Abstract
Toxigenic Vibrio cholerae is responsible for 1.4 to 4.3 million cases with about 21,000–143,000 deaths per year. Dominance of O1 and O139 serogroups, classical and El tor biotypes, alterations in CTX phages and the pathogenicity Islands are some of the major features of V. cholerae isolates that are responsible for cholera epidemics. Whole-genome sequencing (WGS) based analyses of single-nucleotide polymorphisms (SNPs) and other infrequent genetic variants provide a robust phylogenetic framework. Recent studies on the global transmission of pandemic V. cholerae O1 strains have shown the existence of eight different phyletic lineages. In these, the classical and El Tor biotype strains were separated as two distinctly evolved lineages. The frequency of SNP accumulation and the temporal and geographical distribution supports the perception that the seventh cholera pandemic (7CP) has spread from the Bay of Bengal region in three independent but overlapping waves. The 2010 Haitian outbreak shared a common ancestor with South-Asian wave-3 strains. In West Africa and East/Southern Africa, cholera epidemics are caused by single expanded lineage, which has been introduced several times since 1970. The Latin American epidemics that occurred in 1991 and 2010 were the result of introductions of two 7CP sublineages. Sublineages representing wave-3 have caused huge outbreaks in Haiti and Yemen. The Ogawa-Inaba serotype switchover in several cholera epidemics are believed to be due to the involvement of certain selection mechanism(s) rather than due to random events. V. cholerae O139 serogroup is phylogenetically related to the 7CP El Tor, and almost all these isolates belonged to the multilocus sequence type-69. Additional phenotypic and genotypic information have been generated to understand the pathogenicity of classical and El Tor vibrios. Presence of integrative conjugative elements (ICE) with antibiotic resistance gene cassettes, clustered regularly interspaced short palindromic repeats-associated protein system and ctxAB promoter based ToxRS expression of cholera toxin (CT) separates classical and El Tor biotypes. With the availability of WGS information, several important applications including, molecular typing, antimicrobial resistance, new diagnostics, and vaccination strategies could be generated.
Introduction
Cholera is caused by pathogenic strains of Vibrio cholerae due to their colonization in the intestinal milieu and secretion of cholera toxin (CT). The clinical consequences of this diarrheal disease include discharge of substantial volumes of watery stool, loss of electrolyte, rapid dehydration that may advance to hypovolemic shock and metabolic acidosis. Death rates due to cholera infection are reported to be as high as 70%, mainly due to the delay in rehydrating the patients. The global burden of cholera is estimated to be between 1.4 and 4.3 million cases with about 21,000–143,000 deaths per year (1). In 2017 alone, 34 countries reported a total of 1,227,391 cases and 5,654 deaths (2).
Although there are more than 200 serogroups of V. cholerae, epidemics of cholera are caused by two serogroups i.e., O1 and O139. The serogroup O1 is classified into two biotypes, classical and El Tor and each biotype into Ogawa and Inaba serotypes. This disease has marked 200 years, with the first cholera pandemic documented in 1817. The first six pandemics were caused by the classical biotype of V. cholerae serogroup O1. The ongoing seventh cholera pandemic (7CP) is caused by the El Tor biotype, which appeared in Indonesia in 1961 and reached South Asia after 2 years. In the succeeding years, the El Tor vibrios reached Africa (1970s), South-America (1990s), and the Caribbean Islands (2010) (3). In 1992, serogroup O139 emerged in the Indian subcontinent and spread across Asia until mid-2000s (4). However, this serogroup was eventually superseded by O1, which continues to cause cholera today.
Even though the El Tor vibrios are genetically homogenous, WGS (whole-genome sequence) analysis has identified several lineages (L)/transmission types (T). The newly identified El Tor hybrids, variation in the B subunit of the CT encoding gene CT-B subunit (ctxB), polymyxin-B sensitive El Tor vibrios have complicated the biotype differentiation. Recent endemic and epidemic cholera in Asia and Africa are increasingly being attributed to atypical El Tor variants with genetically variable CTX phages, rstR genotypes and single-nucleotide polymorphisms (SNPs) throughout the genome of V. cholerae (5–7).
The conventional methods such as serotyping, phage typing, antibiogram etc., provide information to distinguish V. cholerae isolates involved in outbreaks, but they are inadequate for the clonal definition. In the pre-genomic era, application and interpretation of the results produced using various molecular typing methods had no standardized clonal description and had low discrimination for phylogenetic inference. In order to overcome these complications, WGS based phylogenetic analysis and comparative genome analysis have been used to study the population structure and evolution of V. cholerae as well as their epidemiological significance. Herein, we aim to review the outcomes of the recent genomics of V. cholerae focusing on the global epidemiology of cholera and subtle genetic changes.
Features Considered in The Evolution of V. Cholerae
Genomic comparison of V. cholerae isolates is able to provide critical evidence in understanding the emergence and spread of epidemic cholera. In this, the Bayesian approach (3, 8) supports to time the important nodes with strong temporal signal. The population structure on the other hand might be easily studied by a maximum likelihood approach. WGS-based Bayesian phylogeographic analysis is now being widely used to detect the prototypes of spread, derivation time of species, diversification rates and its time estimation. Using this model, several thousands of WGS of V. cholerae O1 genomes have been analyzed and several lineages identified.
V. cholerae O1 and O139 isolates were originally categorized in 8 different phyletic lineages (L1 to L8). The classical and El Tor clades are placed in distinct lineages, L1 and L2, respectively and separated by 20,000 SNPs (3). The lineage L3 represents USA Gulf Coast isolates; L5, L6, and L8 are with typical El Tor/O139 vibrios and isolates of L4 and L7 lineages have distinct genetic backbones (3). Six different stages in the molecular evolution of the V. cholerae El Tor biotype, from the earliest available isolates (1930s) to the pandemic strain, have also been described (9). The 7CP first emerged in Indonesia and repeatedly spread from the Bay of Bengal to different parts of the world in the form of three waves as determined by the maximum-likelihood phylogenetic tree based on SNP differences across the core genome of V. cholerae (3). It took almost 50 years for the pathogen to adapt itself from wave-1 to wave-3 strains of today.
Based on the amino acid positioning in the CT-B of V. cholerae, several CT genotypes have been reported (7) (Table 1). The CT phage (CTX) is a lysogenic filamentous phage that harbors the CT encoding genes (ctxAB). In the evolution of V. cholerae, the CTX has undergone several changes, mainly due to recombination between two major types of CTX (CTXcla and CTX-1) and a satellite phage (RS1) along with point mutations in ctxB. Different components and types of CTX phages are shown in Figure 1. RS1 has genes that permit phage DNA replication (rstA), integration (rstB), regulation (rstR), and rstC, whose function is unspecified (10, 11). CTX phages are classified mostly by rstR and ctxB genotype and further subdivided by SNPs throughout the V. cholerae genome. CTXcla and CTXUSGulf has the classical type of rstR and CT genotype-1 (ctxB1). CTXAUS also has a classical type of rstR but with CT genotype-2 (ctxB2). The recorded genetic differences in the rstA and rstB of various CTX in 7CP are summarized in Table 2. The type of lineage, CT genotype and CTX phage type are collectively used in defining the wave pattern or clade. The CTX phage arrays (TLC-truncated CTX-CTXΦB3, or TLC-CTXΦB3-CTXθB3-RS1), specifically detected in Latin American isolates were not reported in El Tor, altered El Tor, or El Tor variants from Asia, Africa, and Haiti (12).
