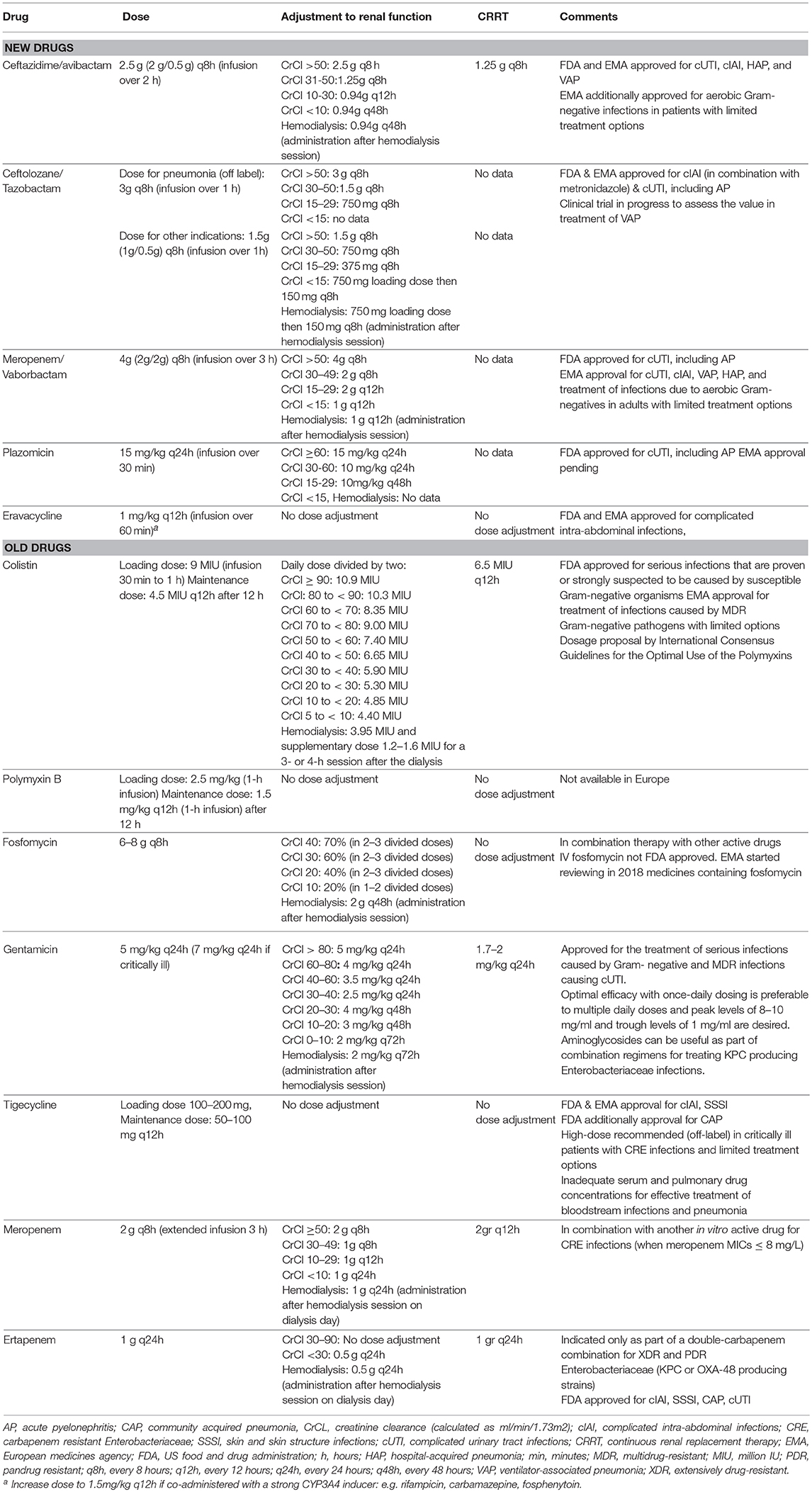
Table 2. Intravenous novel and older antimicrobial agents against MDR and XDR Gram-negative pathogens (1, 8, 24–26, 59, 64, 99, 110, 113–122, 136).
When combination treatment is considered, aminoglycosides have been combined with almost all classes of antibiotics (88). Synergy of aminoglycosides with carbapenems has produced encouraging in vitro results in CRE isolates with resistance to carbapenems (124, 125). Further clinical evidence is important on these observations.
Carbapenem-Containing Combinations and Double Carbapenem Combination
Before the launching of the new combinations of beta-lactams/beta-lactamase inhibitors with activity against KPC producers, carbapenems have been used as a life-saving therapeutic approach in infections caused by strains producing carbapenemases, despite the apparent paradox (25, 76). High doses and extended infusions were used (particularly for meropenem), along with other active in vitro agents. Studies from Greece showed that combinations including meropenem in the treatment of patients with CRKP BSIs had the lowest mortality when the MIC of the pathogen was ≤8 mg/L (19.3 vs. 35.5% in patients with carbapenem MICs > 9 mg/L) (25). Data from Italy, in a study of 36 BSIs by KPC-KP showed that meropenem-containing regimens had a significantly lower 30-days mortality rates when the isolate's meropenem MIC was 8 mg/L or less (15.8 vs. 35.2% when the MIC was at >16 mg/L) (76). Although these data derived from small observational studies, they were verified in larger cohorts. Subsequent Italian cohorts of 661 CRKP infections of various sources and 595 BSIs, respectively, highlighted the importance of high-dose meropenem combinations in achieving successful clinical outcomes with isolate's MIC against meropenem up to 16 mg/L (26, 126). Non-carbapenem regimens were associated with worse outcomes across all these studies, particularly when they consisted of monotherapies (25, 76, 127).
Interestingly, multidrug carbapenem-containing combinations were most effective in patients at high risk for mortality (i.e., those in septic shock (25). Based on the experience gathered from KPC-KP infections in Italy and Greece, in a position document, 13 experts from these two countries concluded that “administration of high-dose (e.g., 2 g every hours), prolonged infusion meropenem could be beneficial in KPC-KP infections if MIC is ≤8 mg/L,” whereas for MICs up to 32–64 mg/L, “meropenem administration should be considered if therapeutic drug monitoring is available to monitor optimal drug exposure” (8). With the advent of the novel antibiotics with activity against KPC-KP, carbapenems are no longer preferable to a beta lactam antibiotic against which the isolate is susceptible. However, early studies have demonstrated similar effects of carbapenem-containing regimens on mortality in severe infections caused by VIM-1 producing K. pneumoniae, provided that the carbapenem MIC of the isolate was <4 mg/L (127). Since we don't have new antibiotics active against MBL-producers, experience acquired so far could be still life-saving against XDR isolates, particularly when the underlying mechanism of resistance is VIM or the isolate coproduces KPC and VIM. Further evaluation is needed and rapid communication of carbapenem MICs to the treating physicians, irrespective of the mechanism of resistance produced by the recovered pathogen.
Double carbapenem combination (DCC) has emerged as a salvage treatment option against XDR and PDR CRE, based on a preliminary concept study by Bulik and Nicolau on the combination of ertapenem and doripenem (128). They're in vitro and in vivo observations, suggested that ertapenem acts as a “suicide inhibitor of KPCs” due to its higher affinity to the enzyme, leaving the other carbapenem intact and active against the isolate (128). Further in vitro studies showed that only KPC and not MBL enzymes respond to this maneuver and various carbapenem combinations were proven successful in vitro (129). These observations were verified in small observational studies with inherent biases (lack of randomization and controls) (23, 130–132). A recent publication summarized the experience among 211 patients treated with this modality in observational studies (133). The most common combination was ertapenem (1 g q24h IV, infused over 30 min) plus meropenem (2 g q8 h, 3 h infusion). The administration of other antibiotics in almost one third of cases (mostly colistin) obscures the evaluation of DCC per se, however, a clinical success rate of 70% with all-cause mortality of 26% seems promising in a population of critically ill patients (50% bacteraemic). It is noteworthy that in a study encompassing 27 patients, the majority of whom received only DCC, PDR infections had a successful clinical and microbiological outcome in 78.5% (131). In a similar study in which the authors preferred triple drug combination with colistin, similar clinical efficacy was shown (75%) (132). In the single study employing a case control approach, 48 patients receiving DCC were compared to 96 control patients who were administered standard treatment (i.e., colistin, tigecycline, aminoglycoside) for documented infections by CPKP. DCC was associated with significantly lower 28-days mortality (47.9 vs. 29.2%, p = 0.04) and was shown to have a protective effect on mortality in the multivariate analysis (OR: 0.33, 0.13–0.87) (133).
Administration of a double carbapenem (with or without colistin) seems a promising salvage treatment in infections caused by PDR isolates harboring serine-carbapenemases. In vitro data showed synergistic activity of DCC against OXA-48 producing MDR and XDR K. pneumoniae (134). Finally, two kidney transplant recipients were reportedly cured with DCC plus oral fosfomycin from urinary tract infections caused by NDM-harboring Enterobacteriaceae (135). Further randomized controlled trials are needed to assess DCC as salvage treatment in infections by XDR pathogens exhibiting various mechanisms of resistance to carbapenems.
New Antibiotics
Ceftolozane-Tazobactam
Ceftolozane/tazobactam (C/T) is a combination of a β-lactamase inhibitor in use for decades, with a new oxyimino-cephalosporin with structural similarity to ceftazidime. It possesses a potent activity against P. aeruginosa, due to the inherent high affinity of ceftolozane for the penicillin-binding proteins that are essential for these species (113, 136). Common mechanisms of resistance in P. aeruginosa (Table 1) such as overexpression of efflux pumps, loss of the outer membrane porin OprD and AmpC overproduction do not affect ceftolozane, whereas development of resistance to C/T requires several mutations leading to an overexpression of AmpC enzymes and structural modifications (113). Tazobactam offers stability against most ESBLs (136). C/T is being hydrolyzed by carbapenemases (KPC, VIM, NDM, GES) with possible exception OXA-48 against which the combination may retain some activity (113). In vitro studies have shown an excellent in vitro activity against P. aeruginosa and Enterobacteriaceae. Of interest for ICU patients is also a demonstrated in vitro susceptibility against Burkholderia spp. and Stenotrophomonas maltophilia isolates with relatively low MICs, but no substantial susceptibility against A. baumannii (113, 137, 138). C/T has considerable activity against Streptococcus spp (except for S. pneumoniae), no activity against staphylococci and enterococci and minimal activity against anaerobes (113, 137–139). Time kill studies have verified that C/T is the most potent antibiotic against susceptible P. aeruginosa, whereas carbapenems were more cidal against Enterobacteriaceae (140). Recent data from 53 ICU VAP infections by P. aeruginosa showed that C/T exhibited the most potent in vitro activity compared to ceftazidime/avibactam, imipenem and ciprofloxacin (141).
C/T is approved in the US and Europe for cIAIs and cUTIs based on two phase 3 registration trials which showed non-inferiority of C/T (with metronidazole) vs. meropenem in cIAIs and vs. levofloxacin in cUTIs (142, 143). Results from a phase 3 clinical trial in HAP requiring mechanical ventilation and VAP (ASPECT-NP) are anticipated to support the approval of the relevant indication. Double C/T dosage (3 g every 8 h for normal renal function) was employed in the trial of nosocomial pneumonia, based on PK/PD studies showing that with this dosage there is more than 90% probability of PD target attainment for pathogens with MIC ≤8 mg/L, well below the breakpoint of susceptibility for P. aeruginosa which was set by both CLSI and EUCAST at 4 mg/L (144, 145). Dosage in impaired renal function is displayed in Table 2 (146–149). Approved dosage corresponds to target attainment for pathogens with an MIC of 4 mg/L in patients with normal renal function or augmented renal clearance (146) and 8 mg/L in patients with impaired renal clearance. Despite this, data from the phase 3 trial investigating cIAIs showed that patients with moderate renal impairment (CrCl 30–50 mL/min) had lower cure rates in the C/T plus metronidazole arm compared to the meropenem arm (48 vs. 69.2%, respectively) (142). Following these reports daily monitoring of renal function in patients with unstable renal function and re-adjustment of C/T dosage according to creatinine clearance measurement is strongly recommended. Although the labeled duration of the infusion is 1 h, a 4–5-hours extended infusion may provide better probability of PD target attainment for pathogens with elevated MICs (147, 150).
Currently available data on the clinical effectiveness of C/T for respiratory infections and VAP derive from case series and case-reports with variable dosage regimens and in combination with other antibiotics. An overall clinical success of 61.4% among patients with P. aeruginosa pneumonia treated with C/T is estimated (151–156), whereas a clinical efficacy of 71% was reported a single study of infections by MDR P. aeruginosa (152). Clinical failures have been associated with an MIC higher than 4 mg/L and the use of the lower dosage regimen (157). Of concern are reports of resistance to C/T during therapy of P. aeruginosa infections (152, 155, 158). A recent retrospective report on 101 infections by P. aeruginosa (50% of which XDR), showed for ceftolozane/tazobactam (35% in combination) an overall clinical success of 83.2%. Continuous renal replacement therapy and sepsis were identified as factors associated with lower success rates (159).
The clinical effectiveness of C/T against ESBL-producing Enterobacteriaceae, was estimated in a secondary analysis of the clinical trials of cIAIs and cUTIs; clinical cure rates of 97.4% were reported for C/T in E. coli or K. pneumoniae infections, compared with 82.6 and 88.5% for levofloxacin and meropenem, respectively (114).
C/T is a valuable addition to our armamentarium, as the most potent antibiotic against P. aeruginosa infections with additional activity against ESBL-producers. The main place in therapy of C/T would be the empirical or definitive treatment of infections suspected or caused by P. aeruginosa (Figure 3), in which it could serve as a beta-lactam backbone of treatment. In settings where carbapenemase production is low among P. aeruginosa strains, C/T can provide a reliable empirical coverage (160). In patients with various risk factors for MDR (hematologic malignancy, prolonged hospitalization, prior ICU admission, previous receipt of other anti-pseudomonal agents etc.), C/T can be used as part of an empirical antibiotic regimen, along with a second agent, to ensure adequate coverage (136, 161). Pending the results of the phase 3 study of nosocomial pneumonia, C/T will probably hold an important role in the reiteration of guidelines for VAP caused by P. aeruginosa (162). In infections by CR-P. aeruginosa without carbapenemase production, C/T should be the drug of choice. C/T holds also an important role against ESBL-producing Enterobacteriaceae, where it represents a reliable a carbapenem-sparing option.
Ceftazidime-Avibactam
Avibactam, a novel non-β-lactam, β-lactamase inhibitor, restores the activity of ceftazidime against the majority of β- lactamases (ESBLs and carbapenemases, including KPCs– Ambler Class A, AmpC —Class C and oxacillinase OXA-48—Class D), resulting in extended spectrum of CAZ-AVI combination against a wide range of MDR bacteria (115, 163). Notably, avibactam is not able to inhibit strains producing metallo-β-lactamases (MBL—Class B), as well as many of the Class D enzymes (115). CAZ-AVI is FDA and EMA approved for the treatment of complicated intra-abdominal infections (cIAIs), complicated urinary tract infections (cUTIs), hospital-acquired, and ventilator associated pneumonia (HAP/VAP) and (EMA only) infections due to aerobic Gram-negative organisms in adult patients with limited treatment options (116, 164).
