 Junnan Shi
Junnan Shi Xianwen Chen
Xianwen Chen Hao Hu
Hao Hu Carolina Oi Lam Ung
Carolina Oi Lam Ung- 1State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Taipa, Macao SAR, China
- 2Centre for Pharmaceutical Regulatory Sciences, University of Macau, Taipa, Macao SAR, China
- 3Department of Public Health and Medicinal Administration, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR, China
Introduction: The purpose of developing and adopting regulatory science (RS) for drug regulatory authorities (DRAs) is to enhance regulatory capacity by advancing the scientific approach for the evaluation of health-related products. While many DRAs around the world advocate the concept of RS, the implementation approaches of RS vary according to local needs and have not been systemically examined. This study aimed to systematically identify the evidence about how RS was developed, adopted, and advanced by the selected DRAs, and analyzed and compared the implementation experiences of RS development under the guidance of an implementation science framework.
Methods: Documentary analysis of government documents and a scoping literature review were conducted, and data analysis was performed under the guidance of the PRECEDE-PROCEED Model (PPM). DRAs in the United States, the European Union, Japan, and China had officially launched RS initiatives and were therefore selected as the target countries in this study.
Results: There is no common consensus on the definition of RS among the DRAs. However, these DRAs shared the same goal of developing and adopting RS, which was used to develop new tools, standards, and guidelines that could improve the effectiveness and efficiency of the risk and benefit assessment of the regulated products. Each DRA had decided its own priority areas for RS development and thus set specific objectives that might be technology-based (e.g., toxicology and clinical evaluation), process-based (e.g., partnership with healthcare systems and high-quality review/consultation services), or product-based (e.g., drug-device combination products and innovative emerging technologies). To advance RS, considerable resources had been allocated for staff training, advancing information technology and laboratory infrastructure, and funding research projects. DRAs also took multifaceted approaches to expand scientific collaborations through public–private partnerships, research funding mechanisms, and innovation networks. Cross-DRA communications were also reinforced through horizon scanning systems and consortiums to better inform and assist the regulatory decision-making process. The output measurements might be scientific publications, funded projects, DRAs interactions, and evaluation methods and guidelines. Improved regulatory efficiency and transparency leading to benefits to public health, patient outcomes, and translation of drug research and development as the key primary outcomes of RS development were anticipated but not yet clearly defined.
Conclusion: The application of the implementation science framework is useful for conceptualizing and planning the development and adoption of RS for evidence-based regulatory decision-making. Continuous commitment to the RS development and regular review of the RS goals by the decision-makers are important for DRAs to meet the ever-changing scientific challenges in their regulatory decision-making process.
1. Introduction
The acceleration of innovation is catalyzing the development of increasingly complex pharmaceutical products and emerging technologies, such as cell and gene therapies, drug–device combinations, artificial intelligence, and digital health. These come with vast opportunities to promote, maintain, and protect human health but also some significant challenging regulatory issues to national drug regulatory authorities (DRAs) (1, 2). Beyond science and technological innovation, as shown by the COVID-19 pandemic, drug regulation has seen challenges in engaging collaborative effort and formulating regulatory flexibility in advancing responses to unexpected public health threats (3). Regulators play an integral role in the complex ecosystem of the pharmaceutical system and often face the challenges of meeting societal expectations to provide patients with timely access to new treatment options while maintaining stakeholder trust by evidently upholding high regulatory practice standards (4).
Regulatory science (RS) is a scientific discipline that embraces a large variety of activities and outputs, from new techniques and products to methodological standards and guidance (5). In order to address the above challenges, major national DRAs have proposed and adopted this cutting-edge discipline. Successful outcomes had been demonstrated by the development of new tools, standards, and approaches (3, 6, 7).
The development of RS helps to bridge “law and regulation” and “science” more effectively in the interests of addressing healthcare needs and promoting innovation (8). For example, the backbone of the FDA's regulatory decision-making process is formed with policy, law, and science supplemented with guidance or standards to facilitate law implementation. By having regulatory measures overseeing the whole product life cycle, regulatory experiences and challenges accumulated lead to the development of new information and knowledge, and continuous improvement and revision of laws and regulations driven by advances in science and innovation (Figure 1A). By adopting RS, it helps the FDA make their regulatory practices in convergence with scientific opinions and identify the “best available science” to achieve improvements in the quality, consistency, and objectivity of regulatory evaluation more timely and effectively (Figure 1B).
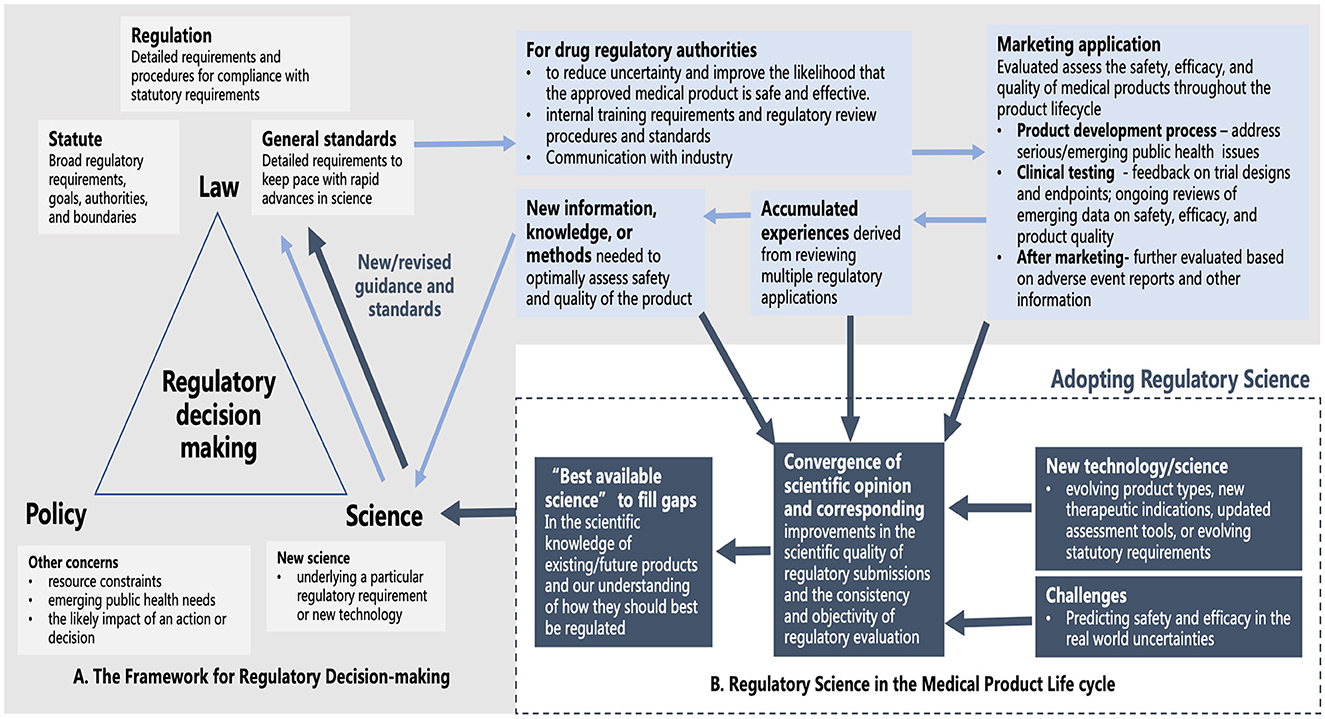
Figure 1. (A) The backbone of the FDA's regulatory decision-making process and the development of new information and knowledge based on the oversight of the medical product life cycle. (B) An additional source of scientific input for improving the quality, consistency, and objectivity of regulatory evaluation with the adoption of RS (Pale blue arrow: how new scientific knowledge is generated based on regulatory practice to support the regulatory decision-making process; Navy blue arrow: additional generation and uptake of scientific input to better inform regulatory decision-making upon adoption of RS).
