 Gwenddolen Kettenburg1
Gwenddolen Kettenburg1 Amy Kistler2
Amy Kistler2 Hafaliana Christian Ranaivoson3,4
Hafaliana Christian Ranaivoson3,4 Vida Ahyong2Angelo Andrianiaina3Santino Andry5Joseph L. DeRisi2
Vida Ahyong2Angelo Andrianiaina3Santino Andry5Joseph L. DeRisi2 Anecia Gentles6Vololoniaina Raharinosy4
Anecia Gentles6Vololoniaina Raharinosy4 Tsiry Hasina Randriambolamanantsoa4Ny Anjara Fifi Ravelomanantsoa3
Tsiry Hasina Randriambolamanantsoa4Ny Anjara Fifi Ravelomanantsoa3 Cristina M. Tato2
Cristina M. Tato2 Philippe Dussart4
Philippe Dussart4 Jean-Michel Heraud4,7†
Jean-Michel Heraud4,7† Cara E. Brook1*†
Cara E. Brook1*†- 1Department of Ecology and Evolution, University of Chicago, Chicago, IL, United States
- 2Chan Zuckerberg Biohub, San Francisco, CA, United States
- 3Department of Zoology and Animal Biodiversity, University of Antananarivo, Antananarivo, Madagascar
- 4Virology Unit, Institut Pasteur de Madagascar, Antananarivo, Madagascar
- 5Department of Entomology, University of Antananarivo, Antananarivo, Madagascar
- 6Odum School of Ecology, University of Georgia, Athens, GA, United States
- 7Virology Department, Institut Pasteur de Dakar, Dakar, Senegal
Bats are natural reservoirs for both Alpha- and Betacoronaviruses and the hypothesized original hosts of five of seven known zoonotic coronaviruses. To date, the vast majority of bat coronavirus research has been concentrated in Asia, though coronaviruses are globally distributed; indeed, SARS-CoV and SARS-CoV-2-related Betacoronaviruses in the subgenus Sarbecovirus have been identified circulating in Rhinolophid bats in both Africa and Europe, despite the relative dearth of surveillance in these regions. As part of a long-term study examining the dynamics of potentially zoonotic viruses in three species of endemic Madagascar fruit bat (Pteropus rufus, Eidolon dupreanum, Rousettus madagascariensis), we carried out metagenomic Next Generation Sequencing (mNGS) on urine, throat, and fecal samples obtained from wild-caught individuals. We report detection of RNA derived from Betacoronavirus subgenus Nobecovirus in fecal samples from all three species and describe full genome sequences of novel Nobecoviruses in P. rufus and R. madagascariensis. Phylogenetic analysis indicates the existence of five distinct Nobecovirus clades, one of which is defined by the highly divergent ancestral sequence reported here from P. rufus bats. Madagascar Nobecoviruses derived from P. rufus and R. madagascariensis demonstrate, respectively, Asian and African phylogeographic origins, mirroring those of their fruit bat hosts. Bootscan recombination analysis indicates significant selection has taken place in the spike, nucleocapsid, and NS7 accessory protein regions of the genome for viruses derived from both bat hosts. Madagascar offers a unique phylogeographic nexus of bats and viruses with both Asian and African phylogeographic origins, providing opportunities for unprecedented mixing of viral groups and, potentially, recombination. As fruit bats are handled and consumed widely across Madagascar for subsistence, understanding the landscape of potentially zoonotic coronavirus circulation is essential for mitigation of future zoonotic threats.
Introduction
In the past 20 years, bat-derived coronaviruses SARS-CoV, MERS-CoV, and SARS-CoV-2 have been responsible for two deadly epidemics and the ongoing COVID-19 pandemic (1–4). These coronaviruses (CoVs) are members of the Betacoronavirus genus, which, along with genus Alphacoronavirus, are primarily associated with bat hosts (1–4); the remaining CoV genera, Gammacoronavirus and Deltacoronavirus, are typically hosted by birds (5). The Betacoronavirus group can be further broken down into five subgenera: Sarbecovirus [hosted by bats in family Rhinolophidae (6, 7)], Merbecovirus [hosted by bats in family Vespertilionidae (8–10)], Nobecovirus [hosted by bats in family Pteropodidae (11–13)], and Hibecovirus [hosted by bats in family Hipposideridae (14–16)]. The fifth Betacoronavirus subgenus, Embecovirus, is primarily associated with rodents and bovids, though a few bat hosts have been documented (17, 18). Since the emergence of SARS-CoV in 2002, there has been increasing interest in surveying potential hosts of coronaviruses and contributing new virus sequences to public databases, with most effort focused on sampling bats from Asia (19–28), the continent of origin for both the SARS-CoV epidemic and the SARS-CoV-2 pandemic. Recently, more concerted efforts have arisen to survey the landscape of bat-borne coronaviruses in other regions of the world, including Africa and Europe (11, 12, 29–33).
The family Coronaviridae is considered one of the most likely viral taxa to switch host species (34, 35) in part because many CoVs utilize well-conserved cell surface receptors presented on a wide variety of mammalian host cells. The zoonotic Sarbecoviruses, SARS-CoV and SARS-CoV-2, for example, use the human cell surface receptor Angiotensin-converting enzyme 2 (ACE2) to gain entry into human cells (36, 37), while many Merbecoviruses interact with the well-conserved vertebrate host cell receptor dipeptidyl peptidase 4 (DPP4) (38). MERS-CoV has an evolutionary history with bat origins, and closely-related viruses have been previously identified in bats from the families Vespertillionidae, Nycteridae, Emballonuridae and Molossidae throughout Africa, Asia, and Europe. More recently, human cases of MERS-CoV have arisen in the human and camel populations through recent host switching events (39–42). As only a fraction of the Alpha- and Betacoronavirus diversity projected to circulate in wild bat hosts has been already described (41), it is possible that many CoVs capable of zoonotic emergence remain uncharacterized. Because CoVs are known to recombine with other CoVs, or more rarely, with other viral groups (43–48), there is additional concern that naturally-circulating CoVs presently unable to infect humans may acquire this ability in the future. Several factors, which have been reviewed at length elsewhere (4, 34, 49), contribute to the propensity for CoV recombination, including the large CoV genome size supported by a unique proofreading mechanism in the RNA-dependent RNA polymerase (RdRp) (50–53), as well as a ‘copy choice’ template switching mechanism of RNA replication whereby RdRp physically detaches from one RNA template during replication and reattaches to an adjacent template, thus facilitating recombination in cases where multiple viruses may be coinfecting the same cell (54).
Madagascar is an island country in southeastern Sub-Saharan Africa, located in the Indian Ocean, ~400 km off the coast from Mozambique. Madagascar has been isolated from the African continent for over 170 million years and all surrounding landmasses for over 80 million years, allowing for the evolution of a unique and highly endemic floral and faunal assemblage (55). The country is home to 51 species of bat (56), two-thirds of which are endemic and boast long evolutionary divergence times with sister species on both the African and Asian continents (57–59). A growing body of work has characterized the landscape of potentially zoonotic viruses in Madagascar bats, identifying evidence of circulating infection (through RNA detection or serology) with henipaviruses, filoviruses, lyssaviruses, and coronaviruses (12, 33, 60, 61). Previously, coronavirus surveillance efforts have identified Alphacoronavirus RNA in the Malagasy insectivorous bat, Mormopterus jugularis, and Betacoronavirus RNA in the subgenus Nobecovirus (12, 33) in all three endemic Malagasy fruit bat species: Pteropus rufus, Eidolon dupreanum, and Rousettus madagascariensis (12, 33). In addition to Madagascar, Nobecoviruses have been previously characterized from Pteropodidae fruit bats across Asia and in both East and West Africa (27, 30, 62–65). Though Nobecoviruses are not known to be zoonotic, previous research has described widespread circulation throughout Asia of a recombinant Nobecovirus which carries an orthoreovirus p10 gene insertion (27, 65, 66), highlighting the capacity for this viral subgenus to undertake rapid shifts in genomic organization which could lead to expanded host range. As both E. dupreanum and R. madagascariensis are known to co-roost with each other, and with several species of insectivorous bat (67), CoV recombination is a distinct concern in the Madagascar system. Though no Rhinolophus spp. bats, the typical host for ACE2-using Sarbecoviruses, inhabit Madagascar, the island is home to four species of Hipposiderid bat (56), which host the Sarbecovirus-adjacent and understudied Hibecoviruses, as well as several species of Vespertilionid bat, the most common hosts for the zoonotic Merbecoviruses.
Human-bat contact rates are high in some regions of Madagascar—including the region in which we conducted our field research—where bats are consumed for subsistence and frequently roost in close proximity to human settlements or natural tourist attractions (68–72). Moreover, in addition to the natural CoV diversity described in Malagasy bats, several human coronaviruses are known to circulate widely in the human population in Madagascar, including the common cold-causing Embecoviruses, HCoV-OC43 and HCoV-HKU1, and, more recently, the zoonotic Sarbecovirus, SARS-CoV-2 (73–75). As spillback of SARS-CoV-2 into wildlife hosts and possible recombination with wildlife viruses remains a global concern (16), characterization of the genetic diversity of bat-borne coronaviruses in Madagascar and elsewhere in Africa is a critical public health priority. Here, we contribute and characterize three full genome sequences of two novel Nobecoviruses, derived from R. madagascariensis and P. rufus hosts. We define five distinct Nobecovirus clades in global circulation across Asia and Africa and assess these new Nobecoviruses for their past and future capacity for recombination.
Materials and Methods
Bat Sampling
As part of a long-term study characterizing the seasonal dynamics of potentially zoonotic viruses in wild fruit bats in Madagascar, monthly captures of Malagasy pteropodid bats were carried out at species-specific roost sites in the Districts of Moramanga and Manjakandriana, Madagascar between 2018 and 2019 (P. rufus: Ambakoana roost, −18.513 S, 48.167 E; E. dupreanum: Angavobe cave, −18.944 S, 47.949 E; Angavokely cave = −18.933 S, 47.758 E; R. madagascariensis: Maromizaha cave, −18.9623 S, 48.4525 E). In brief, bats were captured in nets hung in the tree canopy (P. rufus) or over cave mouths (E. dupreanum, R. madagascariensis) at dusk (17:00–22:00) and dawn (03:00–07:00), removed from nets, and processed under manual restraint following methods that have been previously described (61, 76, 77). All animals were identified to species, sex, and age class (juvenile vs. adult), and fecal, throat, and urine swabs were taken from each individual, collected into viral transport medium, and frozen on site in liquid nitrogen. Post-sampling, swabs were transported to −80°C freezers for long-term storage in the Virology Unit at Institut Pasteur de Madagascar. In total, 2156 bats (P. rufus: 167 females, 184 males; E. dupreanum: 495 females, 421 males; R. madagascariensis: 416 females, 473 males) were captured across the course of our long-term field study, though only fecal samples from a subset of individuals (see ‘Results’) were analyzed in part with the coronavirus research outlined here. Of those captures, 84 P. rufus were juveniles and 267 adults, 108 E. dupreanum were juveniles and 810 adults, and 126 R. madagascariensis were juveniles and 767 adults. Additional details relevant to our time series are included in the results.
This study was carried out in strict accordance with research permits obtained from the Madagascar Ministry of Forest and the Environment (permit numbers 019/18, 170/18, 007/19) and under guidelines posted by the American Veterinary Medical Association. All field protocols employed were pre-approved by the UC Berkeley Animal Care and Use Committee (ACUC Protocol # AUP-2017-10-10393), and every effort was made to minimize discomfort to animals.
