 Petra Marešová
Petra Marešová Blanka Klímová
Blanka Klímová Jan Honegr
Jan Honegr Kamil Kuča
Kamil Kuča Wan Nur Hidayah Ibrahim
Wan Nur Hidayah Ibrahim Ali Selamat
Ali Selamat- 1Faculty of Informatics and Management, University of Hradec Kralove, Hradec Kralove, Czechia
- 2Biomedical Research Centrum, University Hospital Hradec Kralove, Hradec Kralove, Czechia
- 3Faculty of Computing, Universiti Teknologi Malaysia & Media and Game Innovation Centre of Excellence (MaGICX), Universiti Teknologi Malaysia, Kuala Lumpur, Malaysia
Objective: Medical device development, from the product's conception to release to market, is very complex and relies significantly on the application of exact processes. This paper aims to provide an analysis and summary of current research in the field of medical device development methodologies, discuss its phases, and evaluate the associated legislative and risk aspects.
Methods: The literature search was conducted to detect peer-reviewed studies in Scopus, Web of Science, and Science Direct, on content published between 2007 and November 2019. Based on exclusion and inclusion criteria, 13 papers were included in the first session and 11 were included in the second session. Thus, a total of 24 papers were analyzed. Most of the publications originated in the United States (7 out of 24).
Results: The medical device development process comprises one to seven stages. Six studies also contain a model of the medical device development process for all stages or for just some of the stages. These studies specifically describe the concept stage during which all uncertainties, such as the clinical need definition, customer requirements/needs, finances, reimbursement strategy, team selection, and legal aspects, must be considered.
Conclusion: The crucial factor in healthcare safety is the stability of factors over a long production time. Good manufacturing practices cannot be tested on individual batches of products; they must be inherently built into the manufacturing process. The key issues that must be addressed in the future are the consistency in the classification of devices throughout the EU and globally, and the transparency of approval processes.
Introduction
Each year, a considerable amount of medical technologies are developed (1), and billions of crowns are invested in their development to meet the increasing demand for medical technology innovation (MDI). Research shows that extensive implementation of healthcare services worldwide is heavily dependent on medical technologies. According to the healthcare use statistics provided by Organization for Economic Co-operation and Development (OECD) (2), numbers of medical technologies are constantly rising. As a result, more healthcare technology needs to be developed (3). Innovative processes in the pharmaceutical industry appear every 10–20 years, while medical technology becomes outdated within months. Thus, new medical device development processes, which meet the needs of contemporary drug treatments, are currently being investigated and developed.
Nevertheless, there are only a few technologies and resources that penetrate the market. Medical device development (MDD) is expensive and risky. High risk of technology failure in the market leads to the question: Would it be appropriate to create a process or guide to assess healthcare technology at the beginning of the development process so that the development process and future impacts can be addressed on time? (4).
Currently, around 88% of corporations that develop medical device technologies are not able to provide considerable returns for their investors (5). Companies mainly concentrate on regulatory approval targets, without careful scheduling that considers establishing a less costly and more sustainable process (6). Therefore, well-prepared and well-thought launch strategies that capture inefficiencies in a timely manner and lower total costs are crucial in ensuring a successful product development process and satisfying stakeholder requirements.
Product development, from conception to release to market, is a very complex process (7, 8). It significantly relies on the application of exact processes that enable developers to optimally stage development, testing, validation, verification, and market release (9).
Current MDD processes have to respond to several process challenges (10, 11); projects seldom advance as scheduled, and often modifications are introduced during the course of project development and implementation (12). These processes do not respect the current legislative changes that are taking place at the European Union (EU) level. Risk analysis is mostly separately addressed, with respect to specific phases of MDD.
The MDD process has been satisfactorily described in literature; however, there is a lack of comprehensive models that would support design teams with different experiences and backgrounds. In general, published studies in this area either address the MDD with a specific focus on regulations (9, 13, 14) or provide proposals for various approaches to MDD (9, 15, 16).
This paper provides an analysis and summary of current research in the field of MDD methodologies, discusses the phases of MDD, and evaluates associated legislative and risk aspects.
Methods
Research Strategy
The systematic review is based on PRISMA guidelines (17, 18). The databases searched (by authors P.M. and W.N.) included Scopus (2007–2019) and Web of Science (2007–2019). In addition, legislative documents on the Research Topic, as well as the websites of medical companies dealing with the phases of MDD were explored. Keywords included the following collocations: “medical device AND process AND development” in Web of Knowledge and Scopus. Few more studies were found searching with the more specific keyword groups “medical device AND process AND development AND investment evaluation” and “medical device AND stage development.” A Boolean operator procedure was used in the search. The database was searched from 1 October 2019 until 20 November 2019.
Research Questions
To achieve the objective of this review, the main research questions (RQ) were derived as follows:
RQ1 What are the phases of the MDD process?
RQ2 What are the regulation needs related to MDD?
RQ3 What are the risk factors in MDD?
RQ4 With which phases of the MDD process are regulation needs and risk factors associated?
Article Selection and Data Collection
The article selection process was divided into two sessions, and combined with an analysis, as shown in Figure 1. In the first session, we searched for publications between 2007 and 2017, and in the second session, we searched for articles published between 2017 and 2019.