Current evidence from literature about the development of RS mainly focused on the regulation of specific pharmaceutical product categories. For example, the International Neonatal Consortium (INC) addressed the need for measurement and assessment of clinical outcomes in neonates using cell-based therapies through public–private partnerships that shared data and expertise to advance RS (9). The Global Coalition for Regulatory Science Research (GCRSR) provided an overview of new tools and methodologies for regulatory bodies from various countries around the globe in nanotechnology and more particularly nanotechnology-based products (10). In addition, a research project on the harmonization and evaluation of regulations for pharmaceuticals and medical devices by the Japan Agency for Medical Research and Development has taken regulatory consideration of the shared data and information required for interoperable medical devices (11). Japanese scholars summarized the development experience of the ethical Kampo products with new dosage forms and herbal medicines that used Kampo extracts as active pharmaceutical ingredients, illustrating the importance of RS for the development of new drugs for natural products (12).
While most of the current literature explains how RS can lead to technological innovation or quality improvement, little research had been attended to the development and adoption process. In order to provide a tool for decision-makers for developing and adopting RS to support regulatory decision-making, the main objective of this study aimed to systematically identify the evidence about how RS was developed, adopted, and advanced by the selected DRAs, and analyzed and compared the implementation experiences of RS development under the guidance of the commonly accepted PRECEDE-PROCEED model derived from implementation science. It is anticipated that the results of this study will help inform DRAs and academia about how to promote the systematic and innovative application of RS under the guidance of a theoretical framework.
2. Materials and methods
This study adopted a critical review approach to obtain publicly available information about developing and adopting RS by different DRAs from government and official websites, as well as related literature from electronic databases. The collected data were extracted, compiled, and comparatively analyzed under the guidance of the PRECEDE-PROCEED model.
2.1. Theoretical framework
To obtain more evidence from theory to practical application, implementation science has been commonly applied to guide the process of developing and adopting RS to facilitate scientific regulatory decision-making (13). The theories, frameworks, and research of implementation science can be used to lead RS to the more specific focused field of pharmaceutical regulation (14). In particular, the PRECEDE-PROCEED model (PPM) has been fully demonstrated in many practices as a theoretical framework for the planning and evaluation of public health programs (15). It provides a visual display of a program to help guide the regulator's system thinking of the program development. PPM provides continuous efforts for the pre-intervention of goals and post-intervention monitoring and quality improvement. It allows the regulators to logically think about the desired endpoints and work “backwards” to achieve the goal (16). The outcome-oriented character promotes better policy decision-making to improve public health (17, 18).
As shown in Figure 2, in the PPM model, the PRECEDE phase consists of five steps. Steps 1 and 2: to determine the social needs of RS and then set priorities and goals of DRAs; Step 3: to determine the behavioral indicators (e.g., individuals or groups) of DRAs and environmental indicators (e.g., economic or physical) of the whole health system that have an impact on RS; Step 4: to determine the predisposing (e.g., knowledge or values), reinforcing (e.g., attitudes of health peers), and enabling factors (e.g., programs or policies) involved in the educational or ecological that have an impact on RS; Step 5: to determine the administrative and policy components of RS. The PROCEED phase consists of four steps to achieve the evaluation aim of monitoring and continuous quality improvement. Step 6: to evaluate the actual implementation (input) of developing and adopting RS; Step 7: to evaluate the process of advancing RS; Step 8: to evaluate the impact of advancing RS (output or short-term impact, if applicable); Step 9: to evaluate the outcomes of developing and adopting RS (social impact or long-term impact, if applicable).
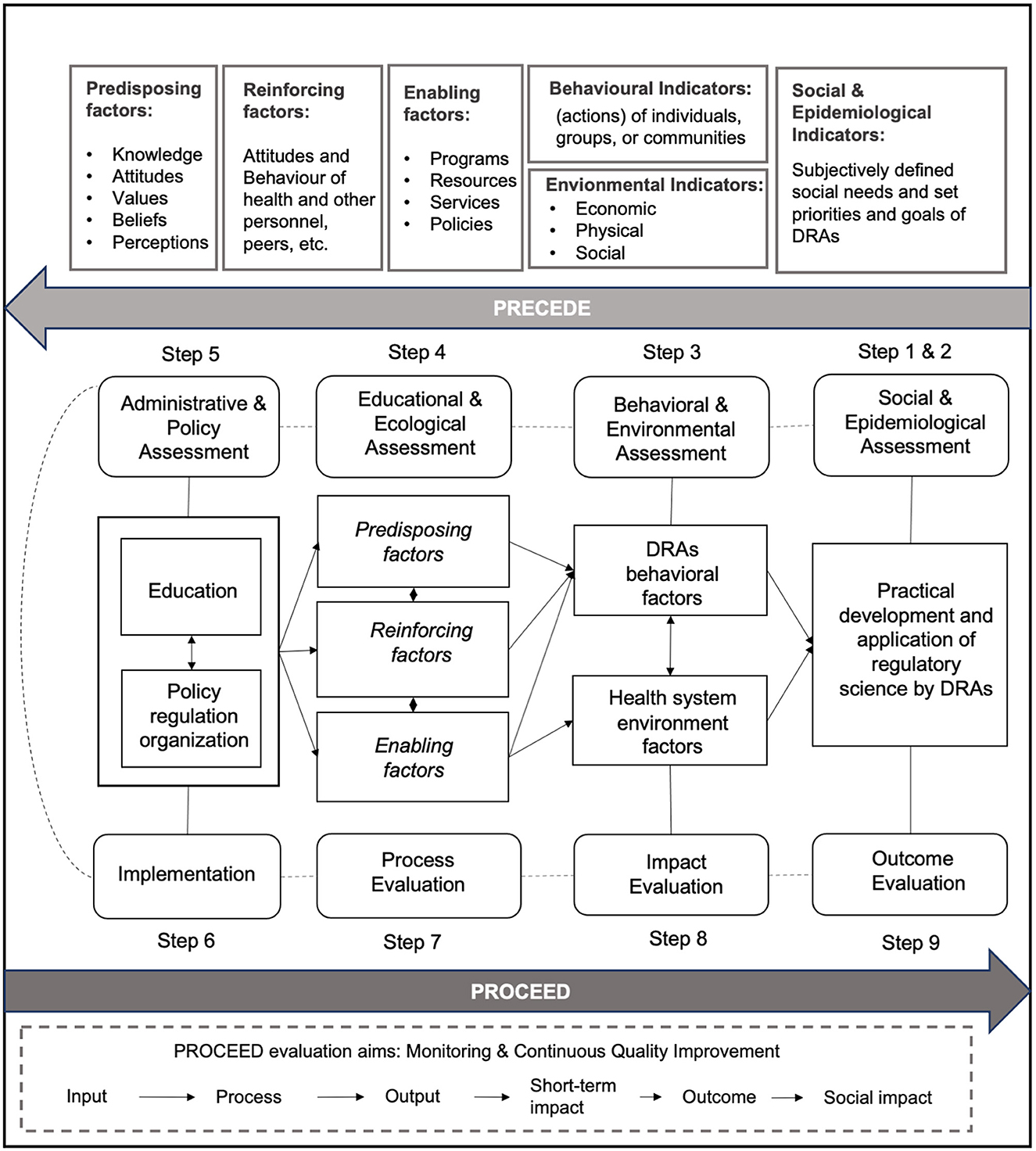
Figure 2. Overview of the PRECEDE-PROCEED model (PPM) applied to the implementation of regulatory science by DRAs.