Table 1
| CT-genotype (ctxB) | Nucleotide position | Amino acid position | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 58 | 72 | 83 | 101 | 106 | 115 | 138 | 165 | 203 | 20 | 24 | 28 | 34 | 36 | 39 | 46 | 55 | 68 | |
| 1 | C | A | A | A | A | C | T | A | C | H | Q | D | H | T | H | F | K | T |
| 2 | C | A | A | A | A | C | G | A | C | H | Q | D | H | T | H | L | K | T |
| 3 | C | A | A | A | A | T | T | A | T | H | Q | D | H | T | T | F | K | I |
| 4 | C | A | A | A | A | T | T | A | C | H | Q | D | H | T | Y | F | K | T |
| 5 | C | A | C | A | A | C | T | A | C | H | Q | A | H | T | H | F | K | T |
| 6 | C | A | A | C | A | T | T | A | C | H | Q | D | P | T | Y | F | K | T |
| 7 | A | A | A | A | A | C | T | A | C | N | Q | D | H | T | H | F | K | T |
| 8 | C | C | C | A | A | C | T | A | C | H | H | A | H | T | H | F | K | T |
| 9 | C | A | A | A | A | C | G | C | C | H | Q | D | H | T | H | L | N | T |
| 10 | C | A | A | C | A | T | T | A | T | H | Q | D | P | T | Y | F | K | I |
| 11 | C | A | A | C | A | C | T | A | C | H | Q | D | P | T | H | F | K | T |
| 12 | C | A | A | A | G | T | G | C | C | H | Q | D | H | A | Y | L | N | T |
CT-genotypes of V. cholerae O1/O139.
Figure 1
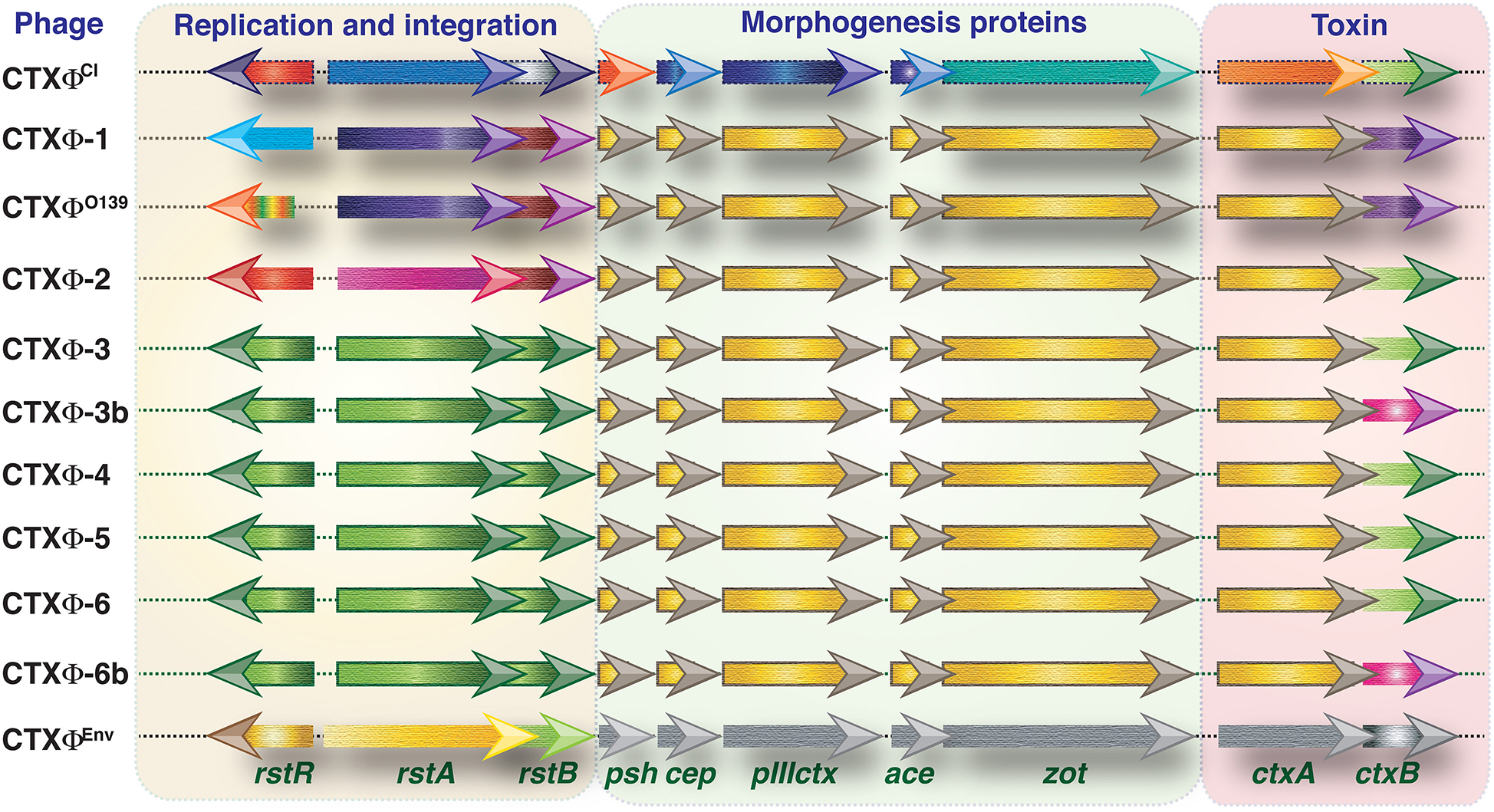
Genetic structure of CTX phages. Different classes of CTXϕs present in the genome of toxigenic V. cholerae strains. Except CTXϕEnv all other phages are from the clinical isolates of V. cholerae. Types of CTXϕ are classified mainly based on their origin of isolation. Functions associated with replication and integration, morphogenesis and toxin production are clustered into three segments. Arrows indicate different open reading frames (not shown to scale). Directions of arrows indicate direction of transcriptions. Similar color indicates identical DNA sequences. The difference in color indicates variances in DNA sequences.