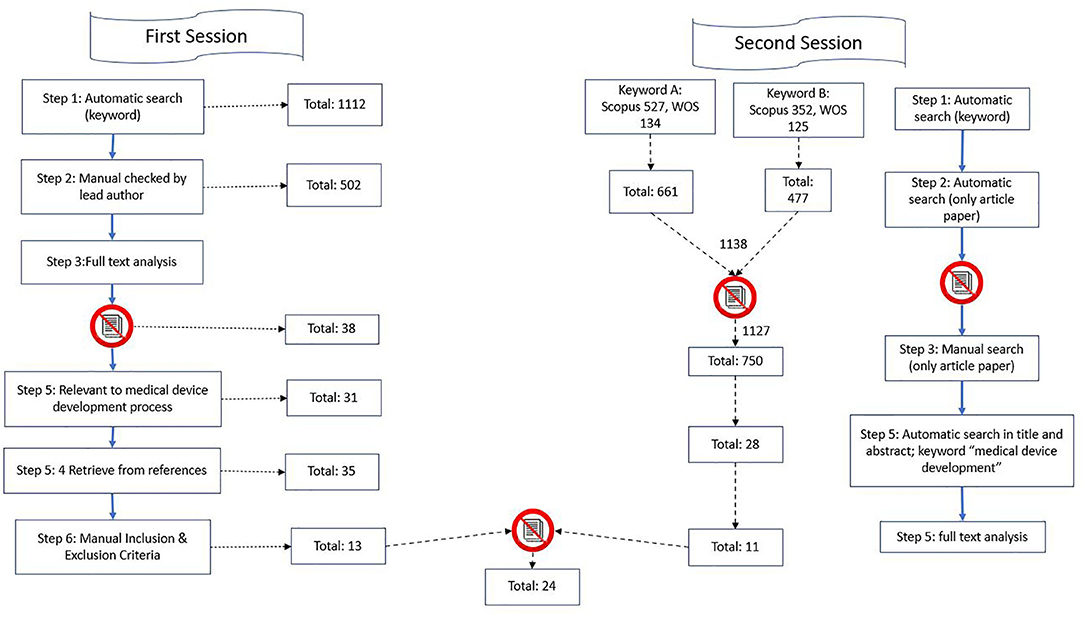
Figure 1. Illustration of search strategy.
The First Session
From the database/journal searches, 1,112 titles/abstracts were retrieved. The titles and abstracts of identified studies were checked by the lead author (J.K.) for relevance. Subsequently, the search was performed again, and it focused on the occurrence of at least one keyword in the title or abstract to significantly narrow down the selection. It provided the authors with a relevant entry-level file base. A total of 82 studies were found. The search procedure is illustrated below. As the search findings in:
Table 1 shows, most of the studies (19) were generated by the keyword string “medical device AND process AND development” in Web of Knowledge and Scopus. A few more studies were found by searching for a more specific keyword group: “medical device AND process AND development AND investment evaluation,” as well as by the search string “medical device AND stage development.”
Table 1. An overview of distribution of publications found in the first session.
In cases of uncertainty, the full text of studies were checked for relevance. After removing duplicates and the titles/abstracts that were unrelated to the stages of the development process, we detected 38 peer-reviewed studies written in English. We included original articles and reviews. Of these, only 31 articles were relevant to the MDD process. These studies were investigated in full by BK and PM, with guidance from PM. Four more studies were detected from the references of the retrieved studies; thus, 35 articles were considered against our study's inclusion and exclusion criteria. On the basis of the criteria, 13 studies were included in the final analysis.
The Second Session
For articles published between 2017 and 2019, the search started with potential keywords based on the trends of the publication (Table 2). Two keywords were used as the main keywords that best corresponded to the objective of this research. Then, the details and abstract of the publication were extracted, and we agreed to narrow down the selection to articles from the database. Only articles that had the string “medical device development” in their title or abstracts were selected. A total of 28 papers were detected to fulfill the criteria. Thereafter, we performed manual full-text analyses, leaving 12 papers after the inclusion and exclusion criteria check. These 12 papers were combined with the 13 papers that we extracted from the first session. The whole process for the first and second sessions is illustrated in Figure 1.

Table 2. An overview of publication distributions for the second session.
Analysis
A combination of reviews and original studies were analyzed. Studies were selected on the basis of the following inclusion criteria:
I1 The publication date of the article is between 2007 and 2017.
I2 Reviewed full-text studies in scientific journals in English.
I3 The aim of the research is to suggest MDD processes.
I4 The study results proposed MDD processes or specifications associated with existing referenced phases of MDD.
I5 The study aimed to provide an overview of existing approaches in relation to risks and valid legislation.
The studies with the following attributes were gradually excluded from the analysis:
E1 The article was not written in English.
E2 The article did not mention the main string (“medical device development”) in its title or abstract.
E3 The article did not concern the research topic. For example,
• Cosgrove et al. (20) focused on a framework of key performance indicators to identify reductions in energy consumption in a medical device production facility;
• Songkajorn and Thawesaengskulthai (3) concentrated on one specific country and the development of medical devices according to the country's legislation;
• Cho and Kim (21) and Shaw (22) aimed at risk analysis;
• Songkajorn and Thawesaengskulthai (3) included incomplete data about the stages of MDD;
• Vaezi et al. (23) focused on the exploration of medical manufacturers' beliefs
• attitudes toward user involvement in the medical device design and development (24);
• Bruse et al. (25) focused on data analysis of image processing that will assist clinicians in decision making during MDD;
• Ciubuc et al. (26) focused on theoretical and experimental approaches to the detection of dopamine.
• The article described the development of healthcare software [e.g., (27, 28)].
E4 The distribution of publications based on their origin is shown in Table 3.

Table 3. The distribution of the publication based on origin.
Text Analysis
During the review process, text analyses were performed to assist the reviewer's decision. We used VOSviewer software to extract the relation between the co-occurrence of keywords before we decided on the keywords to be used in our search. Figures 2, 3 show the mapping of keyword co-occurrence for keywords A and B for the analysis of the second session (publications, 2017–2019).
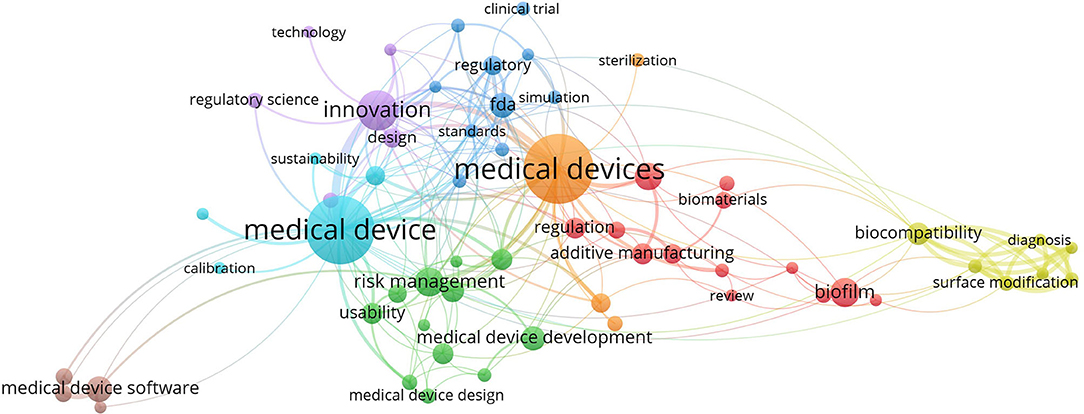
Figure 2. Keyword cluster for the second session; keyword A.