RNA Extraction
RNA was extracted from a randomly selected subset of fecal, throat, and urine swab samples in the Virology Unit at the Institut Pasteur de Madagascar, with each sample corresponding to a unique individual from the field dataset. Samples undergoing mNGS corresponded to individuals captured in Feb-Apr, Jul-Sep and December 2018 or in January 2019. Water controls were extracted in conjunction with samples on each unique extraction day. Extractions were conducted using the Zymo Quick DNA/RNA Microprep Plus kit (Zymo Research, Irvine, CA, USA), according to the manufacturer's instructions and including the step for DNAse digestion. Post-extraction, RNA quality was checked on a nanodrop to ensure that all samples demonstrated 260/280 ratios exceeding 2 and revealed quantifiable concentrations. Resulting extractions were stored in freezers at −80°C, then transported on dry ice to the Chan Zuckerberg Biohub (San Francisco, CA, USA) for library preparation and metagenomic Next Generation Sequencing (mNGS).
Library Preparation and mNGS
Four randomly selected samples from each of three bat species underwent additional quantification using an Invitrogen Qubit 3.0 Fluorometer and the Qubit RNA HS Assay Kit (ThermoFisher Scientific, Carlsbad, CA, USA). After quantification, all total RNA samples, along with water samples from Madagascar extractions, were manually arrayed into 96 well plates to automate high throughput mNGS library preparation. Based on the initial quantitation, a 2 μL aliquot from each plated sample was diluted 1:9 on a Bravo liquid handling platform (Agilent, Santa Clara, CA, USA). A 5 μL aliquot from each diluted sample was arrayed into a 384 well plate for input into the mNGS library prep. Samples derived from fecal, throat, and urine swab samples were arrayed on distinct 384 well plates for separate sequencing runs. Additional unrelated total RNA samples (a dilution series of total RNA isolated from cultured HeLa cells) and a set of local lab water samples were included on each 384 well plate to serve as library preparation controls. Input RNA samples in the 384 well plate were transferred to a GeneVac EV-2 (SP Industries, Warminster, PA, USA) to evaporate the samples to enable miniaturized mNGS library preparation with the NEBNext Ultra II RNA Library Prep Kit (New England BioLabs, Beverly, MA, USA). Library preparation was performed per the manufacturer's instructions, with the following modifications: 25 pg of External RNA Controls Consortium Spike-in mix (ERCCS, Thermo-Fisher) was added to each sample prior to RNA fragmentation; the input RNA mixture was fragmented for 8 min at 94°C prior to reverse transcription; and a total of 14 cycles of PCR with dual-indexed TruSeq adapters was applied to amplify the resulting individual libraries. An initial equivolume library pool was generated, and the quality and quantity of that pool was assessed via electrophoresis (High-Sensitivity DNA Kit and Agilent Bioanalyzer; Agilent Technologies, Santa Clara, CA, USA), real-time quantitative polymerase chain reaction (qPCR) (KAPA Library Quantification Kit; Kapa Biosystems, Wilmington, MA, USA), and small-scale sequencing (2 x146 bp) on an iSeq platform (Illumina, San Diego, CA, US). Subsequent equimolar pooling of individual libraries from each plate was performed prior to performing large-scale paired-end sequencing (2 × 146 bp) run on the Illumina NovaSeq sequencing system (Illumina, San Diego, CA, USA). The pipeline used to separate the sequencing output of the individual libraries into FASTQ files of 146bp paired-end reads is available on GitHub at https://github.com/czbiohub/utilities.
CZID
Raw reads from Illumina sequencing were host-filtered, quality-filtered, and assembled on the CZID (v3.10, NR/NT 2019-12-01) platform, a cloud-based, open-source bioinformatics platform designed for microbe detection from metagenomic data (78), using a host background model of “bat” compiled from all publicly available full-length bat genomes in GenBank at the time of sequencing. Samples were deemed “positive” for coronavirus infection if CZID successfully assembled at least two contigs with an average read depth >2 reads/nucleotide that showed significant nucleotide or protein BLAST alignment(s) (alignment length >100 nt/aa and E-value <0.00001 for nucleotide BLAST/ bit score >100 for protein BLAST) to any CoV reference present in NCBI NR/NT database (version 12-01-2019). To verify that no positives were missed from CZID, all non-host contigs assembled in CZID underwent directed, offline BLASTn and BLASTx (79) against a reference database constructed from all available full-length nucleotide and protein reference sequences for Alpha- and Betacoronavirus available in NCBI Virus (last access: August 15, 2021). Step-by-step instructions for our offline BLAST protocol can be accessed in our publicly available GitHub repository at: https://github.com/brooklabteam/Mada-Bat-CoV/.
Genome Annotation and BLAST
Three full genome-length Nobecovirus contigs returned from CZID (two from R. madagascariensis and one from P. rufus) were aligned with Nobecovirus homologs from NCBI (see ‘Phylogenetic Analysis’) and annotated in the program Geneious Prime (2020.0.5). We then used NCBI BLAST and BLASTx to query identity of our full length recovered genomes and their respective translated proteins to publicly available sequences in NCBI (79). We queried identity to reference sequences for four previously described Nobecovirus strains (accession numbers: MG762674 (Rousettus bat coronavirus HKU9), NC_030886 (Rousettus bat coronavirus RoBat-CoV GCCDC1), MK211379 (Rhinolophus affinis coronavirus BtRt-BetaCoV/GX2018), and NC_048212 (Eidolon helvum bat coronavirus), as well as to the top BLAST hit overall. Finally, we aligned representative sequences from each major Nobecovirus clade and visually examined the region of p10 orthoreovirus insertion from the RoBat-CoV GCCDC1 lineage in the newly described sequences from Madagascar.
Phylogenetic Analysis
Contigs returned from CZID were combined with publicly available coronavirus sequences in NCBI to perform phylogenetic analysis. We carried out four major phylogenetic analyses, building (a) a full-genome Betacoronavirus maximum likelihood (ML) phylogeny, (b) a Betacoronavirus ML phylogeny corresponding to a conserved 259 bp fragment of the RNA-dependent RNA polymerase (RdRp) gene encapsulated in the CoV Orf1b, (c) four amino acid ML phylogenies derived from translated nucleotides corresponding to the spike (S), envelope (E), matrix (M), and nucleocapsid (N) proteins from a subset of full length genomes, and (d) a time-resolved Bayesian phylogeny corresponding to all available full genome Nobecovirus sequences in NCBI virus, which we computed in BEAST2 (80). Detailed methods for the construction of each phylogeny are available at https://github.com/brooklabteam/Mada-Bat-CoV/.
Briefly, our full genome ML phylogeny was comprised of 122 unique NCBI records, corresponding to all available full genome sequences with bat hosts under NCBI taxon IDs, Betacoronavirus (694002), unclassified Betacoronavirus (696098), Betacoronavirus sp. (1928434), unclassified Coronaviridae (1986197), or unclassified Coronavirinae (2664420) (107 records), in addition to all full genome Betacoronavirus (694002) reference sequences with a non-bat host (14 records), plus one Gammacoronavirus outgroup (accession number NC_010800). The full genome phylogeny additionally included three full length Madagascar Nobecovirus sequences returned from CZID (two from R. madagascariensis and one from P. rufus), which are described in this paper for the first time, for a total of 125 unique sequences.
Our Betacoronavirus RdRp ML phylogeny consisted of an overlapping subset of a 259 bp fragment in the center of the RdRp gene that has been previously described in Madagascar fruit bats (12) (7 records), in addition to the same RdRp fragment extracted from near-full length Nobecovirus sequences on NCBI Virus (17 records) and full length reference sequences for other Betacoronavirus subgenera available in NCBI Virus (17 records). This phylogeny also included two NCBI Virus RdRp Nobecovirus fragments, in addition to seven Madagascar Nobecovirus sequences encompassing the RdRp fragment of interest, which were returned from the assembly in CZID (four from R. madagascariensis, two from P. rufus, and one from E. dupreanum). Finally, we included the RdRp fragment of our Gammacoronavirus outgroup, for a total of 51 unique sequences.
Our amino acid phylogenies consisted of S, E, M, and N gene extractions from a subset of the same representative set of near-full genome length sequences used in the RdRp analysis: the same 17 full-length Betacoronavirus reference sequences, 17 near full-length Nobecovirus sequences, and the one Gammacoronavirus outgroup, in addition to our three full genome Madagascar sequences derived from R. madagascariensis and P. rufus. Gene extractions were carried out using annotation tracks reported with each accession number in NCBI or, in cases where annotations were unavailable, genes were manually annotated and extracted in Geneious Prime based on alignment to homologs. After nucleotide extraction, genes were translated prior to alignment.
Finally, our Bayesian time-resolved Nobecovirus phylogeny consisted of all available full genome Nobecovirus sequences in NCBI virus; because recombination events can muddle molecular clock estimation in phylogenetics, we constructed two different versions of this timetree, one including 18 Nobecoviruses sequences only for which no past history of recombination has been described, and a second which added two additional sequences from the Nobecovirus GCCDC1 clade known for its p10 orthoreovirus insertion.
After compiling sequences for each phylogenetic analysis, sequence subsets for the full-length, RdRp, and four amino acid phylogenies were aligned in MAFFT v.7 (81, 82) using default parameter values. Alignments were checked manually for quality in Geneious Prime, and the RdRp alignment was trimmed a fragment (259 bp) conserved across all sequences in the subset. All sequence subsets and alignment files are available for public access in our GitHub repository: https://github.com/brooklabteam/Mada-Bat-CoV/.
After quality control, alignments were sent to Modeltest-NG (83) to assess the best fit nucleotide or amino acid substitution model appropriate for the data. All alignments for ML analysis (full genome, 259 bp RdRp fragment, and amino acid protein sequences) were then sent to RAxML-NG (84) to construct the corresponding phylogenetic trees. Following best practices outlined in the RAxML-NG manual, twenty ML inferences were made on each original alignment and bootstrap replicate trees were inferred using Felsenstein's method (85), with the MRE-based bootstopping test applied after every 50 replicates (86). Bootstrapping was terminated once diagnostic statistics dropped below the threshold value and support values were drawn on the best-scoring tree.
We constructed the Bayesian timetree using the Bayesian Skyline Coalescent (87) model in BEAST2 (80), assuming a constant population prior, and the best fit nucleotide substitution model as indicated by ModelTest-NG. Sampling dates corresponded to collection date as reported in NCBI Virus; in cases where only year was reported, we assumed a collection date of 15-July for the corresponding year. We tested trees using both an uncorrelated exponentially distributed relaxed molecular clock (UCED) and a strict clock but ultimately reported results from the strict clock assumptions, as similar results were inferred from both. Markov chain Monte Carlo (MCMC) sample chains were run for 600 million iterations, convergence was checked using TRACER v1.7 (88), and trees were averaged after 10% burn-in using TreeAnnotater v2.6.3 (89) to visualize mean posterior densities at each node.
All resulting phylogenies (both ML and Bayesian) were visualized in R v.4.0.3 for MacIntosh, using the package ‘ggtree’ (90).
Recombination Analysis
Full length Nobecovirus sequences derived from CZID were analyzed for any signature of past recombination. First, the ORF1a, ORF1b, S, NS3, E, M, N, and NS7 genes from the P. rufus Nobecovirus sequence, the longest R. madagascariensis Nobecovirus sequence (OK067320), and two full genome representative sequences from the HKU9 (NC_009021) and E. helvum African lineages (NC_048212) were extracted, translated, and concatenated. Concatenated, translated sequences were then aligned in MAFFT v.7 (81, 82) using default parameter values. Nobecovirus sequences corresponding to the RoBat-CoV GCCDC1 (27, 65) and BtRt-BetaCoV/GX2018/BtCoV92 (62, 63) genotypes were omitted from recombination analyses because inserted genes and/or genetic material upstream from the nucleocapsid in the corresponding genomes interfered with the alignment.
After alignment, genomes were analyzed for amino acid similarity in the program pySimplot (91), using the P. rufus and, subsequently, the R. madagascariensis genome as query sequences, the HKU9 and Eidolon helvum African Nobecovirus clades as references, and the corresponding Madagascar sequence as the alternative. Analyses were carried out using a window size of 100 aa and a step size of 20 aa.