Table 2
| CTX type | rstR type | ctxB | rstAsequence | rstBsequence | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 27 | 162 | 183 | 258 | 927 | 933 | 942 | 74-76 | 87 | 93 | 105 | 189 | 360 | 364 | 366-368 | 371-372 | 379 | 381 | |||
| CTX-1 | El Tor | 3 | C | C | C | G | T | C | G | GTA | A | T | G | A | A | C | ACC | TT | T | A |
| CTX-3 | El Tor | 1 | C | C | C | G | C | T | T | GTA | A | T | G | A | A | C | ACC | TT | T | A |
| CTX-3b | El Tor | 7 | C | C | C | G | C | T | T | GTA | A | T | G | A | A | C | ACC | TT | T | A |
| CTX-4 | El Tor | 1 | C | C | C | G | C | T | T | Δ | A | T | G | A | A | C | ACC | TT | T | A |
| CTX-5 | El Tor | 1 | C | C | C | G | C | T | T | Δ | T | C | A | A | A | C | ACC | TT | T | A |
| CTX-6 | El Tor | 1 | C | C | C | G | C | T | T | Δ | T | C | A | G | A | C | ACC | TT | T | A |
| CTX-6b | El Tor | 7 | C | C | C | G | C | T | T | Δ | T | C | A | G | A | C | ACC | TT | T | A |
| CTX-2 | Class | 1 | T | T | A | C | T | C | G | Δ | T | C | G | A | A | C | ACC | TT | T | A |
Differences in the sequence of rstA and rstB in various types of CTX phages detected in 7PC V. cholerae.
Δ, deletion of nucleotides. Similar color indicate identical nucleotides in each CTX type.
Horizontal gene transfer (HGT) plays a major role in microbial evolution, allowing microbes to acquire new genes and phenotypes. V. cholerae O139 serogroup might have evolved genetically from V. cholerae serogroup O1 through the alteration of its somatic (O) antigen structure by means of mutations in the O antigen region of El Tor strain or acquisition of El Tor virulence genes by an unknown non-O1 (e. g., O22) V. cholerae (13, 14). SNPs based phylogenomic tree has placed the several O139 isolates into a close cluster, indicating their discrete derivation from the O1 serogroup (15). Despite the presence of two CTX types, (CTX-2 and CTX-3) the O139 isolates descent into one cluster in the wave-2 (3, 16).
Other than the core genome, evaluation of insertion or deletion of bases (indels) and gene elements by HGT allow more resolution to differentiate genetically related isolates. This is because V. cholerae is naturally capable of internalizing the DNA from the environment (17). HGT and recombination events made a large impact on the transmission of virulence factors, antimicrobial resistance genes and on serogroup/serotype modifications in V. cholerae (serogroup O1 to O139; serotype Ogawa with AB antigen to Inaba with AC antigen). A rare Hikojima serotype has also been described that expresses all three antigens (A, B, and C). Evidence indicates that the Hikojima serotype is a transient serotype when V. cholerae O1 undergoes serotype switching from Ogawa to Inaba (18).
Two genomic regions of the Vibrio seventh pandemic island, VSP-I and VSP-II were found to be unique to the El Tor and O139 vibrios (19, 20). In El Tor vibrios, VSP-II is a part of a novel genomic island (GEI) encompassing VC0490-VC0516. Several variants in this VSP-II have been reported from many countries (12, 21–24). The wave-1 West African–South American (WASA) clade is differentiated from all other V. cholerae by gaining novel VSP-II genes (20) and a genomic island (WASA-1) (3). VSP-II gene variants (deletions of VC0495-VC0498, VC0495-VC0500, or VC0495-VC0512) were found in wave 3 7CP isolates. These deletions might have been driven by ISVch4, which was inserted in VC0495 in early wave-3 isolates (Table 3). WASA-1 genomic island was also identified in African and Latin American isolates during the late 1980s (T1, LAT-1). V. cholerae O1 isolated between 2007 and 2014 from Kolkata were divided into lineage 1, which had VSP-IIB and ctxB1, and lineage 2 that contained VSP-IIC and ctxB7 (24).
Table 3
| Wave/sub-lineage | MRCA in years | Spread from | Spread into | Features | References |
|---|---|---|---|---|---|
| Wave-1 | 1938–61 | Indonesia | Indian S | ctxB3, CTX-1 with rstRElTor on chrom-1, no ICE SXT/R391 | (3) |
| 1967–71 | Indian SC | South-East Asia | |||
| 1975–85 | Indian SC | East Asia | |||
| 1967–89 | Indian SC | Mozambique | |||
| 1969–81 | Indian SC | Angola | |||
| 1973–78 | Indian SC | Middle East | |||
| 1974–75 | Indian SC | East-Europe | |||
| 1973–85 | Indian SC | Ethiopia | |||
| 1981–85 | Angola | US-Gulf coast Latin America | |||
| Wave-2 | 1990–93 | Indian SC | South-East Asia | ctxB1 and tandem repeat of CTX-2 on chrom-2, ctxB4 to ctxB6 in O139, ICE SXT/R391 | (3) |
| 1992–02 | South-East Asia | East Africa | |||
| Wave-3 | 1989–97 | Indian SC | Nairobi Tanzania Djibou | ctxB1 or ctxB7 rstRElTor in CTX3 to CTX6, ICEVchInd5 | (3) |
| 2003–07 | Indian SC | South-East Asia | |||
| 2005–09 | Indian SC | Haiti | |||
| T1 (wave-1) | 1970–75 | Middle East | North Africa, West Africa, East Africa | ctxB3, VSP-II var-WASA, WASA-1 | (31) |
| T2 (wave-1) | 1989–91 | West Africa | Latin America | ctxB3, VSP-II, WASA-1 | |
| T3 (wave-1) | 1970 | Middle East | East Africa | ctxB3, VSP-II | |
| T4 (wave-1) | 1970–78 | Middle East | East Africa | ctxB3, VSP-II | |
| T5 (wave-1) | 1970–72 | Middle East | East Africa, Central Africa | ctxB3, VSP-II, IncA/C plasmid | |
| T6 (wave-1) | 1986–89 | Middle East | East Africa | ctxB3, VSP-II | |
| T7 (wave-2) | 1982–84 | East Asia | North Africa, West Africa | ctxB3, VSP-II var-Cameroon, | |
| T8 (wave-2) | 1994–98 | Middle East | East Africa, South Africa | ctxB1, ICEVchBan9 | |
| T9 (wave-3) | 1988–91 | South-East Asia | West Africa | ctxB1, VSP-II (VC0495::ISVch4), ICEVchInd5 | |
| T10 (wave-3) | 1991–95 | South-East Asia | East Africa, Central Africa | ctxB1, VSP-II (ΔVC0495-VC0498), ICEVchInd5 | |
| T11 (wave-3) | 2001 | South-East Asia | East Africa, South Africa | ctxB1, VSP-II (ΔVC0495–VC0512), ICEVchInd5 | |
| T12 (wave-3) | 2007 | South-East Asia | West Africa | ctxB7, (ΔVC0495–VC0512), ICEVchInd5 | |
| T13 (wave-3) | 2015–16 | Indian SC | East Africa and Yemen | ctxB7, tcpACIRS101 VSP-II (ΔVC0495–VC0512), ICEVchInd5/Ban5 | (31) |
| LAT-1 (wave-1) | 1989 | Western and Central Africa | Latin America | ctxB3, VSP-II var-WASA, WASA-1 | (7) |
| LAT-2 (wave-1) | 1987–1989 | South/South-East Asia | Central America | ctxB1 in Chrom-2 | |
| LAT-3 (wave-3) | 2010 | South Asia | Haiti, Latin America | ctxB7, VSP-II (ΔVC0495-VC0512), ICEVchInd5/Ban5 |
Inferred origin and transmission events of V. cholerae O1 with cholera waves, phylogenetic sublineages of introduction events (T), and Latin American transmission (LAT).