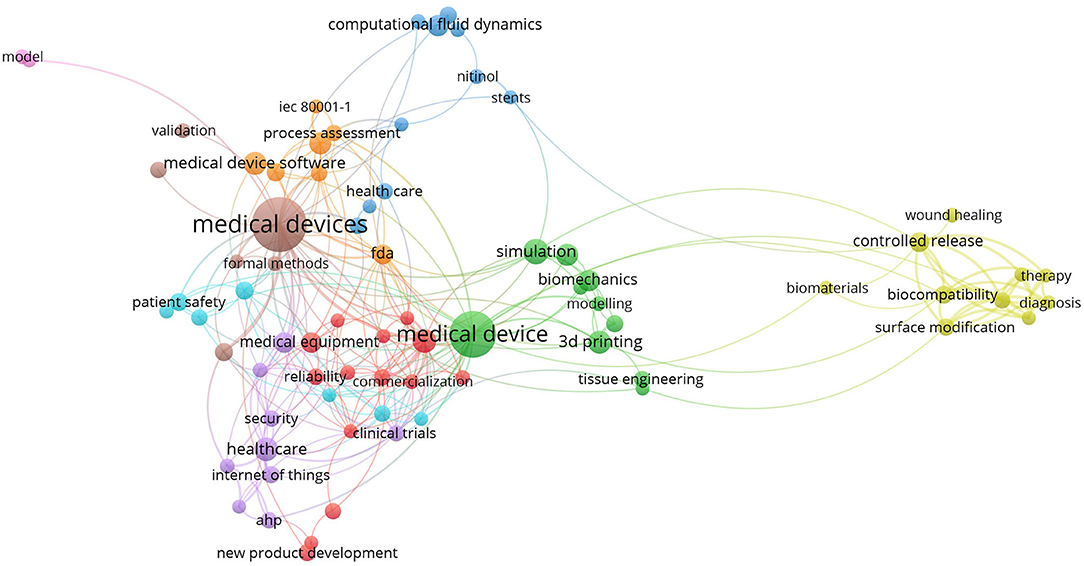
Figure 3. Keyword cluster for the second session; keyword B.
Keyword Clusters
Figure 4 shows the mapping of terms that co-occurred in the title and abstract during Step 5 of both sessions. A total of 61 non-duplicate publications were retrieved (35 from the first session and 28 from the second).

Figure 4. Title/abstract text-mapping in Step 5 (search strategy procedure).
In the abovementioned figures, one can observe the areas that are solved in the publications. After excluding the topics and areas that are directly related to the technical solutions of MDD (e.g., represented by biomaterial, diagnosis, therapy, computational fluid dynamics), two main areas of study remained: regulatory/legislative aspects and risk/risk management. These two areas are further specified as they are related to the stages of MDD.
Results
We detected a total of 13 research studies on the topic. Five of them originated in the United States, four in the United Kingdom, one in Canada, one in Thailand, and one in Portugal. One study was of multiple origin, i.e., USA, Canada, and Denmark. According to these studies, the MDD process comprises one to seven stages. Six studies (3, 9, 29–32) also contained a model of the MDD process for all stages or just some of the stages. The findings of the selected studies, especially the stages of the development of new medical devices, and presence or absence of relevant legislative aspects and risks, are summarized in Table 4 below. The columns are ordered according to the alphabetical name of the first author of the selected study. To minimize bias or systematic error, every time we combined the published paper, the duplication check is performed automatically using JaBref's software and manually based on author, title, and DOI. Other than that, each author also plays a role in checking either the classification of term or the content of each table, interpreting the real content published. Other than that, we are analyzing all the published articles and explain the process step-by-step in this article.
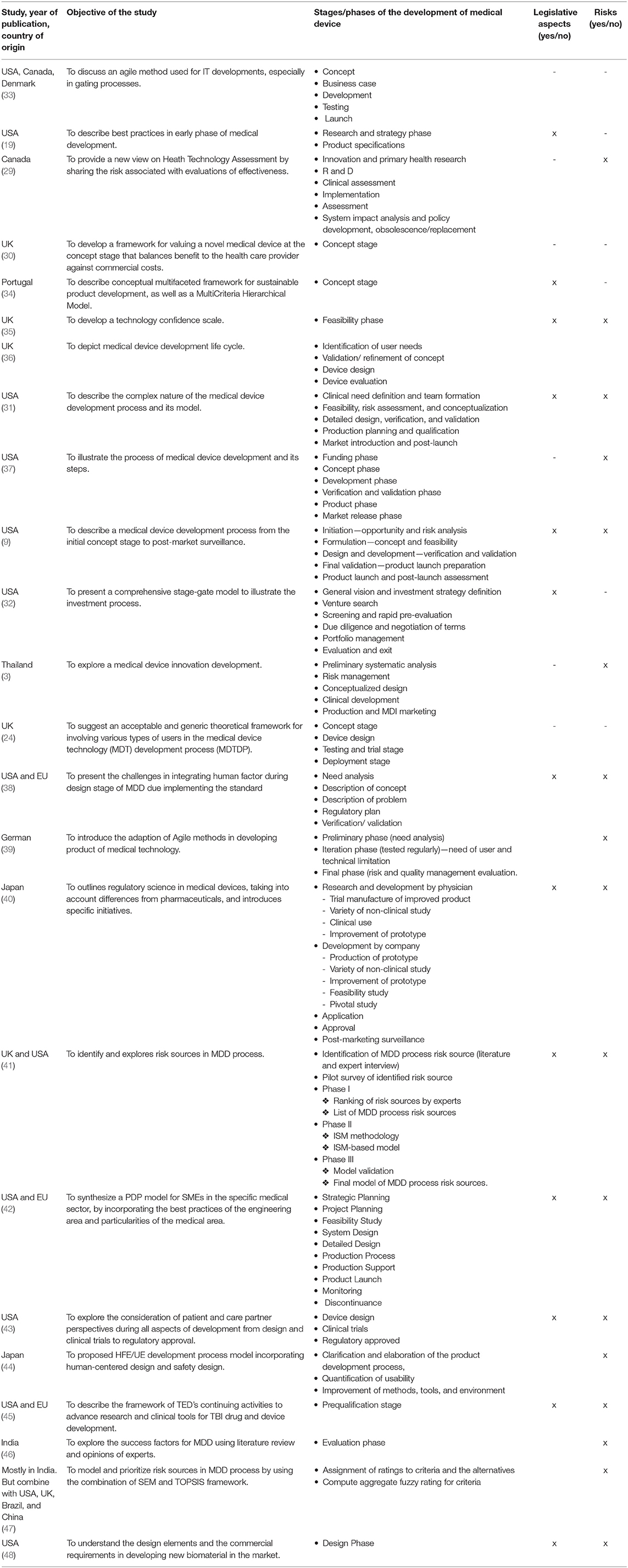
Table 4. Stages of MDD, an overview of the findings from the selected research studies.