Next, all three full length nucleotide sequences of Madagascar Nobecovirus genomes were aligned with grouped full genome sequences corresponding to the two disparate Nobecovirus lineages: the HKU9 lineage (EF065514-EF065516, HM211098-HM211100, MG693170, NC_009021, MG762674) and the E. helvum African lineage (MG693169, MG693171-MG693172, NC_048212). As before, alignment was conducted in MAFFT v.7 (81, 82) using default parameter values.
After alignment, genomes were analyzed for recombination in the program SimPlot (v.3.5.1). Nucleotide similarity plots were generated using the P. rufus and, subsequently, the R. madagascariensis genomes as query sequences, the HKU9 and Eidolon helvum African Nobecovirus clades as references, and the corresponding Madagascar sequence as the alternative. Bootscan analyses were conducted on the same alignment, using the same query and reference inputs. Both nucleotide similarity and Bootscan analyses were carried out using a window size of 200 bp and a step size of 20 bp.
Nucleotide Sequence Accession Numbers
All three annotated full-length genome sequences (two from R. madagascariensis, one from P. rufus), plus four additional RdRp gene fragment sequences (two from R. madagascariensis, one from P. rufus, and one from E. dupreanum) were submitted to NCBI and assigned accession numbers OK020086-OK020089 (RdRp fragments) and OK067319-OK067321 (full genomes).
Results
Prevalence of CoV Sequence Detection in Field Specimens
RNA from 285 fecal, 143 throat, and 196 urine swab samples was prepped into libraries and submitted for Illumina sequencing. In 28/285 (9.82%) fecal specimens and in 2/196 (1.00%) urine specimens, at least two contigs with an average read depth >2 reads/nucleotide, and nucleotide or protein-BLAST alignments to any CoV reference sequence in NCBI were identified via CZID analysis. Because the prevalence detected in the urine samples was low, it is likely attributable to field contamination with fecal excrement upon urine swab collection, as bats often excrete both substances simultaneously under manual restraint. None of the 143 throat swabs assayed demonstrated evidence of CoV infection.
Prevalence in feces varied slightly across species, with 4/44 (9.1%) P. rufus specimens, 16/145 (11.0%) E. dupreanum specimens, and 8/96 (8.3%) R. madagascariensis specimens sequencing CoV positive. Juveniles demonstrated higher CoV prevalence than adults for P. rufus and E. dupreanum but not for R. madagascariensis. Juvenile vs. adult prevalence was 3/15 (20%) vs. 1/29 (3.5%) for P. rufus, 5/13 (38.5%) vs. 11/132 (8.3%) for E. dupreanum, and 0/13 (0%) vs. 8/83 (9.6%) for R. madagascariensis (Figure 1A). Prevalence varied seasonally across all three species, peaking coincidentally in adult and juvenile populations for P. rufus and E. dupreanum, with the highest prevalence for all three species observed during the wet season months of February-April when late-stage juveniles are present in the population, following each species' annual birth pulse (Figure 1B).
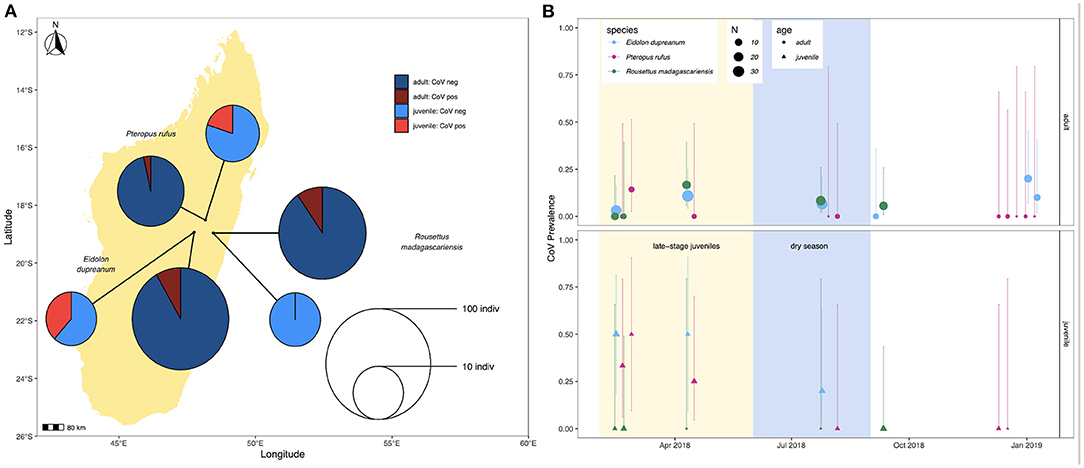
Figure 1. (A) Map of sampling sites for P. rufus, E. dupreanum, and R. madagascariensis in the Districts of Moramanga and Manjakandriana, Madagascar (P. rufus: Ambakoana roost; E. dupreanum: Angavobe/Angavokely caves; R. madagascariensis: Maromizaha cave). Pie charts correspond to coronavirus prevalence in juveniles vs. adults across all three species: 3/15 (20%) vs. 1/29 (3.5%) for P. rufus, 5/13 (38.5%) vs. 11/132 (8.3%) for E. dupreanum, and 0/13 (0%) vs. 8/83 (9.6%) for R. madagascariensis. Pie circle size corresponds to sample size on a log-10 scale. (B) Seasonal variation in adult (circle) vs. juvenile (triangle) CoV prevalence by species, from sites depicted in (A). Color corresponds to species and point size to sampling number, as indicated in the legend. Background shading corresponds to the season in which late-stage juveniles are present in the population (yellow) preceding the dry season (lightblue). Lines represent 95% confidence intervals.
Genome Annotation and BLAST
Three full or near-full CoV genome length contigs were recovered from CZID for Nobecoviruses derived from R. madagascariensis (two genomes: 28,980 and 28,926 bps in length) and P. rufus (one genome: 29,122 bps in length). In all three genomes, we successfully identified ORF1ab (including RdRp) and structural proteins S (spike), E (envelope), M (matrix), and N (nucleocapsid), in addition to accessory genes NS3, NS7a, and NS7b (Figure 2A). In keeping with convention outlined in (65), the accessory genes, NS7a and NS7b, were so named based on nucleotide alignment and amino acid identity to homologous proteins in previously described Nobecoviruses.
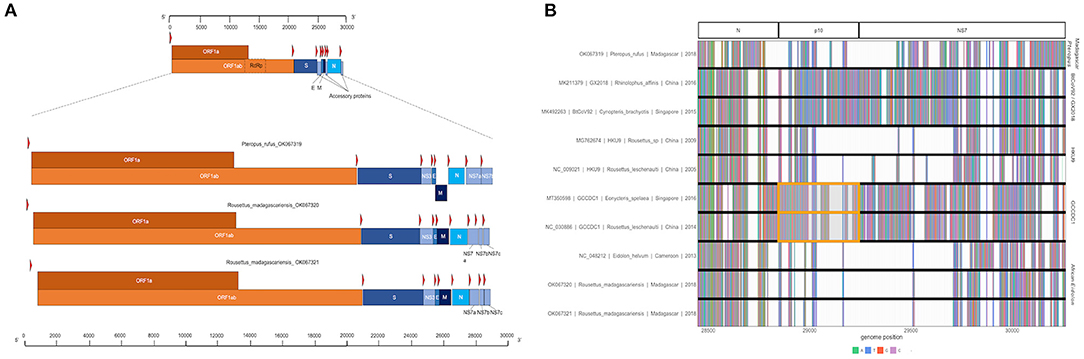
Figure 2. (A) Genome structure of novel Nobecoviruses derived from P. rufus and R. madagascariensis fruit bats. TRS locations are highlighted by red arrows, and genes are distinguished by color, with orange corresponding to ORF1a and ORF1b, and various shades of blue to structural proteins S, E, M, and N. Accessory genes NS3, NS7a, NS7b, and NS7c (R. madagascariensis genomes only) are depicted in powder blue. (B) Multiple sequence alignment of representative sequences from the five main Nobecovirus clades, spanning nucleotide positions 28449-30263. This region includes part of the N gene for all sequences and spans the region of p10 orthoreovirus insertion in GCCDC1 lineage (orange highlight), through the NS7 gene region to the 3' end of each genome.
BLAST analysis of the full genome indicated that the P. rufus Nobecovirus sequence is highly divergent, demonstrating only 72.87–73.54% identity to all previously described Nobecovirus clades, with the top blast association to E. helvum Nobecovirus lineages (Supplementary Table 1). Additionally, Nobecovirus genomes derived from R. madagascariensis demonstrated high identity (~95%) to E. helvum Nobecovirus lineages circulating in Africa. BLASTx analysis of individual genes from viruses derived from both Madagascar species demonstrated the highest identity with previously described Nobecovirus sequences in the Orf1ab region (which includes RdRp) for both P. rufus and R. madagascariensis viruses (76.1% identity for P. rufus Nobecovirus to E. helvum bat coronavirus and ~99% identity for R. madagascariensis Nobecovirus to E. helvum bat coronavirus). By contrast, both P. rufus and R. madagascariensis Nobecovirus genomes demonstrated substantial divergence from all known homologs in the S gene, where only 46.93–86.9% identity was observed. The P. rufus Nobecovirus was similarly divergent in the N gene, though R. madagascariensis Nobecoviruses demonstrated high (~91%) identity to CoV genotypes from E. helvum in this region.
In general, BLASTx queries of NS7 accessory proteins in both R. madagascarienis and P. rufus Nobecovirus demonstrated ~40–91% amino acid identity to already-characterized Nobecovirus proteins (Supplementary Table 1). Genomes derived from R. madagascariensis appeared slightly more complex than those derived from P. rufus, allowing for annotation of one additional accessory gene, NS7c, which has been characterized in recombinant Nobecovirus sequences of the RoBat-CoV GCCDC1 lineage (27, 65). Curiously, BLASTx query of the NS7a accessory protein in the P. rufus genome showed no identity to any previously described Nobecovirus protein; rather, the highest scoring protein alignment (31.25% identity, 1e-06 E-value) of the NS7a translation encompassed 40% of the query (query coverage was located at the 3' end of the query length) and corresponded to an arachnid Low-Density Lipoprotein Receptor-Related Protein 1 (LRP-1) (Supplementary Table 2). As LRP-1 is involved in the mammalian innate immune response (92), we hypothesized that this putative novel ORF could be a viral gene involved in immune antagonism. To check the integrity of our de novo assembly in NS7a, we mapped the deduplicated raw reads from mNGS to the full genome P. rufus Nobecovirus contig generated by CZID (78) (Supplementary Figure 1). We confirmed >200x read coverage across the region corresponding to the putative NS7a accessory protein, with good representation of both forward and reverse-facing reads across the length of the protein, as well as the intergenic regions preceding and succeeding it. We were also able to identify a putative Transcription Regulatory Sequence (TRS) preceding this gene (Table 1), further validating our confidence that P. rufus Nobecovirus NS7a represents a real though highly divergent protein.