2.2. Data retrieval and collection
The United States, the European Union, Japan, and China were targeted for evaluation because they all have officially released regulatory science programs. The data collection was conducted in December 2022 to search the publicly available documents and other related literature. Publicly available documents and reports were found from the government and official websites of the corresponding DRAs: National Medical Products Administration (NMPA) in China https://www.nmpa.gov.cn/, Food and Drug Administration (FDA) in the U.S. https://www.fda.gov/, European Medicines Agency (EMA) in the European Union https://www.ema.europa.eu/en, and the Pharmaceuticals and Medical Devices Agency in Japan (PMDA) http://www.pmda.go.jp/. Additionally, academic and gray literature was conducted using Google Scholar and PubMed. Each search used the following terms: [(regulatory science) OR (translational science) OR (regulation) OR (regulatory agenc*) OR (regulatory authorit*)] AND [(United States) OR (European Union) OR (Japan) OR (China) OR (FDA) OR (EMA) OR (PMDA) OR (NMPA)]. The search was conducted separately by two of the authors (JS and XC), and the documents and materials to be included for further analysis were cross-checked and confirmed by two other authors (HH and COLU).
2.3. Data extraction and analysis
The PPM provides structures to guide the extraction of data related to specific objectives and influencing factors before the implementation of intervention as well as a continuous quality improvement after the intervention is implemented. For eligible data, two authors (JS and XC) extracted and evaluated information independently and any disagreements were resolved by discussion or reaching an agreement with the third author (COLU). The two phases of PRECEDE and PROCEED in the PPPM were combined to form the complete theoretical data extraction. The authors (JS, COLU, and HH) adjusted construct definitions applicable to develop and adopt RS by adapting from PRECEDE-PROCEED steps defined by the framework. After adjudicating these applications for RS, the authors' team then refined the definitions accordingly. In addition, the construct definitions were adjusted for adoption in the context of RS in consultation with experienced researchers in drug regulation.
3. Results
As an overview, based on the PRECEDE section of the PPM, we sorted out the information on developing and adopting RS from PRECEDE to PROCEED of DRAs in selected countries. To advance RS, considerable resources allocated for staff training, advancing information technology and laboratory infrastructure, and funding research projects were also identified. DRAs also took multifaceted approaches to expand scientific collaborations through public–private partnerships, research funding mechanisms, and innovation networks. Cross-DRAs communications were also reinforced through a horizon scanning system and consortium to better inform and assist the regulatory decision-making process.
3.1. Step 1 and Step 2: social and Epidemiological diagnosis
As an overview, based on the PRECEDE section of the PPM, we sorted out the information on developing and adopting RS from PRECEDE to PROCEED of DRAs in selected countries. To advance RS, considerable resources had been allocated for staff training, advancing information technology and laboratory infrastructure, and funding research projects. DRAs also took multifaceted approaches to expand scientific collaborations through public–private partnerships, research funding mechanisms, and innovation networks. Cross-DRAs communications were also reinforced through a horizon scanning system and consortium to better inform and assist the regulatory decision-making process. For DRAs who opted to use a theoretical framework to effectively apply RS to scientific decision-making of the full life cycle of pharmaceutical products, identifying and assessing the various societal needs and priorities that influenced regulators to apply RS became an important first step toward achieving this goal.
3.1.1. Social needs
Entering the 21st century, DRAs faced a series of public health challenges, including new industrial transformation and globalization brought about by technological innovation, new changes, and challenges caused by the rapid development of knowledge and research in the field of basic science. More specifically, gaps between advanced therapy medicinal products (ATMP) and the actual application of treatments to patients remained unsolved, and early communication between HTA agencies or key stakeholders of drug companies and government regulators was often found insufficient. Regulatory review standards might easily lag behind the technical needs of evaluating clinical trials. For the yield of reliable and high-quality real-world data, appropriate regulatory frameworks and platforms were still developing for data collection and analysis (19, 20).
3.1.2. Set priorities and goals
In recent years, FDA had always been committed to develop, evaluate, and manufacture novel medical products or technologies through strategic planning of advancing regulatory science (8, 21). The EMA and PMDA were equally committed to advancing RS to create a regulatory environment for pharmaceutical products that supports innovation and the development of solutions to meet human needs (22, 23). China had carried out a comprehensive reform of the drug regulatory system and launched the regulatory science action plan (RSAP) to achieve more scientific and effective drug regulation (24).
Due to the differences in the national circumstances and the history of drug regulation reform, the definitions, visions, and priorities of RS set by different DRAs also differed as shown in Table 1. Regarding RS definitions, the FDA and the PMDA noted the same acceptance of RS and highlighted that RS was a scientific discipline with the goal to provide new tools and technology to speed up the approval of medical products, while the EMA emphasized that RS was a scientific discipline covering the regulatory decision-making throughout the medicine lifecycle. The visions of the FDA, EMA, and PMDA were more global in scope, focusing on the advancement of all humankind and international regulatory development. On the other hand, NMPA's vision was more concerned with meeting the demands of recent advances in regulatory policies, focusing on the reform and innovation of the drug review and approval system, and closely following the frontier of international regulatory innovation.
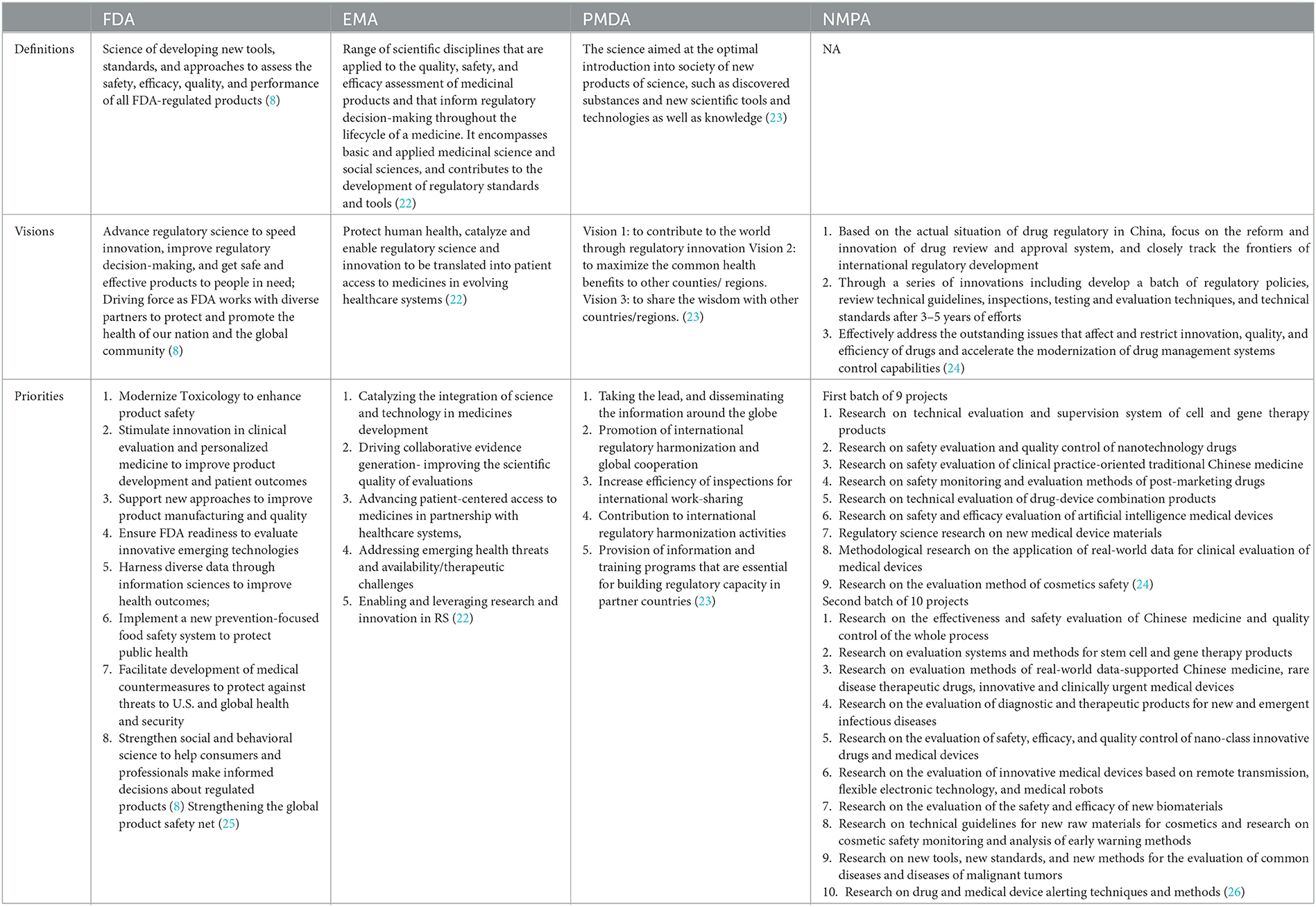
Table 1. Definitions, visions, and priorities of RS in selected DRAs.