The findings in Table 4 show that there is no agreement on the number of stages required for the development of medical devices. The number of product life cycle stages usually ranges from four to six. The study by Ocampo and Kaminski (42) suggests three stages—pre-development, development, and post-development (the PDP model). The specific medical device development is written in Table 4. Some studies, such as that of Girling et al. (30), Hede et al. (34), and Johnson and Moultrie (35), focus only on one stage of the MDD process, especially the concept stage during which all uncertainties such as the clinical need definition, customer requirements and needs, finances, reimbursement strategy, team selection, or legal aspects must be considered. The most representative studies are that of Medina et al. (31) and Pietzsch et al. (9), which include all key phases of the MDD life cycle, as well as legal aspects and risk factors; other studies are less detailed, and their model often lacks case studies [e.g., (32)]. Although Privitera et al. (38) reviews 18 medical devices as case studies and the reviews include legislative and risk analysis aspects, the research focuses only on the design stage of the MDD process. This is also true for the study by Panescu (37), whose descriptions of individual stages are very generic. He does not formulate a way to implement individual activities but only lists the activities and the order in which they should be performed. Moreover, the stages are not connected to specific legislative conditions or the type of medical device according to its level of risk.
According to Pietzsch et al. (9), the comprehensive MDD life cycle comprises five phases. Before the commencement of Phase 1, a clinical needs analysis must be conducted. Sometimes, this phase is referred to as Phase 0. Furthermore, preliminary market analysis must be conducted to check whether there is a satisfactory market opportunity for this clinical need and whether the new product is compatible with the company's strategy and ability to successfully commercialize this product. This phase is followed by Phase 1, with several important steps. These include a financial review and market analysis or competitive assessment that focuses on needs assessment and validation, demographics analysis, and SWOT analysis. These are followed by the legal intellectual property (IP) analysis and the regulatory review. The final step is to develop a business plan. In Phase 2, a cross-functional team is formed to formulate the concept, evaluate feasibility, and develop a design plan. Models and prototypes are made, and an initial design for manufacturing is developed. In addition, regulatory and reimbursement strategies from Phase 1 are further specified in this phase to comply with new requirements. In Phase 3, verification and validation tests are conducted to ensure that the quality of the device meets set standards and customer needs. In addition, regulatory and reimbursement activities continue in this phase. In Phase 4, formal design prints are made, and preparations are commenced for a medical device launch. The key step in this phase in the United States is the receipt of regulatory approval/clearance from the FDA. Phase 5 includes the product launch and post-market monitoring. If the device appears to succeed, it is distributed for widespread clinical use. Post-market activities involve post-market monitoring, quality audits, clinical validation, and the constant improvement of products and processes. Medina et al.'s (31) MDD stages resemble those of Pietzsch et al. (9). However, in comparison with Pietzsch et al.'s, they form the cross-functional team earlier on in Phase 1, while product launch preparation is a part of Phase 5.
Generally, the abovementioned linear stage-gate processes of the chosen authors have been used for almost three decades and been pivotal contributions for the medical device industry (49), because they are both conceptual and functional. Furthermore, they acknowledge that MDI is a manageable process (49).
Nevertheless, for the general model to be at least partially usable as a best practice, it must be updated to link to valid legislation, related risks, and valid changes in the management system of individual activities related to the audit trends and development of modern technologies, which affect most business activities.
Discussion
The findings of the selected studies on the Research Topic show that the comprehensive MDD life cycle comprises five phases: opportunity and risk analysis phase, concept and feasibility phase, verification and validation phase, product launch preparation phase, and product launch and post-launch assessment phase. These individual MDD phases are linear and separated by gates that are characterized by certain set criteria that must be met before MDD can proceed further. That is why the whole MDD process is also called a linear stage-gate process, which is the most commonly used process in the development and innovation of medical devices.
However, Goldenberg and Gravagna (6) identified several gaps in the traditional stage-gate product development process. They point out that the stage-gate approach is linear, without a full life cycle plan and that companies, especially smaller ones, mainly focus on regulatory approval milestones than on providing significant returns to potential stakeholders. They suggest implementing an integrated customer engagement roadmap approach that identifies all stakeholder requirements/needs and device-specific marketing messages for product differentiation. Furthermore, detailed information on budget, timeline for data studies, and communications and marketing is included. Overall, a global launch strategy is implemented.
In addition, Cooper and Sommer (33) proposed the hybrid “agile-stage-gate” approach, which can be integrated into the traditional stage-gate model for the following benefits:
• It is built on customer needs in a cost-effective way.
• It reacts quickly to needs.
• It copes with uncertainty and ambiguity that are typical of innovative developments.
• It deals with resourcing issues more directly.
Furthermore, the sources of risks that can threaten the whole MDD process, in terms of price, timing, and quality, should be carefully considered to avoid failure. The key issue is meeting user needs. As far as the legislation aspects are concerned, the key issues are consistency in the classification of devices in the EU countries, as well as the transparency of the approval process worldwide.