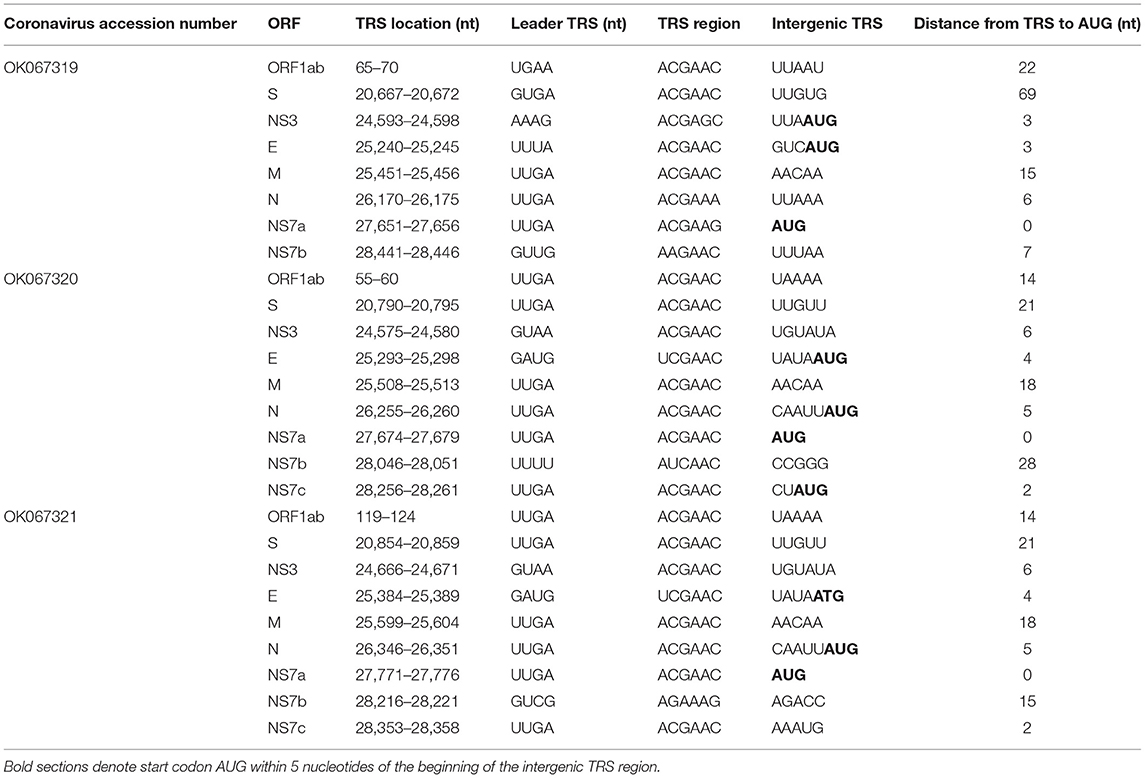
Table 1. Putative Transcription Regulatory Sequences in novel Nobecoviruses from Madagascar fruit bats.
The p10 orthoreovirus insertion within the RoBat-CoV GCCDC1 Nobecovirus lineage was not observed in either Nobecovirus genomes from R. madagascariensis or P. rufus. Nonetheless, examination of the multiple sequence alignment of representative sequences of all Nobecovirus clades in this region demonstrated the presence of some variable genetic material downstream from the N gene and upstream from the NS7a gene in the divergent P. rufus Nobecovirus genome (Figure 2B). Nobecoviruses clustering in the BtRt-BetaCoV/GX2018 - BtCoV92 lineage also carry a unique coding sequence in this region, highlighting the dynamic nature of the 3' end of the CoV genome (63).
In addition to the identification of both canonical and novel ORFs described above, we also observed non-coding TRS elements preceding all the major proteins in all three Nobecovirus genomes (Table 1). Many of these correspond to the 5'-ACGAAC-3' six bp core motif common to many Betacoronaviruses, including SARS-CoV and previously described in Nobecoviruses of the GCCDC1 and GX2018/BtCoV92 lineages (62, 65, 93). For most genes, these TRS elements were located a short distance upstream from the corresponding gene (Table 1). Elements identified in the two R. madagascariensis genomes were largely comparable, suggesting that these two sequences could represent slight variations in the same virus lineage. Some putative TRS elements, including that preceding P. rufus NS7a, showed variation from the 5'-ACGAAC-3' core motif, with some recapitulating the 5'-AAGAA-3' motif common to SARS-CoV-2 (94). TRS variations may be indicative of variation in gene expression across individual bats and/or species.
Phylogenetic Analysis
Phylogenetic analysis of full length Betacoronavirus genomes confirmed that both P. rufus and R. madagascariensis genomes cluster in the Nobecovirus subgenus of the Betacoronaviruses, with the divergent P. rufus forming its own distinct clade and both R. madagascariensis genomes grouping with the previously described E. helvum reference sequence from Cameroon (64) (Figure 3A). We observed distinct groupings of five main Nobecovirus lineages in our phylogeny: (a) the largely Asian-derived HKU9 sequences, (b) the African E. helvum-derived sequences (now including new R. madagascariensis Nobecovirus genomes), (c) the recombinant GCCDC1 genomes, (d) the BtRt-BetaCoV/GX2018 and BtCoV92 genomes described respectively from China and Singapore, and (e) the divergent P. rufus genome contributed here from Madagascar. Intriguingly, the P. rufus genome groups ancestral to all other Nobecoviruses, followed by the E. helvum/R. madagascariensis African lineage, with the Asian genotypes forming three distinct (and more recent) clades corresponding to genotypes HKU9, GCCDC1, and GX2018 – BtCoV92. This finding suggests that the evolutionary origins of the Nobecovirus clade likely predate the recent divergence of P. rufus from sister Pteropus spp. taxa (though see results for Bayesian timetree below), which are primarily Asian in their distribution. To date, no other Nobecovirus sequences have yet been described from any Pteropus spp. bats; additional sampling across other Pteropus lineages will be needed to confirm our hypothesis of a Pteropus genus origin for this Betacoronavirus clade. Further phylogenetic analysis of a 259 bp fragment of the RdRp gene reconfirmed these groupings and suggested the presence of at least two distinct genetic variants within the P. rufus lineage (Figure 3B). One RdRp fragment derived from feces of the third Malagasy fruit bat, E. dupreanum, grouped within the E. helvum – R. madagascariensis African Nobecovirus lineage, consistent with previous reporting (12). Characterization of the full length genome of this virus will be needed to clarify whether it represents a genetic variant of or a distinct genotype from the R. madagascariensis virus. Phylogenetic analysis of the RdRp fragment allowed for inclusion of one partial Nobecovirus sequence derived from E. helvum bats in Kenya (HQ728482), which also grouped within the E. helvum – R. madagascariensis African clade, confirming the distribution of this genotype across West and East Africa and into the South-Western Indian Ocean Islands. Notably, one partial Cameroonian E. helvum sequence (MG693170) clustered with HKU9 sequences from Asia, rather than within the E. helvum – R. madagascariensis African clade. These findings suggest that both “African” and “Asian” Nobecovirus lineages are likely broadly geographically distributed.
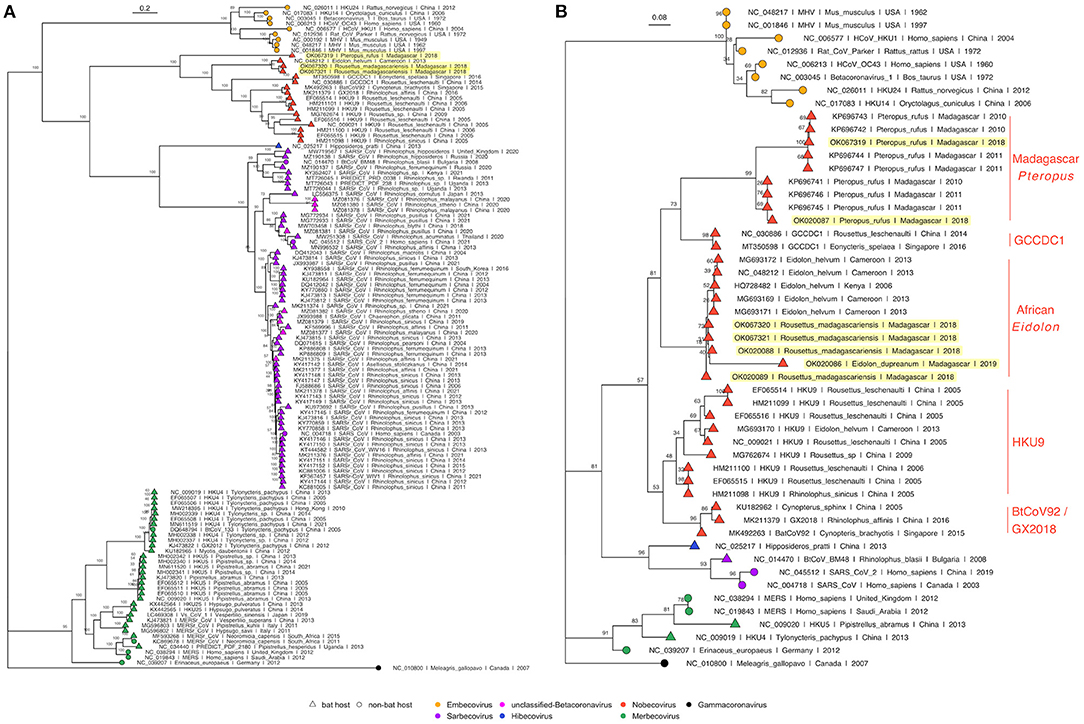
Figure 3. (A) Maximum Likelihood phylogeny of full genome Betacoronavirus sequences, (RAxML-NG, GTR+I+G4) and (B) RdRp phylogeny of a 259bp fragment of Betacoronavirus Orf1b (RAxML-NG, TVM+I+G4) (83, 84). Bootstrap support values computed using Felsenstein's method (85) are visualized on tree branches. In both (A,B), novel Madagascar sequences are highlighted in yellow, and tip points are colored by Betacoronavirus subgenus, corresponding to the legend. Tip shape indicates whether the virus is derived from a bat (triangle) or non-bat (circle) host. Both trees are rooted in turkey Gammacoronavirus, accession number NC_010800. Branch lengths are scaled by nucleotide substitutions per site, corresponding to the scalebar given in (A,B).
Amino acid phylogenies computed from translated protein alignments of the S, E, M, and N Betacoronavirus structural genes (Supplementary Figure 2) further confirmed evolutionary relationships suggested in Figure 3. S, M, and N gene phylogenies demonstrated distinct groupings of five main Nobecovirus lineages outlined above, while in the E gene phylogeny, the P. rufus sequence grouped adjacent to the single Cameroonian-derived E. helvum sequence within the HKU9 clade. However, we note that bootstrap values were extremely low in this E gene phylogeny, suggesting that additional sampling is needed to confirm these evolutionary relationships.
Results from our Bayesian timetree analysis (Figure 4; Supplementary Figure 3) recapitulated ML support for the five distinct Nobecovirus subclades but indicated a time to MRCA for the entire Nobecovirus clade of ~300 years ago (1695; 95% HPD: 1643–1748) (Figure 4), with the African Eidolon lineage (which included our R. madagascariensis sequences) branching off ~200 years ago (1827; 95% HPD: 1797–1857). Surprisingly, both of these viral divergence times post-date the estimated radiation of Madagascar host bats from sister species (58, 59), suggesting that Malagasy bat populations may support substantial viral exchange with bats from surrounding islands and the African continent. Additional sampling of Nobecovirus lineages in Asian-distributed Pteropus spp. bats will be crucial to ascertaining whether the evolutionary origins of the Nobecovirus subgenus could date back any further than that suggested by samples highlighted here. Evolutionary relationships were preserved in phylogenies which included GCCDC1 orthoreovirus recombinant sequences, though inclusion of these sequences diminished certainty surrounding evolutionary divergence times, resulting in broad HPD distributions (Supplementary Figure 3).