As for the priorities, each of these four DRAs had decided their own priority areas for RS development and thus set specific objectives which might be technology-based (e.g., toxicology and clinical evaluation), process-based (e.g., partnership with healthcare systems and high-quality consultation services) or product-based (e.g., drug–device combination products). The FDA emphasized the adoption of RS to improve the assessment of the safety of medical products and promote product innovation. The convergence of regulation and innovation was the major focus of the EMA. The PMDA highlighted the need to improve the efficiency of global convergence and regulatory activities. As for NMPA, it is committed to effectively addressing the outstanding issues that affect and restrict the innovation, quality, and efficiency of drugs.
3.2. Step 3: behavioral and environmental diagnosis
The development path of medical products (including drugs, biological products, and medical devices) had been increasingly challenging, inefficient, and costly. Innovators in this context tended to focus on medical products with high potential market returns; however, developing products that address critical public health needs, rare diseases, and personalized treatments was increasingly challenging. Without enough new tools and concepts created with applied science, developers or scientists could only use old scientific tools and ideas to evaluate candidate medical products. After investing a lot of time and resources, most of the R&D products that entered clinical trials failed. For successful product candidates, the cumbersome review approach and process made the road to market lengthy, costly, and inefficient. This was particularly evident in the reports of Critical Path Initiatives published by the FDA. As reported, the number of new drug and biologics applications submitted by companies to the FDA fell sharply, and the number of applications for innovative medical devices also decreased, but the costs of pharmaceutical product development had soared rapidly since the beginning of this century (21). Although similar reports published by other DRAs, such as the EMA, PMDA, or NMPA, were not identified, regulatory challenges were common across those agencies driving the development of the adoption of RS (27–29).
3.3. Step 4: educational and ecological diagnosis
Regulators' perceptions and attitudes toward the current medical product development process and the existing deficiencies of scientific tools and ideas from regulatory authorities constituted predisposing factors. DRAs reinforced and enabled behavioral changes by expanding collaboration with the larger scientific community and issued related actions, plans, and policies. The FDA was found to be the only DRA that had a traceable, well-reported track record of how RS was developed, adopted, and advanced. As such, the FDA's experiences were used primarily to inform this part of the result. In this study, we traced back to the FDA's Critical Path Action Report and Checklist Report to explore the education and ecological diagnostics that led to RS (21, 30–32).
3.3.1. Predisposing factors
Advancing biomedical technologies had brought hope to prevent, treat, and even cure more diseases. However, regulators and stakeholders were increasingly concerned that new basic science discoveries might not quickly yield more safety, effective, and affordable pharmaceutical products for patients. At the same time, with the costs and difficulties of medical product innovation continuing to grow, there was a concern that the development of advanced therapies would be stagnated and declined.
Advanced product development science was needed by drug regulators to address these concerns. The lag in the process of medical product development urgently required a new product development toolkit that incorporated a robust scientific and technological approach to improve predictability and efficiency along the Critical Path, such as computer-based predictive models and new clinical review techniques. A knowledge base was also needed to be built to grasp ideas from biomedical research and reliable insights into the pathway in patients.
3.3.2. Reinforcing factors
Working together to address the above concerns and challenges has received a positive response from many accomplished scientists from academia, government, and industry. A coordinated effort by the stakeholders is needed to apply the new biomedical science to medical product development in order to promote the modernizing of the Critical Path. When serious issues arise during development or common problems persisted, FDA was devoted to seek collaboration from a wider range of internal and external scientists and refocused its efforts, in order to enhance support for critical programs and ensure that the most important issues were addressed.
3.3.3. Enabling factors
FDA's standard-setting process based on the best science facilitated the efficient development of safe and effective new medical therapies. In 2004, FDA planned a Critical Path Initiative to identify and prioritize the most pressing development issues and areas that offer the greatest opportunities for rapid development and public health benefits in the Critical Path White Paper. FDA engaged all stakeholders in the Critical Path work and created a list of Critical Path opportunities to focus its internal efforts to ensure that the most important problems and critical programs are addressed. The Critical Path Initiative—Report on Key Achievements in 2009 illustrated the diversity and complexity of Critical Path work and emphasized the public health outcomes (32). This series of initiatives and policies enabled the development of RS.
3.4. Step 5: administrative and policy assessment
The RS in FDA kept pace with advanced technologies. The FDA protected consumers by applying the best science to regulatory activities, and each of its medical product centers had taken steps to begin implementing regulatory processes, policies, and internal organizational improvements to address and coordinate regulatory challenges for complex innovative products. In addition, the Office of Regulatory Science and Innovation (ORSI) provided excellence and innovation in strategic leadership, collaboration, coordination, and infrastructure development, ensuring that the FDA continued to have a strong regulatory science foundation (33–36). In advance of its mission to protect and promote public health, FDA began a full institutional reorganization in early 2019 to meet the challenges of rapid innovation in the industry. The FDA's restructuring aligned with the agency's multiple entities to promote regulatory strategic priorities and the specific frameworks of RS in different medical product centers.
To name an example, as shown in Figure 3, FDA set up offices of regulatory science, regulatory policy, or regulatory operations in different centers of medical products to facilitate the development of RS internationally. In addition, FDA also established collaborations externally to advance RS. For example, the Centers of Excellence in Regulatory Science and Innovation (CERSI) represented the collaboration between the DRA and external academic institutions to promote cutting-edge scientific research (37) that addressed the needs in drug regulation, and the Partnerships to Advance Innovation and Regulatory Science (PAIRS) were set to acquire the expertise and material resources from different sectors to support scientific and clinical assessments conducted in the offices of CDRH to better promote public health (38). As demonstrated by the actions taken by the FDA, a logical and scientific regulatory system was kept in place to strategically embrace external scientific input and foster internal organizational synergy.

Figure 3. Organization frameworks of RS in different FDA medical product centers. OCHEN, Office of Cardiology, Hematology, Endocrinology and Nephrology; OII, Office of Immunology and Inflammation; OID, Office of Infectious Diseases; ON, Office of Neuroscience; ONPD, Office of Nonprescription Drugs; OOD, Office of Oncologic Diseases; ORPURM, Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine; OTBB, Office of Therapeutic Biologics and Biosimilars; COA, Clinical Outcome Assessments; Navy blue box, external cooperation; Pale blue box, internal cooperation.