Risk Aspects
Individual MDD phases are closely connected with risks (50–54) that the individual steps bring about. For example, developing a new medical device is quite costly and risky (36); its success significantly relies on the application of accurate processes (9). Product designers and developers attempt to reduce these risks; however, tough competition encourages them to investigate the sources of risks during the MDD process, which can threaten the MDD process in terms of price, timing, and quality (38, 41). Aguwa et al. (55) reported that medical technology is quite unsuccessful (90%) during the first prototype test, which should be carefully considered by any MDD company. Some researchers have evaluated risks in medical device design. Privitera et al. (38) indicated the integration of human factors as one of the methods to reduce risks during the design stage of the MDD process; however, challenges exist because of the implementation of standards. These challenges can be solved if both parties, medical device developers and users, cooperate. Schmuland et al. (56) provided practical ideas to allow medical device manufacturers to evaluate residual risk of their devices. Risk analysis (ISO 14971) and failure analysis (FMEA) were combined by Chan et al. (57) to ensure device quality in the design phase of the MDD process, with a case study of a ventilation breathing circuit. Rane and Kirkire (41) summarized the key risks into three main groups: user-related sources of risks, internal sources of risks, and third party-related sources of risks. User-related risks include poor translation of user requirements or unmet user needs/requirements. Internal risks are due to the lack of application of adequate standards to check device performance; poor consideration of the effect of labeling and packaging; or poor communication among device developers, end users, and marketing. Third party-related sources of risks may include lack of training for end users; improper or poor assessment of progress by reviewers; and poor planning for regulatory and clinical approvals and tests. Their findings indicate that the most important source of risks is unmet user needs, which means that user needs should be met to successfully market any device.
The detection of risks and their sources in the MDD process plays a significant role, because it can prevent a lot of adverse effects of the use of medical devices by end users, save a lot of time on design and development of the medical device, and reduce costs during the MDD process. Therefore, the MDD process should be critically planned and modeled to decrease the number of risks and their severity.
Legislative Aspects
Global harmonization in the field of medical device regulation is following the pathway set by the pharmaceutical industry at the turn of the 1980s (58). In 1989, regulatory bodies of the United States, EU, and Japan came to the conclusion that it would be more effective for the industry to develop universal standards for all aspects of drug development, manufacturing, and pharmacovigilance, with the aim to bring more safety to the process of drug manufacturing (Table 5).
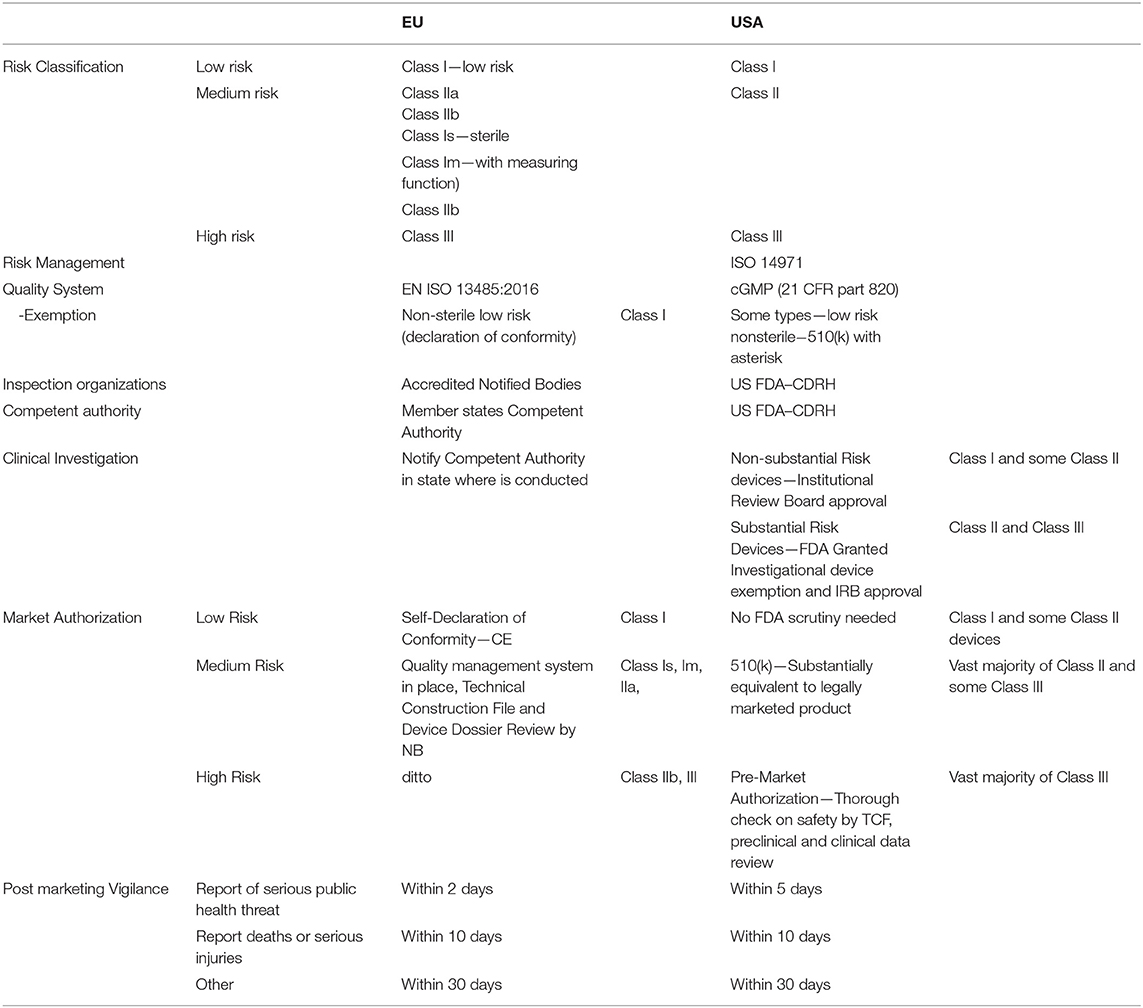
Table 5. The regulation in the European Union and in the United States of America.
Therefore, the International Council1 for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) was founded. Since then, almost all important pharmaceutical markets have closely similar legislation that stems from ICH guidelines. ICH plays a crucial role in adopting novel policies for the safety of pharmaceuticals.