The International Network For Optimal Resistance Monitoring (INFORM) global surveillance program from 2012 to 2015 demonstrated 99.4 and 98.5% susceptibility to CAZ-AVI for all Enterobacteriaceae isolates and for meropenem-non-susceptible, MBL-negative isolates, respectively (16, 17). In a recent surveillance study of 394 clinical isolates of CR-KP from Greece, CAZ-AVI inhibited 99.6% of KPC and 100% of OXA-48-like-producing isolates (40); data from US were similar, showing 98.7% in vitro susceptibility of CAZ-AVI against KPC-producing K. pneumoniae isolates (165). Although only few studies have differentiated activity of CAZ-AVI against KPC subtypes, emerging data indicate that KPC-3-producing strains have higher MICs than KPC-2 producers. In a recent study KPC-3 strains exhibited a ceftazidime-avibactam MIC of 8 μg/ml; furthermore KPC-3 possessed 30-fold greater hydrolytic activity against ceftazidime than KPC-2 (166, 167). According to European Committee on Antimicrobial Susceptibility Testing (EUCAST) (51) and Clinical Laboratory Standards Institute (CLSI) (98). Minimum Inhibitory Concentration (MIC) breakpoints for CAZ-AVI is ≤8 mg/L for both Enterobacteriaceae and P. aeruginosa species.
KPC-producing strains may harbor additional resistance mechanisms, including overexpression of efflux-pump, mutations in porin genes, production of multiple carbapenemases and de novo mutations in the blaKPC-3 gene (168–170). Emerging resistance to CAZ-AVI has been attributed to the expression of KPC variants with substitutions in the omega-loop; this knowledge has fueled the research for a new active formulation of β-lactamase inhibitors, that will potentially overcome this new resistance mechanism (171, 172). These KPC variants seem to lose their carbapenemase activity and exhibit a meropenem susceptible phenotype (173). Unfortunately, restored meropenem susceptibility is not permanent, since the isolate after exposure to meropenem rapidly returns to the initial KPC-expressing status along with the emergence of porin mutations (174). In some early reports, resistance of K. pneumoniae to CAZ-AVI (MIC ≥16 mg/L) was observed with short courses of therapy, especially after monotherapy, reflecting the need for vigilance in clinical practice (168). As a result of these reports, some experts suggest combination treatments in order to prevent emergence of resistance (8).
In vitro activity of CAZ-AVI against P. aeruginosa varies across studies, owing to the different underlying mechanisms of resistance, compared to Enterobacteriaceae (175). Data from recent studies showed higher in vitro activity of CAZ-AVI compared to ceftazidime, piperacillin-tazobactam or meropenem when tested against both MDR and XDR, P. aeruginosa strains (175–177). A susceptibility rate as high as 85.1% for meropenem-non-susceptible P. aeruginosa has been reported, rendering CAZ-AVI the second most potent in vitro agent after colistin; however, in other studies in vitro activity around 50% was shown for isolates resistant to ceftazidime and meropenem (16, 17, 176, 177). Differences in susceptibility among strains of P. aeruginosa to β-lactam agents, including CAZ-AVI, may be attributed to a variable prevalence of the metallo-β-lactamases, such as VIM-2 (176–178). CAZ-AVI combination is only slightly active against A. baumannii (due to avibactam's vulnerability to OXA-type carbapenemases frequently carried by A. baumannii species). Marginal activity against anaerobic Gram-negative bacteria mandates addition of metronidazole in intraabdominal infections; finally, CAZ-AVI is inactive against Gram-positive cocci (173, 176).
PK/PD studies for both ceftazidime and avibactam revealed linear pharmacokinetics, glomerular excretion and human protein binding 10 and 8%, respectively (116, 164, 179, 180). As demonstrated by PK/PD studies in humans, ceftazidime and avibactam equally penetrate into human bronchial epithelial lining fluid (ELF), with AUC values in ELF ~30% of those in plasma (181). The recommended dosage of CAZ-AVI is 2/0.5 g every 8 h by intravenous infusion over 120 min. Dose adjustment to impaired renal function is shown in Table 2 (116, 164).
Two identical, randomized Phase III trials (RECLAIM I and II) compared CAZ-AVI plus metronidazole with meropenem in patients with cIAIs, and proved non-inferiority (182). In the subgroup of patients with impaired renal function, clinical cure rates were higher with meropenem, arguing for caution with dosage regimens in patients with unstable renal function. Subgroup analysis of patients with ceftazidime-resistant bacterial infections, showing clinical cure rates similar to meropenem (83.0 vs. 85.9%, respectively) (16, 17). In line were the outcomes reported from the RECLAIM III, which enrolled 432 patients from Asia (183). The REPROVE study, enrolling 879 patients with nosocomial pneumonia, including VAP, proved non-inferiority of CAZ-AVI compared to meropenem; again, efficacy of CAZ-AVI against ceftazidime-non-susceptible pathogens was comparable to meropenem (184). Two Phase III trials (RECAPTURE I, II), proved non-inferiority of CAZ-AVI to doripenem in patients with cUTIs (185). REPRISE study, a prospective, pathogen-directed (ceftazidime-resistant, Enterobacteriaceae or P. aeruginosa), open-label, phase 3 trial, demonstrated the efficacy and safety of treatment with CAZ-AVI vs. best available therapy in cUTIs or cIAIs (186), establishing CAZ-AVI as a potential alternative to carbapenems in ceftazidime-resistant strains.
An excellent safety and tolerability profile has been shown for CAZ-AVI, with mild main adverse events, including hypersensitivity reactions, headache, nausea, constipation, and diarrhea (164, 183, 187). Physicians should take into consideration the possibility of positive Coombs direct test without documented haemolysis (3.2–20.8%) (182–186).
Post market studies showed that CAZ-AVI was mostly administered as treatment for CRE infections, with clinical response rates between 55–85%, relapse rates of 23% and a mortality rate of 10–39.5% (170, 187–194). Real life data render CAZ-AVI a “game changer” in the treatment of KPC-producers. Compared to colistin, in a prospective study of 38 patients treated with CAZ-AVI vs. 99 treated with colistin, the first group had a 64% probability of a better outcome at 30 days of treatment (193). Another single-center observational study in KPC-bacteraemic patients receiving CAZ-AVI vs. other regimens, showed clinical success more frequently with CAZ-AVI in terms of higher corresponding rates and mortality (190). In addition, a promising role for CAZ-AVI was elucidated from a retrospective analysis of the compassionate use program of CAZ-AVI from Italy; among patients with CRKP bacteraemic infections those treated with CAZ-AVI had significantly lower mortality compared with patients receiving other regimes (192). Furthermore, in multivariable analysis of 208 patients with CRKP BSI, receipt of CAZ-AVI was the only independent predictor of survival, whereas septic shock, neutropenia, Charlson score index >3. and recent mechanical ventilation were independent predictors of mortality (192). Patient's severity of disease, as displayed by the INCREMENT CPE score > 7 (193) was demonstrated as risk factor of mortality in another study, whereas pneumonia and renal replacement therapy have been elucidated as factors associated with treatment failure of CAZ-AVI treatment (190). Finally, the results of a recently published meta-analysis encompassing 12 studies and 4,951 patients, further support that CAZ-AVI is as effective as carbapenems with equivalent safety. In particular, significantly increased cure rates were achieved with CAZ-AVI in infections from resistant causative microorganisms (RR = 1.61; 95% CI, 1.13–2.29); reduced mortality was also reported (RR = 0.29; 95% CI, 0.13–0.63). Similar results were reported for cUTIs and BSI (195).
Summarizing, CAZ-AVI stands out as one of the most important additions in our armamentarium, as the first marketed fixed combination with activity against KPC and OXA producers. Post market reports, reflecting real-life use are very encouraging, in terms of safety, clinical response, and survival. Currently, we do not have enough evidence to relay on monotherapy with CAZ-AVI, since previous reports on CRE infections from observational studies consistently favored combination treatments (26, 57). Expert-driven recommendations issued by relevant Italian and Greek ID Societies, support combination treatment (with an aminoglycoside including plazomicin, or fosfomycin, tigecycline, colistin). However, in non- life-threatening infections, monotherapy could be considered as part of a definitive treatment (8). The prerequisite conditions for monotherapy are illustrated in Figure 1. A proposed algorithm for the optimization of treatments of CRE is shown in Figure 2. Companion drugs of CAZ-AVI can be selected based on the antibiogram and the required PK/PD parameters in the infectious focus (8). The broad spectrum covering also ESBL-producing Enterobacteriaceae and significant proportions of P. aeruginosa, makes CAZ-AVI a strong component of empiric regimens in patients with risk factors for MDR infections. When designing an adequate empiric regimen, the probability of MBL-producing Enterobacteriaceae or A. baumannii should be balanced according to local epidemiological data and covered with a second antibiotic (an aminoglycoside, colistin, tigecycline, or fosfomycin). We believe that empiric use of CAZ-AVI should be reserved for patients with strong risk factors for infections by KPC- or OXA-48- producers and audited by antibiotic stewardship teams to avoid irrational use of this antibiotic combination (Figure 2).
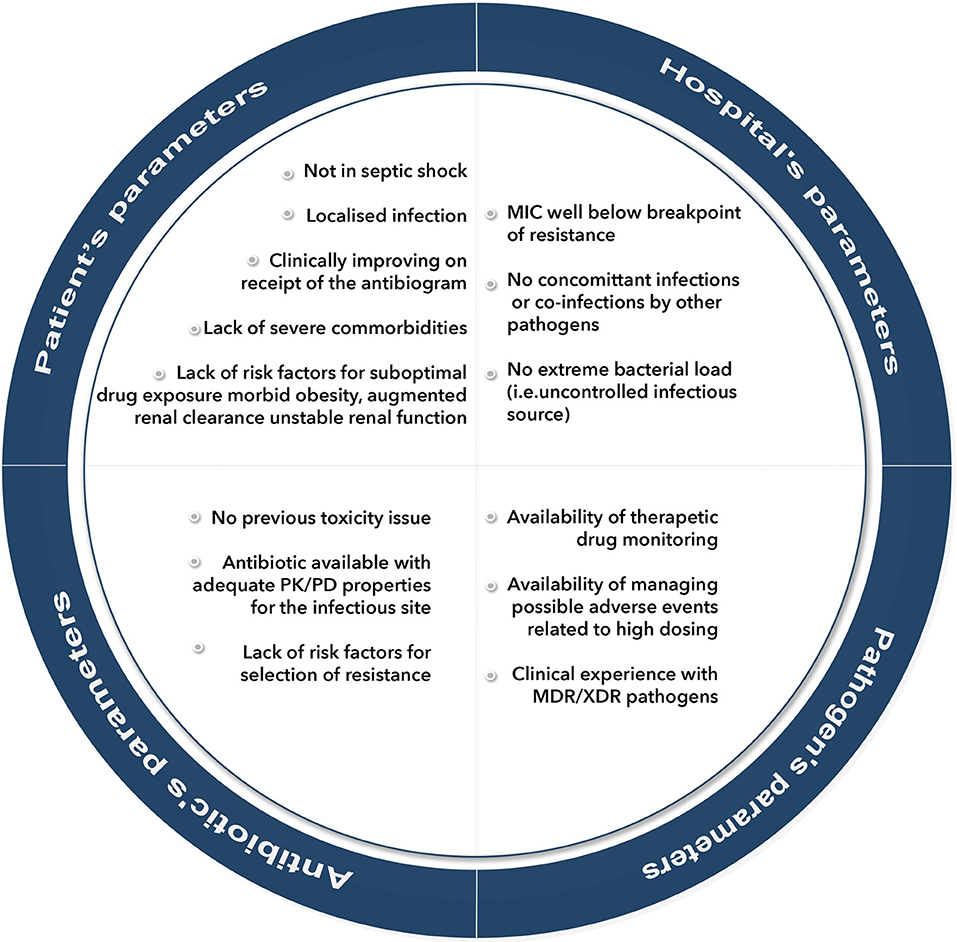
Figure 1. Prerequisite conditions for selecting monotherapy as definitive treatment of infections by extensively-drug-resistant (XDR) pathogens. PK/PD, pharmacokinetic/pharmacodynamic; MDR, multi-drug -resistant; MIC, minimum inhibitory concentration; XDR, extensively-drug-resistant.

Figure 2. How to optimize treatment of Carbapenem-Resistant Enterobacteriaceae (CRE). CAZ-AVI, ceftazidime avibactam; CRE, carbapenem-resistant Enterobacteriaceae; MER/VAB, meropenem vaborbactam; MIC, minimum inhibitory concentration. ≠ OXA-48 is permissive only for CAZ-AVI. *Components of the combination will be based on: (i) epidemiology data (for empirical regimen); (ii) pharmacokinetic/pharmacodynamic considerations relating to the source of infection; (iii) lower MIC (if possible, avoidance of antibiotics with borderline susceptibility). ∧ Selection of CAZ-AVI or MER/VAB in definitive treatment precludes demonstrated in vitro susceptibility and absence of detected metallo-beta lactamase mechanism of resistance; for MER/VAB absence of OXA as well. Δ Higher MICs against meropenem (up to 64 mg/L) may require higher doses and therapeutic drug monitoring.
Meropenem-vaborbactam
Varobactam, previously referred as RPX7009, is a boronic acid, non-β-lactam β-lactamase inhibitor with no antibacterial activity. The addition of varobactam to meropenem, restored the activity of meropenem against KPC-producing strains due to its high affinity for serine proteases. Nevertheless, vaborbactam possesses minimal or no activity against carbapenemases of Amber class B (metallo-β-lactamases/MBLs) (196–198). More specifically, vaborbactam inhibits various class A carbapenemases (KPC-2, KPC-3, KPC-4, BKC-1, FRI-1, and SME-2), class A ESBLs (CTXM, SHV, and TEM), and class C cephalosporinases (CMY, P99), while its activity against class D carbapenemases (OXA- 48-like) is minimal (199, 200). The combination provided no additional in vitro activity against P. aeruginosa, Acinetobacter spp. and Stenotrophomonas maltophilia compared to meropenem alone (199).
Meropenem/vaborbactam became the first available carbapenem/ β-lactamase inhibitor for clinical use. It has shown an excellent tolerability profile and no safety concerns, with main adverse events headache, diarrhea and catheter site complications, while mild lethargy was observed in the highest-dose group (201, 202). No seizures were reported from clinical trials, however, concomitant administration of valproic acid, and other drugs medications that may compete with meropenem for active tubular secretion, such as probenecid, is not advisable (117).
In a study that evaluated the activity of meropenem/varobactam against Gram-negative strains, including MDR and XDR Enterobacteriaceae, meropenem-vaborbactam at concentration ≤2 mg/L inhibited 133 of the 135 KPC-producing isolates, while all isolates were inhibited at ≤8 mg/L (199).
In a similar study, meropenem/vaborbactam inhibited 99.0% of KPC-positive isolates of Enterobacteriaceae at ≤4 mg/L and compared to CAZ-AVI and tigecycline had equivalent—if no superior—in vitro activity (meropenem-vaborbactam at MIC90: 1 mg/L, was four times more potent than CAZ-AVI and at least 64-fold greater than meropenem alone) (203). In another in vitro study of CRE, MICs of meropenem/vaborbactam were elevated for isolates producing MBLs (MIC, 16->64 mg/L); isolates with decreased expression of porin OmpK37 or hyperexpression of the AcrAB-TolC efflux system had also increased MICs(16 mg/L) (198). Interestingly, in vitro studies showed similar MIC distributions of meropenem/vaborbactam for isolates producing KPC-2 and KPC-3 (203).