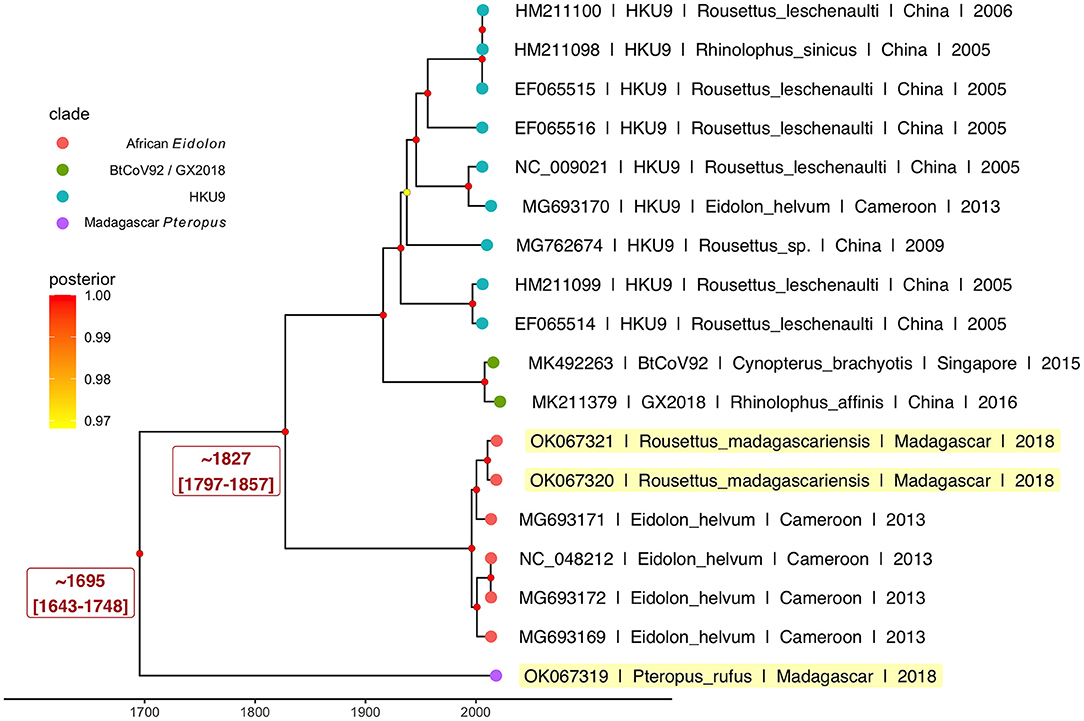
Figure 4. Bayesian phylogeny to estimate time to MRCA for novel P. rufus Nobecovirus subclade. Plot depicts output of 600 million runs of a strict molecular clock Bayesian Skyline Coalescent model (GTR+I+G4) as implemented in BEAST2 (80, 87). Four of the five major Nobecovirus subclades are depicted based on colored tip points, and the mean posterior estimates from averaging of all 600 billion trees after removal of 10% burn-in are visualized by color at the corresponding node. The dates of estimated time to MRCA for the P. rufus Nobecovirus subclade and the African Eidolon subclades are highlighted in red text.
Recombination Analysis
SimPlot analysis confirmed the evolutionary distinctiveness of the P. rufus Nobecovirus genome, which showed <70% amino acid similarity and <50% nucleotide similarity to HKU9, E. helvum, and R. madagascariensis genotypes across the majority of its genome length (Figures 5A,B). Consistent with BLAST results, the P. rufus Nobecovirus genome demonstrated the highest similarity to previously described sequences in the Orf1b region, which includes RdRp. The R. madagascariensis Nobecoviruses, by contrast, showed >90% amino acid and nucleotide similarity to the E. helvum African lineage throughout Orf1ab, but both P. rufus and R. madagascariensis sequences diverged from all other reference genomes in the first half of the spike protein, which corresponds to the S1 subunit and includes the receptor binding domain that mediates viral entry into host cells (95). Further divergence for both P. rufus and R. madagascariensis Nobecoviruses was observed in the N structural protein and in the NS7 accessory genes. Bootscan analysis further confirmed these findings, showing that the P. rufus Nobecovirus clusters with HKU9 lineages across Orf1ab, NS3, E, and M genes but demonstrates evidence of recombination with E. helvum – R. madagascariensis African lineages in the S (particularly S1), N, and NS7 genes (Figure 5C). Similarly, bootscanning demonstrated that R. madagascariensis Nobecoviruses group with the E. helvum lineage across Orf1ab, NS3, E, and M but show evidence of recombination with HKU9 and P. rufus Nobecovirus in S (again, particularly S1), N, and NS7 genes (Figure 5C), thus highlighting the dynamic nature of these regions of the Nobecovirus genome.
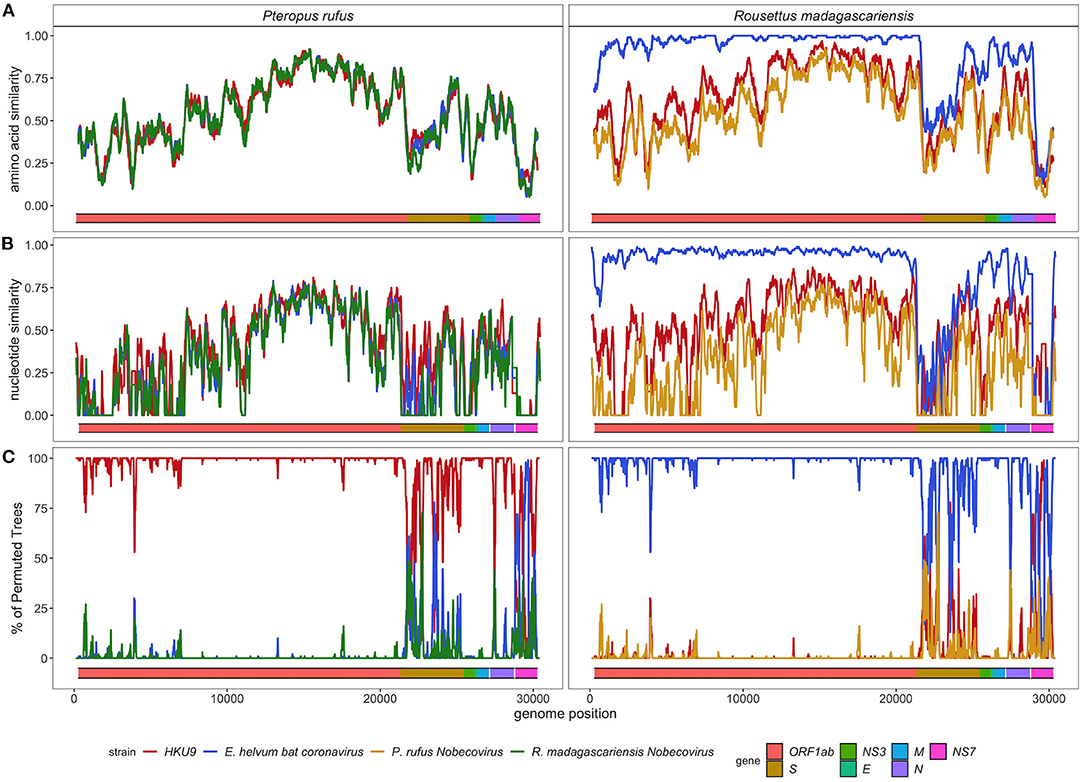
Figure 5. (A) Amino acid similarity, (B) nucleotide similarity and (C) Bootscan plots computed in pySimplot (91) (A) and SimPlot (v.3.5.1) (B,C), using a query sequence of P. rufus (left) and R. madagascariensis (right)-derived Nobecovirus sequences. (A) Amino acid similarity plots compares P. rufus Nobecovirus and R. madagascariensis MIZ240 against one HKU9 (NC_009021) and one E. helvum bat CoV (NC_048212) sequence and against each other. Nucelotide similarity and bootscan plots compare P. rufus Nobecovirus and both R. madagascariensis Nobecovirus sequences against grouped reference sequences corresponding to HKU9 (EF065514-EF065516, HM211098-HM211100, MG693170, NC_009021, MG762674) and Eidolon helvum Africa-derived (MG693169, MG693171-MG693172, NC_048212) Nobecovirus lineages. Line color indicates similarity (A,B) and bootscan grouping (C) of the query sequence with the corresponding Nobecovirus genotype, along disparate regions of the CoV genome, as indicated by the colored bar at the bottom of each plot. Amino acid similarity plots (A) were generated using a window size of 100aa and a step size of 20aa. Nucleotide similarity and bootscan plots (B,C) were generated using a window size of 200bp and a step size of 20bp.
Discussion
Here, we contribute three full-length genome sequences and four RdRp fragments to public NCBI repositories; these sequences correspond to at least two novel Nobecoviruses derived from wild Malagasy fruit bats, Pteropus rufus and Rousettus madagascariensis, with evidence of additional genetic variants circulating in Eidolon dupreanum, as well. Phylogenetic analyses suggest that previously described Nobecoviruses can be grouped into five general clades: (a) the HKU9 lineage of largely Asian origins, (b) the mostly African-distributed lineage derived from E. helvum bats (which contains our R. madagascariensis and E. dupreanum sequence contributions), (c) the recombinant GCCDC1 lineage, which has been previously reported from China and Singapore (27, 65), (d) the BtRt-BetaCoV/GX2018 – BtCoV92 lineage, also known from China and Singapore (62, 63), and (e) a novel, divergent clade corresponding to the newly-described P. rufus genome. This new P. rufus Nobecovirus appears to be ancestral to all previously-described Nobecovirus sequences with a time to MRCA dating back ~300 years and likely post-dating the late Pleistocene divergence of the P. rufus host from other Southwest Indian Ocean Island pteropodids (58). The relatively recent branching of the ancestral P. rufus Nobecovirus sequence and the nested grouping of the R. madagascariensis sequences within the African Eidolon subclade suggest that substantial viral genetic exchange takes place between fruit bats in Madagascar, in the surrounding Southwest Indian Ocean Islands, and on the African continent. Thirty-eight of Madagascar's 49 bat species are endemic, while two species exhibit ranges that include the Comoros and Réunion islands (respectively, Scotophilus borbonicus and Miniopterus aellini), and nine species can be found broadly distributed across Africa and, in some cases, parts of Asia and Europe (e.g., Pipistrellus kuhlii, Mops midas) (67). Additional coronavirus sampling in some of these more cosmopolitan insectivorous bat species will shed light on their role in diversification and distribution of Nobecoviruses more broadly. Notably, all three Malagasy fruit bat species in the family Pteropodidae—from which Nobecoviruses are thought to be sourced—are endemic to Madagascar and not known to disperse outside the island.
Importantly, though largely characterized in Asia, HKU9 Nobecovirus genotypes have been identified in West Africa (Cameroon) (64), and E. helvum lineages have been characterized across both West (Cameroon) (64) and East (Kenya) (30, 96) Africa, as well as on one Indian Ocean island (Madagascar). These findings suggest that different Nobecovirus clades may be more broadly geographically distributed than has been previously recognized. To our knowledge, no Nobecoviruses have been identified from the southern extension of the pteropodid fruit bat range in Australia, or from any Pteropus spp. bat in Asia; characterization of any CoVs infecting these bats, which are known to host important, zoonotic henipaviruses (97) and lyssaviruses (93), would do much to enhance our understanding of the phylogeography of the Nobecovirus clade. Madagascar represents a unique phylogeographic melting pot, with flora and fauna—and corresponding viruses—of both African and Asian descent (55), offering opportunities for mixing of largely disparate viral groups. This mixing is important in light of the CoV penchant for recombination, which can allow viruses from one clade to gain function through acquisition of genetic material from another, thus facilitating rapid changes in host range (43–48). Indeed, recombination events have been implicated in the cross-species emergence of most zoonotic coronaviruses (44–48, 98), and viral acquisition of an ACE2-compatible receptor binding domain—often mediated through recombination—has been a critical step in the expansion of bat Sarbecovirus ranges to include human hosts (98–101). As bats can host multiple coronaviruses at once and often roost together, there are ample opportunities for recombination events to occur (7, 102).