EMA had established special committees for each relevant area to support scientific decision-making in related areas, such as the Committee for Medicinal Products for Human Use (CHMP), the Pharmacovigilance Risk Assessment Committee (PRAC), the Committee for Medicinal Products for Veterinary Use (CVMP), the Committee for Orphan Medicinal Products (COMP), the Committee on Herbal Medicinal Products (HMPC), the Committee for Advanced Therapies (CAT), and the Pediatric Committee (PDCO) (39).
On 14 May 2012, Japan established the Scientific Board as a high-level advisory body to discuss the scientific nature of drug and medical device review (40). The board strengthened the cooperation and exchanges between PMDA and scientists from universities and research institutions and helped PMDA to incorporate the latest scientific knowledge into its services, thereby improving PMDA's review and safety measures and promoting the construction of RS. On 1 April 2018, PMDA established the Center for Regulatory Science to work on solving and simplifying scientific issues, improving audit quality and safety measures, and initiating discussions with each stakeholder by providing information on RS (41). Through the efforts of the Center, PMDA furthered the development of product and post-market safety measures, while actively participating in global regulatory affairs to enable the development of rational medicine in the future (42).
In 2013, the Department of Science, Technology, and Standards of the former State Food and Drug Administration (SFDA) held the first research project initiation meeting on drug regulatory science in Beijing, and the China Society for Drug Regulatory (CSDR) was formally established, which played an active role in promoting the scientific development of drug regulation in China (43). In 2015, the State Council issued the “Opinions on Reforming the Review and Approval System for Drugs and Medical Devices”, and the former SFDA decided to use Peking University as a platform to apply for the establishment of the Asia-Pacific Economic Cooperation (APEC) Regulatory Science Center of Excellence (CoA) (44).
3.5. Step 6: implementation
This step explored the implementation of RS in the selected DRAs. Figure 4 depicts the different implementation processes of RS in the FDA, EU, Japan, and China. Since 2004, the FDA had been issuing Critical Path reports and launching Critical Path Initiatives to help accelerate medical product development and review. The regulatory challenges reported in the Critical Path Initiative gradually became a major driver of innovation for the FDA. FDA issued a series of reports that analyze the scientific progress, challenges, and achievements from 2007 to 2009 (45, 46). In 2010, the report Advancing Regulatory Science for Public Health first proposed the basic structure of RS. Since then, FDA's regulatory science shifted from the analysis stage to the strategy formation stage (47). In August of the same year, the FDA released the Advancing Regulatory Science at FDA Strategic Plan, which proposed eight priority areas to advance regulatory science and thoroughly modernize science and technology used in the development and evaluation of pharmaceutical products and planned to transform the regulatory concept of products such as the development, evaluation, manufacturing, and application of medical products by building the Critical Path Model (48). In 2013, the FDA further released the Strategy and Implementation Plan for Advancing Regulatory Science for Medical Products white paper and added a ninth priority area (49). In 2020, recognizing that the science and technology underpinning FDA-regulated products evolved significantly since 2011, FDA formed an agency-wide committee to develop an efficient way to communicate its RS activities. The committee developed the report Advancing Regulatory Science at FDA: Focus Areas of Regulatory Science (FARS) to identify and communicate areas FDA has identified as needing continued targeted investment in RS research to fulfill FDA's regulatory and public health mission (50, 51).

Figure 4. The implementation process of RS in the different countries.
EMA and PMDA also published their strategic plans for RS. On 19 December 2018, EMA first published the EMA Regulatory Science Strategic Plan 2025 (Draft for Comment), which defined RS and informed regulatory decision-making throughout the lifecycle of medicine (52). The strategic plan covered five strategic goals and provided core recommendations and actions based on the goals. The overall top five core recommendations were fostering innovation in clinical trials, promoting the use of high-quality real-world data (RWD) in decision-making, reinforcing patients' relevance in evidence generation, contributing to HTA's preparedness and downstream decision-making for innovative medicines, and supporting developments in precision medicine, biomarkers, and “omics.” After continued consultation and multi-stakeholder participation, in March 2020, the EMA Management Committee officially released the “EMA 2025 Regulatory Science Strategic Plan” (22).
In June 2015, PMDA released the International Pharmaceutical Regulatory Harmonization Strategy—Regulatory Science Initiative (hereinafter referred to as the “Initiative”). The initiative mentioned that RS is the foundation of PMDA's activities (23). It was also in this year that China's State Council issued the “Opinions on Reforming the Review and Approval System for Drugs and Medical Devices,” which marked the launch of the comprehensive authorization reform plan. Since then, the Chinese government has successively issued hundreds of policies, initiatives, and announcements in order to achieve a more scientific and effective drug regulatory system. The NMPA officially released the first batch of the regulatory science action plan (RSAP) in 2019 and the second batch of RSAP key projects in 2021 (24, 26).
3.6. Step 7: process evaluation
In the process of advancing RS, all the selected DRAs focused on staff training and education, as well as internal and external communication and cooperation. The full description could be seen in Table 2. The successful advancement of RS in the FDA focused on its strong regulatory science culture and infrastructure, with an emphasis on government agency partnerships, staff training and professional development, direct funding mechanisms, and public–private partnerships (PPP). The EU supported member state regulators through the establishment of Innovation Networks and created a joint horizon scanning system to provide policymakers, healthcare providers, and patients with access to emerging medical technologies that have a significant impact. At the same time, the EU actively promoted Good Regulatory Practices (GRP) to facilitate coordination and cooperation among multiple forces. PMDA placed great emphasis on streamlining approval time, funding support, and big data utilization. Through the implementation of the “SAKIGAKE” strategy, the “Sakigake designation system and “Senkuteki Iyakuhin” (legislation of system) were typical examples of PMDA accelerating the evaluation of innovative drugs (72, 73). NMPA focused on expanding the education and training and building the RS talent pool and had since cooperated with universities and research institutes to establish a number of RS research centers to promote talent training and scientific research for regulatory capacity building.

Table 2. Process evaluation of RS in selected DRAs.
3.7. Step 8: impact evaluation
Through the continuous efforts of regulatory bodies, the application of RS had a great impact on different procedures. Currently, advances in the adoption of FDA medical product RS included integrating new science into the regulatory process, more professional training, and infrastructure building to enhance the evaluation of RS and advocate the adoption of new science and technology. For the EU EMA, it increased the interaction with the pharmaceutical industry throughout the drug development lifecycle by supporting patient involvement throughout the drug research and development process. By incorporating RS, the EMA also translated scientific regulation into better processes through a process for writing guidelines. PMDA proactively created guidelines leading to international regulatory harmonization and established different institutional departments in specialized fields.
RS in China started later than previous DRAs, but through two sets of RSAP, RS has gradually become a mature discipline in China. Since 2019, NMPA had issued a series of new systems, tools, standards, and methods for drug review and supervision to promote the innovation of drug regulation systems and improve the regulatory capacity. NMPA relied on universities to establish various special research projects, focusing on the key and difficult issues of the regulation of drugs, medical devices, and cosmetics. NMPA actively held and participated in domestic and international regulatory activities to build a platform for common exchanges and discussions among regulatory authorities, industry, and academia. Table 3 describes the detailed information.