Meropenem/varobactam obtained FDA approval for cUTIs including pyelonephritis (117), after completion of a phase 3 study (TANGO I) demonstrating non-inferiority compared to piperacillin-tazobactam in cUTIs, including pyelonephritis (204). The currently tested dosage, approved for clinical use is 2gr−2gr every 8 h and is mainly excreted in urine (202). Another multicentre phase 3 rial (TANGO II), compared meropenem/varobactam to best available treatment (mono/combination therapy with polymyxins, carbapenems, aminoglycosides, tigecycline; or ceftazidime-avibactam alone) in infections caused by CRE—including HAP, VAP, cIAIs, cUTIs, and bacteraemia (205). The study enrolled 77 patients, 47 of whom fulfilled criteria for primary analysis. The study showed increased clinical cure rates 65.6 (21/32) and 33.3% (5/15) [95% confidence interval (CI) of difference, 3.3% to 61.3%; P = 0.03)] at End of Treatment and decreased mortality 15.6 (5/32) and 33.3% (5/15) (95% CI of difference,−44.7 to 9.3%). Equally important is the reduced nephrotoxicity in the arm of meropenem/varbobactam. Exploratory risk-benefit analyses of composite clinical failure or nephrotoxicity favored meropenem-vaborbactam vs. best available treatment (31.3 [10/32] vs. 80.0% [12/15]; 95% CI of difference, −74.6 to −22.9%; P < 0.001) (205). Based on these additional studies, in November 2018 EMA approved meropenem-varbobactam for use in “adult patients with CIAI, cUTI, HAP, VAP, bacteraemia that occurs in association with any of these infections, and infections due to aerobic gram-negative organisms where treatment options are limited” (206). TANGO III trial assessing meropenem-vaborbactam vs. piperacillin/tazobactam in patients with HAP and VAP, estimated to finish in 2020 was withdrawn (207).
Scarce published data exists on real-life experience with meropenem/vaborbactam, which is extremely important to define its real position in clinical practice. Among its major advantages is the potent in vitro activity against KPC-producers and the low potential for resistance development. It represents an important player in the battle against CRE mediated by KPC production.
Plazomicin
Plazomicin is a next-generation semisynthetic aminoglycoside. As all aminoglycosides, it acts by inhibiting bacterial protein synthesis. Plazomicin is active against MDR Enterobacteriaceae, due to its stability against strains that express aminoglycoside-modifying enzymes (208). However, it is vulnerable to ribosomal ribonucleic acid (rRNA) methyltransferase enzymes which were already identified in Enterobacteriaceae, P. aeruginosa, and A. baumannii, particularly among Enterobacteriaceae harboring NDM-1 carbapenemases. These enzymes confer broad-spectrum resistance to all aminoglycosides, including plazomicin; their identification before plazomicin's introduction into clinical practice is of concern (209–212). A major advantage of plazomicin is its dose-dependent bactericidal activity. Its antibacterial spectrum includes Gram-negative bacteria, such as a wide range of Enterobacteriaceae (including CRE, ESBL, and MDR isolates) irrespective of resistance to currently available aminoglycosides (209, 211, 213). Collections of clinically important KPC-producers with resistance to aminoglycosides were inhibited by plazomicin displaying an MIC90 of ≤2 or 4 mg/L (210, 213). In a recent in vitro study of 300 CRE K. pneumoniae isolates, susceptibility to plazomicin was 87.0% (≤2 mg/L) with MIC50/MIC90 of 0.5/4 mg/L; rRNA methyltransferases (mostly rmtB) were found in an alarming 8% of isolates, never exposed to plazomicin (40).
Although plazomicin is less potent in vitro against non-fermenters compared to Enterobacteriaceae, OXA-producing A. baumannii resistant to other aminoglycosides may be susceptible to plazomicin (211, 212). Tested against 82 isolates of CRE, plazomicin showed 79% in vitro susceptibility with MICs ≤2 mg/L, including isolates producing metallo-β-lactamases type VIM or IMP but not NDM-1, due to the co-production of ribosomal methyltransferases (209). Anti-MRSA activity offers an extra advantage when plazomicin is part of empirical regimens in severely ill patients in settings with MRSA prevalence, despite resistance to previous generation aminoglycosides (211). Plazomicin demonstrated in vitro activity with MIC90 of ≤4mg/L against isolates of polymyxin-resistant Enterobacteriaceae, including mcr-1 producing isolates (214).
Plazomicin displays a linear and dose-proportional pharmacokinetic profile, with an elimination half time (t1/2) of 4 h and lack of accumulation indicative of once daily therapy. Therapeutic dosage of plazomicin was set at 15 mg/kg (118, 215).
EPIC (Evaluating Plazomicin in cUTI), was the first phase 3 registration trial (NCT02486627), in which plazomicin compared to meropenem in cUTIs met the non-inferiority endpoint (216). The second phase 3, CARE descriptive trial (ClinicalTrials.gov Identifier NCT01970371), published in January 2019, compared plazomicin vs. colistin as part of a definitive combination regimen in serious infections due to CRE excluding NDM-producers (217). CARE enrolled 39 patients with a variety of infections including 29 bloodstream infections (BSI), and 8 HAP/VAP; the study was prematurely terminated due to low enrolment. Patients received plazomicin 15 mg/kg every 24 h or colistin, in both arms combined with a second agent (tigecycline or meropenem). Using a composite endpoint, patients who received plazomicin had fewer primary end-point events [4 of 17 patients (24%) compared to 10 of 20 patients (50%) who received colistin (difference, −26 percentage points; 95% confidence interval [CI], −55 to 6). The same applied for bloodstream infections but not for VAP/HAP, probably due to the low number of patients with the latter infection. Less deaths were observed at day 14 among patients who received plazomicin-based treatment, and this trend continued through day 60. The group of plazomicin had fewer adverse events and fewer patients with clinically significant increase in serum creatinine at any point of the trial, demonstrating a safer profile against the comparator colistin (217–219).
Plazomicin was granted FDA approval in June 2018 for patients 18 years of age or older with the indication of cUTIs, including pyelonephritis caused by the following susceptible microorganism(s): Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, and Enterobacter cloacae. In the drug's SPC it is advised to be reserved as salvage approach for cUTI patients who have limited or no alternative treatment options. Potential nephrotoxicity, ototoxicity, neuromuscular blockade, and fetal harm have been included in a boxed warning despite scarce data from clinical trials (119, 219). EMA approval is pending.
In summary, plazomicin a novel aminoglycoside with minor toxicity issues and a challenging antimicrobial spectrum holds promise in the battle against difficult to treat organisms. Its favorable lung penetration make it a candidate for treatment regimens of VAP, particularly as part of empiric regimens or as definitive combination treatment where monotherapy is not advised. Conceptually, aminoglycosides are not used as monotherapy; in that sense, plazomicin could be a perfect companion to the new beta-lactam-beta-lactamase inhibitors. As part of an initial empiric regimen, depending on local epidemiological data it could replace colistin in pulmonary infections due to the poor pharmacokinetics of the latter in the lung, given as companion to a tailored beta-lactam backbone (Figures 2, 3). More data on combinations and particularly on monotherapy need to be compiled. Aminoglycosides hold a pivotal role in the treatment of UTIs, with the potential of monotherapy; plazomicin, with demonstrated low rates of renal toxicity could be an option for a targeted monotherapy in XDR pathogens causing UTIs. Such an approach would spare other advanced antimicrobials.
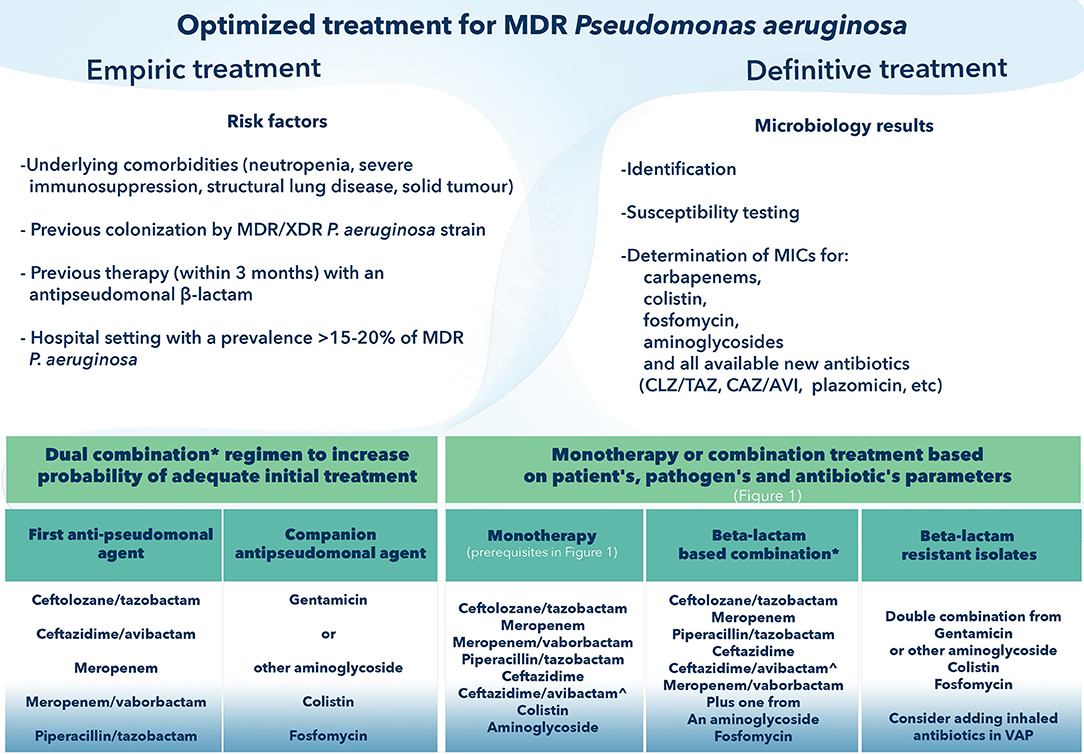
Figure 3. How to optimize treatment of Multi-drug-resistant Pseudomonas aeruginosa. CAZ-AVI, ceftazidime avibactam; CLZ/TAZ, ceftolozane tazobactam; MDR, Multi-drug resistant; MIC, minimum inhibitory concentration; XDR, extensively drug-resistant; VAP, ventilator-associated pneumonia. *Components of the combination will be based on: (i) epidemiology data (for empirical regimen); (ii) pharmacokinetic/pharmacodynamic considerations relating to the source of infection; (iii) lower MIC (if possible, avoidance of antibiotics with borderline susceptibility). ∧ Selection of CAZ-AVI in definitive treatment precludes demonstrated in vitro susceptibility and absence of detected metallo-beta lactamase mechanism of resistance.
Eravacycline
Eravacycline is a synthetic fluorocycline, with similarities to tigecycline in mechanism of action, structure and antibacterial spectrum, including: Gram-negative bacilli (P. aeruginosa- excluded), regardless of resistance to other antibiotic classes (ESBL and KPC- producing Enterobacteriaceae, MDR A. baumannii) and Gram-positives (staphylococci, including methicillin-resistant Staphylococcus aureus/MRSA; enterococci, including Vancomycin-resistant Enterococcus/VRE) (220, 221). Potential advantages over tigecycline feature a more potent in vitro activity for both Gram-positive cocci (2- to 4 fold) and Gram-negative bacilli (2- to 8 fold) (220, 222), an excellent oral bioavailability, a lower potential for drug interactions and a superior activity in biofilm (220).
PK/PD studies support once daily administration, whereas higher serum concentrations and better tolerability are some more advantages compared to its ancestor (220, 221, 223). In a phase 1 study of healthy volunteers, eravacycline achieved in the ELF concentrations greater than plasma by 6-fold and in the alveolar macrophages by 50-fold, indicating the potential for treatment of MDR bacteria causing pneumonia (224).
In vitro studies of eravacycline against E. coli resistant to 3rd generation cephalosporines and fluoroquinolones showed a potent in vitro activity with an MIC90 of 0.5 mg/L (221, 222). A recent in vitro study compared eravacycline and commonly used antibiotics against a collection of 284 CRAB isolates possessing an acquired OXA or a metallo-beta-lactamase, or expressing an up-regulated intrinsic OXA-51-like enzyme (225). Eravacycline was demonstrated as the most potent in vitro agent vs. all comparators (reference beta- lactams, aminoglycosides, colistin, tetracyclines, sulbactam and fluoroquinolones) displaying the lowest MIC50/90 values of 0.5/1 mg/L, respectively. Colistin resistance in this collection was 13.3% with relevant MIC50/90 values of 1/4 mg/L, respectively. No correlation was found between blaOXA genes and eravacycline MICs, indicating an excellent candidate in the treatment of CRAB infections.
IGNITE1 and IGNITE4 (Investigating Gram-negative Infections Treated with Eravacycline) were randomized, double-blind, double-dummy, multi-center Phase 3 clinical trials in cIAIs, in which eravacycline administered at a dose of 1 mg/kg IV q12h was compared to ertapenem (1 g IV every 24 h) and meropenem (1 g IV every 8 h), respectively. In both studies non-inferiority criteria were met (226, 227); adverse events were low for a drug of this class, with nausea, vomiting, and diarrhea reported at 5, 4, and 3%, respectively (227). Based on these data eravacycline was approved for cIAIs by both FDA and EMA in 2018 (120, 121).
IGNITE 2 and 3 were phase 3 randomized, multi-center, double-blind, clinical trials in cUTIs, in which eravacycline administered at a dose of 1.5 mg/kg IV once daily was compared to levofloxacin (750 mg every 24 h) and ertapenem (1 g every 24 h), respectively. Non-inferiority criteria were not met and UTI indication was withdrawn (122). Although failure may be attributed to the lower dose employed in cUTIs compared to the cIAI trial, there are additional concerns about clinical efficacy of tetracycline class in UTIs.
In light of the available in vitro and in vivo data, eravacycline holds promise as a key player in the treatment of infections caused by A. baumannii being the first launched antibiotic with potent activity against this notorious pathogen since decades. Real life data will determine its real impact on patients severely ill and with multiple co-morbidities. Keeping in mind the paradigm of tigecycline for which data from healthy volunteers did not translate successfully in critically ill patients, and the failure of eravacycline in pivotal RCTs in UTIs, caution is required. When this new drug will enter clinical practice, surveillance for resistance and PK/PD studies in special populations (i. e. mechanically ventilated patients or on CRRT), are of utmost importance to ensure optimal patient outcomes (70, 71).
How to Integrate New and Old Antibiotics in Clinical Practice
In this comprehensive review we tried to appraise currently available treatments for MDR and XDR pathogens. Our ultimate aim was to provide hints on how to integrate recently marketed antibiotics in the challenges of clinical practice, particularly referring to CRE, MDR P. aeruginosa and CRAB. It is of great importance to emphasize that the novel beta-lactam/beta-lactamase inhibitor combinations, such as those containing avibactam and vaborbactam, are promising to be the best options for the treatment of CRE where carbapenem resistance is mediated by KPC. In vitro all these inhibitors are active against Enterobacteriaceae producing ESBL, KPC-2, KPC-3, and AmpC, whereas only avibactam inhibits certain class D β-lactamases, mainly OXA-48. Although they have been subjected to a rigorous program of development, their “real life” efficacy against CRE remains unascertained because clinical trials design were not focused on the treatment of infections caused by XDR and PDR pathogens (115). As far as CAZ-AVI is concerned, an almost 3-years in-market experience has brought in light some important studies comparing its efficacy vs. colistin. The first study employing a prospective design, highlighted the probability of better outcome of 64% compared to colistin (193), whereas a second retrospective study demonstrated CAZ-AVI as the only variable associated with survival (192). These data indicate that CAZ-AVI may represent the most appropriate so far choice of CRE mediated by KPC or OXA-48. Further to the activity against OXA enzymes, CAZ-AVI's important antipseudomonal activity is an advantage for empirical regimens in high risk patients and in mixt infections. The addition of aztreonam, still marketed individually, could expand CAZ-AVI's spectrum to MBL-producers (228, 229). On the other hand, meropenem-vaborbactam seems to have a lesser potential for selection of resistance among KPC-producers than CAZ-AVI (230, 231), while demonstrating similar efficacy against KPC-2 and KPC-3 (203). In addition, meropenem possesses excellent anti-anaerobe coverage and can stand alone in the treatment of intraabdominal infections without the addition of metronidazole. Fortunately, so far, no cross resistance is reported between these two antibiotic combinations (203). The compilation of real-life data for meropenem-vaborbactam will enable us to understand better its true position in the treatment of CRE infections.