Present Status of Classical V. Cholerae O1
Globally, the 7CP is dominated by the El Tor vibrios. Existence of V. cholerae O1 classical biotype was reported from Bangladesh during 1961–1968 and 1982–1992 (25), Mexico during 1991–1997 (26) and Thailand in 2000 (27). This indicates that the classical biotype actually persisted longer than previously thought. The Bangladesh classical vibrios isolated in two events remained clonally distinct (25). WGS based phylogenetic analysis displayed that classical biotype isolates in Mexico are part of the classical lineage, and not derived from the Gulf Coast lineage (8).
Cholera Waves Triggered by El Tor Vibrios
Based on several molecular features, V. cholerae has been classified into three waves of cholera (3). The features of each wave are as follows:
Wave-1
The wave-1 spanned 1961–1999. Isolates belonging to this wave contains CTX-1 that harbors rstRElTor on chromosome 1, CT genotype-3 (ctxB3), but not the ICE of the SXT/R391 family. V. cholerae O1 of this wave may have ascended with the addition of TLC element and then by CTX-1 and RS1 element. Wave-1 spread from Indonesia to the Bay of Bengal region in the Indian subcontinent and then to East Africa and South-West Americas (Table 3).
Wave-2
Wave-2 isolates were dominant between 1978 and 1984. Isolates of this wave were first identified in the Indian subcontinent. These isolates have ctxB1 and tandem repeat of CTX-2 on chromosome 2. Some of them may contain CTX-1 and/or RS1 on chromosome 1. Wave-2 spread to East Asia and Africa (Table 3). CT genotypes-4 to 6 (ctxB4-ctB6) were detected in V. cholerae O139 isolated from Bangladesh during 1999–2005 (28). Isolates of this wave may or may not contain R391 family ICE-SXT element (3).
Wave-3
The wave-3 isolates harbor CT-genotype 1 (1991–2010) or CT-genotype 7 (2010~). Wave-3 isolates mainly have rstRElTor:TLC:RS1:CTX3 to CTX6. Based on the genetic differences, these isolates are subcategorized into three or more sub-clades. The wave-3 clade with ctxB7 allele has emerged from Kolkata, India in 2006 (29) and continues to spread globally, as seen in the Haiti, Yemen and other recent cholera epidemics. V. cholerae isolated from a patient during an outbreak of cholera in Mariupol, Ukraine, in 2011 also contained the ctxB7 (30). Almost all the V. cholerae isolates belonging to wave-3 contain ICE of the SXT/R391 family, mostly the ICEVchInd5.
Global Transmission of Cholera
Historically, cholera has been considered to have originated in Asia and spread from there. All the three waves began with Asiatic cholera. China has played an important role in the earliest global transmission of 7CP after its appearance in Indonesia (31). V. cholerae O1 had entered China four times from South Asia (1975–2004). From China, cholera transmission has occurred in South Asia (three times during 1967–1999), Indonesia (1960) and Southeast Asia (2007) (31). Based on the phylogenetic analysis, V. cholerae representing the cholera wave-3 have been subdivided into several clades (Table 3). This includes T1–T12 from African countries (32) LAT-1 to LAT-3 from Latin Americas (7) and T13 from East Africa and Yemen (33). Several monophyletic clades in V. cholerae O1 isolated in China (31), in these, clade 3.A links African epidemic isolates that may be imported from South Asia, clades 3.B contains isolates representing Haiti outbreak as well as from South Asia. In China, cholera outbreaks in 2008 and 2010 were caused by isolates that represented clade 3.B from South Asia (31).
The causative pathogen of the Haiti 2010 outbreak was from a Nepal cluster in 3.C, which is consistent with previous reports (34, 35). An isolate from Cameroon in 2010 was assumed to be a close genetic relative of the Haiti outbreak strain (31). However, phylogeny based on the SNPs within the orthologous core genes revealed a closer relationship between Haitian and Indian isolates than with those from Cameroon (36). Cholera outbreaks have been reported in Russia during 1990–2001, mostly from Stavropol, Republic of Dagestan, Vladivostok, and Yuzhno-Sakhalinsk. V. cholerae O1 El Tor isolates from Siberia and the Far East during 1990s belonged to wave-2 and-3 with CT-genotype 1 (37). A new CTX prophage variant (CTX-2a) linking the traits of CTX-2 and CTX-1 was identified. Phylogenetic analysis distinguished the common ancestors of Russian isolates from Bangladesh and Turkey (37).
During 2016–2018, Yemen faced the largest documented cholera epidemic with 1,103,683 cases and 2,385 deaths (38). Yemen cholera epidemic originated from a recently emerged 7CP wave-3 clade, which contains the ctxB7. This clade has been identified as sublineage T12 in West Africa in 2008, Haiti in 2010 and as T13 in East Africa between 2013 and 2014 before its spread in Yemen (33). Phylogenetic analysis revealed that the V. cholerae O1 isolates from Yemen are different from those in circulation in the Middle East countries where cholera was previously identified. The Yemen isolates are similar to isolates (T13) found in the Eastern African countries, such as Kenya, Tanzania and Uganda in 2015–2016 (33). V. cholerae that belongs to sub-lineage T13 includes the presence of a variant tcpA (tcpACIRS101) and a deletion within VSP-II (VC0495–VC0512) (33).
Cholera in the Americas
Cholera was not reported from Latin America for nearly 100 years. Cholera cases representing small outbreaks have been reported in 1970s from the Gulf Coast (Louisiana and Texas) of the United States and in Mexico during early 1980s (39). The Latin American cholera epidemic began in January 1991 along the Pacific coast of Peru. In the subsequent 6 years, cholera spread swiftly to Ecuador, Colombia, Chile, Brazil, the US, Mexico, Guatemala, Bolivia, and El Salvador, infecting 1.2 million people with 12,000 deaths until 1997 (40). WGS-based maximum likelihood phylogeny analysis identified the epidemic clone as Latin American transmission (LAT-1) sublineage containing ctxB3. This sublineage corresponded to the T2 sublineage described by Weill et al. (33). Western Africa experienced cholera outbreaks caused by T2 during the 1980s and hence this region would have been the source for the LAT-1/T2 sublineage. This sublineage was still found during 2004–2010 in Mexico and it harbored a truncated CTXφ duplication, which is unique among the 7CP strains (41).
The introduction of the LAT-2 sublineage to Mexico might be from South Asia or China or introduction via Eastern Europe. Introduction of wave-2 second sub-lineage (LAT-2) may have occurred during 1987–1989, parallel with that of the LAT-1 introduction. In 2010, the 7CP was introduced into Haiti, affecting about 800,000 with 9,600 deaths (42). The Haitian isolates of V. cholerae O1 were distinguished as LAT-3, has its origin from the Asia region. Even though V. cholerae O1 biotype El Tor was identified in most of the cholera cases, the classical biotype was identified in Mexico until 1997 (26, 41). Previous investigations identified 11 different lineages in Latin American V. cholerae O1 isolates. Some of these include, the Gulf Coast lineage (43), ribotypes MX1 to MX3 lineages in Mexico (44), endemic Latin American 1 (ELA-1) lineage from Brazil and Argentina (45, 46). These results display that the Latin American cholera epidemics were due to multiple introductions and not derived from indigenous local lineages. In addition, ctxB3, ctxB1, and ctxB7 were identified in LAT-1, LAT-2, and LAT-3, respectively.