The Global Harmonization Task Force (GHTF), founded in 1992, was replaced in 2011 by the International Medical Device Regulators Forum (IMDRF). In an ever-changing global market, focus on harmonization is needed to achieve the desired level of safety of medical devices. During the 1980s, almost no regulation of medical devices existed. Since the 1990s, some regulations have emerged mainly in the United States and the EU, as well as in the East Asia region, mainly in Japan and Taiwan. Since the beginning of the new millennium, one can observe convergence in the regulation of the medical devices industry owing to the work of the GHTF. However, a global world needs global approaches. That is why the IMDRF came to life. The two largest markets for medical devices are Europe and North America. Regulatory requirements converge on both sides of the Atlantic; yet, American rules had been stricter compared to European rules—until the recent approval of the new medical devices regulation (MDR) by the European Parliament. The rules concerning medical devices had been much more relaxed in the EU; however, after the large-scale scandal involving Poly Implant Prothèse (PIP)-manufactured breast implants, the European Union embarked on the path leading to the approval of the MDR. As thorough as it is, it is still inferior to Title 21, Part 812 and 820 of the Code of Federal Regulations set by the United States, also referred to as current good manufacturing practices (cGMP).
In the EU, the key role is versed on the so-called “accredited notified bodies” that are privately held for-profit companies. Their nature poses a great risk for the whole system. Since there is only a limited number of such bodies (gradually decreasing), and because of the mandatory re-evaluation of all medical devices approved in the EU common market, there will be shortages of available capacity for re-evaluation. Simultaneously, notified bodies would probably be less willing to inspect small companies, which make only a few types of medical devices and tend to be generally less prepared for the transition to novel regulations, because it will be much profitable to inspect large companies with diverse portfolios and better prepared paperwork.
Another limiting factor is the relatively large number of such bodies compared to the situation in the United States where all inspectors are employees of the Food and Drug Administration (FDA), and partially the Center for Devices and Radiological Health (CDRH). It seems plausible that there could be significant differences between the level of scrutiny among bodies based in distant parts of the EU. As such, the key factor of proposed regulation could be endangered by this flaw. Another issue that is addressed by the MDR is post-marketing vigilance of medical devices. The novel regulations impose the duty of post-marketing follow-up for all devices marketed in the EU.
Since the beginning of the PIP breast implant scandals, there has been a steady shift in the perception of how to achieve this goal within the industry. Before the MDR came into effect, the focus had been more on the safety of individual products. Thus, almost all effort was put to releasing the product by obtaining the CE mark. However, as a lesson learned from the pharmaceutical industry, safety should be achieved primarily by setting up a rigorous framework of rules for the whole product life cycle. A quick overview of the regulations in the EU and the United States could be seen in Table 5. The EU and the United States were chosen because other states are modeling regulations after theirs. For further information on the topic, readers are kindly referred to the reviews by Gupta and Thomke (10) and Ocampo and Kaminski (42), which discuss the global regulation aspects of medical devices.
In Japan, as stated in Niimi (40), the risks are divided into four classes: Class I, Class II, Class III, and Class IV, where the highest risk is in Class III and Class IV, which are for highly controlled medical devices and need the approval from the minister and a review by the Pharmaceutical and Medical Devices Agency (PMDA).
Conclusion
Regarding the phases involved in MDD, and the related regulations and risk factors, the results indicate that the general model applied in the MDD process should follow the well-established linear stage-gate process, which is conceptual and manageable from the perspective of innovation. Nevertheless, the model should include recently suggested approaches such as implementing an integrated customer engagement roadmap. In addition, the model must respond to current valid legislation processes, their changes, and related risks, as well as to the valid changes in the management system of individual activities related to the audit trends and development of modern technologies, which affect most business activities. The crucial factor in healthcare safety (59, 60) is the stability of factors over a long production time. Good manufacturing practices cannot be tested on individual batches of products; they must be inherently built into the manufacturing process. This is the goal that medical device regulations and cGMP are trying to achieve. The key issues that must be addressed in the future are consistency in the classification of devices throughout the EU and globally, and the transparency of the approval processes.
Strengths and Limitations of this Study
• This review presents in-depth specifications of the stages of the medical device development process and the associated risks, which are not described in organizational or managerial research. It provides a general point of view as opposed to large numbers of case studies.
• Research findings are strategically important for healthcare development, because they clearly state the requirements for medical device development and offer a way for researchers to apply this specific process in general managerial research.
• This study is limited in the sense that it cannot cover all consequences of changes in legislative aspects.
Author Contributions
PM and KK suggested the design of the study. WI wrote the methodology. WI and PM searched the databases. PM, AS, BK, JH, and KK prepared the tables, wrote the manuscript, and reviewed the paper. All authors approved this version of the paper.
Funding
This study was supported by the research project The Czech Science Foundation (GACR) 2017 No. 17-03037S Investment evaluation of medical device development run at the Faculty of Informatics and Management, University of Hradec Kralove, Czech Republic.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the author KK.
Abbreviations
MD, Medical device; MDD, Medical device development; FDA, Food and Drug Administration; CDRH, Center for Devices and Radiological Health; GHTF, The Global Harmonization Task Force; MDR, International Medical Device Regulators Forum; MDR, Medical Devices Regulation; cGMP, Current Good Manufacturing Practices; FMEA, Failure analysis; TOPSIS, Technique for Order Performance by Similarity to Ideal Solution; SEM, Structural Equation Modeling.
Footnotes
1. ^Between 1990 and 2015, it was known as the International Conference on Harmonization.
References
1. Kuca K, Maresova P, Penhaker M, Selamat A. The potential of medical device industry in technological and economical context. Therap Clin Risk Manag. (2015) 11:1505–14. doi: 10.2147/TCRM.S88574
2. Health - OECD Data. citován 19. zárí 2018]. Available online at: http://data.oecd.org/health.htm (accessed September 19, 2018).
3. Songkajorn Y, Thawesaengskulthai N. Medical device innovation development process. Int J Innov Technol Manag. (2014) 11:1450027. doi: 10.1142/S0219877014500278
4. Rome BN, Kramer DB, Kesselheim AS. Approval of high-risk medical devices in the US: implications for clinical cardiology. Curr Card Rep. (2014) 16:489. doi: 10.1007/s11886-014-0489-0
5. NVCA Research Resources. Patient Capital, National Venture Capital Research Resources. Available online at: https://nvca.org/research/research-resources/ (accessed September 19, 2018).