The disadvantage to be seriously considered by the clinician is the inability of both CAZ-AVI and meropenem-vaborbactam to inhibit metallo-β-lactamases (MBLs) i.e., VIM, IMP, and NDM, as well as A. baumannii (115). The knowledge of the epidemiology of CRE infections in each country is of major importance. OXA-48 like enzymes are more prevalent in France, Spain and Belgium, whereas NDM-1 is most frequently isolated in Southeast Asia countries and India, whereas regional or inter-regional spread of NDM in Europe has been reported in Belgium, Denmark, France, Romania, Poland, Turkey, and Greece (11). In terms of empiric therapy of CRE infections, taking into consideration the epidemiological profile of each region, agents active against both serine β-lactamases and MBLs are indicated. The predominance of KPC could indicate the administration of either CAZ-AVI or meropenem-vaborbactam. However, in case of the emergence of MBLs the combination with colistin, should be considered to cover both possibilities. Figure 2 provides an algorithm for optimal integration of new and old antibiotics in the treatment of CRE.
It is evident that combination empiric regimens are mandatory in areas with MDR prevalence. The big question, is whether new antibiotics against CRE would abide the need of combination in definitive treatments, for which data from observational studies are clearly in favor, being contradicted by data from meta-analyses (8, 25, 26, 45, 232, 233). Factors elucidated as independent predictors of mortality or treatment failure of CAZ-AVI treatment are septic shock, neutropenia, high comorbidity scores, recent ventilation, pneumonia, and receipt of renal replacement therapy outlining a critical role for combination with another active in vitro antibiotic in these cases (190, 192, 193). However, the latter position is arbitrary and based mostly on experience before the new beta-lactamase combinations requiring targeted and prospective randomized trials. Figure 1 summarizes conditions that must be met in order to decide monotherapy in the treatment of MDR pathogens, based on the existing literature and expert driven recommendations (8, 26, 57, 58, 95, 234).
When P. aeruginosa is considered, combination treatment is the rule in the empirical phase (58). In definite treatment, data from meta-analyses found no benefit for combination treatments, however clinicians hardly accept monotherapy in patients with major risk factors for mortality, in bacteraemic infections and septic shock (233, 234). The launching of ceftolozane-tazobactam expands beta-lactam activity in CR strains, providing a potent backbone; colistin, fosfomycin and plazomicin could act as companion agents (Figure 3). CAZ-AVI as well may contribute as a backbone antipseudomonal agent in the new era (161).
When CRAB is prevalent, colistin or tigecycline are the only currently available treatments (69). Launching of eravacycline holds promise, however real-life data from CRAB infections and critically ill patients are lacking. Other newer agents against CRAB i.e., cefiderocol are welcomed and high clinical cure rates are expected (7, 9). Regarding CRAB resistant to colistin, one unanticipated finding from the exploratory subgroup analysis of the AIDA study revealed lower mortality among patients with colistin-resistant isolates when treated with colistin monotherapy over combination of colistin -meropenem, with a possible explanation being the loss of fitness and virulence relative to colistin-susceptible strains (232). Indeed, cumulative data have shown that combination treatments against A. baumanni might be less necessary compared to CRE, to ensure optimal outcomes (233, 234).
In summary, the novel combinations are not to be considered “panacea” for the ongoing crisis in the therapy of XDR Gram-negative bacteria and colistin is still considered as possessing a fundamental position for the treatment of CRE in combination (particularly in areas where MBL predominate) as well as for the treatment of CRPA (in many cases being the only in vitro active drug) as well as CRAB. CAZ-AVI and meropenem-vaborbactam can be used as backbones in the treatment of CRE and under circumstances, they could be used as definitive monotherapy. Similarly, ceftolozane-tazobactam could be seen as an ideal beta-lactam backbone for the treatment of MDR P. aeruginosa, as well as a stand-alone antibiotic, when monotherapy is sought and conditions fulfilled. Plazomicin could absorb some volume of colistin prescriptions, both in empiric and definitive treatment of difficult-to-treat pathogens, subject to local epidemiological evidence. Finally, fosfomycin as a companion antibiotic for P. aeruginosa and CRE infections, deserves a greater attention, as it has no cross resistance with other antibiotic classes, may retain susceptibility against a variety of resistance mechanisms to other antibiotics and possesses negligible toxicity. Lessons learnt from the pauci-antibiotic era, make vigilance for emergence of resistance a priority. Furthermore, it is important to minimize use of these new precious antibiotics as empiric treatments and focus on microbiologic documentation of infections; this will ensure their longevity in our armamentarium.
Author Contributions
GP: conception, writing, and supervising of the manuscript. IK, SL, KP, and VR: literature review, writing parts of the manuscript. All authors have approved the final version.
Conflict of Interest Statement
GP has received speaker's honoraria and/or grants from Angelini, MSD, Pfizer. KP has received speaker's honoraria from MSD and Pfizer.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Giamarellou H, Poulakou G. Multidrug-resistant Gram-negative infections: what are the treatment options? Drugs. (2009) 69:1879–901. doi: 10.2165/11315690-000000000-00000
2. Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, et al. Multidrug-resistant, extensively drug-resistant and pan drug-resistant bacteria: an inter-national expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. (2012) 18:268–81. doi: 10.1111/j.1469-0691.2011.03570.x
3. Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. (2013) 13:785–96. doi: 10.1016/S1473-3099(13)70190-7
4. World Health Organization (WHO). Antimicrobial Resistance: No Action Today, No Cure Tomorrow. Geneva, Switzerland: WHO Press (2011). Available online at: https://www.who.int/world-health-day/2011/en (accessed February 19, 2019)
5. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. (2009) 48:1–12. doi: 10.1086/595011
6. James O. Neill, review on antimicrobial resistance, tackling drug-resistant infections globally (2015). Available online at: https://amr-review.org/sites/default/files/_0.pdf (accessed February 20, 2019).
7. Poulakou G, Lagou S, Karageorgopoulos DE, Dimopoulos G. New treatments of multidrug-resistant Gram-negative ventilator-associated pneumonia. Ann Transl Med. (2018) 6:423. doi: 10.21037/atm.2018.10.29
8. Bassetti M, Giacobbe DR, Giamarellou H, Viscoli C, Daikos GL, Dimopoulos G, et al. Critically Ill patients study group of the european society of clinical microbiology and infectious disease (ESCMID); Hellenic Society of Chemotherapy (HSC) and Società Italiana di Terapia Antinfettiva (SITA). Management of KPC-producing Klebsiella pneumoniae infections. Clin Microbiol Infect. (2018) 24:133–44. doi: 10.1016/j.cmi.2017.08.030
9. Bassetti M, Poulakou G, Ruppe E, Bouza E, Van Hal SJ, Brink A. Antimicrobial resistance in the next 30 years, humankind, bugs and drugs:a visionary approach. Intensive Care Med. (2017) 43:1464–75. doi: 10.1007/s00134-017-4878-x
10. Grundmann H, Glasner C, Albiger B, Aanensen DM, Tomlinson CT, Andrasević AT, et al. European survey of carbapenemase-producing Enterobacteriaceae (EuSCAPE) working group. Occurrence of carbapenemase-producing Klebsiella pneumoniae and Escherichia coli in the European survey of carbapenemase-producing Enterobacteriaceae (EuSCAPE): a prospective, multinational study. Lancet Infect Dis. (2017) 17:153–63. doi: 10.1016/S1473-3099(16)30257-2
11. Miriagou V, Cornaglia G, Edelstein M, Galani I, Giske CG, Gniadkowski M, et al. Acquired carbapenemases in Gram-negative bacterial pathogens: detection and surveillance issues. Clin Microbiol Infect. (2010) 16:112–122. doi: 10.1111/j.1469-0691.2009.03116.x
12. Queenan AM, Bush K. Carbapenemases: the versatile beta-lacta-mases. Clin Microbiol Rev. (2007) 20:440–58. doi: 10.1128/CMR.00001-07
13. Ruppé É, Woerther PL, Barbier F. Mechanisms of antimicrobial resistance in Gram-negative bacilli. Ann Intensive Care. (2015) 5:61. doi: 10.1186/s13613-015-0061-0
14. Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. (2016) 16:161–8. doi: 10.1016/S1473-3099(15)00424-7
15. Karaiskos I, Souli M, Galani I, Giamarellou H. Colistin: still a lifesaver for the 21st century? Expert Opin Drug Metab Toxicol. (2017) 13:59–71. doi: 10.1080/17425255.2017.1230200
16. Kazmierczak KM, de Jonge BLM, Stone GG, Sahm DF. In vitro activity of ceftazidime/avibactam against isolates of Enterobacteriaceae collected in European countries: INFORM global surveillance 2012-15. J Antimicrob Chemother. (2018) 73:2782–8. doi: 10.1093/jac/dky266
17. Kazmierczak KM, de Jonge BLM, Stone GG, Sahm DF. In vitro activity of ceftazidime/avibactam against isolates of Pseudomonas aeruginosa collected in European countries: INFORM global surveillance 2012–15. J Antimicrob Chemother. (2018) 73:2777–81. doi: 10.1093/jac/dky267
18. Plachouras D, Karvanen M, Friberg LE, Papadomichelakis E, Antoniadou A, Tsangaris I, et al. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by gram-negative bacteria. Antimicrob Agents Chemother. (2009) 53:3430–6. doi: 10.1128/AAC.01361-08
19. Karaiskos I, Friberg LE, Pontikis K, Ioannidis K, Tsagkari V, Galani L, et al. Colistin population pharmacokinetics after application of a loading dose of 9 MU colistin methanesulfonate in critically Ill patients. Antimicrob Agents Chemother. (2015) 59:7240–48. doi: 10.1128/AAC.00554-15
20. Nation RL, Garonzik SM, Thamlikitkul V, Giamarellos-Bourboulis EJ, Forrest A, Paterson DL, et al. Dosing guidance for intravenous colistin in critically-ill patients. Clin Infect Dis. (2017) 64:565–71. doi: 10.1093/cid/ciw839
21. Dalfino L, Puntillo F, Ondok MJ, Mosca A, Monno R, Coppolecchia S, et al. Colistin-associated acute kidney injury in severely ill patients: a step toward a better renal care? a prospective cohort study. Clin Infect Dis. (2015) 61:1771–7. doi: 10.1093/cid/civ717
22. Nation RL, Garonzik SM, Li J, Thamlikitkul V, Giamarellos-Bourboulis EJ, Paterson DL, et al. Updated US and european dose recommendations for intravenous colistin: how do they perform? Clin Infect Dis. (2016) 62:552–8. doi: 10.1093/cid/civ964
23. Karaiskos I, Antoniadou A, Giamarellou H. Combination therapy for extensively-drug resistant gram-negative bacteria. Expert Rev Anti Infect Ther. (2017) 15:1123–40. doi: 10.1080/14787210.2017.1410434
24. Tsuji BT, Pogue JM, Zavascki AP, Paul M, Daikos GL, Forrest A, et al. International consensus guidelines for the optimal use of the polymyxins: endorsed by the American College of Clinical Pharmacy (ACCP), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Infectious Diseases Society of America (IDSA), International Society for Anti-infective Pharmacology (ISAP), Society of Critical Care Medicine (SCCM), and Society of Infectious Diseases Pharmacists (SIDP). Pharmacotherapy. (2019) 39:10–39. doi: 10.1002/phar.2209
25. Daikos GL, Tsaousi S, Tzouvelekis LS, Anyfantis I, Psichogiou M, Argyropoulou A, et al. Carbapenemase-producing Klebsiella pneumoniae bloodstream infections: lowering mortality by antibiotic combination schemes and the role of carbapenems. Antimicrob Agents Chemother. (2014) 58:2322–8. doi: 10.1128/AAC.02166-13
26. Tumbarello M, Trecarichi EM, De Rosa FG, Giannella M, Giacobbe DR, Bassetti M, et al. Infections caused by KPC-producing Klebsiella pneumoniae:differences in therapy and mortality in a multicentre study. J Antimicrob Chemother. (2015) 70:2133–43. doi: 10.1093/jac/dkv086
27. Durante-Mangoni E, Signoriello G, Andini R, Mattei A, De Cristoforo M, Murino P, et al. Colistin and rifampicin compared with colistin alone for the treatment of serious infections due to extensively drug-resistant Acinetobacter baumannii: a multicenter, randomized clinical trial. Clin Infect Dis. (2013) 57:349–58. doi: 10.1093/cid/cit253
28. Sirijatuphat R, Thamlikitkul V. Preliminary study of colistin versus colistin plus fosfomycin for treatment of carbapenem-resistant Acinetobacter baumannii infections. Antimicrob Agents Chemother. (2014) 58:5598–601. doi: 10.1128/AAC.02435-13
29. Paul M, Daikos GL, Durante-Mangoni E, Yahav D, Carmeli Y, Benattar YD, et al. Colistin alone versus colistin plus meropenem for treatment of severe infections caused by carbapenem-resistant Gram-negative bacteria: an open-label, randomised controlled trial. Lancet Infect Dis. (2018) 18:391–400. doi: 10.1016/S1473-3099(18)30099-9
30. Dinh A, Saliomon J, Bru JP, Bernard L. Fosfomycin: efficacy against infections caused by multidrug-resistant bacteria. Scand J Infect Dis. (2012) 44:182–9. doi: 10.3109/00365548.2011.616221
31. Pontikis K, Karaiskos I, Bastani S, Dimopoulos G, Kalogirou M, Katsiari M, et al. Outcomes of critically ill intensive care unit patients treated with fosfomycin for infections due to pandrug-resistant and extensively drug-resistant carbapenemase-producing Gram-negative bacteria. Int J Antimicrob Agents. (2014) 43:52–9. doi: 10.1016/j.ijantimicag.2013.09.010
32. Albur MS, Noel A, Bowker K, MacGowan A. The combination of colistin and fosfomycin is synergistic against NDM-1-producing Enterobacteriaceae in In vitro pharmacokinetic/pharmacodynamic model experiments. Int J Antimicrob Agents. (2015) 46:560–7. doi: 10.1016/j.ijantimicag.2015.07.019
33. Matzi V, Lindenmann J, Porubsky C, Kugler SA, Maier A, Dittrich P, et al. Extracellular concentrations of fosfomycin in lung tissue of septic patients. J Antimicrob Chemother. (2010) 65:995–8. doi: 10.1093/jac/dkq070
34. Pfausler B, Spiss H, Dittrich P, Zeitlinger M, Schmutzhard E, Joukhadar C. Concentrations of fosfomycin in the cerebrospinal fluid of neurointensive care patients with ventriculostomy-associated ventriculitis. J Antimicrob Chemother. (2004) 53:848–52. doi: 10.1093/jac/dkh158
35. Sauermann R, Karch R, Langenberger H, Kettembach J, Mayer-Helm B, Petsch M, et al. Antibiotic abscess penetration: fosfomycin levels measured in pus and simulated concentration-time profiles. Antimicrob Agents Chemother. (2005) 49:4448–54. doi: 10.1128/AAC.49.11.4448-4454.2005
36. Florent A, Chichmanian R-M, Cua E, Pulcini C. Adverse events associated with intravenous fosfomycin. Int J Antimicrob Agents. (2011) 37:82–3. doi: 10.1016/j.ijantimicag.2010.09.002
37. Grabein B, Graninger W, Rodríguez Baño J, Dinh A, Liesenfeld DB. Intravenous fosfomycin – Back to the future. Systematic review and meta-analysis of the clinical literature. Clin Microbiol Infect. (2017) 23:363–72. doi: 10.1016/j.cmi.2016.12.005
38. Vardakas KZ, Legakis NJ, Triarides N, Falagas ME. Susceptibility of contemporary isolates to fosfomycin: a systematic review of the literature. Int J Antimicrob Agents. (2016) 47:269–85. doi: 10.1016/j.ijantimicag.2016.02.001
39. Díez-Aguilar M, Morosini MI, Del Campo R, García-Castillo M, Zamora J, Cantóna R. In vitro activity of fosfomycin against a collection of clinical Pseudomonas aeruginosa isolates from 16 spanish hospitals: establishing the validity of standard broth microdilution as susceptibility testing method. Antimicrob Agents Chemother. (2013) 57:5701–3. doi: 10.1128/AAC.00589-13
40. Galani I, Karaiskos I, Karantani I, Papoutsaki V, Maraki S, Papaioannou V, et al. Epidemiology and resistance phenotypes of carbapenemase-producing Klebsiella pneumoniae in Greece, 2014–2016. Eurosurveillance. (2018) 23:pii=1700775. doi: 10.2807/1560-7917.ES.2018.23.30.1700775
41. Flamm RK, Rhomberg PR, Watters AA, Sweeney K, Ellis-Grosse EJ, Shortridge D. Activity of fosfomycin when tested against US contemporary bacterial isolates. Diagn Microbiol Infect Dis. (2019) 93:143–6. doi: 10.1016/j.diagmicrobio.2018.08.010
42. Karageorgopoulos DE, Miriagou V, Tzouvelekis LS, Spyridopoulou K, Daikos GL. Emergence of resistance to fosfomycin used as adjunct therapy in KPC Klebsiella pneumoniae bacteraemia: report of three cases. J Antimicrob Chemother. (2012) 67:2777–9. doi: 10.1093/jac/dks270
43. Michalopoulos A, Virtzili S, Rafailidis P, Chalevelakis G, Damala M, Falagas ME. Intravenous fosfomycin for the treatment of nosocomial infections caused by carbapenem-resistant Klebsiella pneumoniae in critically ill patients: a prospective evaluation. Clin Microbiol Infect. (2010) 16:184–6. doi: 10.1111/j.1469-0691.2009.02921.x
44. Ellis-Grosse EJ, Skarinsky D, Das A, Eckburg PB. Phenotypic antibiotic resistance in ZEUS: a multi-center, randomized, double-blind phase 2/3 Study of ZTI-01 versus Piperacillin-Tazobactam (P-T) in the treatment of patients with Complicated Urinary Tract Infections (cUTI) including Acute Pyelonephritis. In: ID Week. San Diego, CA (2017).