African Cholera
The 7CP first emerged during 1970, in the Middle East and West Africa with more than 150,000 cases and 20,000 deaths (47, 48). After these initial outbreaks, cholera became endemic in many African countries. During 1970–2011, Democratic Republic of the Congo, Mozambique, Nigeria and United Republic of Tanzania were affected by large number of cholera cases (260,000–390,000) and deaths (11,000–25,000) (49). Previous molecular studies could not give substantial evidence about the origin and spread of V. cholerae from these outbreaks. The genomic approach has identified several transmission events of cholera from South Asia to Africa (T1, T3–T13) and from Africa to America (T2) or to Asia (T13) (32). Of these, T1–T5, T6–T8, T9–12 belongs to wave-1, wave-2, and wave-3, respectively (32). T1 occurred during the 1970s in South/East Asia and trailed to Middle East and Russia.
In Europe, only imported cases of cholera have been reported in the recent past. The European T1 isolates of early 1970s had its origin in West or North Africa. T2 (1989–1991) was responsible for the spread of cholera from West Africa to Peru for the Latin American epidemic during 1990s (Table 3). In Northeastern Africa, T3 (1970), T4 (1970–1978) and T5 (1970–1972) were associated with several outbreaks originating from the Middle East. T5 was linked with large outbreaks in Rwanda in 1994. The T6 sublineage was also imported from the Middle East to the Horn of Africa/East Africa during 1986–1989. The South Asia origin T7 (1982–1984) was detected in isolates from several outbreaks in North and West Africa. T8 sublineage from the Middle East was associated with outbreaks in South Africa in 2001–2002. Zimbabwe outbreaks in 2008–2009 were linked with T8 and T11. Most of the African countries were affected by T9–T12 during 1990–2014 and these sub-lineages originated in South Asia.
Alterations in Antimicrobial Resistance
As an adjunct to rehydration therapy, antibiotics have been used in the treatment of cholera to shorten the duration of diarrhea and to limit the bacterial spread. Over the years, resistance to many useful antibiotics developed, which include chloramphenicol, furazolidone, trimethoprim-sulfamethoxazole, nalidixic acid, tetracycline, and fluoroquinolones (50). Antibiotic resistance profiles have also been used to designate the V. cholerae strains in the epidemiology of cholera. Since the 1960s, many resistance phenotypes of V. cholerae have been reported in Asian regions. In Africa, the first antibiotic-resistant V. cholerae isolates was described in Tanzania during 1979 (32). The genetic basis of antibiotic resistance is mostly due to the existence of genes conferring drug resistance in the plasmids, chromosomal mutations and ICEs of the SXT/R391 family. Incompatibility (Inc) group A/C plasmids are stably maintained in V. cholerae that emerged during the 7CP (51). Some isolates of T1, T5, T6, and T10 V. cholerae from West/East Africa isolates were shown to contain a multidrug resistance encoding IncA/C plasmid (32).
ICEs are mobile genetic elements integrated into the bacterial genome. In V. cholerae, the R391 family ICEs play a crucial role in acquiring antimicrobial resistance genes and are widely used as one of the markers of clonal expansion of the pathogen. The ICE-SXT element was first reported in V. cholerae serogroup O139 from India that emerged during 1993 (52). Based on the WGS phylogenetic analysis of wave-2 7CP isolates, the calculated acquisition time of SXT element was between 1978 and 1984, much earlier to its detection in the O139 serogroup (53). This interpretation corresponds with the discovery of SXT in a V. cholerae O1 isolate from Vietnam during 1990 that was devoid of any resistance gene cassettes (54). Resistance to 2,4-diamino-6, 7-diisopropylpteridine (vibriostatic agent O/129) has been strongly correlated with chloramphenicol, streptomycin, trimethoprim-sulfamethoxazole resistance in V. cholerae O1 isolated during 1989–1990 in Kolkata, India (55). However, the role of ICE-SXT/R391 was not determined in these isolates.
SXT/R391 ICE has been detected in all most all the isolates that represented wave-2 and wave-3 (3). SXTs are reported to display polymorphism, with their genetic backbone and resistance encoding genes to chloramphenicol (floR), streptomycin (strA and strB), trimethoprim (dfrA18 and dfrA1), sulfamethoxazole (sul2), and tetracycline (tetA and tetR). The type of SXT ICE element is specific to geographical regions. More than 30 ICEs have been grouped within the SXT/R391 family of clinical and environmental V. cholerae isolates (56). Worldwide, ICEVchInd5/ICEVchBan5 is very common among V. cholerae. In Africa, sublineages T9 to T13 that originated from South Asia carried SXT/R391 with ICEVchInd5/ICEVchBan5 (32, 33). SXT/R391 ICE was absent in the epidemic V. cholerae O1 isolated in Latin America in the 1990s (3, 12). Deletion of ICEVchInd5/ICEVchBan5-like SXT/R391-ICE including the variable region-III that encodes resistance to streptomycin (strA and strB), chloramphenicol (floR), and sulfonamides (sul2) has also been found in the recent Yemen isolates (33). This trend has been reported in several other 7CP wave-3 isolates (32, 57).
Conventionally, polymyxin-B susceptibility testing is used to differentiate classical and El Tor biotypes, in which the latter is always resistant. Polymyxin-B susceptible V. cholerae O1 El Tor isolates emerged in 2012 Kolkata and spread to other parts of India (58, 59), East Africa and Yemen in 2016 (33). A non-synonymous SNV in vprA (VC1320) i.e., change in the VprA (D89N) was found for this polymyxin susceptible conversion among El Tor V. cholerae isolates (33). It is possible to determine the AMR encoding genes using the WGS. However, adequate evidence to support clinical decision-making based on the genome based antibiotic susceptibility testing is still not available (60).
The Other Important Attributes of V. Cholerae
Most of the 7PC isolates of O1 and O139 serogroups belonged to multilocus sequence type ST69 whereas V. cholerae O1 classical variants belonged to ST73 (27, 61, 62). Serotype changing of V. cholerae O1 from Ogawa to Inaba and back to Ogawa has been seen in many cholera endemic regions. This change is associated with mutational disruption of the methyltransferase encoded in the serotype-determining wbeT, which is part of the O1 somatic antigen. Shift in serotypes of Ogawa and Inaba was previously thought due to unknown selection mechanisms based on modifications of the O1 antigen or in the wbeT (63, 64). WGS analysis of African isolates of Inaba serotype showed alterations in the wbeT gene, including premature stop codons caused by SNPs or short indels, non-synonymous SNPs, insertion sequences, integrations, deletions and disruption of the start codon (32).