6. Goldenberg SJ, Gravagna J. A real-world perspective: building and executing an integrated customer engagement roadmap that bridges the gaps in traditional medical device development processes. J Med Market. (2018) 16:41–9. doi: 10.1177/1745790418770598
7. Fisher GJ, Qualls WJ. A framework of interfirm open innovation: relationship and knowledge based perspectives. J Bus Industr Mark. (2018) 33:240–50. doi: 10.1108/JBIM-11-2016-0276
8. Sharma A, Jha S. Innovation from emerging market firms: what happens when market ambitions meet technology challenges? J Busin Indust Market. (2016) 31:507–18. doi: 10.1108/JBIM-12-2014-0265
9. Pietzsch JB, Shluzas LA, Paté-Cornell ME, Yock PG, Linehan JH. Stage-gate process for the development of medical devices. J Med Dev. (2009) 3:14–21. doi: 10.1115/1.3148836
10. Gupta B, Thomke S. An exploratory study of product development in emerging economies: evidence from medical device testing in India. R&D Manag. (2018) 48:485–501. doi: 10.1111/radm.12324
11. Lubowitz JH, Brand JC, Rossi MJ. Medical device and pharmaceutical industry employees as medical research publication authors. Arthroscopy. (2018) 34:2745–7. doi: 10.1016/j.arthro.2018.05.008
12. Augustýnek M, Laryš D, Kubíček J, Marešová P, Kuča K. Use effectiveness of medical devices: a case study on the deployment of ultrasonographic devices. Therap Innov Regul Sci. (2018) 52:499–506. doi: 10.1177/2168479017739291
13. Maisel WH. Medical device regulation: an introduction for the practicing physician. Am Coll Phys. (2004) (140):296–302. doi: 10.7326/0003-4819-140-4-200402170-00012
14. Hamrell MR. An overview of the US regulatory environment for drug-device and biologic-device combination products. Drug Inform J. (2006) 40:23–32. doi: 10.1177/009286150604000104
15. Aitchison GA, Hukins DWL, Parry JJ, Shepherd DET, Trotman SG. A review of the design process for implantable orthopedic medical devices. Open Biomed Eng J. (2009) 3:21–7. doi: 10.2174/1874120700903010021
16. Alexander K, Clarkson PJ. A validation model for the medical devices industry. J Eng Design. (2002) 13:197–204. doi: 10.1080/09544820110108890
17. Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. (2009). 62:1006–12. doi: 10.1016/j.jclinepi.2009.06.005
18. Moher D, Liberati A, Tetzlaff J, Altman DG, Group TP. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. (2009) 6:e1000097. doi: 10.1371/journal.pmed.1000097
19. Fearis K, Petrie A. Best practices in early phase medical device development: Engineering, prototyping, and the beginnings of a quality management system. Surgery. (2017) 161:571–5. doi: 10.1016/j.surg.2016.08.052
20. Cosgrove J, Littlewood J, Wilgeroth P. Development of a framework of key performance indicators to identify reductions in energy consumption in a medical devices production facility. Int J Amb Energy. (2018) 39:202–10. doi: 10.1080/01430750.2017.1278718
21. Cho K-T, Kim S-M. Selecting medical devices and materials for development in Korea: the analytic hierarchy process approach. Int J Health Plann Manag. (2003) 18:161–74. doi: 10.1002/hpm.703
22. Shaw B. Innovation and new product development in the UK medical equipment industry. Int J Technol Manag. (1998) 15:433. doi: 10.1504/IJTM.1998.002620
23. Vaezi J, Nekoomanesh M, Khonakdar HA, Jafari SH, Aghjeh MR. Dynamic mechanical thermal analysis and rheological properties of synthesized polypropylene reactor blends using homogeneous binary metallocene catalyst. Polymer-Plastics Technol Eng. (2017) 56:1898–907. doi: 10.1080/03602559.2017.1295313
24. Shah SGS, Robinson I, AlShawi S. Developing medical device technologies from users' perspectives: A theoretical framework for involving users in the development process. Int J Technol Assess Health Care. (2009) 25:514–21. doi: 10.1017/S0266462309990328
25. Bruse JL, Zuluaga MA, Khushnood A, McLeod K, Ntsinjana HN, Hsia T-Y, et al. Detecting clinically meaningful shape clusters in medical image data: metrics analysis for hierarchical clustering applied to healthy and pathological aortic arches. IEEE Trans Biomed Eng. (2017) 64:2373–83. doi: 10.1109/TBME.2017.2655364
26. Ciubuc JD, Bennet KE, Qiu C, Alonzo M, Durrer WG, Manciu FS. Raman computational and experimental studies of dopamine detection. Biosensors-Basel. (2017). 7:43. doi: 10.3390/bios7040043
27. Özcan-Top Ö, McCaffery F. A Lightweight Software Process Assessment Approach Based on MDevSPICE® for Medical Device Development Domain. Cham: Springer (2017). p. 578–88. doi: 10.1007/978-3-319-64218-5_48
28. Özcan-Top Ö, McCaffery F. How does scrum conform to the regulatory requirements defined in MDevSPICE®? Commun Comp Inform Sci. (2017) 770:257–68. doi: 10.1007/978-3-319-67383-7_19
29. Ferrusi IL, Ames D, Lim ME, Goeree R. Health technology assessment from a canadian device industry perspective. J Am Coll Radiol. (2009) 6:353–9. doi: 10.1016/j.jacr.2009.01.013
30. Girling A, Young T, Brown C, Lilford R. Early-stage valuation of medical devices: the role of developmental uncertainty. Value Health. (2010) 13:585–91. doi: 10.1111/j.1524-4733.2010.00726.x
31. Medina LA, Kremer GEO, Wysk RA. Supporting medical device development: a standard product design process model. J Eng Design. (2013) 24:83–119. doi: 10.1080/09544828.2012.676635
32. Soenksen LR, Yazdi Y. Stage-gate process for life sciences and medical innovation investment. Technovation. (2017) 62–63:14–21. doi: 10.1016/j.technovation.2017.03.003
33. Cooper RG, Sommer AF. The agile-stage-gate hybrid model: a promising new approach and a new research opportunity. J Product Innov Manag. (2016) 33:513–26. doi: 10.1111/jpim.12314
34. Hede S, Nunes MJL, Ferreira PFV, Rocha LA. Incorporating sustainability in decision-making for medical device development. Technol Soc. (2013) 35:276–93. doi: 10.1016/j.techsoc.2013.09.003