45. Gutiérrez-Gutiérrez B, Salamanca E, de Cueto M, Hsueh PR, Viale P, Paño-Pardo JR, et al. Effect of appropriate combination therapy on mortality of patients with bloodstream infections due to carbapenemase-producing Enterobacteriaceae (INCREMENT): a retrospective cohort study. Lancet Infect Dis. (2017) 17:726–734. doi: 10.1016/S1473-3099(17)30228-1
46. European Medicines Agency. List of the Names, Pharmaceutical Forms, Strengths of the Medicinal Products, Routes of Administration, Marketing Authorisation Holders in the Member States for Fosfomycin-Containing Medicinal Products. (2018). Avaliable online at: https://www.ema.europa.eu/documents/referral/fosfo (accessed January 12, 2019).
47. Nabriva Therapeutics. Nabriva Therapeutics Strengthens Antibiotics Leadership Position With Acquisition of Zavante Therapeutics. (2018). Avaliable online at: http://investors.nabriva.com/news-releases/news-release-details/nabriva-therapeutics-strengthens-antibiotics-leadership-position (accessed February 13, 2019).
48. van Mens SP, ten Doesschate T, Kluytmans-van den Bergh MFQ, Mouton JW, Rossen JWA, Verhulst C, et al. Fosfomycin Etest for Enterobacteriaceae: Interobserver and interlaboratory agreement. Int J Antimicrob Agents. (2018) 52:678–81. doi: 10.1016/j.ijantimicag.2018.06.014
49. Camarlinghi G, Parisio EM, Antonelli A, Nardone M, Coppi M, Giani T, et al. Discrepancies in fosfomycin susceptibility testing of KPC-producing Klebsiella pneumoniae with various commercial methods. Diagn Microbiol Infect Dis. (2019) 93:74–6. doi: 10.1016/j.diagmicrobio.2018.07.014
50. Clinical and Laboratory Standards Instiute. Performance Standards for Antimicrobial Susceptibility Testing ; Twenty-Second Informational Supplement. 298. Document M100-S22. Vol 32. Wayne, PA (2012).
51. European Committee on Antimicrobial Susceptibility Testing. EUCAST Clinical MIC Breakpoints. Available online at: www.eucast.org. (accessed February 07, 2019)
52. Fomicyt 40mg/ml powder for solution for infusion. Public assessment report. Mutual recognition Procedure. Medicines and Healthcare products. Regulatory Agents. (2017). 1–19. Available online at: http://www.mhra.gov.uk/home/groups/par/documents/websiteresources/con579887.pdf
53. Kumar A, Roberts D, Wood KE, Light B, ParRillo JE, Sharma S, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. (2006) 34:1589–96. doi: 10.1097/01.CCM.0000217961.75225.E9
54. Papadomichelakis E, Kontopidou F, Antoniadou A, Poulakou G, Koratzanis E, Kopterides P, et al. Screening for resistant gram-negative microorganisms to guide empiric therapy of subsequent infection. Intensive Care Med. (2008) 34:2169–75. doi: 10.1007/s00134-008-1247-9
55. Parker S, Lipman J, Koulenti D, Dimopoulos G, Roberts JA. What is the relevance of fosfomycin pharmacokinetics in the treatment of serious infections in critically ill patients? A systematic review. Int J Antimicrob Agents. (2013) 42:289–93. doi: 10.1016/j.ijantimicag.2013.05.018
56. Parker SL, Frantzeskaki F, Wallis SC, Diakaki C, Giamarellou H, Koulenti D, et al. Population pharmacokinetics of fosfomycin in critically Ill patients. Antimicrob Agents Chemother. (2015) 59:6471–6. doi: 10.1128/AAC.01321-15
57. Daikos GL, Markogiannakis A, Souli M, Tzouvelekis LS. Bloodstream infections caused by carbapenemase-producing Klebsiella pneumoniae: a clinical perspective. Expert Rev Anti Infect Ther. (2012) 10:1393–404. doi: 10.1586/eri.12.138
58. Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the infectious diseases society of america and the american thoracic society. Clin Infect Dis. (2016) 63:575–82. doi: 10.1093/cid/ciw504
59. Zhao M, Bulman ZP, Lenhard JR, Satlin MJ, Kreiswirth BN, Walsh TJ, et al. Pharmacodynamics of colistin and fosfomycin: a “treasure trove” combination combats KPC-producing Klebsiella pneumoniae. J Antimicrob Chemother. (2017) 72:1985–90. doi: 10.1093/jac/dkx070
60. Yu W, Shen P, Bao Z, Zhou K, Zheng B, Ji L, et al. In vitro antibacterial activity of fosfomycin combined with other antimicrobials against KPC-producing Klebsiella pneumoniae. Int J Antimicrob Agents. (2017) 50:237–41. doi: 10.1016/j.ijantimicag.2017.03.011
61. Souli M, Galani I, Boukovalas S, Gourgoulis MG, Chryssouli Z, Kanellakopoulou K, et al. In vitro interactions of antimicrobial combinations with fosfomycin against KPC-2-producing Klebsiella pneumoniae and protection of resistance development. Antimicrob Agents Chemother. (2011) 55:2395–7. doi: 10.1128/AAC.01086-10
62. Samonis G, Maraki S, Karageorgopoulos DE, Vouloumanou EK, Falagas ME. Synergy of fosfomycin with carbapenems, colistin, netilmicin, and tigecycline against multidrug-resistant Klebsiella pneumoniae, Escherichia coli, and Pseudomonas aeruginosa clinical isolates. Eur J Clin Microbiol Infect Dis. (2012) 31:695–701. doi: 10.1007/s10096-011-1360-5
63. Berçot B, Poirel L, Dortet L, Nordmann P. In vitro evaluation of antibiotic synergy for NDM-1-producing Enterobacteriaceae. J Antimicrob Chemother. (2011) 66:2295–7. doi: 10.1093/jac/dkr296
64. Yu W, Luo Q, Shi Q, Huang C, Yu X, Niu T, et al. In vitro antibacterial effect of fosfomycin combination therapy against colistin-resistant Klebsiella pneumoniae. Infect Drug Resist. (2018) 11:577–85. doi: 10.2147/IDR.S160474
65. Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. (1999) 43:1003–12. doi: 10.1128/AAC.43.5.1003
66. Kollef MH, Ricard J-D, Roux D, Francois B, Ischaki E, Rozgonyi Z, et al. A randomized trial of the amikacin fosfomycin inhalation system for the adjunctive therapy of Gram-negative ventilator-associated pneumonia: IASIS trial. Chest. (2017) 151:1239–46. doi: 10.1016/j.chest.2016.11.026
67. PerdigãoNeto LV, Oliveira MS, Martins RCR, Marchi AP, Gaudereto JJ, da Costa LATJ, et al. Fosfomycin in severe infections due to genetically distinct pan-drug-resistant Gram-negative microorganisms: synergy with meropenem. J Antimicrob Chemother. (2019) 74:177–81. doi: 10.1093/jac/dky406
68. Albiero J, Sy SKB, Mazucheli J, Caparroz-Assel SM, Costa BB, Alves JL, et al. Pharmacodynamic evaluation of the potential clinical utility of fosfomycin and meropenem in combination therapy against KPC-2-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. (2016) 60:4128–39. doi: 10.1128/AAC.03099-15
69. Garnacho-Montero J, Dimopoulos G, Poulakou G, Akova M, Cisneros JM, De Waele J, et al. European society of intensive care medicine. Task force on management and prevention of Acinetobacter baumannii infections in the ICU. Intensive Care Med. (2015) 41:2057–75. doi: 10.1007/s00134-015-4079-4
70. Giamarellou H, Poulakou G. Pharmacokinetic and pharmacodynamic evaluation of tigecycline. Expert Opin Drug Metab Toxicol. (2011) 7:1459–70. doi: 10.1517/17425255.2011.623126
71. Burkhardt O, Rauch K, Kaever V, Hadem J, Kielstein JT, Welte T. Tigecycline possibly underdosed for the treatment of pneumonia: a pharmacokinetic viewpoint. Int J Antimicrob Agents. (2009) 34:101–2. doi: 10.1016/j.ijantimicag.2009.01.015
72. Koomanachai P, Kim A, Nicolau DP. Pharmacodynamic evaluation of tigecycline against Acinetobacter baumannii in a murine pneumonia model. J Antimicrob Chemother. (2009) 63:982–7. doi: 10.1093/jac/dkp056
73. Routsi C, Kokkoris S, Douka E, Ekonomidou F, Karaiskos I, Giamarellou H. High-dose tigecycline-associated alterations in coagulation parameters in critically ill patients with severe infections. Int J Antimicrob Agents. (2015) 45:90–3. doi: 10.1016/j.ijantimicag.2014.07.014
74. Falagas ME, Vardakas KZ, Tsiveriotis KP, Triarides NA, Tansarli GS. Effectiveness and safety of high-dose tigecycline-containing regimens for the treatment of severe bacterial infections. Int J Antimicrob Agents. (2014) 44:1–7. doi: 10.1016/j.ijantimicag.2014.01.006
75. Bassetti M, Poulakou G, Giamarellou H. Is there a future for tigecycline? Intensive Care Med. (2014) 40:1039–45. doi: 10.1007/s00134-014-3343-3
76. Tumbarello M, Viale P, Viscoli C, Trecarichi EM, Tumietto F, Marchese A, et al. Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K.pneumoniae: importance of combination therapy. Clin Infect Dis. (2012) 55:943–50. doi: 10.1093/cid/cis588
77. Pournaras S, Vrioni G, Neou E, Dendrinos J, Dimitroulia E, Poulou A, et al. Activity of tigecycline alone and in combination with colistin and meropenem against Klebsiella pneumoniae carbapenemase (KPC)-producing Enterobacteriaceae strains by time-kill assay. Int J Antimicrob Agents. (2011) 37:244–7. doi: 10.1016/j.ijantimicag.2010.10.031
78. Stein C, Makarewicz O, Bohnert JA, Pfeifer Y, Kesselmeier M, Hagel S, et al. Three dimensional checkerboard synergy analysis of colistin, meropenem, tigecycline against multidrug-resistant clinical Klebsiella pneumonia isolates. PLoS ONE. (2015) 10:e0126479. doi: 10.1371/journal.pone.0126479
79. Betts JW, Phee LM, Hornsey M, Woodford M, Wareham DW. In vitro and in vivo activities of tigecycline-colistin combination therapies against carbapenem-resistant Enterobacteriaceae. Antimicrob Agents Chemother. (2014) 58:3541–6. doi: 10.1128/AAC.02449-14
80. Hagihara M, Housman ST, Nicolau DP, Kuti JL. In vitro pharmacodynamics of polymyxin B and tigecycline alone and in combination against carbapenem-resistant Acinetobacter baumannii. Antimicrob Agents Chemother. (2014) 58:874–9. doi: 10.1128/AAC.01624-13
81. Toledo PV, Aranha AA Jr, Arend LN, Ribeiro V, Zavascki AP, Tuon FF. Activity of antimicrobial combinations against KPC-2-producing Klebsiella pneumoniae in a rat model and time-kill assay. Antimicrob Agents Chemother. (2015) 59:4301–10. doi: 10.1128/AAC.00323-15
82. Di Carlo P, Gulotta G, Casuccio A, Pantuso G, Raineri M, Farulla C, et al. KPC-3 Klebsiella pneumoniae ST258 clone infection in postoperativeabdominal surgery patients in an intensive care setting: analysis of a case seriesof 30 patients. BMC Anesthesiol. (2013) 13:13 doi: 10.1186/1471-2253-13-13
83. De Pascale G, Montini L, Pennisi M, Bernini V, Maviglia R, Bello G, et al. High dose tigecycline in critically ill patients with severe infections due to multidrug-resistant bacteria. Crit Care. (2014) 18:R90 doi: 10.1186/cc13858
84. Geng TT, Xu X, Huang M. High-dose tigecycline for the treatment of nosocomial carbapenem-resistant Klebsiella pneumoniae bloodstream infections: a retrospective cohort study. Medicine. (2018) 97:e9961 doi: 10.1097/MD.0000000000009961
85. Ni W, Han Y, Liu J, Wei C, Zhao J, Cui J, et al. Tigecycline treatment for carbapenem-resistant Enterobacteriaceae infections: a systematic review and meta-analysis. Medicine. (2016) 95:e3126 doi: 10.1097/MD.0000000000003126
86. Ramirez J, Dartois N, Gandjini H, Yan JL, Korth-Bradley J, McGovern PC. Randomized phase 2 trial to evaluate the clinical efficacy of two high-dosage tigecycline regimens versus imipenem-cilastatin for treatment of hospital-acquired pneumonia. Antimicrob Agents Chemother. (2013) 57:1756–62. doi: 10.1128/AAC.01232-12
87. Abdelraouf K, Kim A, Krause KM, Nicolau DP. In vivo efficacy of plazomicin alone or in combination with meropenem or tigecycline against Enterobacteriaceae isolates exhibiting various resistance mechanisms in an immunocompetent murine septicemia model. Antimicrob Agents Chemother. (2018) 62:e01074–1118. doi: 10.1128/AAC.01074-18
88. Zavascki AP, Klee BO, Bulitta JB. Aminoglycosides against carbapenem resistant Enterobacteriaceae in the critically ill: the pitfalls of aminoglycoside susceptibility. Expert Rev Anti Infect Ther. (2017) 15:519–26 doi: 10.1080/14787210.2017.1316193
89. Doi Y, Wachino JI, Arakawa Y. Aminoglycoside resistance: the emergence of acquired 16S ribosomal RNA methyltransferases. Infect Dis Clin North Am. (2016) 30:523–37. doi: 10.1016/j.idc.2016.02.011
90. Almaghrabi R, Clancy CJ, Doi Y, Hao B, Chen L, Shields RK, et al. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycosidemodifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob Agents Chemother. (2014) 58:4443–51. doi: 10.1128/AAC.00099-14
91. Bremmer DN, Clancy CJ, Press EG, Almaghrabi R, Chen L, Doi Y, Nguyen MH, et al. KPC-producing Klebsiella pneumoniae strains that harbor AAC(6′)-Ib exhibit intermediate resistance to amikacin. Antimicrob Agents Chemother. (2014) 58:7597–600. doi: 10.1128/AAC.03831-14
92. Monaco M, Giani T, Raffone M, Arena F, Garcia-Fernandez A, Pollini S, et al. Colistin resistance superimposed to endemic carbapenem-resistant Klebsiella pneumoniae: a rapidly evolving problem in Italy, November 2013–April 2014. Euro Surveill. (2014) 19:20939. doi: 10.2807/1560-7917.ES2014.19.42.20939
93. Alexander BT, Marschall J, Tibbetts RJ, Neuner EA, Dunne WM Jr, Ritchie DJ. Treatment and clinical outcomes of urinary tract infections caused by KPC-producing Enterobacteriaceae in a retrospective cohort. Clin Ther. (2012) 34:1314–23. doi: 10.1016/j.clinthera.2012.05.002
94. Satlin MJ, Kubin CJ, Blumenthal JS, Cohen AB, Furuya EY, Wilson SJ, et al. Comparative effectiveness of aminoglycosides, polymyxin B, and tigecycline for clearance of carbapenem resistant Klebsiella pneumoniae from urine. Antimicrob Agents Chemother. (2011) 55:5893–9. doi: 10.1128/AAC.00387-11
95. Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev. (2012) 25:682–707. doi: 10.1128/CMR.05035-11
96. Rello J, Solé-Lleonart C, Rouby JJ, Chastre J, Blot S, Poulakou G, et al. Use of nebulized antimicrobials for the treatment of respiratory infections in invasively mechanically ventilated adults: a position paper from the European Society of Clinical Microbiology and Infectious Diseases. Clin Microbiol Infect. (2017) 23:629–39. doi: 10.1016/j.cmi.2017.04.011
97. Poulakou G, Siakallis G, Tsiodras S, Arfaras-Melainis A, Dimopoulos G. Nebulized antibiotics in mechanically ventilated patients: roadmap and challenges. Expert Rev Anti Infect Ther. (2017) 15:211–29. doi: 10.1080/14787210.2017.1268052
98. Clinical Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing. 28th Informational Supplement M100. Wayne, PA: CLSI (2018).