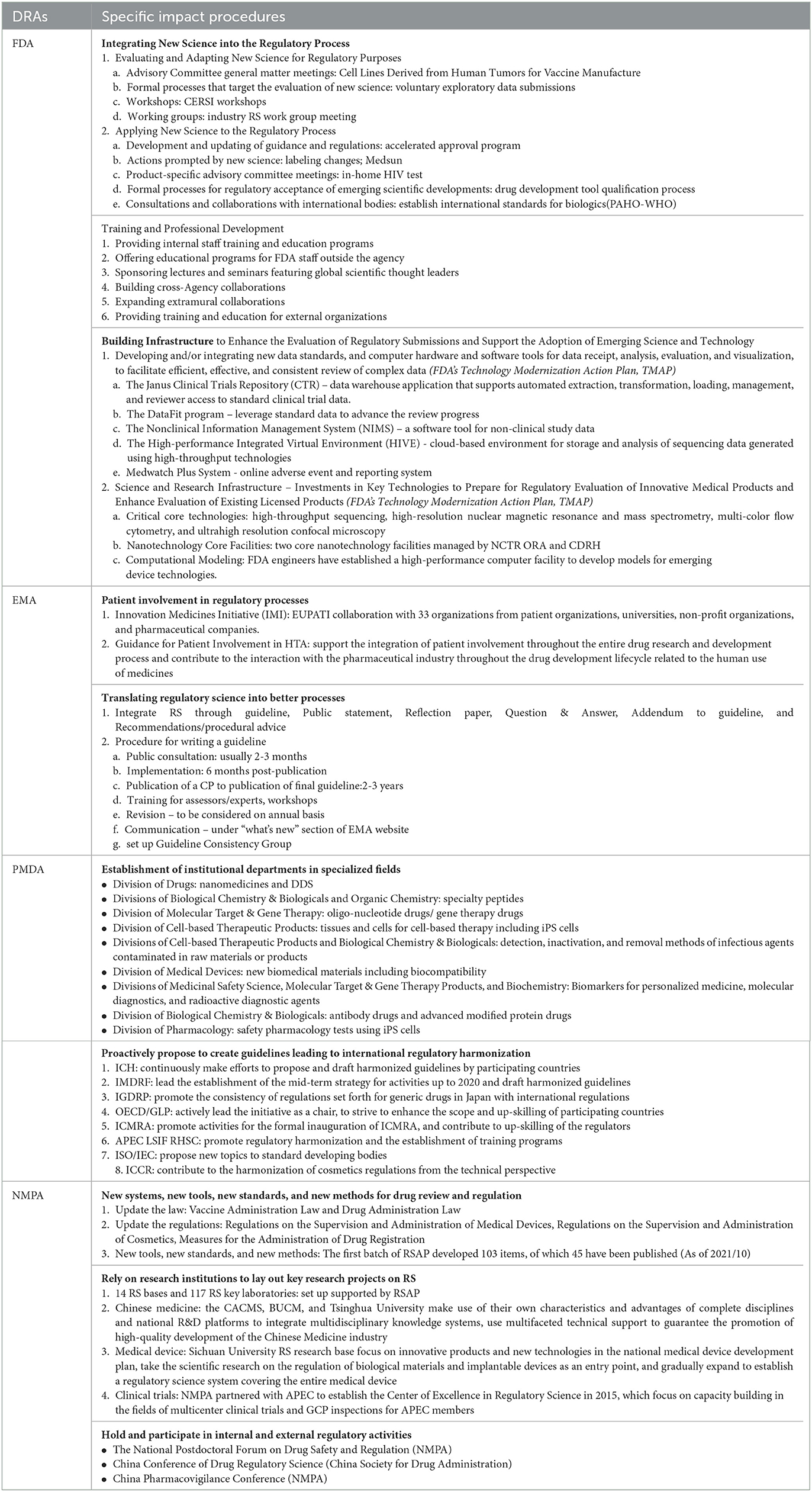
Table 3. Impact evaluation of RS in selected DRAs.
3.8. Step 9: outcome evaluation
As early as 2013, the FDA issued a clear indication in the white paper “Strategy and Implementation Plan for Advancing Regulatory Science of Pharmaceutical Products” for measuring outcomes in advancing and adopting RS in the United States, which was also a unique point of the FDA in the development of RS. FDA measured the metrics about the process of advancing RS through internal RS activities and external communications and collaboration. Three main metrics were used to measure the effectiveness of the impact of RS, including evaluating scientific training and professional development, integrating new science into the regulatory process, and building infrastructure to evaluate emerging science and technology. Enhanced staff capacity increased regulatory efficiency and transparency, and improved infrastructure in which translation of science and technology was anticipated as the key primary outcomes of the RS development. Other DRAs had not yet formally documented specific metrics to measure the outcomes of advancing the RS.
The FDA launched the Sentinel Initiative in 2008. In September 2014, it gradually transitioned from a mini-sentinel pilot phase to the full Sentinel System, which was used to monitor the safety of regulated products as a national electronic system and had developed the world's largest multi-site distributed data dedicated to the safety of medical products. In 2019, the FDA released the “Sentinel System: FIVE-YEAR STRATEGY 2019–2023,” which aimed to guide the path of the Sentinel System from 2019 to 2023. The main direction was to strengthen and expand the foundation of the Sentinel System (i.e., data, infrastructure, operations, and technology). The progress had also seen the enhancement of the security analytics capabilities of the Sentinel System by leveraging advanced technologies in data science and signal detection. At the same time, leveraging the Sentinel System to accelerate access and broaden the use of real-world data to generate real-world evidence was also reported. Finally, the stakeholder ecosystem of the Sentinel System was further expanded in pursuit of more effective coordination of national resources (58).
FDA's advances in RS could be implemented in many ways to streamline the device development process. The FDA Center for Devices and Radiological (CDRH) released the report “Regulatory Science in FDA's CDRH: a Vital Framework for Protecting and Promoting Public Health,” which focused on advancing the RS of medical devices and fostering innovation by substantially reducing the cost and time required for medical devices while feeding back accurate information to evaluate their safety, quality, and performance (74).
4. Discussion
This review analyzed how RS was developed, adopted, and advanced by the FDA, EMA, PMDA, and NMPA. By comparing their experiences systematically, an implementation science framework specific to RS development was also developed that is pragmatic and comprehensive for reference by other DRAs and research institutes. Evidence about the adoption, advancement, and monitoring of RS by the studied DRAs was presented to rationalize the formation of the model. The DRAs in this study acknowledged the basic principles of the RS discipline in general terms and identified their respective priorities and specific objectives for the development of RS according to their needs and priorities. Regulatory workforce capacity building and internal and external scientific exchange and collaboration were the primary means most commonly employed by DRAs to advance and adopt RS. The practical outcomes of adopting RS in terms of regulatory performance turning into benefits to public health, patient outcomes, and translation of drug research and development should be further defined to allow monitoring and evaluation for continuous improvement in the implementation of RS. Continuous government commitment and key stakeholder engagement and collaboration are essential for the dynamic development of RS to better prepare DRAs for the mounting challenges of drug regulation.
4.1. The vital role of the government in advancing RS
In recent years, there has been a meaningful shift in the view of professionals and the government in its use of research evidence for policy decision-making (75). Evidence-informed policymaking has been widely considered in a variety of fields, including the healthcare arena, social science, and business management. The policymakers make use of this approach, which ensures that decision-making is well-informed by the best available research evidence, for systematic and transparent access to evidence as a prerequisite for the process of policymaking (76, 77). Adopting RS to obtain scientific evidence and apply it in the decision-making process has been used by many governments (78–80). It has been found that providing regulators with the resources, knowledge, and evidence they need for the decision-making process helps to improve their identification of the most pressing practical social needs and strengthen their regulatory capacity (81, 82). Practically speaking, the development of technological and industrial developments often puts forward higher challenges and requirements for regulation, thereby promoting the reform of the legal system, including the 2019 coronavirus pandemic, the breakthrough developments of advanced therapy medicinal products, and the digital transformation of the healthcare industry.