35. Johnson J, Moultrie J. Technology confidence in early stage development of medical devices. Int J Innov Sci. (2012) 4:57–70. doi: 10.1260/1757-2223.4.2.57
36. Martin JL, Barnett J. Integrating the results of user research into medical device development: insights from a case study. BMC Med Inform Dec Making. (2012) 12:74. doi: 10.1186/1472-6947-12-74
37. Panescu D. Medical device development. In: 2009 Annual International Conference of the IEEE Engineering in Medicine and Biology Society. Minneapolis, MN: IEEE (2009). p. 5591–4. doi: 10.1109/IEMBS.2009.5333490
38. Privitera MB, Evans M, Southee D. Human factors in the design of medical devices – Approaches to meeting international standards in the European Union and USA. Appl Ergonom. (2017) 59:251–63. doi: 10.1016/j.apergo.2016.08.034
39. Gerber C, Goevert K, Schweigert-Recksiek S, Lindemann U. Agile development of physical products—A case study of medical device product development. Smart Innov Syst Technol. (2019) 135:823–34. doi: 10.1007/978-981-13-5977-4_69
40. Niimi S. Practice of regulatory science (Development of medical devices). Yakugaku Zasshi. (2017) 137:431–7. doi: 10.1248/yakushi.16-00244-3
41. Rane SB, Kirkire MS. Interpretive structural modelling of risk sources in medical device development process. Int J Syst Assur Eng Manag. (2017) 8:451–64. doi: 10.1007/s13198-015-0399-6
42. Ocampo JU, Kaminski PC. Medical device development, from technical design to integrated product development. J Med Eng Technol. (2019) 43:287–304. doi: 10.1080/03091902.2019.1653393
43. Hurst FP, Chianchiano D, Upchurch L, Fisher BR, Flythe JE, Castillo Lee C, et al. Stimulating patient engagement in medical device development in kidney disease: a report of a kidney health initiative workshop. Am J Kidney Dis. (2017) 70:561–9. doi: 10.1053/j.ajkd.2017.03.013
44. Nagao T, Misumi K, Ikeda D. Study of HFE/UE process model in medical device development. Adv Intell Syst Comput. (2019) 824:139–46. doi: 10.1007/978-3-319-96071-5_15
45. Manley GT, Mac Donald CL, Markowitz AJ, Stephenson D, Robbins A, Gardner RC, et al. The traumatic brain injury endpoints development (TED) initiative: progress on a public-private regulatory collaboration to accelerate diagnosis and treatment of traumatic brain injury. J Neurotr. (2017) 34:2721–30. doi: 10.1089/neu.2016.4729
46. Kirkire MS, Rane SB. Evaluation of success factors for medical device development using grey DEMATEL approach. J Modell Manag. (2017) 12:204–23. doi: 10.1108/JM2-09-2015-0062
47. Kirkire MS, Rane SB, Singh SP. Integrated SEM-FTOPSIS framework for modeling and prioritization of risk sources in medical device development process. Benchm Int J. (2018) 25:178–200. doi: 10.1108/BIJ-07-2016-0112
48. Harris JJ, Lu S, Gabriele P. Commercial challenges in developing biomaterials for medical device development. Polymer Int. (2018) 67:969–74. doi: 10.1002/pi.5590
49. Cooper RG. Stage-gate systems: A new tool for managing new products. Business Horizons. (1990) 33:44–54. doi: 10.1016/0007-6813(90)90040-I
50. Gilmartin C, Arbe-Barnes EH, Diamond M, Fretwell S, McGivern E, Vlazaki M, et al. Varsity medical ethics debate 2018: constant health monitoring - the advance of technology into healthcare. Phil Ethics Hum Med. (2018) 13:12. doi: 10.1186/s13010-018-0065-0
51. Ho M, Saha A, McCleary KK, Levitan B, Christopher S, Zandlo K, et al. A framework for incorporating patient preferences regarding benefits and risks into regulatory assessment of medical technologies. Value Health. (2016) 19:746–50. doi: 10.1016/j.jval.2016.02.019
52. Ivlev I, Kneppo P, Bartak M. Multicriteria decision analysis: a multifaceted approach to medical equipment management. Technol Econ Dev Econ. (2014) 20:576–89. doi: 10.3846/20294913.2014.943333
53. Klar E. Medical Device Regulation as current challenge for the legally safe introduction of new technologies. Der Chirurg. (2018) 89:755–9. doi: 10.1007/s00104-018-0705-3
54. Millson MR, Wilemon D. Impact of new product development (NPD) proficiency and NPD entry strategies on product quality and risk. R&D Manag. (2008) 38:491–509. doi: 10.1111/j.1467-9310.2008.00534.x
55. Aguwa CC, Monplaisir L, Sylajakumari PA. Rules modification on a Fuzzy-based modular architecture for medical device design and development. IIE Transact Healthcare Syst Eng. (2012) 2:50–61. doi: 10.1080/19488300.2012.666630
56. Schmuland C. Value-added medical-device risk management. IEEE Trans Device Mater Reliab. (2005) 5:488–93. doi: 10.1109/TDMR.2005.857860
57. Chan SL, Ip WH, Zhang WJ. Integrating failure analysis and risk analysis with quality assurance in the design phase of medical product development. Int J Prod Res. (2012) 50:2190–203. doi: 10.1080/00207543.2011.565084
58. Maresova P, Kacetl J. Legislative and ethical aspects of introducing new technologies in medical care for senior citizens in developed countries. Clin Inter Aging. (2016) 11:977–84. doi: 10.2147/CIA.S104433
59. Maresova P, Cerna L. Patients:attitudes to the use of modern technologies in the treatment of diabetes. Patient Prefer Adher. (2016) 10:1869–79. doi: 10.2147/PPA.S118040
Keywords: medical devices, development, stages, risks, legislations
Citation: Marešová P, Klímová B, Honegr J, Kuča K, Ibrahim WNH and Selamat A (2020) Medical Device Development Process, and Associated Risks and Legislative Aspects-Systematic Review. Front. Public Health 8:308. doi: 10.3389/fpubh.2020.00308
Received: 08 January 2020; Accepted: 05 June 2020;
Published: 30 July 2020.
Edited by:
Lana Nezic, University of Banja Luka, Bosnia and HerzegovinaReviewed by:
Vesna Jaćević, National Poison Control Center, SerbiaBojan Markovic, University of Belgrade, Serbia
Copyright © 2020 Marešová, Klímová, Honegr, Kuča, Ibrahim and Selamat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamil Kuča, a2FtaWwua3VjYUB1aGsuY3o=