99. Roger C, Nucci B, Louart B, Friggeri A, Knani H, Evrard A, et al. Impact of 30 mg/kg amikacin and 8 mg/kg gentamicin on serum concentrations in critically ill patients with severe sepsis. J Antimicrob Chemother. (2016) 71:208–12. doi: 10.1093/jac/dkv291
100. Roger C, Nucci B, Molinari N, Bastide S, Saissi G, Pradel G, et al. Standard dosing of amikacin and gentamicin in critically ill patients results in variable and subtherapeutic concentrations. Int J Antimicrob Agents. (2015) 46:21–7. doi: 10.1016/j.ijantimicag.2015.02.009
101. De Montmollin E, Bouadma L, Gault N, Mourvillier B, Mariotte F, Chemam S, et al. Predictors of insufficient amikacin peak concentration in critically ill patients receiving a 25 mg/kg total body weight regimen. Intensive Care Med. (2014) 40:998–1005. doi: 10.1007/s00134-014-3276-x
102. Galvez R, Luengo C, Cornejo R, Korsche J, Romero C, Tobar E, et al. Higher than recommended amikacin loading doses achieve pharmacokinetic targets without associated toxicity. Int J Antimicrob Agents. (2011) 38:146–51. doi: 10.1016/j.ijantimicag.2011.03.022
103. Taccone FS, Laterre PF, Spapen H, Dugernier T, Delattre I, Layeux B, et al. Revisiting the loading dose of amikacin for patients with severe sepsis and septic shock. Crit Care. (2010) 14:R53. doi: 10.1186/cc8945
104. Burdet C, Pajot O, Couffignal C, Armand-Lefevre L, Foucrier A, Laouenan C, et al. Population pharmacokinetics of single-dose amikacin in critically ill patients with suspected ventilator-associated pneumonia. Eur J Clin Pharmacol. (2015) 71:75–83. doi: 10.1007/s00228-014-1766-y
105. Brasseur A, Hites M, Roisin S, Cotton F, Vincent JL, De Backer D, et al. A high-dose aminoglycoside regimen combined with renal replacement therapy for the treatment of MDR pathogens: a proof-of-concept study. J Antimicrob Chemother. (2016) 71:1386–94. doi: 10.1093/jac/dkv491
106. Gonzalez-Padilla M, Torre-Cisneros J, Rivera-Espinar F, Pontes-Moreno A, López-Cerero L, Pascual A, et al. Gentamicin therapy for sepsis due to carbapenem-resistant and colistin-resistant Klebsiella pneumoniae. J Antimicrob Chemother. (2015) 70:905–13. doi: 10.1093/jac/dku432
107. Shields RK, Clancy CJ, Press EG, Nguyen MH. Aminoglycosides for treatment of bacteremia due to carbapenem-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother. (2016) 60:3187–92. doi: 10.1128/AAC.02638-15
108. Van Duin D, Cober E, Richter SS, Perez F, Kalayjian RC, Salata RA, et al. Impact of therapy and strain type on outcomes in urinary tract infections caused by carbapenem-resistant Klebsiella pneumoniae. J Antimicrob Chemother. (2015) 70:1203–11. doi: 10.1093/jac/dku495
109. Vidal L, Gafter-Gvili A, Borok S, Fraser A, Leibovici L, Paul M. Efficacy and safety of aminoglycoside monotherapy: systematic review and meta-analysis of randomized controlled trials. J Antimicrob Chemother. (2007) 60:247–57. doi: 10.1093/jac/dkm193
110. Paquette F, Bernier-Jean A, Brunette V, Ammann H, Lavergne V, Pichette V, et al. Acute kidney injury and renal recovery with the use of aminoglycosides: a large retrospective study. Nephron. (2015) 131:153–60. doi: 10.1159/000440867
111. Stankowicz MS, Ibrahim J, Brown DL. Once-daily aminoglycoside dosing: an update on current literature. Am J Health Syst Pharm. (2015) 72:1357–64. doi: 10.2146/ajhp140564
112. Ong LZ, Tambyah PA, Lum LH, Low ZJ, Cheng I, Murali TM, et al. Aminoglycoside-associated acute kidney injury in elderly patients with and without shock. J Antimicrob Chemother. (2016) 71:3250–7. doi: 10.1093/jac/dkw296
113. Moyá B, Zamorano L, Juan C, Ge Y, Oliver A. Affinity of the new cephalosporin CXA-101 to penicillin-binding proteins of Pseudomonas aeruginosa. Antimicrob Agents Chemother. (2010) 54:3933–7. doi: 10.1128/AAC.00296-10
114. Popejoy MW, Paterson DL, Cloutier D, Huntington JA, Miller B, Bliss CA, et al. Efficacy of ceftolozane/tazobactam against urinary tract and intra-abdominal infections caused by ESBL-producing Escherichia coli and Klebsiella pneumoniae: a pooled analysis of Phase 3 clinical trials. J Antimicrob Chemother. (2017) 72:268–72. doi: 10.1093/jac/dkw374
115. Karaiskos I, Galani I, Souli M, Giamarellou H. Novel β-lactam-β-lactamase inhibitor combinations: expectations for the treatment of carbapenem-resistant Gram-negative pathogens. Expert Opin Drug Metab Toxicol. (2019) 15:133–49. doi: 10.1080/17425255.2019.1563071
116. AstraZeneca. Summary of Product Characteristics: European Medicines Agency SPC for ceftazidime avibactam (Zavicefta™),. (2016). Available online at: https://www.medicines.org.uk/emc/medicine/33061 (accessed January 10, 2019).
117. Vabomere® (meropenem/vaborbactam for injection) [prescribing information]. FDA. (2017). Available online at: www.accessdata.fda.gov/ drugsatfda_docs/label/2017/209776lbl.pdf (accessed March 10, 2019).
118. Karaiskos I, Souli M, Giamarellou H. Plazomicin: an investigational therapy for the treatment of urinary tract infections. Expert Opin Investig Drugs. (2015) 24:1501–11. doi: 10.1517/13543784.2015.1095180
119. FDA Approval of Plazomicin (ZEMDRI™). (2018). Avaliable online at: https://www.fda.gov/Drugs/InformationOnDrugs/ucm612925.htm (accessed December 20, 2018).
120. Tetraphase, Announces FDA Approval of Eravacyline for the Treatment of cIAIs,. Avaliable online at: https://ir.tphase.com/news-releases/news-release-details/tetraphase-pharmaceuticals-announces-fda-approval-xeravatm (accessed January 27, 2018).
121. Tetraphase Announces EMA Approval of Eravacyline for the Treatment of cIAIs. (2018). Avaliable online at: https://ir.tphase.com/news-releases/news-release-details/tetraphase-pharmaceuticals-announces-adoption-commission (accessed March 5, 2019).
122. Tetraphase Announces Non-Inferiority not Met in IGNITE 3 Trial,. (2018). Avaliable online at: https://ir.tphase.com/news-releases/news-release-details/tetraphase-announces-top-line-results-ignite3-phase-3-clinical (accessed March 5, 2019).
123. Allou N, Bouteau A, Allyn J, Snauwaert A, Valance D, Jabot J, et al. Impact of a high loading dose of amikacin in patients with severe sepsis or septic shock. Ann Intensive Care. (2016) 6:106. doi: 10.1186/s13613-016-0211-z
124. Hirsch EB, Guo B, Chang KT, Cao H, Ledesma KR, Singh M, et al. Assessment of antimicrobial combinations for Klebsiella pneumoniae carbapenemase-producing K. Pneumoniae. J Infect Dis. (2013) 207:786–93. doi: 10.1093/infdis/jis766
125. Le J, McKee B, Srisupha-Olarn W, Burgess DS. In vitro activity of carbapenems alone and in combination with amikacin against KPC-producing Klebsiella pneumoniae. J Clin Med Res. (2011) 3:106–10. doi: 10.4021/jocmr551w
126. Giannella M, Trecarichi EM, Giacobbe DR, De Rosa FG, Bassetti M, Bartoloni A, et al. Italian Study Group on Resistant Infections of the Societa‘ Italiana Terapia Antinfettiva (ISGRI-SITA). Effect of combination therapy containing a high-dose carbapenem on mortality in patients with carbapenem-resistant Klebsiella pneumoniae bloodstream infection. Int J Antimicrob Agents. (2018) 51:244–8. doi: 10.1016/j.ijantimicag.2017.08.019
127. Daikos GL, Petrikkos P, Psichogiou M, Kosmidis C, Vryonis E, Skoutelis A, et al. Prospective observational study of the impact of VIM-1 metallo-beta-lactamase on the outcome of patients with Klebsiella pneumoniae bloodstream infections. Antimicrob Agents Chemother. (2009) 53:1868–73. doi: 10.1128/AAC.00782-08
128. Bulik CC, Nicolau DP. Double-carbapenem therapy for carbapenemase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. (2011) 55:3002–4. doi: 10.1128/AAC.01420-10
129. Poirel L, Kieffer N, Nordmann P. In vitro evaluation of dual carbapenem combinations against carbapenemase-producing Enterobacteriaceae. J Antimicrob Chemother. (2016) 71:156–61. doi: 10.1093/jac/dkv294
130. Giamarellou H, Galani L, Baziaka F, Karaiskos I. Effectiveness of a double-carbapenem regimen for infections in humans due to carbapenemaseproducing pandrug-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother. (2013) 57:2388–90. doi: 10.1128/AAC.02399-12
131. Oliva A, Scorzolini L, Castaldi D, Gizzi F, De Angelis M, Storto M, et al. Double-carbapenem regimen, alone or in combination with colistin, in the treatment of infections caused by carbapenem-resistant Klebsiella pneumoniae (CR-Kp). J Infect. (2017) 74:103–6. doi: 10.1016/j.jinf.2016.10.002
132. Souli M, Karaiskos I, Masgala A, Galani I, Barmpouti E, Giamarellou H. Double-carbapenem combination as salvage therapy for untreatable infections by KPC-2-producing Klebsiella pneumoniae. Eur J Clin Microbiol Infect Dis. (2017) 36:1305–15. doi: 10.1007/s10096-017-2936-5
133. De Pascale G, Martucci G, Montini L, Panarello G, Cutuli SL, Di Carlo D, et al. Double carbapenem as a rescue strategy for the treatment of severe carbapenemase-producing Klebsiella pneumoniae infections: a two-center, matched case control study. Crit Care. (2017) 21:173. doi: 10.1186/s13054-017-1769-z
134. Galani I, Nafplioti K, Chatzikonstantinou M, Souli M. In vitro evaluation of double-carbapenem combinations against OXA-48-producing Klebsiella pneumoniae isolates using time-kill studies. J Med Microbiol. (2018) 67:662–668. doi: 10.1099/jmm.0.000725
135. Rosa R, Rudin SD, Rojas LJ, Hujer AM, Perez-Cardona A, Perez F, et al. Double carbapenem and oral fosfomycin for the treatment of complicated urinary tract infections caused by blaNDM -harboring Enterobacteriaceae in kidney transplantation. Transpl Infect Dis. (2018) 20:e12795. doi: 10.1111/tid.12795
136. Giacobbe DR, Bassetti M, De Rosa FG, Del Bono V, Grossi PA, Menichetti F, et al. Ceftolozane/tazobactam:place in therapy. Expert Rev Anti Infect Ther. (2018) 16:307–20. doi: 10.1080/14787210.2018.1447381
137. Livermore DM, Mushtaq S, Meunier D, Hopkins KL, Hill R, Adkin R, et al. Activity of ceftolozane/tazobactam against surveillance and “problem” Enterobacteriaceae, Pseudomonas aeruginosa and non-fermenters from the British Isles. J Antimicrob Chemother. (2017) 72:2278–89. doi: 10.1093/jac/dkx136
138. Cabot G, Bruchmann S, Mulet X, Zamorano L, Moya B, Juan C, et al. Pseudomonas aeruginosa ceftolozane-tazobactam resistance development requires multiple mutations leading to overexpression and structural modification of AmpC. Antimicrob Agents Chemother. (2014) 58:3091–9. doi: 10.1128/AAC.02462-13
139. Farrell DJ, Sader HS, Flamm RK, Jones RN. Ceftolozane/tazobactam activity tested against Gram-negative bacterial isolates from hospitalised patients with pneumonia in US and European medical centres 2012. Int J Antimicrob Agents. (2014) 43:533–9. doi: 10.1016/j.ijantimicag.2014.01.032
140. Noel AR, Bowker KE, Attwood M, MacGowan AP. Antibacterial effect of ceftolozane/tazobactam in combination with amikacin against aerobic Gram-negative bacilli studied in an In vitro pharmacokinetic model of infection. J Antimicrob Chemother. (2018) 73:2411–7. doi: 10.1093/jac/dky225
141. Pérez A, Gato E, Pérez-Llarena J, Fernández-Cuenca F, Gude MJ, Oviaño M, et al. High incidence of MDR and XDR Pseudomonas aeruginosa isolates obtained from patients with ventilator-associated pneumonia in Greece, Italy and Spain as part of the MagicBullet clinical trial. J Antimicrob Chemother. (2019) 74:1244–52. doi: 10.1093/jac/dkz030
142. Solomkin J, Hershberger E, Miller B, Popejoy M, Friedland I, Steenbergen J, et al. Ceftolozane/Tazobactam plus metronidazole for complicated intra-abdominal infections in an era of multidrug resistance: results from a randomized, double-blind, phase 3 trial (ASPECT-cIAI). Clin Infect Dis. (2015) 60:1462–71. doi: 10.1093/cid/civ097
143. Wagenlehner FM, Umeh O, Steenbergen J, Yuan G, Darouiche RO. Ceftolozane-tazobactam compared with levofloxacin in the treatment of complicated urinary-tract infections, including pyelonephritis: a randomised, double-blind, phase 3 trial (ASPECT-cUTI). Lancet. (2015) 385:1949–56. doi: 10.1016/S0140-6736(14)62220-0
144. Chandorkar G, Huntington JA, Gotfried MH, Rodvold KA, Umeh O. Intrapulmonary penetration of ceftolozane/tazobactam and piperacillin/tazobactam in healthy adult subjects. J Antimicrob Chemother. (2012) 67:2463–9. doi: 10.1093/jac/dks246
145. Xiao AJ, Miller BW, Huntington JA, Nicolau DP. Ceftolozane/tazobactam pharmacokinetic/ pharmacodynamic-derived dose justification for phase 3 studies in patients with nosocomial pneumonia. J Clin Pharmacol. (2016) 56:56–66. doi: 10.1002/jcph.566
146. Xiao AJ, Caro L, Popejoy MW, Huntington JA, Kullar R. PK/PD target attainment with ceftolozane/tazobactam using monte carlo simulation in patients with various degrees of renal function, including augmented renal clearance and end-stage renal disease. Infect Dis Ther. (2017) 6:137–48. doi: 10.1007/s40121-016-0143-9