Expression of CT in classical and El Tor vibrios depends on many factors. More than 500 genes (13%) have been found to be differentially expressed in classical and El Tor biotypes. Biofilm formation, chemotaxis, transport of amino acids/peptides and iron encoding genes that support environmental survival are expressed at a higher level in the El Tor biotype, whereas expression of vieSAB genes encoding ctxA transcription is more in the classical biotype leading to its higher virulence (65). ToxRS contribute in the activation of the toxT promoter, which encodes another transcriptional activator and triggers other virulence genes. In classical V. cholerae isolates, the ToxRS directly activates ctxAB in a ToxT-independent manner in the presence of bile acids (66). This mechanism is not found in El Tor vibrios, as there is a difference in the number of heptad repeats (TTTTGAT) in the upstream regions.
Clustered regularly interspersed short palindromic repeats-associated proteins (CRISPR-Cas) are microbial nuclease systems in the chromosome that are involved in defense against entering nucleic acids/bacteriophages. Bacteria also resist phages by hosting phage-inducible chromosomal islands (PICI) which prevent phage reproduction. In classical biotype, CRISPR-Cas is widely present (67), whereas most of the El Tor vibrios lack this system (68). Many clinical El Tor isolates from Bangladesh were found to encode a phage-inhibitory chromosomal island, known as a PICI-like element (PLE) (67). PLE-mediated inhibition of phage replication is common in V. cholerae O1 El Tor (69). A CRISPR array consists of short, direct repeats divided by several short sequences called spacers. The spacer sequences of the CRISPR arrays carried 100% sequence homology with different regions of the PLE (67).
Conclusions
Comparative genomic analysis of V. cholerae might give answer to several challenging and historical questions that could only be speculated previously. Based on the available scientific information/evidence, action plans have been set to achieve a 90% reduction in cholera deaths in 20 endemic countries by 2030 (70). WGS will form a critical component in accomplishing the sustained developmental goal (SDG) with several important applications including molecular typing, antimicrobial resistance, new diagnostics and vaccination strategies.
Statements
Author contributions
TR conceptualized and wrote the initial draft. AM and F-XW contributed critical interpretation of the content. AG, BD, and GN critically appraised and revised the manuscript. All the authors read and approved the final manuscript. AG is J. C. Bose Chair Professor of the National Academy of Sciences, India (NASI).
Funding
This study was supported in part by the Department of Biotechnology (DBT), Government of India (Grant BT/MB/THSTI/HMC-SFC/2011).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
WHO. Cholera. World Health Organization fact sheet (2017). Available online at: http://www.who.int/mediacentre/factsheets/fs107/en/ (Accessed December 26, 2018).
2.
World Health Organization (WHO). Cholera 2017. Weekly Epidemiol Rec. (2018) 93:489–500.
3.
MutrejaAKimDWThomsonNRConnorTRLeeJHKariukiSet al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. (2011) 477:462–5. 10.1038/nature10392
4.
RamamurthyTYamasakiSTakedaYNairGB. Vibrio cholerae O139 Bengal: odyssey of a fortuitous variant. Microbes Infect. (2003) 5:329–44. 10.1016/S1286-4579(03)00035-2
5.
NguyenBMLeeJHCuongNTChoiSYHienNTAnhDDet al. Cholera outbreaks caused by an altered Vibrio cholerae O1 El Tor biotype strain producing classical cholera toxin B in Vietnam in 2007 to 2008. J Clin Microbiol. (2009) 47:1568–71. 10.1128/JCM.02040-08
6.
GrimCJHasanNATavianiEHaleyBChunJBrettinTSet al. Genome sequence of hybrid Vibrio cholerae O1 MJ-1236, B-33, and CIRS101 and comparative genomics with V. cholerae. J Bacteriol. (2010) 192:3524–33. 10.1128/JB.00040-10
7.
KimEJLeeCHNairGBKimDW. Whole-genome sequence comparisons reveal the evolution of Vibrio cholerae O1. Trends Microbiol. (2015) 23:479–89. 10.1016/j.tim.2015.03.010
8.
DommanDQuiliciMLDormanMJNjamkepoEMutrejaAMatherAEet al. Integrated view of Vibrio cholerae in the Americas. Science. (2017) 358:789–93. 10.1126/science.aao2136
9.
HuDLiuBFengLDingPGuoXWangMet al. Origins of the current seventh cholera pandemic. Proc Natl Acad Sci USA. (2016) 113:E7730–9. 10.1073/pnas.1608732113
10.
WaldorMKMekalanosJJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. (1996) 272:1910–14. 10.1126/science.272.5270.1910
11.
DavisBMMoyerKEBoydEFWaldorMK. CTX prophages in classical biotype Vibrio cholerae: functional phage genes but dysfunctional phage genomes. J Bacteriol. (2000) 182:6992–8. 10.1128/JB.182.24.6992-6998.2000
12.
ChoiSYRashedSMHasanNAAlamMIslamTSadiqueAet al. Phylogenetic diversity of Vibrio cholerae associated with endemic cholera in Mexico from 1991 to 2008. mBio. (2016) 7:e02160. 10.1128/mBio.02160-15
13.
JohnsonJASallesCAPanigrahiPAlbertMJWrightACJohnsonRJet al. Vibrio cholerae O139 synonym Bengal is closely related to Vibrio cholerae El Tor but has important differences. Infect Immun. (1994) 62:2108–10.
14.
BikeEMBunschotenAEGouwRDMooiFRGenesis of the novel epidemic Vibrio cholerae O139 strain: evidence for horizontal transfer of genes involved in polysaccharide synthesis. EMBO J. (1995) 14:209–16. 10.1002/j.1460-2075.1995.tb06993.x
15.
DutilhBEThompsonCCVicenteACMarinMALeeCSilvaGGet al. Comparative genomics of 274 Vibrio cholerae genomes reveals mobile functions structuring three niche dimensions. BMC Genomics. (2014) 15:654. 10.1186/1471-2164-15-654
16.
FaruqueSMMekalanosJJ. Pathogenicity islands and phages in Vibrio cholerae evolution. Trends Microbiol. (2003) 11:505–10. 10.1016/j.tim.2003.09.003
17.
MeibomKLBlokeschMDolganovNAWuCYSchoolnikGK. Chitin induces natural competence in Vibrio cholerae. Science. (2005) 310:1824–27. 10.1126/science.1120096
18.
ManningPAStroeherUHMoronaR. Molecular basis for O-antigen biosynthesis in Vibrio cholerae O1: Ogawa–Inaba switching. In: WachsmuthIKOlsvikØ editor. Vibrio cholerae and Cholera: Molecular to Global Perspectives. Washington, DC: American Society of Microbiologists Press (1994). p. 77–94.
19.
DziejmanMBalonEBoydDFraserCMHeidelbergJFMekalanosJJ. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci USA. (2002) 99:1556–61. 10.1073/pnas.042667999
20.
O'SheaYAFinnanSReenFJMorrisseyJPO'GaraFBoydEF. The Vibrio seventh pandemic island-II is a 26.9 kb genomic island present in Vibrio cholerae El Tor and O139 serogroup isolates that shows homology to a 43.4 kb genomic island in V. vulnificus. Microbiology. (2004) 150:4053–63. 10.1099/mic.0.27172-0
21.