Nobecoviruses are not known to be zoonotic and have been thus far described exclusively infecting fruit bats hosts of the Old World bat family, Pteropodidae. Nonetheless, the Nobecovirus subgenus demonstrates a CoV-characteristic tendency to recombine, as evidenced by circulation of the widespread GCCDC1 lineage in Asia, which carries a p10 gene insertion derived from an orthoreovirus between the N structural protein and NS7a accessory protein toward the 3' end of the genome (65). This finding highlights CoVs' capacity to undergo both homologous (within-clade) and heterologous (out-of-clade) recombination. Since bats are known to maintain co-infections with many viruses simultaneously (103, 104), this recombinatory potential could allow CoVs to rapidly acquire new genetic material from coinfections, including receptor binding or host immune evasion capabilities that may expand host range. The orthoreovirus insertion within the GCCDC1 virus genome was not detected among the CoVs in our dataset, though, anecdotally, mNGS of fecal and throat samples collected in our sampling did identify evidence of orthoreovirus infection in several throat swabs derived from R. madagascariensis bats, highlighting the potential for recombination opportunities between these two viral groups. Notably, recombination analyses suggested substantial selection has taken place in this region of both R. madagascariensis and P. rufus-derived Nobecoviruses. Selection at the 3' end of the CoV genome may modulate viral replication ability, since several regulatory sequences and accessory genes (e.g., NS7) are defined in this region (94). Viral replication ability may be further impacted by variation in TRS motifs, which regulate expression of corresponding genes. We identified putative TRS sequences corresponding to all structural and non-structural genes identified in all three contributed Nobecovirus genomes; while the majority of these TRS motifs recapitulated the well-conserved 5'-ACGAAC-3' Betacoronavirus core sequence (62, 65), variation in a subset of genes across species and individuals (e.g., differing motifs between two R. madagascariensis-derived genomes) may correspond to variation in gene expression.
Recombination potential is a particular cause for concern in cases where viruses that lack the ability to infect human cells may acquire this zoonotic capacity through genetic exchange with other viruses coinfecting the same host. Indeed, the original SARS-CoV is believed to have acquired its capacity to bind human ACE2 through a recombination event with ACE2-using Sarbecoviruses in the disparate SARS-CoV-2 clade (98). Sarbecoviruses, in particular, are known to recombine frequently, giving rise to new genetic variants, in regions where different species of Rhinolophid bat hosts co-roost and share viruses (7). Cave-resident Malagasy fruit bats, E. dupreanum and R. madagascariensis, are known to co-roost with each other and with several species of insectivorous bat (67), which could facilitate Nobecovirus recombination. The observed similarity in Nobecovirus sequences derived from E. dupreanum and R. madagascariensis (which cluster in the same lineage), as compared with disparate sequences derived from tree-roosting P. rufus, suggest that some CoV genetic exchange may have already taken place between bats with overlapping habitats. To date, zoonotic potential has not been demonstrated for any previously described Nobecoviruses, and Rhinolophid bats associated with ACE2 usage are not resident in Madagascar. Nonetheless, bats in family Vespertilionidae, the family most commonly associated with zoonotic Merbecoviruses (8–10), are widespread in Madagascar, and Mormopterus jugularis, a known Molossidae bat host for Alphacoronaviruses of undetermined zoonotic potential (33), has been described co-roosting with R. madagascariensis (105). Bootscan analyses identified signatures of recombination in the S1 subunit of both P. rufus and R. madagascariensis Nobecovirus spike proteins, suggesting that this region of the genome, which modulates host range through cell surface receptor binding, may be under selective pressure. In addition to facilitating direct bat-to-human spillover, recombination can also play an important role in facilitating cross-species emergence via intermediary bridge hosts: both SARS-CoV-1 and MERS-CoV demonstrated a critical role for intermediate hosts (respectively, palm civets and camels) in their evolutionary history of zoonotic emergence (106–108). Given the high endemicity and biodiversity characteristic of Madagascar's mammalian fauna, the island abounds with opportunities for CoV recombination with unique viruses in unique hosts.
In addition to posing risk for future zoonoses, Nobecoviruses derived from wild Madagascar fruit bats could provide unprecedented genetic material for recombination to existing human coronaviruses already in circulation across the island—most notably SARS-CoV-2 (75). At the time of this writing, SARS-CoV-2 infections remain widespread and vaccination limited across Madagascar (109). Previous work has assessed the risk of reverse zoonosis, or ‘spillback’ of SARS-CoV-2 from human to bat populations in the United States (16), concluding that high human caseloads and frequent human-bat contact rates in research settings pose both conservation risks to naïve bat populations presented with a novel pathogen, as well as human health risks presented by the possible establishment of secondary wildlife reservoirs for SARS-CoV-2 capable of sourcing future epidemics or the generation of unique viral variants through human-wildlife virus recombination (16). Bat-human contact rates are higher, on average, in Madagascar than in the US, as bats are consumed across the island for subsistence and frequently found roosting in human establishments or human-adjacent habitats (68–72). SARS-CoV-2 has already demonstrated its capacity for successful reverse zoonosis and adaptation to non-human hosts, in the case of farmer-sourced infections of mink in Finland (110), underscoring the legitimacy of these concerns. Notably, spillback is less likely to be an issue in regions where animals are killed upon capture for consumption (vs. transported live), as is often the case in Madagascar (72).
Prevalence of coronavirus RNA by sequence detection in fecal samples averaged around 10% across all three Malagasy fruit bat species examined in our study, consistent with CoV prevalence reported in wild bat species elsewhere (12, 33). One previous study of CoV circulation in Madagascar fruit bats reported much lower prevalence of infection in E. dupreanum and R. madagascariensis-derived fecal specimens, respectively 1/88 (1.1%) and 0/141 (0%), as compared with a 13/88 (14.8%) prevalence in P. rufus-derived feces (12). As in our study, this previous work found no positive infections in throat swabs, supporting a gastrointestinal tropism for CoVs in this fruit bat system, in contrast to the respiratory infections more commonly observed in humans (though humans do shed SARS-CoV-2 gastrointestinally, as well). Currently, very little is known concerning the cell receptors of Nobecoviruses and their tissue tropism. One previous study demonstrated how bat coronavirus HKU9 (along with MERS-CoV) gained cell entry via the ER chaperone activity cell membrane receptor GRP78, but the study did not describe the distribution of this receptor beyond demonstrating co-expression with DPP4 (111). Future co-sampling of both throat and fecal swabs for bat coronaviruses will shed light on their tropism and shedding across different tissues.
One previous study in the West Indian Ocean provided more information about CoV prevalence in Madagascar bats, with 6/45 (13.3%) R. madagascariensis fecal specimens testing CoV positive, as compared to 10/63 (15.9%) M. jugularis specimens, 4/44 (9.1%) Triaenops menamena specimens, and 2/21 (9.5%) Mops midas specimens (33). Consistent with previous findings (32, 41, 112, 113), we observed the highest prevalence of CoV infection in P. rufus and E. dupreanum juveniles. We hypothesize that the absence of juvenile infection identified in R. madagascariensis bats in our study could be due to the staggered nature of the birth pulse for these three species: Madagascar fruit bats birth in three successive birth pulse waves, led by P. rufus in October, and followed by E. dupreanum in November and R. madagascariensis in December and January (114). As the bulk of juvenile R. madagascariensis bats sampled in our study were captured in February, it is possible that most were still too young to be CoV-positive [perhaps under protection from inherited maternal immunity (61)]. By the time of the second R. madagascariensis sampling in April, juveniles would have been large enough to be erroneously classed as adults, as size range variation is more limited in small R. madagascariensis bats as compared with the two other Malagasy fruit bat species (67). Our observations are consistent with previous records indicating that juvenile and subadult bats show enhanced CoV shedding after weaning and the loss in maternal immunity in other systems (115, 116).
Our work emphasizes the importance of longitudinal ecological studies in identifying viral shedding events in transiently-infected wildlife hosts across multiple age and reproductive classes. Enhanced future surveillance efforts will be useful in pinpointing the exact seasonality of peak CoV shedding events, and mitigation efforts for both zoonotic and reverse zoonotic risks should be focused on limiting human-bat contact (in particular, the government-sanctioned hunting seasons) during these periods. Our study highlights the enhanced evolutionary and functional virological inference that can be derived from full genome sequences, detected by unbiased metagenomic sequencing. Characterization of these genomes provides the basis for basic virology experiments to follow, such as pseudovirus or reverse genetics experiments aimed at understanding host receptor utilization. More thorough studies documenting the seasonal dynamics of bat-borne CoVs, which elucidate genetic variation within and between species that share habitats in wild populations will be essential to understanding CoV recombination, host shifting, and zoonotic potential. Replication of such studies across the global range of both coronaviruses and their bat hosts, in particular in understudied regions of Africa, is needed to assess the landscape of future zoonotic risks and present opportunities for intervention and mitigation.
Data Availability Statement
The sequences presented in the study are deposited in NCBI, accession numbers OK020086-OK020089 and OK067319-OK067321.
Ethics Statement
The animal study was reviewed and approved by UC Berkeley Animal Care and Use Committee and Madagascar Ministry of Forest and the Environment under guidelines posted by the American Veterinary Medical Association.
Author Contributions
CB conceived of the project and acquired the funding, in collaboration with J-MH, PD, JD, and CT. Field samples were collected and RNA extracted by AA, SA, AG, HR, TR, NR, and CB. AK led the mNGS, with support from VA, HR, TR, and CB. GK and CB analyzed the resulting data and co-wrote the original draft of the manuscript, which all authors edited and approved.