147. Oliver WD, Heil EL, Gonzales JP, Mehrotra S, Robinett K, Saleeh P, et al. Ceftolozane-tazobactam pharmacokinetics in a critically Ill patient on continuous venovenous hemofiltration. Antimicrob Agents Chemother. (2015) 60:1899–901. doi: 10.2174/97816810814031150101
148. Bremmer DN, Nicolau DP, Burcham P, Chunduri A, Shidham G, Bauer KA. Ceftolozane/tazobactam pharmacokinetics in a critically Ill adult receiving continuous renal replacement therapy. Pharmacotherapy. (2016) 36:30–3. doi: 10.1002/phar.1744
149. Kuti JL, Ghazi IM, Quintiliani R Jr, Shore E, Nicolau DP. Treatment of multidrug-resistant Pseudomonas aeruginosa with ceftolozane/tazobactam in a critically ill patient receiving continuous venovenous haemodiafiltration. Int J Antimicrob Agents. (2016) 48:342–3. doi: 10.1016/j.ijantimicag.2016.06.005
150. Natesan S, Pai MP, Lodise TP. Determination of alternative ceftolozane/tazobactam dosing regimens for patients with infections due to Pseudomonas aeruginosa with MIC values between 4 and 32 mg/L. J Antimicrob Chemother. (2017) 72:2813–6. doi: 10.1093/jac/dkx221
151. Castón JJ, De la Torre Á, Ruiz-Camps I, Sorli ML, Torres V, Torre-Cisneros J. Salvage therapy with ceftolozane-tazobactam for multidrug-resistant Pseudomonas aeruginosa infections. Antimicrob Agents Chemother. (2017) 61:pii e02136–16. doi: 10.1128/AAC.02136-16
152. Haidar G, Philips NJ, Shields RK, Snyder D, Cheng S, Potoski BA, et al. Ceftolozane-tazobactam for the treatment of multidrug-resistant Pseudomonas aeruginosa infections: clinical effectiveness and evolution of resistance. Clin Infect Dis. (2017) 65:110–20. doi: 10.1093/cid/cix182
153. Munita JM, Aitken SL, Miller WR, Perez F, Rosa R, Shimose LA, et al. Multicenter evaluation of ceftolozane/tazobactam for serious infections caused by carbapenem-resistant Pseudomonas aeruginosa. Clin Infect Dis. (2017) 65:158–61. doi: 10.1093/cid/cix014
154. Escolà-Vergé L, Pigrau C, Los-Arcos I, Arevalo A, Vinado B, Campany D, et al. Ceftolozane/tazobactam for the treatment of XDR Pseudomonas aeruginosa infections. Infection. (2018) 46:461–8. doi: 10.1007/s15010-018-1133-5
155. Katchanov J, Asar L, Klupp E-M, Both A, Rothe C, Konig C, et al. Carbapenem-resistant Gram-negative pathogens in a German university medical center: prevalence, clinical implications and the role of novel β-lactam/β-lactamase inhibitor combinations. PLoS ONE. (2018) 13:e0195757. doi: 10.1371/journal.pone.0195757
156. Xipell M, Paredes S, Fresco L, Bodro M, Marco F, Martinez JA, et al. Clinical experience with ceftolozane/tazobactam in patients with serious infections due to resistant Pseudomonas aeruginosa. J Glob Antimicrob Resist. (2018) 13:165–70. doi: 10.1016/j.jgar.2018.01.010
157. Kullar R, Wagenlehner FM, Popejoy MW, Long J, Yu B., Goldstein EJ. Does moderate renal impairment affect clinical outcomes in complicated intra-abdominal and complicated urinary tract infections? Analysis of two randomized controlled trials with ceftolozane/tazobactam. J Antimicrob Chemother. (2017) 72:900–5. doi: 10.1093/jac/dkw486
158. Plant AJ, Dunn A, Porter RJ. Ceftolozane-tazobactam resistance induced in vivo during the treatment of MDR Pseudomonas aeruginosa pneumonia. Expert Rev Anti Infect Ther. (2018) 16:367–8. doi: 10.1080/14787210.2018.1473079
159. Bassetti M, Castaldo N, Cattelan A, Mussini C, Righi E, Tascini C, et al. Ceftolozane/tazobactam for the treatment of serious Pseudomonas aeruginosa infections: a multicentre nationwide clinical experience. Int J Antimicrob Agents. (2018) 53:408–415. doi: 10.1016/j.ijantimicag.2018.11.001
160. Goodlet KJ, Nicolau DP, Nailor MD. In vitro comparison of ceftolozane-tazobactam to traditional beta-lactams and ceftolozane-tazobactam as an alternative to combination antimicrobial therapy for Pseudomonas aeruginosa. Antimicrob Agents Chemother. (2017) 61:e01350–17. doi: 10.1128/AAC.01350-17
161. Bassetti M, Vena A, Russo A, Croxatto A, Calandra T, Guery B. Rational approach in the management of Pseudomonas aeruginosa infections. Curr Opin Infect Dis. (2018) 31:578–86. doi: 10.1097/QCO.0000000000000505
162. Torres A, Niederman MS, Chastre J, Ewig S, Fernandez-Vandellos P, Hanberger H, et al. Summary of the international clinical guidelines for the management of hospital-acquired and ventilator-acquired pneumonia. ERJ Open Res. (2018) 4:e00028–2018. doi: 10.1183/23120541.00028-2018
163. Keepers TR, Gomez M, Celeri C, Nichols WW, Krause KM. Bactericidal activity, absence of serum effect, and time-kill kinetics of ceftazidime-avibactam against beta-lactamase producing Enterobacteriaceae and Pseudomonas aeruginosa. Antimicrob Agents Chemother. (2014) 58:5297–305. doi: 10.1128/AAC.02894-14
164. Allergan Summary of Product Characteristics Food and Drug Administration Ceftazidime avibactam (Avycaz™). Available online at: https://www.allergan.com/assets/pdf/avycaz_pi (accessed January 10, 2019).
165. Sader HS, Castanheira M, Flamm RK. Antimicrobial activity of ceftazidime-avibactam when tested against gram-negative bacteria isolated from patients hospitalized with pneumonia in U.S. Medical Centers, 2011 to 2015. Antimicrob Agents Chemother. (2017) 61:e02083–16. doi: 10.1128/AAC.02083-16
166. Alba J, Ishii Y, Thomson K, Moland ES, Yamaguchi K. Kinetics study of KPC-3, a plasmid-encoded class A carbapenem-hydrolyzing beta-lactamase. Antimicrob Agents Chemother. (2005) 49:4760–2. doi: 10.1128/AAC.49.11.4760-4762.2005
167. Levasseur P, Girard AM, Miossec C, Pace J, Coleman K. In vitro antibacterial activity of the ceftazidime-avibactam combination against Enterobacteriaceae, including strains with well-characterized β-lactamases. Antimicrob Agents Chemother. (2015) 59:1931–4. doi: 10.1128/AAC.04218-14
168. Shields RK, Clancy CJ, Hao B, Chen L, Press EG, Iovine NM, et al. Effects of Klebsiella pneumoniae carbapenemase subtypes, extended-spectrum β-lactamases, and porin mutations on the In vitro activity of ceftazidime-avibactam against carbapenem-resistant K. pneumoniae. Antimicrob Agents Chemother. (2015) 59:5793–7. doi: 10.1128/AAC.00548-15
169. Levasseur P, Girard AM, Lavallade L, Miossec C, Pace J, Coleman K. Efficacy of a ceftazidime-avibactam combination in a murine model of septicemia caused by Enterobacteriaceae species producing AmpC or extended-spectrum beta-lactamases. Antimicrob Agents Chemother. (2014) 58:6490–5. doi: 10.1128/AAC.03579-14
170. Temkin E, Torre-Cisneros J, Beovic B, Benito N, Giannella M, Gilarranz R, et al. Ceftazidime-avibactam as salvage therapy for infections caused by carbapenem-resistant organisms. Antimicrob Agents Chemother. (2017) 61:e01964–16. doi: 10.1128/AAC.01964-16
171. Barnes MD, Winkler ML, Taracila MA, Page MG, Desarbre E, Kreiswirth BN, et al. Klebsiella pneumoniae Carbapenemase-2 (KPC-2), substitutions at ambler position Asp179, and resistance to ceftazidime-avibactam: unique antibiotic-resistant phenotypes emerge from β-lactamase protein engineering. MBio. (2017) 8:e00528–17. doi: 10.1128/mBio.00528-17
172. Compain F, Arthur M. Impaired inhibition by avibactam and resistance to the ceftazidime-avibactam combination due to the D179Y substitution in the KPC-2 β-lactamase. Antimicrob Agents Chemother. (2017) 61:e00451–17. doi: 10.1128/AAC.00451-17
173. Haidar G, Clancy CJ, Shields RK, Hao B, Cheng S, Nguyen MH. Mutations in blaKPC-3 that confer ceftazidime-avibactam resistance encode novel KPC-3 variants that function as extended-spectrum β-lactamases. Antimicrob Agents Chemother. (2017) 61:e02534–16. doi: 10.1128/AAC.02534-16
174. Shields RK, Nguyen MH, Press EG, Chen L, Kreiswirth BN, Clancy CJ. In vitro selection of meropenem resistance among ceftazidime-avibactam-resistant, meropenem-susceptible Klebsiella pneumoniae isolates with variant KPC-3 carbapenemases. Antimicrob Agents Chemother. (2017) 61:e00079–17. doi: 10.1128/AAC.00079-17
175. Levasseur P, Girard AM, Claudon M, Goossens H, Black MT, Coleman K, et al. In vitro antibacterial activity of the ceftazidime-avibactam (NXL104) combination against Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. (2012) 56:1606–8. doi: 10.1128/AAC.06064-11
176. Flamm RK, Sader HS, Farrell DJ, Jones RN. Ceftazidime-avibactam and comparator agents tested against urinary tract isolates from a global surveillance program 2011. Diagn Microbiol Infect Dis. (2014) 80:233–8. doi: 10.1016/j.diagmicrobio.2014.07.005
177. Sader HS, Castanheira M, Mendes RE, Flamm RK, Farrell DJ, Jones RN. Ceftazidime- avibactam activity against multidrug-resistant Pseudomonas aeruginosa isolated in U.S. medical centers in 2012 and 2013. Antimicrob Agents Chemother. (2015) 59:3656–9. doi: 10.1128/AAC.05024-14
178. Castanheira M, Costello SE, Woosley LN, Deshpande LM, Davies TA, Jones RN. Evaluation of clonality and carbapenem resistance mechanisms among Acinetobacter baumannii-Acinetobacter calcoaceticus complex and Enterobacteriaceae isolates collected in European and Mediterranean countries and detection of two novel β-lactamases, GES-22 and VIM-35. Antimicrob Agents Chemother. (2014) 58:7358–66. doi: 10.1128/AAC.03930-14
179. Tominaga N, Edeki T, Li J, Learoyd M, Bouw MR, Das S. Phase I study assessing the safety, tolerability, and pharmacokinetics of avibactam and ceftazidime-avibactam in healthy Japanese volunteers. J Infect Chemother. (2015) 21:551–8. doi: 10.1016/j.jiac.2015.04.006
180. Merdjan H, Tarral A, Das S, Li J. Phase 1 study assessing the pharmacokinetic profile and safety of avibactam in patients with renal impairment. J Clin Pharmacol. (2017) 57:211–8. doi: 10.1002/jcph.793
181. Nicolau DP, Siew L, Armstrong J, Li J, Edeki T, Learoyd M, et al. Phase 1 study assessing the steady-state concentration of ceftazidime and avibactam in plasma and epithelial lining fluid following two dosing regimens. J Antimicrob Chemother. (2015) 70:2862–9. doi: 10.1093/jac/dkv170
182. Mazuski JE, Gasink LB, Armstrong J, Broadhurst H, Stone GG, Rank D, et al. Efficacy and safety of ceftazidime-avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infection:results from a randomized, controlled, double-blind, phase 3 program. Clin Infect Dis. (2016) 62:1380–9. doi: 10.1093/cid/ciw133
183. Qin X, Tran BG, Kim MJ, Wang L, Nguyen DA, Chen Q, et al. A randomised, double-blind, phase 3 study comparing the efficacy and safety of ceftazidime/avibactam plus metronidazole versus meropenem for complicated intra-abdominal infections in hospitalised adults in Asia. Int J Antimicrob Agents. (2017) 49:579–88. doi: 10.1016/j.ijantimicag.2017.01.010
184. Torres A, Zhong N, Pachl J, Timsit JF, Kollef M, Chen Z, et al. Ceftazidime-avibactam versus meropenem in nosocomial pneumonia, including ventilator-associated pneumonia (REPROVE): a randomised, double-blind, phase 3 non-inferiority trial. Lancet Infect Dis. (2018) 18:285–95. doi: 10.1016/S1473-3099(17)30747-8
185. Wagenlehner FM, Sobel JD, Newell P, Armstrong J, Huang X, Stone GG, et al. Ceftazidime-avibactam versus doripenem for the treatment of complicated urinary tract infections, including acute pyelonephritis: RECAPTURE, a phase 3 randomized trial program. Clin Infect Dis. (2016) 63:754–62. doi: 10.1093/cid/ciw378
186. Carmeli Y, Armstrong J, Laud PJ, Newell P, Stone G, Wardman A, et al. Ceftazidime-avibactam or best available therapy in patients with ceftazidime resistant Enterobacteriaceae and Pseudomonas aeruginosa complicated urinary tract infections or complicated intra-abdominal infections (REPRISE): a randomised, pathogen-directed, phase 3 study. Lancet Infect Dis. (2016) 16:661–73. doi: 10.1016/S1473-3099(16)30004-4
187. Shields RK, Potoski BA, Haidar G, Hao B, Doi Y, Chen L, et al. Clinical outcomes, drug toxicity and emergence of ceftazidime-avibactam resistance among patients treated for carbapenem-resistant Enterobacteriaceae infections. Clin Infect Dis. (2016) 63:1615–8. doi: 10.1093/cid/ciw636
188. King M, Heil E, Kuriakose S, Bias T, Huang V, El-Beyrouty, et al. Multicenter study of outcomes with ceftazidime-avibactam in patients with carbapenem-resistant Enterobacteriaceae infections. Antimicrob Agents Chemother. (2017) 61:e00449–17 doi: 10.1128/AAC.00449-17
189. Shields RK, Nguyen MH, Chen L, Press EG, Kreiswirth BN, Clancy CJ. Pneumonia and renal replacement therapy are risk factors for ceftazidime-avibactam treatment failures and resistance among patients with carbapenem-resistant Enterobacteriaceae infections. Antimicrob Agents Chemother. (2018) 62:e02497–517. doi: 10.1128/AAC.02497-17
190. Shields RK, Nguyen MH, Chen L, Press EG, Potoski BA, Marini RV, et al. Ceftazidime-avibactam is superior to other treatment regimens against carbapenem-resistant Klebsiella pneumoniae bacteremia. Antimicrob Agents Chemother. (2017) 61:e00883–917. doi: 10.1128/AAC.00883-17