NusrinSGilAIBhuiyanNASafaAAsakuraMLanataCFet al. Peruvian Vibrio cholerae O1 El Tor strains possess a distinct region in the Vibrio seventh pandemic island-II that differentiates them from the prototype seventh pandemic El Tor strains. J Med Microbiol. (2009) 58:342–54. 10.1099/jmm.0.005397-0
22.
BhuiyanNANusrinSAnsaruzzamanMIslamASultanaMAlamMet al. Genetic characterization of Vibrio cholerae O1 strains isolated in Zambia during 1996–2004 possessing the unique VSP-II region of El Tor variant. Epidemiol Infect. (2012) 140:510–18. 10.1017/S0950268811000926
23.
TavianiEGrimCJChoiJChunJHaleyBHasanNAet al. Discovery of novel Vibrio cholerae VSP-II genomic islands using comparative genomic analysis. FEMS Microbiol Lett. (2010) 308:130–37. 10.1111/j.1574-6968.2010.02008.x
24.
ImamuraDMoritaMSekizukaTMizunoTTakemuraTYamahiroTet al. Comparative genome analysis of VSP-II and SNPs reveals heterogenic variation in contemporary strains of Vibrio cholerae O1 isolated from cholera patients in Kolkata, India. PLoS Negl Trop Dis. (2017) 11:e0005386. 10.1371/journal.pntd.0005386
25.
FaruqueSMAbdul AlimARRahmanMMSiddiqueAKSackRBAlbertMJ. Clonal relationships among classical Vibrio cholerae O1 strains isolated between 1961 and 1992 in Bangladesh. J Clin Microbiol. (1993) 31:2513–16.
26.
AlamMNusrinSIslamABhuiyanNARahimNDelgadoGet al. 2010. Cholera between 1991 and 1997 in Mexico was associated with infection by classical, El Tor, and El Tor variants of Vibrio cholerae. J Clin Microbiol. (2010) 48:3666–74. 10.1128/JCM.00866-10
27.
SiriphapALeekitcharoenphonPKaasRSTheethakaewCAarestrupFMSutheinkulOet al. Characterization and genetic variation of Vibrio cholerae isolated from clinical and environmental sources in Thailand. PLoS ONE. (2017) 12:e0169324. 10.1371/journal.pone.0169324
28.
BhuiyanNANusrinSAlamMMoritaMWatanabeHRamamurthyTet al. Changing genotypes of cholera toxin (CT) of Vibrio cholerae O139 in Bangladesh and description of three new CT genotypes. FEMS Immunol Med Microbiol. (2009) 57:136–41. 10.1111/j.1574-695X.2009.00590.x
29.
NahaAPazhaniGPGangulyMGhoshSRamamurthyTNandyRKet al. Development and evaluation of a PCR assay for tracking the emergence and dissemination of Haitian variant ctxB in Vibrio cholerae O1 strains isolated from Kolkata, India. J Clin Microbiol. (2012) 50:1733–36. 10.1128/JCM.00387-12
30.
SmirnovaNIKrasnovYMAgafonovaEYShchelkanovaEYAlkhovaZVKutyrevVV. Whole-genome sequencing of Vibrio cholerae O1 El Tor strains isolated in ukraine. (2011) and Russia (2014). Genome Announc. (2017) 5:e01640-16. 10.1128/genomeA.01640-16
31.
DidelotXPangBZhouZMcCannANiPLiDet al. The role of China in the global spread of the current cholera pandemic. PLoS Genet. (2015) 11:e1005072. 10.1371/journal.pgen.1005072
32.
WeillF-XDommanDNjamkepoETarrCRauzierJFawalNet al. Genomic history of the seventh pandemic of cholera in Africa. Science. (2017) 358:785–89. 10.1126/science.aad5901
33.
WeillF-XDommanDNjamkepoEAlmesbahiAANajiMANasherSSet al. A recent seventh pandemic Vibrio cholerae O1 El Tor lineage identified as the cause of the 2016-2017 cholera outbreak in Yemen. Nature. (2019) 565:230–33. 10.1038/s41586-018-0818-3
34.
HendriksenRSPriceLBSchuppJMGilleceJDKaasRSEngelthalerDMet al. Population genetics of Vibrio cholerae from Nepal in 2010: evidence on the origin of the Haitian outbreak. mBio. (2011) 2:e00157-11. 10.1128/mBio.00157-11
35.
ChinCSSorensonJHarrisJBRobinsWPCharlesRCJean-CharlesRRet al. The origin of the Haitian cholera outbreak strain. N Engl J Med. (2011) 364:33–42. 10.1056/NEJMoa1012928
36.
ReimerARVan DomselaarGStroikaSWalkerMKentHTarrCet al. Comparative genomics of Vibrio cholerae from Haiti, Asia, and Africa. Emerg Infect Dis. (2011) 17:2113–21. 10.3201/eid1711.110794
37.
MironovaLVGladkikhASPonomarevaASFeranchukSIBochalginN?BasovEAet al. Comparative genomics of Vibrio cholerae El Tor strains isolated at epidemic complications in Siberia and at the Far East. Infect Genet Evol. (2018) 60:80–8. 10.1016/j.meegid.2018.02.023
38.
CamachoABouheniaMAlyusfiRAlkohlaniANajiMAMdeRadiguès Xet al. Cholera epidemic in Yemen, 2016-18: an analysis of surveillance data. Lancet Glob Health. (2018) 6:e680–90. 10.1016/S2214-109X(18)30230-4
39.
BlakePAWachsmuthKDavisBRBoppCAChaikenBPLeeJV. Toxigenic Vibrio cholerae O1 strain from Mexico identical to United States isolates. Lancet. (1983) 2:912. 10.1016/S0140-6736(83)90894-2
40.
KumateJSepúlvedaJGutiérrezG. Cholera epidemiology in Latin America and perspectives for eradication. Bull Inst Pasteur. (1998) 96:217–26. 10.1016/S0020-2452(99)80002-5
41.
AlamMRashedSMMannanSBIslamTLizarraga-PartidaMLDelgadoGet al., 2014. Occurrence in Mexico, 1998-2008, of Vibrio cholerae CTX El Tor carrying an additional truncated CTX prophage. Proc Natl Acad Sci USA. (2014) 111:9917–22. 10.1073/pnas.1323408111
42.
ZarocostasJ. Cholera outbreak in Haiti-from 2010 to today. Lancet. (2017) 389:2274–75. 10.1016/S0140-6736(17)31581-7
43.
KaperJBBradfordHBRobertsNCFalkowS. Molecular epidemiology of Vibrio cholerae in the U.S. Gulf Coast. J Clin Microbiol. (1982) 16:129–34.
44.
Lizárraga-PartidaMLQuiliciML. Molecular analyses of Vibrio cholerae O1 clinical strains, including new nontoxigenic variants isolated in Mexico during the Cholera epidemic years between 1991 and 2000. J Clin Microbiol. (2009) 47:1364–71. 10.1128/JCM.00720-08
45.
CoelhoAAndradeJRVicenteACSallesCA. New variant of Vibrio cholerae O1 from clinical isolates in Amazonia. J Clin Microbiol. (1995) 33:114–18.
46.
PichelMRivasMChinenIMartínFIbarraCBinszteinN. Genetic diversity of Vibrio cholerae O1 in Argentina and emergence of a new variant. J Clin Microbiol. (2003) 41:124–34. 10.1128/JCM.41.1.124-134.2003
47.