4.2. Dynamic evaluation of the development of RS using implementation science framework
To be able to deliver drug regulators with expectations for advancing RS, a systems thinking approach guided by implementation science might offer a clear roadmap that helps translate the outcomes of RS into the formulation of new technique guidelines, new tools, or new scientific methods (83). The adoption of PPM is of particular importance in facilitating the translation of concepts into action for the sustainable development of RS (14). Addressing the gaps in regulatory behaviors and environment is part of a highly complex undertaking involving not just the knowledge, attitudes, and skills of regulators or DRAs but also communication and cooperation with other counterparts throughout the whole health system, as well as different resources, services, policies, and plans in the larger global pharmaceutical markets (84). The use of systems thinking approaches encourages relationship-building across various functionalities of the DRAs so as to achieve a common set of relevant goals and objectives on drug regulation (85).
Predisposing, reinforcing, and enabling factors are unique to a different national context that should be clearly and systematically identified for better planning of RS adoption. The best example is the Critical Path Initiative issued by the FDA which served as a precursor to its Regulatory Science Strategic plan. In the 2004 Critical Path Report, the FDA presented its diagnosis of the scientific challenges underlying the medical product problem and aimed to identify the gaps between the development of medical products and clinical use in Critical Path actions.
As depicted in Figure 5, we summarized the planned RS visions, priorities, and goals of the PRECEDE phase and identified the elements of each step by analyzing the experience of the development and adoption of RS in the US, EU, and Japan through the PROCEED phase. The continuous investment into capacity building of the regulatory agency in terms of hardware, software, laboratory, expertise, as well as collaboration with stakeholders is the key to implementing RS actions. Through the entire process of the implementation science framework, it is not difficult to find that the development of RS is a dynamic process that should be resilient and responsive to new health threats and healthcare needs as well as advancements in technology and innovation.
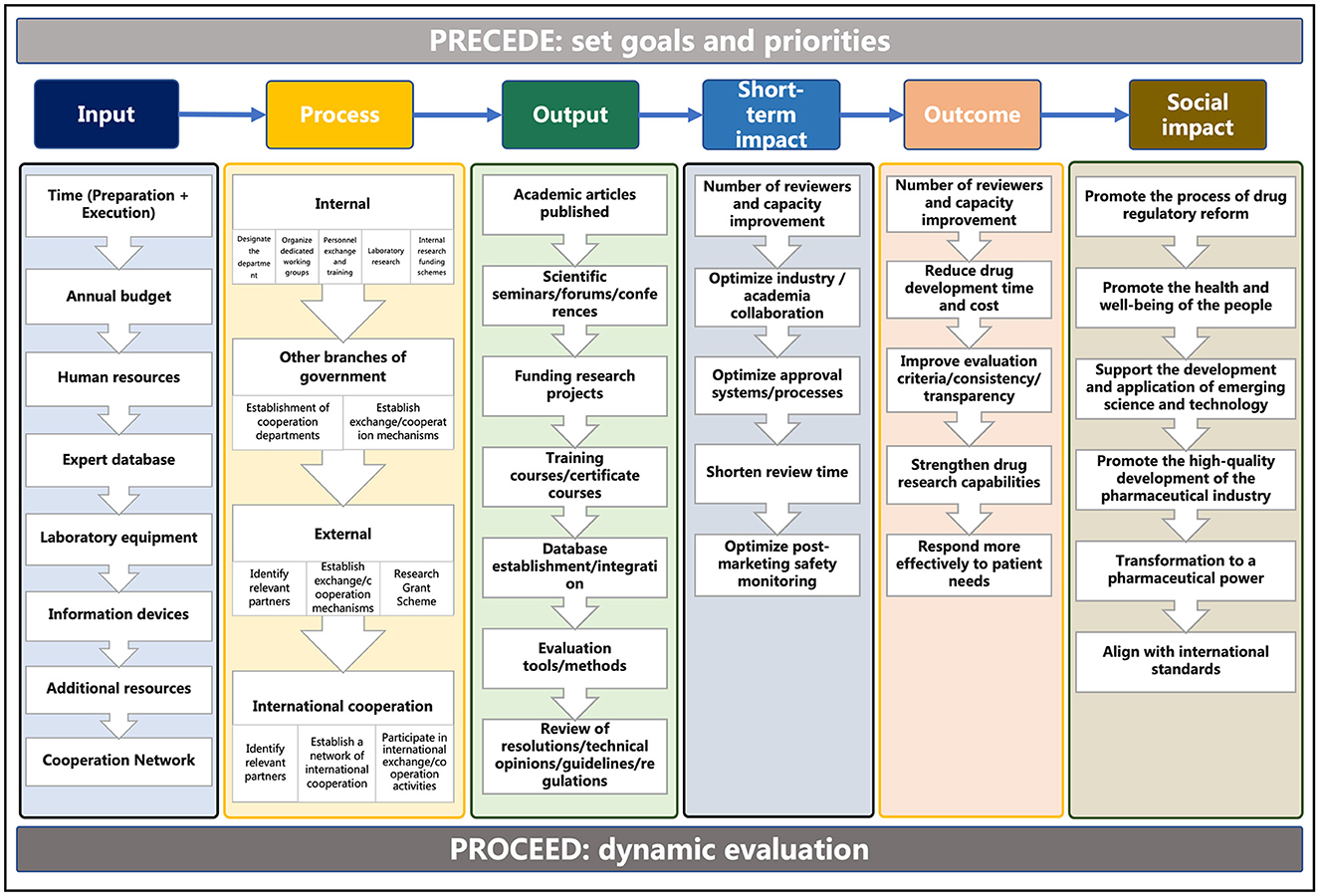
Figure 5. Elements of the development of RS based on the PPM.
The elements of the various stages of regulatory science development summarized based on the PPM framework are also applicable to countries or regions outside of those selected for this study. By analyzing the PRECEDE part, regulatory authorities can clearly identify the comprehensive factors that influence the development of RS, set priorities or specific goals, and then implement RS. By using the PROCEED part to systematically monitor and evaluate the input, implementation process, output, impact, and outcomes of RS. Regulatory authorities are more easily able to identify strengths and weaknesses for continuous quality improvement. In addition, by evaluating the development path of RS in different countries or regions through similar elements, regulatory authorities can engage in cross-comparisons and absorb international advanced experiences, thereby driving the reform process of regulation.
4.3. Continuous investment in regulatory capacity building
The development of RS should be considered in a context that can be evaluated. In the outcome evaluation (PROCEED step 9), only FDA has published metrics for measuring the advancement and adoption of regulatory science. The regulatory management system that measures change and demonstrates any outcomes related to changes in regulatory capacity is essential to support the sustainability of interventions or services. A scientific approach is needed to identify a range of factors that may facilitate the adoption of recommended actions and changes in regulatory practice. To this end, performance measurement knowledge and strategies must be adopted and incorporated into regulatory management systems to increase the effectiveness of interventions, while collecting the benchmarking data needed to establish evidence-based improvements (86).
Benchmarking tools have been increasingly used by DRAs to internally measure the performance of the regulation system in order to identify areas of improvement to achieve sustainable capacity optimization. In addition, the assessment of regulators against the international best counterparts likewise helps to inform actions to standardize regulatory practices. These would continuously improve the level of regulatory performance and international recognition. For instance, the Health Sciences Authority (HSA) of Singapore and the Ministry of Food and Drug Safety of the Republic of Korea have reached the highest level (Maturity Level 4) achievable for regulatory system evaluation against the WHO's GBT (87, 88), which provide a reference point for regulatory action by Asian drug regulators.