191. Castón JJ, Lacort-Peralta I, Martín-Dávila P, Loeches B, Tabarres S, Temkin L, et al. Clinical efficacy of ceftazidime/avibactamversus other active agents for the treatment of bacteremia due to carbapenemase-producing Enterobacteriaceae in hematologic patients. Int J Infect Dis. (2017) 59:118–23. doi: 10.1016/j.ijid.2017.03.021
192. Tumbarello M, Trecarichi EM, Corona A, De Rosa FG, Bassetti M, Mussini C, et al. Efficacy of ceftazidime-avibactam salvage therapy in patients with infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae. Clin Infect Dis. (2019) 68:355–64. doi: 10.1093/cid/ciy492
193. van Duin D, Lok J J, Earley M, Cober F, Richter SS, Perez F, et al.Colistin versus ceftazidime-avibactam in the treatment of infections due to carbapenem-resistant Enterobacteriaceae. Clin Infect Dis. (2018) 66:163–71. doi: 10.1093/cid/cix783
194. Sousa A, Pérez-Rodríguez MT, Soto A, Rodriguez L, Perez-Landeiro A, Martinez-Lamas, et al. Effectiveness of ceftazidime/ avibactam as salvage therapy for treatment of infections due to OXA-48 carbapenemase-producing Enterobacteriaceae. J Antimicrob Chemother. (2018) 73:3170–5. doi: 10.1093/jac/dky295
195. Zhong H, Zhao XY, Zhang ZL, Gu ZC, Zhang C, Gao Y, et al. Evaluation of efficacy and safety of ceftazidime-avibactam in the treatment of Gram-negative bacterial infections: a systematic review and meta-analysis. Int J Antimicrob Agents. (2018) 52:443–50. doi: 10.1016/j.ijantimicag.2018.07.004
196. Hecker SJ, Reddy KR, Totrov M, Hirst GC, Lomovskaya O, Griffith DC, et al. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs. class A serine carbapenemases. J Med Chem. (2015) 58:3682–92. doi: 10.1021/acs.jmedchem.5b00127
197. Lapuebla A, Abdallah M, Olafisoye O, Cortes C, Urban C, Quale J, et al. Activity of meropenem combined with RPX7009, a novel beta-lactamase inhibitor, against Gram-negative clinical isolates in New York City. Antimicrob Agents Chemother. (2015) 59:4856–60. doi: 10.1128/AAC.00843-15
198. Castanheira M, Rhomberg PR, Flamm RK, Jones RN. Effect of the beta-lactamase inhibitor vaborbactam combined with meropenem against serine carbapenemase-producing Enterobacteriaceae. Antimicrob Agents Chemother. (2016) 60:5454–8. doi: 10.1128/AAC.00711-16
199. Castanheira M, Huband MD, Mendes RE, Flamm RK. Meropenem-vaborbactam tested against contemporary gram-negative isolates collected worldwide during 2014, including carbapenem-resistant, kpc producing, multidrug-resistant, and extensively drug-resistant Enterobacteriaceae. Antimicrob Agents Chemother. (2017) 61:e00567–17. doi: 10.1128/AAC.00567-17
200. Lomovskaya O, Sun D, Rubio-Aparicio D, Nelson K, Tsivkovski R, Griffith DC, et al. Vaborbactam: spectrum of beta-lactamase inhibition and impact of resistance mechanisms on activity in Enterobacteriaceae. Antimicrob Agents Chemother. (2017) 61:e01443–17. doi: 10.1128/AAC.01443-17
201. Bassetti M, Righi E, Russo A, Carnelutti A. New antibiotics for pneumonia. Clin Chest Med. (2018) 39:853–69. doi: 10.1016/j.ccm.2018.08.007
202. Griffith DC, Loutit JS, Morgan EE, Durso S, Dudley MN. Phase 1 study of the safety, tolerability, and pharmacokinetics of the b-lactamase inhibitor vaborbactam (RPX7009) in healthy adult subjects. Antimicrob Agents Chemother. (2016) 60:6326–32. doi: 10.1128/AAC.00568-16
203. Hackel MA, Lomovskaya O, Dudley MN, Karlowsky JA, Sahm DF. In vitro activity of meropenem-vaborbactam against clinical isolates of KPC-positive Enterobacteriaceae. Antimicrob Agents Chemother. (2017) 62:e01904–17. doi: 10.1128/AAC.01904-17
204. Kaye KS, Bhowmick T, Metallidis S, Bleasdale SC, Sagan OS, Stus V, et al. Effect of meropenem-vaborbactam vs. piperacillin-tazobactam on clinical cure or improvement and microbial eradication in complicated urinary tract infection: the TANGO I randomized clinical trial. JAMA. (2018) 319:788–99. doi: 10.1001/jama.2018.0438
205. Wunderink RG, Giamarellos-Bourboulis EJ, Rahav G, Mathers AJ, Bassetti M, Vazquez J, et al. Effect and safety of meropenem-vaborbactam versus best-available therapy in patients with carbapenem-resistant Enterobacteriaceae infections: the TANGO II randomized clinical trial. Infect Dis Ther. (2018) 7:439–55. doi: 10.1007/s40121-018-0214-1
206. Website of European Medicines Agency. (2019). Avaliable online at: https://www.ema.europa.eu/en/medicines/human/EPAR/vabomere#authorisation-details-section (accessed March 14, 2019).
207. Clinical Trials Registry. (2019). Avaliable online at: https://clinicaltrials.gov/ct2/show/NCT03006679 (accessed March 14, 2019).
208. Zhanel GG, Lawson CD, Zelenitsky S, Findlay B, Schweizer F, Adam H, et al. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin, and amikacin. Expert Rev Anti Infect Ther. (2012) 10:459–73. doi: 10.1586/eri.12.25
209. Livermore DM, Mushtaq S, Warner M, Zhang JC, Mahatrjan S, Doumith M, et al. Activity of aminoglycosides, including ACHN-490, against carbapenem resistant Enterobacteriaceae isolates. J Antimicrob Chemother. (2011) 66:48–53. doi: 10.1093/jac/dkq408
210. Endimiani A, Hujer KM, Hujer AM, Huger AM, Armstrong ES, Choudhary Y, et al. ACHN-490, a neoglycoside with potent In vitro activity against multidrug-resistant Klebsiella pneumoniae isolates. Antimicrob Agents Chemother. (2009) 53:4504–7. doi: 10.1128/AAC.00556-09
211. Walkty A, Adam H, Baxter M, Denisuik A, Lagace-Wiens P, Karlowsky JA, et al. In vitro activity of plazomicin against 5,015 Gram-negative and Gram-positive clinical isolates obtained from patients in Canadian hospitals as part of the CANWARD study, 2011–2012. Antimicrob Agents Chemother. (2014) 58:2554–63. doi: 10.1128/AAC.02744-13
212. Landman D, Kelly P, Backer M, Babu E, Shah N, Bratu S, et al. Antimicrobial activity of a novel aminoglycoside, ACHN-490, against Acinetobacter baumannii and Pseudomonas aeruginosa from New York City. J Antimicrob Chemother. (2011) 66:332–4. doi: 10.1093/jac/dkq459
213. Galani I, Souli M, Daikos GL, Chrysouli Z, Poulakou G, Psichogiou M, et al. Activity of plazomicin (ACHN-490) against MDR clinical isolates of Klebsiella pneumoniae, Escherichia coli and Enterobacter spp. from Athens, Greece. J Chemother. (2012) 24:191–4. doi: 10.1179/1973947812Y.0000000015
214. Denervaud-Tendon V, Poirel L, Connolly LE, Krause KM, Nordmann P.Plazomicin activity against polymyxin-resistant Enterobacteriaceae, including MCR-1-producing isolates. J Antimicrob Chemother. (2017) 72:2787–91. doi: 10.1093/jac/dkx239
215. Cass RT, Brooks CD, Havrilla NA, Tack K, Borin M, Young D, et al. Pharmacokinetics and safety of single and multiple doses of ACHN-490 injection administered intravenously in healthy subjects. Antimicrob Agents Chemother. (2011) 55:5874–880. doi: 10.1128/AAC.00624-11
216. Results of the EPIC Trial Released in Achaogen's. (2016). Avaliable online at: http://investors.achaogen.com/news-releases/news-release-details/achaogen-announces-positive-results-phase-3-cuti-and-cre, Results of the EPIC trial released in Achaogen's website (accessed December 29, 2018).
217. McKinnell JA, Connoly LE, Pushkin R, Jubb A, Keefe BO, Serio A, et al. Improved Outcomes with Plazomicin (PLZ) Compared with Colistin (CST) in Patients with Bloodstream Infections (BSI) Caused by Carbapenem-resistant Enterobacteriaceae (CRE): results from the CARE Study. Open Forum Infect Dis. (2017) 4:S531–1531. doi: 10.1093/ofid/ofx163.1383
218. Connolly LE, Jubb AM, Keeffe BO, Serio A, Smith A, Gall J, et al. Plazomicin is associated with improved survival and safety compared to colistin in serious carbapenem-resistant Enterobacteriaceae (CRE) infections: results of the CARE study. Presented at the Annual Meeting of the American Society of Microbiology. New Orleans, LA (2017).
219. McKinnell JA, Dwyer JP, Talbot GH, Connolly LE, Friedland I, Smith A, et al. Plazomicin for infections caused by carbapenem-resistant Enterobacteriaceae. N Engl J Med. (2019) 380:791–3. doi: 10.1056/NEJMc1807634
220. Bassetti M, Righi E. Eravacycline for the treatment of intra-abdominal Infections. Expert Opin Investig Drugs. (2014) 23:1575–84. doi: 10.1517/13543784.2014.965253
221. Abdallah M, Olafisoye O, Cortes C, Urban C, Landman D, Quale J. Activity of eravacycline against Enterobacteriaceae and Acinetobacter baumannii, including multidrug-resistant isolates from New York City. Antimicrob Agents Chemother. (2015) 59:1802–5. doi: 10.1128/AAC.04809-14
222. Livermore DM, Mushtaq S, Warner M, Woodford N. In vitro activity of eravacycline against carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii. Antimicrob Agents Chemother. (2016) 60:3840–44. doi: 10.1128/AAC.00436-16
223. Grossman TH, Murphy TM, Slee AM, Lofland D, Sutcliffe JA. Eravacycline (TP-434) is efficacious in animal models of infection. Antimicrob Agents Chemother. (2015) 59:2567–71. doi: 10.1128/AAC.04354-14
224. Connors KP, Housman ST, Pope JS, Russomanno J, Salerno E, Shore E, et al. Phase I, open-label, safety and pharmacokinetic study to assess bronchopulmonary disposition of intravenous eravacycline in healthy men and women. Antimicrob Agents Chemother. (2014) 58:2113–8. doi: 10.1128/AAC.02036-13
225. Seifert H, Stefanik D, Sutcliffe JA, Higgins PG. In-vitro activity of the novel fluorocycline eravacycline against carbapenem non-susceptible Acinetobacter baumannii. Int J Antimicrob Agents. (2018) 51:62–4. doi: 10.1016/j.ijantimicag.2017.06.022
226. Solomkin J, Evans D, Slepavicius A, Lee P, Marsh A, Tsai L, et al.Assessing the efficacy and safety of eravacycline vs ertapenem in complicated intra-abdominal infections in the investigating gram-negative infections treated with eravacycline (IGNITE 1) trial: a randomized clinical trial. JAMA Surg. (2017) 152:224–32. doi: 10.1001/jamasurg.2016.4237
227. Solomkin JS, Gardovskis J, Lawrence K, Montravers P, Sway A, Evans D, et al. IGNITE4: results of a phase 3, randomized, multicenter, prospective trial of eravacycline vs. meropenem in the treatment of complicated intra-abdominal infections. Clin Infect Dis. (2018). doi: 10.1093/cid/ciy1029. [Epub ahead of print].
228. Mojica MF, Ouellette CP, Leber A, Becknell MB, Ardura MI, Perez F, et al. Successful treatment of bloodstream infection due to metallo-β-lactamase-producing Stenotrophomonas maltophilia in a renal transplant patient. Antimicrob Agents Chemother. (2016) 60:5130–4. doi: 10.1128/AAC.00264-16
229. Shaw E, Rombauts A, Tubau F, Padulleso A, Camara J, Lozano T, et al. Clinical outcomes after combination treatment with ceftazidime/avibactam and aztreonam for NDM-1/OXA-48/CTX-M-15-producing Klebsiella pneumoniae infection. J Antimicrob Chemother. (2018) 73:1104–6. doi: 10.1093/jac/dkx496
230. Sabet M, Tarazi Z, Rubio-Aparicio D, Nolan TG, Parkinson J, Lomovskaya O, et al. Activity of simulated human dosage regimens of meropenem and vaborbactam against carbapenem-resistant Enterobacteriaceae in an In vitro hollow-fiber model. Antimicrob Agents Chemother. (2018) 62:e01969–17. doi: 10.1128/AAC.01969-17
231. Sun D, Rubio-Aparicio D, Nelson K, Dudley MN, Lomovskaya O. Meropenem-vaborbactam resistance selection, resistance prevention, and molecular mechanisms in mutants of KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. (2017) 61:e01694–17. doi: 10.1128/AAC.01694-17
232. Dickstein Y, Lellouche J, Dalak Amar MB, Schwartz D, Nutman A, Daitch V, et al. Treatment outcomes of colistin and carbapenem-resistant Acinetobacter baumannii infections: an exploratory subgroup analysis of a randomized clinical trial. Clin Infect Dis. (2018). doi: 10.1093/cid/ciy988. [Epub ahead of print].
233. Zusman O, Altunin S, Koppel F, Dishon Benattar Y, Gedik H, Paul M. Polymyxin monotherapy or in combination against carbapenem-resistant bacteria: systematic review and meta-analysis. J Antimicrob Chemother. (2017) 72:29–39. doi: 10.1093/jac/dkw377
Keywords: ceftazidime avibactam, ceftolozane tazobactam, colistin, combination, monotherapy, carbapenemase producing Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa
Citation: Karaiskos I, Lagou S, Pontikis K, Rapti V and Poulakou G (2019) The “Old” and the “New” Antibiotics for MDR Gram-Negative Pathogens: For Whom, When, and How. Front. Public Health 7:151. doi: 10.3389/fpubh.2019.00151
Received: 18 March 2019; Accepted: 23 May 2019;
Published: 11 June 2019.
Edited by:
George Dimopoulos, University General Hospital Attikon, GreeceReviewed by:
Gennaro De Pascale, Università Cattolica del Sacro Cuore, ItalyAna Afonso, University of São Paulo, Brazil
Copyright © 2019 Karaiskos, Lagou, Pontikis, Rapti and Poulakou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Garyphallia Poulakou, Z3BvdWxha291QGdtYWlsLmNvbQ==