CohenJSchwartzTKlasmerRPridanDGhalayiniHDaviesAM. Epidemiological aspects of cholera El Tor outbreak in a non-endemic area. Lancet. (1971) ii:86–9. 10.1016/S0140-6736(71)92056-3
48.
GoodgameRWGreenoughWB. Cholera in Africa: a message for the West. Ann Intern Med. (1975) 82:101–6. 10.7326/0003-4819-82-1-101
49.
MengelMADelrieuIHeyerdahlLGessnerBD. Cholera outbreaks in Africa. Curr Top Microbiol Immunol. (2014) 379:117–44. 10.1007/82_2014_369
50.
GhoshARamamurthyT. Antimicrobials & cholera: are we stranded?Indian J Med Res. (2011) 133:225–31.
51.
RahalKGerbaudGBouanchaudDH. Stability of R plasmids belonging to different incompatibility groups in Vibrio cholerae El tor. Ann Microbiol. (1978) 129:409–14.
52.
WaldorMKTschapeHMekalanosJJ. 1996. A new type of conjugative transposon encodes resistance to sulfamethoxazole, trimethoprim, and streptomycin in Vibrio cholerae O139. J. Bacteriol. (1996) 178:4157–65. 10.1128/jb.178.14.4157-4165.1996
53.
SpagnolettiMCeccarelliDRieuxAFondiMTavianiEFaniRet al. Acquisition and evolution of SXT-R391 integrative conjugative elements in the seventh-pandemic Vibrio cholerae lineage. mBio. (2014) 5:e01356-14. 10.1128/mBio.01356-14
54.
BaniSMastromarinoPNCeccarelliDLe VanASalviaAMNgo VietQTet al. Molecular characterization of ICEVchVie0 and its disappearance in Vibrio cholerae O1 strains isolated in 2003 in Vietnam. FEMS Microbiol Lett. (2007) 266:42–8. 10.1111/j.1574-6968.2006.00518.x
55.
RamamurthyTPalAPalSCNairGB. Taxonomical implications of the emergence of high frequency of occurrence of 2,4-diamino-6,7-diisopropylpteridine-resistant strains of Vibrio cholerae from clinical cases of cholera in Calcutta, India. J Clin Microbiol. (1992) 30:742–3.
56.
PandeKMendirattaDKVijayashriDThamkeDCNarangP. SXT constin among Vibrio cholerae isolates from a tertiary care hospital. Indian J Med Res. (2012) 135:346–50.
57.
KatzLSPetkauABeaulaurierJTylerSAntonovaESTurnsekMAet al. Evolutionary dynamics of Vibrio cholerae O1 following a single-source introduction to Haiti. mBio. (2013) 4:e00398-13. 10.1128/mBio.00398-13
58.
SamantaPGhoshPChowdhuryGRamamurthyTMukhopadhyayAK. Sensitivity to polymyxin B in El Tor Vibrio cholerae O1 strain, Kolkata, India. Emerg Infect Dis. (2015) 21:2100–2. 10.3201/eid2111.150762
59.
SamantaPSahaRNChowdhuryGNahaASarkarSDuttaSet al. Dissemination of newly emerged polymyxin B sensitive Vibrio cholerae O1 containing Haitian-like genetic traits in different parts of India. J Med Microbiol. (2018) 67:1326–33. 10.1099/jmm.0.000783
60.
EllingtonMJEkelundOAarestrupFMCantonRDoumithMGiskeCet al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST Subcommittee. Clin Microbiol Infect. (2017) 23:2–22. 10.1016/j.cmi.2016.11.012
61.
GreigDRSchaeferUOctaviaSHunterEChattawayMADallmanTJet al. Evaluation of whole-genome sequencing for identification and typing of Vibrio cholerae. J Clin Microbiol. (2018) 56:e00831-18. 10.1128/JCM.00831-18
62.
AnandanSDevanga RagupathiNKMuthuirulandi SethuvelDPThangamaniSVeeraraghavanB. Prevailing clone (ST69) of Vibrio cholerae O139 in India over 10 years. Gut Pathog. (2017) 9:60. 10.1186/s13099-017-0210-0
63.
KarlssonSLThomsonNMutrejaAConnorTSurDAliMet al. Retrospective analysis of serotype switching of Vibrio cholerae O1 in a cholera endemic region shows It is a non-random process. PLoS Negl Trop Dis. (2016) 10:e0005044. 10.1371/journal.pntd.0005044
64.
RijpkemaSGDurraniZRamamurthyTNairGB. Assessing clonality of Vibrio cholerae Inaba isolates by characterization of nonsense mutations in wbeT. J Med Microbiol. (2004) 53:1105–7. 10.1099/jmm.0.45744-0
65.
BeyhanSTischlerADCamilliAYildizFH. Differences in gene expression between the classical and El Tor biotypes of Vibrio cholerae O1. Infect Immun. (2006) 74:3633–42. 10.1128/IAI.01750-05
66.
HungDTMekalanosJJ. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc Natl Acad Sci USA. (2005) 102:3028–33. 10.1073/pnas.0409559102
67.
SeedKDLazinskiDWCalderwoodSBCamilliA. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. (2012) 494:489–91. 10.1038/nature11927
68.
NaserIBHoqueMMNahidMATareqTMRockyMKFaruqueSM. Analysis of the CRISPR-Cas system in bacteriophages active on epidemic strains of Vibrio cholerae in Bangladesh. Sci Rep. (2017) 7:14880. 10.1038/s41598-017-14839-2
69.
O'HaraBJBarthZKMcKitterickACSeedKD. A highly specific phage defense system is a conserved feature of the Vibrio cholerae mobilome. PLoS Genet. (2017) 13:e1006838. 10.1371/journal.pgen.1006838
70.
World Health Organization (WHO). Global Task Force on Cholera Control, Ending Cholera-A Global Roadmap to 2030. (2017). Available online at: http://www.who.int/cholera/publications/global-roadmap.pdf
Summary
Keywords
Vibrio cholerae, seventh cholera pandemic, single nucleotide polymorphism, whole-genome sequence, CTX phage, CT-genotype
Citation
Ramamurthy T, Mutreja A, Weill F-X, Das B, Ghosh A and Nair GB (2019) Revisiting the Global Epidemiology of Cholera in Conjunction With the Genomics of Vibrio cholerae. Front. Public Health 7:203. doi: 10.3389/fpubh.2019.00203
Received
31 January 2019
Accepted
08 July 2019
Published
23 July 2019
Volume
7 - 2019
Edited by
Nirmal Kumar Ganguly, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), India
Reviewed by
Firdausi Qadri, International Centre for Diarrhoeal Disease Research (ICDDR), Bangladesh; Ana Afonso, University of São Paulo, Brazil
Updates
Copyright
© 2019 Ramamurthy, Mutreja, Weill, Das, Ghosh and Nair.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thandavarayan Ramamurthy tramu@thsti.res.in
This article was submitted to Infectious Diseases - Surveillance, Prevention and Treatment, a section of the journal Frontiers in Public Health
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.