Funding
Research was funded by the National Institutes of Health (1R01AI129822-01 grant to J-MH, PD, and CB), DARPA (PREEMPT Program Cooperative Agreement no. D18AC00031 to CB), the Bill and Melinda Gates Foundation (GCE/ID OPP1211841 to CB and J-MH), the Adolph C. and Mary Sprague Miller Institute for Basic Research in Science (postdoctoral fellowship to CB), the Branco Weiss Society in Science (fellowship to CB), and the Chan Zuckerberg Biohub.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge Kimberly Rivera and Sarah Guth for help in the field and the lab and the Virology Unit at the Institut Pasteur de Madagascar and Maira Phelps of the Chan Zuckerberg Biohub (CZB) for logistical support. They additionally thank Angela Detweiler, Michelle Tan, and Norma Neff of the CZB genomics platform for mNGS support and thank the Brook lab at the University of Chicago for helpful contributions to the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2022.786060/full#supplementary-material
References
1. Banerjee A, Kulcsar K, Misra V, Frieman M, Mossman K. Bats and coronaviruses. Viruses. (2019) 11:7–9. doi: 10.3390/v11010041
2. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 580:E7. doi: 10.1038/s41586-020-2202-3
3. Hu B, Ge X, Wang LF, Shi Z. Bat origin of human coronaviruses. Virol J. (2015) 12:1–10. doi: 10.1186/s12985-015-0422-1
4. Ravelomanantsoa NAF, Guth S, Andrianiaina A, Andry S, Gentles A, Ranaivoson HC, et al. The zoonotic potential of bat-borne coronaviruses. Emerg Top Life Sci. (2020) 4:353–69. doi: 10.1042/ETLS20200097
5. Wille M, Holmes EC. Wild birds as reservoirs for diverse and abundant gamma- And deltacoronaviruses. FEMS Microbiol Rev. (2020) 44:631–44. doi: 10.1093/femsre/fuaa026
6. Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. (2010) 84:11336–49. doi: 10.1128/JVI.00650-10
7. Hu B, Zeng LP, Yang XL, Ge XY, Zhang W, Li B, et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. (2017) 13:e1006698. doi: 10.1371/journal.ppat.1006698
8. Anthony SJ, Gilardi K, Menachery VD, Goldstein T, Ssebide B, Mbabazi R, et al. Further evidence for bats as the evolutionary source of middle east respiratory syndrome coronavirus. MBio. (2017) 8:1–13. doi: 10.1128/mBio.00373-17
9. Woo PC, Lau SK, Li KS, Tsang AK, Yuen KY. Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg Microbes Infect. (2012) 1:e35–e35. doi: 10.1038/emi.2012.45
10. Corman VM, Ithete NL, Richards LR, Schoeman MC, Preiser W, Drosten C, et al. Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J Virol. (2014) 88:11297–303. doi: 10.1128/JVI.01498-14
11. Frutos R, Serra-Cobo J, Pinault L, Lopez Roig M, Devaux CA. Emergence of bat-related Betacoronaviruses: hazard and risks. Front Microbiol. (2021) 12:437. doi: 10.3389/fmicb.2021.591535
12. Razanajatovo NH, Nomenjanahary LA, Wilkinson DA, Razafimanahaka JH, Goodman SM, Jenkins RK, et al. Detection of new genetic variants of Betacoronaviruses in endemic frugivorous bats of Madagascar. Virol J. (2015) 12:42. doi: 10.1186/s12985-015-0271-y
13. Woo PCY, Huang Y, Lau SKP, Yuen KY. Coronavirus genomics and bioinformatics analysis. Viruses. (2010) 2:1804–20. doi: 10.3390/v2081803
14. Chen SC, Olsthoorn RCL, Yu CH. Structural phylogenetic analysis reveals lineage-specific RNA repetitive structural motifs in all coronaviruses and associated variations in SARS-CoV-2. Virus Evol. (2021) 7:1–18. doi: 10.1093/ve/veab021
15. Zhou H, Chen X, Hu T, Li J, Song H, Liu Y, et al. A novel bat coronavirus reveals natural insertions at the S1/S2 cleavage site of the Spike protein and a possible recombinant origin of HCoV-19. Curr Biol. (2020) 30:2196–203. doi: 10.1016/j.cub.2020.05.023
16. Olival KJ, Cryan PM, Amman BR, Baric RS, Blehert DS, Brook CE, et al. Possibility for reverse zoonotic transmission of SARS-CoV-2 to free-ranging wildlife: A case study of bats. PLoS Pathogens. (2020) 16:e1008758. doi: 10.1371/journal.ppat.1008758
17. Forni D, Cagliani R, Sironi M. Recombination and positive selection differentially shaped the diversity of Betacoronavirus subgenera. Viruses. (2020) 12:1313. doi: 10.3390/v12111313
18. Llanes A, Restrepo CM, Caballero Z, Rajeev S, Kennedy MA, Lleonart R. Betacoronavirus genomes: How genomic information has been used to deal with past outbreaks and the COVID-19 pandemic. Int J Mol Sci. (2020) 21:4546. doi: 10.3390/ijms21124546
19. Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. (2005) 310:676. doi: 10.1126/science.1118391
20. Hul V, Delaune D, Karlsson EA, Hassanin A, Tey PO, Baidaliuk A, et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat Commun. (2021) 12:6563. doi: 10.1038/s41467-021-26809-4
21. Valitutto MT, Aung O, Tun KYN, Vodzak ME, Zimmerman D, Yu JH, et al. Detection of novel coronaviruses in bats in Myanmar. PLoS ONE. (2020) 15:e0230802. doi: 10.1371/journal.pone.0230802
22. Lau SKP, Li KSM, Huang Y, Shek CT, Tse H, Wang M, et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol. (2010) 84:2808–19. doi: 10.1128/JVI.02219-09
23. Latinne A, Hu B, Olival KJ, Zhu G, Zhang L, Li H, et al. Origin and cross-species transmission of bat coronaviruses in China. Nat Commun. (2020) 11:4235. doi: 10.1038/s41467-020-17687-3
24. Wacharapluesadee S, Duengkae P, Rodpan A, Kaewpom T, Maneeorn P, Kanchanasaka B, et al. Diversity of coronavirus in bats from Eastern Thailand. Virol J. (2015) 12:57. doi: 10.1186/s12985-015-0289-1
25. Zhou H, Ji J, Chen X, Bi Y, Li J, Wang Q, et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell. (2021) 184:4380–91.e14. doi: 10.1101/2021.03.08.434390
26. Lacroix A, Duong V, Hul V, San S, Davun H, Omaliss K, et al. Genetic diversity of coronaviruses in bats in Lao PDR and Cambodia. Infect Genet Evol. (2017) 48:10–8. doi: 10.1016/j.meegid.2016.11.029
27. Paskey AC, Ng JHJ, Rice GK, Chia WN, Philipson CW, Foo RJH, et al. Detection of recombinant Rousettus bat coronavirus GCCDC1 in lesser dawn bats (Eonycteris spelaea) in Singapore. Viruses. (2020) 12:539. doi: 10.3390/v12050539
28. Becker DJ, Albery GF, Sjodin AR, Poisot T, Bergner LM, Chen B, et al. Optimising predictive models to prioritise viral discovery in zoonotic reservoirs. The Lancet Microbe. (2022) 21:00245-7. doi: 10.1016/S2666-5247(21)00245-7
29. Anthony SJ, Ojeda-Flores R, Rico-Chávez O, Navarrete-Macias I, Zambrana-Torrelio CM, Rostal MK, et al. Coronaviruses in bats from Mexico. J Gen Virol. (2013) 94(PART 5):1028–38. doi: 10.1099/vir.0.049759-0
30. Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis. (2009) 15:482–5. doi: 10.3201/eid1503.081013
31. Tao Y, Tong S. Complete genome sequence of a Severe Acute Respiratory Syndrome-related Coronavirus from Kenyan bats. Microbiol Resour Announc. (2019) 8:e00548–19. doi: 10.1128/MRA.00548-19
32. Montecino-Latorre D, Goldstein T, Gilardi K, Wolking D, van Wormer E, Kazwala R, et al. Reproduction of East-African bats may guide risk mitigation for coronavirus spillover. One Health Outlook. (2020) 2:2. doi: 10.1186/s42522-019-0008-8
33. Joffrin L, Goodman SM, Wilkinson DA, Ramasindrazana B, Lagadec E, Gomard Y, et al. Bat coronavirus phylogeography in the Western Indian Ocean. Sci Rep. (2020) 10:6873. doi: 10.1038/s41598-020-63799-7
34. Su S, Wong G, Shi W, Liu J, Lai ACK, Zhou J, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. (2016) 24:490–502. doi: 10.1016/j.tim.2016.03.003
35. Woolhouse MEJ, Haydon DT, Antia R. Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol Evol. (2005) 20:238–44. doi: 10.1016/j.tree.2005.02.009
36. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 588:E6. doi: 10.1038/s41586-020-2951-z
37. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. (2003) 147:120–1. doi: 10.1038/nature02145
38. Raj VS, Mou H, Smits SL, Dekkers DHW, Müller MA, Dijkman R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. (2013) 495:251–4. doi: 10.1038/nature12005
39. Wang Q, Qi J, Yuan Y, Xuan Y, Han P, Wan Y, et al. Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe. (2014) 16:328–37. doi: 10.1016/j.chom.2014.08.009
40. Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, et al. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg Infect Dis. (2013) 19:1697–9. doi: 10.3201/eid1910.130946
41. Anthony SJ, Johnson CK, Greig DJ, Kramer S, Che X, Wells H, et al. Global patterns in coronavirus diversity. Virus Evol. (2017) 3:vex012. doi: 10.1093/ve/vex012
42. Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. (2014) 101:45–56. doi: 10.1016/j.antiviral.2013.10.013
43. Lau SKP, Lee P, Tsang AKL, Yip CCY, Tse H, Lee RA, et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol. (2011) 85:11325-37. doi: 10.1128/JVI.05512-11
44. Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, Lemey P, et al. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol. (2005) 79:1595–604. doi: 10.1128/JVI.79.3.1595-1604.2005
45. Woo PCY, Lau SKP, Huang Y, Tsoi HW, Chan KH, Yuen KY. Phylogenetic and recombination analysis of coronavirus HKU1, a novel coronavirus from patients with pneumonia. Arch Virol. (2005) 150:2299–311. doi: 10.1007/s00705-005-0573-2
46. Sabir JSM, Lam TTY, Ahmed MMM, Li L, Shen Y. E. M, Abo-Aba S, et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science. (2016) 351:81–4. doi: 10.1126/science.aac8608
47. Wang Y, Liu D, Shi W, Lu R, Wang W, Zhao Y, et al. Origin and possible genetic recombination of the Middle East respiratory syndrome coronavirus from the first imported case in China: Phylogenetics and coalescence analysis. mBio. (2015) 6:e01280–15. doi: 10.1128/mBio.01280-15
48. Lau SKP, Feng Y, Chen H, Luk HKH, Yang WH, Li KSM, et al. Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J Virol. (2015) 89:10532–47. doi: 10.1128/JVI.01048-15
49. Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. (2010) 84:3134–46. doi: 10.1128/JVI.01394-09
50. Ogando NS, Ferron F, Decroly E, Canard B, Posthuma CC, Snijder EJ. The curious case of the Nidovirus exoribonuclease: Its role in RNA synthesis and replication fidelity. Front Microbiol. (2019) 10:1813. doi: 10.3389/fmicb.2019.01813
51. Nga PT, de Parquet MC, Lauber C, Parida M, Nabeshima T, Yu F, et al. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog. (2011) 7:e1002215. doi: 10.1371/journal.ppat.1002215
52. Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ. Nidovirales: Evolving the largest RNA virus genome. Virus Res. (2006) 117:17–37. doi: 10.1016/j.virusres.2006.01.017
53. Smith EC, Blanc H, Vignuzzi M, Denison MR. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: Evidence for proofreading and potential therapeutics. PLoS Pathogens. (2013) 9:e1003565. doi: 10.1371/journal.ppat.1003565
54. Lai MMC. RNA recombination in animal and plant viruses. Microbiol Rev. (1992) 56:61–79. doi: 10.1128/mr.56.1.61-79.1992
55. Masters J, Wit M de, Asher RJ. Reconciling the origins of Africa, India and Madagascar with vertebrate dispersal scenarios. Folia Primatologica. (2006) 3200:399–418. doi: 10.1159/000095388
57. Shi JJ, Chan LM, Peel AJ, Lai R, Yoder AD, Goodman SM, et al. deep divergence time between sister species of Eidolon (Pteropodidae) with evidence for widespread panmixia. Acta Chiropterologica. (2014) 16:279–92. doi: 10.3161/150811014X687242
58. Almeida FC, Giannini NP, Simmons NB, Helgen KM. Each flying fox on its own branch: a phylogenetic tree for Pteropus and related genera (Chiroptera: Pteropodidae). Molecular Phylogenetics and Evolution (2014). Available online at: http://linkinghub.elsevier.com/retrieve/pii/S1055790314001092 doi: 10.1016/j.ympev.2014.03.009 (accessed March 23, 2014).
59. Goodman SM, Chan L, Nowak M, Yoder AD. Phylogeny and biogeography of western Indian Ocean Rousettus (Chiroptera : Pteropodidae). J Mammal. (2010) 91:593–606. doi: 10.1644/09-MAMM-A-283.1
60. Reynes JM, Andriamandimby SF, Razafitrimo GM, Razainirina J, Jeanmaire EM, Bourhy H, et al. Laboratory surveillance of rabies in humans, domestic animals, and bats in Madagascar from 2005 to 2010. Adv Prev Med. (2011) 2011:727821. doi: 10.4061/2011/727821
61. Brook CE, Ranaivoson HC, Broder CC, Cunningham AA, Héraud JM, Peel AJ, et al. Disentangling serology to elucidate henipa- and filovirus transmission in Madagascar fruit bats. J Anim Ecol. (2019) 88:1001–16. doi: 10.1111/1365-2656.12985
62. Han Y, Du J, Su H, Zhang J, Zhu G, Zhang S, et al. Identification of diverse bat Alphacoronaviruses and Betacoronaviruses in China provides new insights into the evolution and origin of coronavirus-related diseases. Front Microbiol. (2019) 10:1900. doi: 10.3389/fmicb.2019.01900
63. Lim XF, Lee CB, Pascoe SM, How CB, Chan S, Tan JH, et al. Detection and characterization of a novel bat-borne coronavirus in Singapore using multiple molecular approaches. J Gen Virol. (2019) 100:1363–74. doi: 10.1099/jgv.0.001307
64. Yinda CK, Ghogomu SM, Conceição-Neto N, Beller L, Deboutte W, Vanhulle E, et al. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. (2018) 4:1–15. doi: 10.1093/ve/vey008
65. Huang C, Liu WJ, Xu W, Jin T, Zhao Y, Song J, et al. A bat-derived putative cross-family recombinant coronavirus with a reovirus gene. PLoS Pathogens. (2016) 12:e1005883. doi: 10.1371/journal.ppat.1005883
66. Obameso JO, Li H, Jia H, Han M, Zhu S, Huang C, et al. The persistent prevalence and evolution of cross-family recombinant coronavirus GCCDC1 among a bat population : a two-year follow-up. Sci China Life Sci. (2017) 60:1357–63. doi: 10.1007/s11427-017-9263-6
67. Goodman SM. Les chauves-souris de Madagascar [in French]. Antananarivo, Madagascar: Association Vahatra (2011).
68. Golden CD, Bonds MH, Brashares JS, Rodolph Rasolofoniaina BJ, Kremen C. Economic valuation of subsistence harvest of wildlife in Madagascar. Conserv Biol. (2014) 28:1–10. doi: 10.1111/cobi.12174
69. Randrianandrianina F, Andriafidison D, Amyot F, Ramilijaona O, Ratrimomanarivo F, Racey PA, et al. Habitat use and conservation of bats in rainforest and adjacent human-modified habitats in eastern Madagascar. Acta Chiropterologica. (2006) 8:429–37. doi: 10.3161/1733-5329(2006)8[429:HUACOB]2.0.CO;2
70. Cardiff SG, Ratrimomanarivo FH, Goodman SM. The effect of tourist visits on the behavior of Rousettus madagascariensis (Chiroptera: Pteropodidae) in the caves of Ankarana, northern Madagascar. Acta Chiropterologica. (2012) 14:479–90. doi: 10.3161/150811012X661783
71. Razafindrakoto N, Harwell A, Jenkins R. Bats roosting in public buildings: a preliminary assessment from Moramanga, eastern Madagascar. Madagascar Conserv Dev. (2011) 5:85–8. doi: 10.4314/mcd.v5i2.63136
72. Jenkins RKB, Racey PA. Bats as bushmeat in Madagascar. Madag Conserv Dev. (2008) 3:22–30. doi: 10.4314/mcd.v3i1.44132
73. Razanajatovo NH, Richard V, Hoffmann J, Reynes J, Razafitrimo M, Randremanana RV, et al. Viral etiology of Influenza-like illnesses in Antananarivo, Madagascar, July 2008 to June 2009. PLoS ONE. (2011) 6:e17579. doi: 10.1371/journal.pone.0017579
74. Razanajatovo NH, Guillebaud J, Harimanana A, Rajatonirina S, Ratsima EH, Andrianirina ZZ, et al. Epidemiology of severe acute respiratory infections from hospital-based surveillance in Madagascar, November 2010 to July 2013. PLoS ONE. (2018) 13:1–17. doi: 10.1371/journal.pone.0205124
75. Randremanana R, Andriamandimby S, Rakotondramanga JM, Razanajatovo N, Mangahasimbola R, Randriambolamanantsoa T, et al. The COVID-19 Epidemic in Madagascar: clinical description and laboratory results of the first wave, March-September 2020. Influenza Other Respi Viruses. (2021) 15:1–12. doi: 10.22541/au.161088504.46456502/v1
76. Ranaivoson HC, Héraud JM, Goethert HK, Telford SR, Rabetafika L, Brook CE. Babesial infection in the Madagascan flying fox, Pteropus rufus É. Geoffroy, 1803. Parasit Vectors. (2019) 12:51. doi: 10.1186/s13071-019-3300-7
77. Brook CE, Bai Y, Dobson AP, Osikowicz LM, Ranaivoson HC, Zhu Q, et al. Bartonella spp. in fruit bats and blood-feeding ectoparasites in Madagascar. PLoS Negl Trop Dis. (2015) 9:e0003532. doi: 10.1371/journal.pntd.0003532
78. Kalantar KL, Carvalho T, de Bourcy CFA, Dimitrov B, Dingle G, Egger R, et al. IDseq-An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience. (2021) 9:1–14. doi: 10.1093/gigascience/giaa111
79. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
80. Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, et al. BEAST 25: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. (2019) 15:e1006650. doi: 10.1371/journal.pcbi.1006650
81. Kuraku S, Zmasek CM, Nishimura O, Katoh K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucl Acids Res. (2013) 41(Web Server issue):22–8. doi: 10.1093/nar/gkt389
82. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. (2018) 20:1160–6. doi: 10.1093/bib/bbx108
83. Darriba Di, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. (2020) 37:291–4. doi: 10.1093/molbev/msz189
84. Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. (2019) 35:4453–5. doi: 10.1093/bioinformatics/btz305
85. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. (1985) 39:783–91. doi: 10.1111/j.1558-5646.1985.tb00420.x
86. Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? J Comput Biol. (2010) 17:337–54. doi: 10.1089/cmb.2009.0179
87. Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. (2005) 22:1185–92. doi: 10.1093/molbev/msi103
88. Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol. (2018) 67:901–4. doi: 10.1093/sysbio/syy032
89. Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. (2007) 7:1–8. doi: 10.1186/1471-2148-7-214
90. Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. Ggtree: an R Package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. (2017) 8:28–36. doi: 10.1111/2041-210X.12628
91. Davies J. pySimPlot. GitHub. Available online at: https://github.com/jonathanrd/pySimPlot (accessed September 6, 2021).
92. Wu Y, Zhu X, Li N, Chen T, Yang M, Yao M, et al. CMRF-35–like molecule 3 preferentially promotes TLR9-triggered proinflammatory cytokine production in macrophages by enhancing TNF receptor-associated factor 6 ubiquitination. J Immunol. (2011) 187:4881–9. doi: 10.4049/jimmunol.1003806
93. Xu J, Hu J, Wang J, Han Y, Hu Y, Wen J, et al. Genome organization of the SARS-CoV. Genomics Proteomics Bioinformatics. (2003) 1:226–35. doi: 10.1016/S1672-0229(03)01028-3
94. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. (2020) 181:914–21.e10. doi: 10.1016/j.cell.2020.04.011
95. Li F. Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Res. (2013) 100:246–54. doi: 10.1016/j.antiviral.2013.08.014
96. Tao Y, Shi M, Chommanard C, Queen K, Zhang J, Markotter W, et al. Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. Virology. (2017) 91:1–16. doi: 10.1128/JVI.01953-16
97. Halpin K, Rota P. A Review of hendra virus and nipah virus Infections in man and other animals. In: Sing A, editor. Zoonoses - Infections Affecting Humans and Animals: Focus on Public Health Aspects. Berlin: Springer (2014). pp. 997–1012.
98. Wells HL, Letko M, Lasso G, Ssebide B, Nziza J, Byarugaba DK, et al. The evolutionary history of ACE2 usage within the coronavirus subgenus sarbecovirus. Virus Evol. (2021) 7:veab007. doi: 10.1093/ve/veab007
99. Boni MF, Lemey P, Jiang X, Lam TTY, Perry BW, Castoe TA, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. (2020) 5:1408–17. doi: 10.1038/s41564-020-0771-4
100. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. (2020) 26:450–2. doi: 10.1038/s41591-020-0820-9
101. Starr TN, Zepeda SK, Walls AC, Greaney AJ, Veesler D, Bloom JD. ACE2 binding is an ancestral and evolvable trait of sarbecoviruses. bioRxiv [Preprint]. (2021). doi: 10.1101/2021.07.17.452804
102. Ge XY, Wang N, Zhang W, Hu B, Li B, Zhang YZ, et al. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol Sin. (2016) 31:31–40. doi: 10.1007/s12250-016-3713-9
103. Luis AD, O'Shea TJ, Hayman DTS, Wood JLN, Cunningham AA, Gilbert AT, et al. Network analysis of host–virus communities in bats and rodents reveals determinants of cross-species transmission. Ecol Lett. (2015) 18:1153–62. doi: 10.1111/ele.12491
104. Peel AJ, Wells K, Giles J, Boyd V, Burroughs A, Edson D, et al. Synchronous shedding of multiple bat paramyxoviruses coincides with peak periods of Hendra virus spillover. Emerg Microbes Infect. (2019) 8:1314–23. doi: 10.1080/22221751.2019.1661217
105. Andrianaivoarivelo RA, Ramilijaona OR, Racey PA, Razafindrakoto N, Jenkins RKB. Feeding ecology, habitat use and reproduction of Rousettus madagascariensis Grandidier, 1928 (Chiroptera: Pteropodidae) in eastern Madagascar. Mammalia. (2011) 75:69–78. doi: 10.1515/mamm.2010.071
106. Wong ACP, Lau SKP, Woo PCY. Interspecies jumping of bat coronaviruses. Viruses. (2021) 13:2188. doi: 10.3390/v13112188
107. Song HD, Tu CC, Zhang GW, Wang SY, Zheng K, Lei LC, et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Nat Acad Sci. (2005) 102:2430–5. doi: 10.1073/pnas.0409608102
108. Conzade R, Grant R, Malik M, Elkholy A, Elhakim M, Samhouri D, et al. Reported direct and indirect contact with dromedary camels among laboratory-confirmed MERS-CoV cases. Viruses. (2018) 10:425. doi: 10.3390/v10080425
109. Rasambainarivo F, Ramiadantsoa T, Raherinandrasana A, Randrianarisoa S, Rice BL, Evans M v, et al. Prioritizing COVID-19 vaccination efforts and dose allocation within Madagascar. medRxiv [Preprint]. (2021). doi: 10.1101/2021.08.23.21262463
110. Rabalski L, Kosinski M, Mazur-Panasiuk N, Szewczyk B, Bienkowska-Szewczyk K, Kant R, et al. Zoonotic spill-over of SARS-CoV-2: mink adapted virus in humans. Clin Microbiol Infect. (2021) 12:001. doi: 10.1016/j.cmi.2021.12.001
111. Chu H, Chan CM, Zhang X, Wang Y, Yuan S, Zhou J, et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J Biol Chem. (2018) 293:11709–26. doi: 10.1074/jbc.RA118.001897
112. Wacharapluesadee S, Duengkae P, Chaiyes A, Kaewpom T, Rodpan A, Yingsakmongkon S, et al. Longitudinal study of age-specific pattern of coronavirus infection in Lyle's flying fox (Pteropus lylei) in Thailand. Virol J. (2018) 15:38. doi: 10.1186/s12985-018-0950-6
113. Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, Nkrumah EE, et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg infect Dis. (2013) 19:456–9. doi: 10.3201/eid1903.121503
114. Brook CE, Ranaivoson HC, Andriafidison D, Ralisata M, Razafimanahaka J, Héraud JM, et al. Population trends for two Malagasy fruit bats. Biol Conserv. (2019) 234:165–71. doi: 10.1016/j.biocon.2019.03.032
115. Ruiz-Aravena M, McKee C, Gamble A, Lunn T, Morris A, Snedden CE, et al. Ecology, evolution and spillover of coronaviruses from bats. Nat Rev Microbiol. (2021) 1–16. doi: 10.1038/s41579-021-00652-2
Keywords: Nobecovirus, bat-borne coronavirus, recombination, zoonosis, Madagascar
Citation: Kettenburg G, Kistler A, Ranaivoson HC, Ahyong V, Andrianiaina A, Andry S, DeRisi JL, Gentles A, Raharinosy V, Randriambolamanantsoa TH, Ravelomanantsoa NAF, Tato CM, Dussart P, Heraud J-M and Brook CE (2022) Full Genome Nobecovirus Sequences From Malagasy Fruit Bats Define a Unique Evolutionary History for This Coronavirus Clade. Front. Public Health 10:786060. doi: 10.3389/fpubh.2022.786060
Received: 29 September 2021; Accepted: 17 January 2022;
Published: 11 February 2022.
Edited by:
Seth Judson, University of Washington, United StatesReviewed by:
Roopali Rajput, National Institute of Tuberculosis and Respiratory Diseases, IndiaJames Tristan Gordy, Johns Hopkins University, United States
Copyright © 2022 Kettenburg, Kistler, Ranaivoson, Ahyong, Andrianiaina, Andry, DeRisi, Gentles, Raharinosy, Randriambolamanantsoa, Ravelomanantsoa, Tato, Dussart, Heraud and Brook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cara E. Brook, Y2Jyb29rQHVjaGljYWdvLmVkdQ==
†These authors share senior authorship