4.4. Strengths and limitations
Based on the existing concepts and frameworks for conducting documents and literature retrieval, evaluation, and evidence synthesis, this study shows a systematic view of the process of developing and adopting RS in the FDA, EMA, Japan, and China. The use of an implementation science framework offers some strengths to advance the development of RS. On the one hand, using the evaluation plan through PPM promotes standardization, consistency, and completeness in evaluating the development and adoption of RS, facilitates cross-regional comparisons, and fosters generalizable regulatory knowledge. On the other hand, the framework has great value in building the evidence base for the input and process involved in implementing RS. The evidence can be used to increase the likelihood of output, outcome, and long-term impacts of implementation. The findings of this study could push the elements of developing RS to a wider range of institutions, groups, regions, or countries.
However, there are still some limitations in the study. First, there are many assessment frameworks existing for implementation science, and it is difficult to use one framework to provide a comprehensive evaluation of how things are evolving. The PRECEDE-PROCEED model has the advantage of clearly linking program design and program evaluation metrics to support continuous process improvement. However, the PPM itself has some limitations, including the narrow definition of steps that requires at times more extensive application. In addition, the PPM has traditionally been applied to population-health programs but may be less applicable to some clinical settings. There is no involvement of access to key stakeholders in the PROCEED evaluation phase. Moreover, the bias in the publications that adopt RS in different international authorities persists, which means, no matter what form of developing or advancing RS by governments, the negative evaluation outcomes are rarely or unlikely to be fully reported. At the same time, some of the data that measures regulatory performance is internally confidential and less easily accessible, which might inevitably affect the completeness of the study findings reported in this review.
5. Conclusion
Through the pragmatic application of PPM, the development and adoption of RS can be translated into evidence-based decision-making by DRAs. This study provided a detailed analysis and summary of the elements that needed to be taken into account in the process of advancing RS and helped informed some insights for policymakers when considering whether to adopt RS and how to advance RS. In the future, methodological research on how to measure the outcomes of RS is warranted. The use of benchmarking against international best practices may be useful in providing some insights into different aspects of improved regulatory capacity building for DRAs. Continuous government commitment and key stakeholder engagement and collaboration are essential for the dynamic development of RS to better prepare DRAs for the mounting challenges of drug regulation.
Data availability statement
Publicly available datasets were analyzed in this study.
Author contributions
JS and CU planned and designed the study. JS, XC, CU, and HH were responsible for data management and analysis. JS, XC, and CU drafted the manuscript. CU and HH critically reviewed and revised the manuscript. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of this article.
Funding
This study was funded by the University of Macau (reference numbers: SRG2021-00007- ICMS and MYRG2022-00229-ICMS).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rivera SC, Torlinska B, Marston E, Denniston AK, Oliver K, Hoare S, et al. Advancing UK regulatory science strategy in the context of global regulation: a stakeholder survey. Ther Innov Regul Sci. (2021) 55:646–55. doi: 10.1007/s43441-021-00263-2
2. Sepodes B, Mol P. Editorial: insights in regulatory science 2021. Front Med. (2022) 9:1033558. doi: 10.3389/fmed.2022.1033558
3. Calvert MJ, Marston E, Samuels M, Rivera SC, Torlinska B, Oliver K, et al. Advancing UK regulatory science and innovation in healthcare. J R Soc Med. (2021) 114:5–11. doi: 10.1177/0141076820961776
4. Honig P, Zhang L. Regulation and innovation: role of regulatory science in facilitating pharmaceutical innovation. Clin Pharmacol Ther. (2019) 105:778–81. doi: 10.1002/cpt.1367
5. Kurz X. Advancing regulatory science, advancing regulatory practice. Pharmacoepidemiol Drug Saf. (2017) 722:726. doi: 10.1002/pds.4181
6. Dinda AK. Regulatory science: the need for empowering Indian innovation. Indian J Med Res. (2021) 154:770–5. doi: 10.4103/ijmr.IJMR_1665_19
7. Kondo T, Hayashi Y, Sato J, Sekine S, Hoshino T, Sato D. Evolving vision of regulatory science in the global medical community. Clin Pharmacol Ther. (2020) 107:136–9. doi: 10.1002/cpt.1604
8. Food Drug Administration. (FDA). Advancing regulatory science at FDA: a strategic plan. (2011). Available online at: https://www.fda.gov/media/81109/download (accessed August 20, 2022).
9. Davis JM, Pursley DWM. Cell-based therapies in neonates: the emerging role of regulatory science. Pediatr Res. (2019) 86:145–146. doi: 10.1038/s41390-019-0442-4
10. Allan J, Belz S, Hoeveler A, Hugas M, Okuda H, Patri A, et al. Regulatory landscape of nanotechnology and nanoplastics from a global perspective. Regul Toxicol Pharmacol. (2021) 122:104885. doi: 10.1016/j.yrtph.2021.104885
11. Ishimoto K, Arafune T, Washio T, Haishima Y, Matsumoto K, Uematsu M, et al. Japanese regulatory considerations for interoperability of medical devices. Ther Innov Regul Sci. (2023) 57:104–8. doi: 10.1007/s43441-022-00444-7
12. Goda Y. Regulatory science of natural products. J Nat Med. (2022) 76:732–47. doi: 10.1007/s11418-022-01639-w
13. Sales AE, Wilson PM, Wensing M, Aarons GA, Armstrong R, Flottorp S, et al. Implementation science and implementation science communications: our aims, scope, and reporting expectations. Implement Sci. (2019) 14:77. doi: 10.1186/s13012-019-0922-2
14. Huynh L, Toyserkani G A, Morrato E H. Pragmatic applications of implementation science frameworks to regulatory science: an assessment of FDA risk evaluation and mitigation strategies. (REMS)(2014–2018). BMC Health Serv Res. (2021) 21:779. doi: 10.1186/s12913-021-06808-3
15. Hlaing PH, Sullivan PE, Chaiyawat P. Application of PRECEDE-PROCEED Planning model in transforming the clinical decision making behavior of physical therapists in Myanmar. Front Public Health. (2019) 7:114. doi: 10.3389/fpubh.2019.00114
16. Crosby R, Noar SM. What is a planning model? An introduction to PRECEDE-PROCEED .J Public Health Dent. (2011) 71:S7–15. doi: 10.1111/j.1752-7325.2011.00235.x
17. Binkley CJ, Johnson KW. Application of the PRECEDE-PROCEED planning model in designing an oral health strategy. J theory Pract Dent public Heal. (2013) 1:1–18.
18. Howat P, Jones S, Hall M, Cross D, Stevenson M. The PRECEDE-PROCEED model: application to planning a child pedestrian injury prevention program. Inj Prev. (1997) 3:282–7. doi: 10.1136/ip.3.4.282
19. Tominaga T, Asahina Y, Uyama Y, Kondo T. Regulatory science as a bridge between science and society. Clin Pharmacol Ther. (2011) 90:29–31. doi: 10.1038/clpt.2011.89
20. Hines PA, Gonzalez-Quevedo R, Lambert AIOM, Janssens R, Freischem B, Edo JT, et al. Regulatory science to 2025: an analysis of stakeholder responses to the European medicines agency's strategy. Front Med (Lausanne). (2020) 7:508. doi: 10.3389/fmed.2020.00508
21. Food and Drug Administration. Innovation or Stagnation: Challenges and opportunity on the critical path to new medical products. (2004). Available online at: https://www.fda.gov/science-research/science-and-research-special-topics/UCM077262 (accessed March 2004).
22. European Medicines Agency. EMA regulatory science to 2025–strategic reflection. (2020). Available online at: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf (Accessed March 30, 2020).
23. Pharmaceutical and Medical Devices Agency. International Pharmaceutical Regulatory Harmonization Strategy - Regulatory Science Initiative. (2015). Available online at: https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/dl/150827-01-01.pdf. (accessed August 20, 2022)
24. National Medical Products Administration. The NMPA launched the China Drug Regulatory Science Action Plan. (2019). Available online at: http://www.gov.cn/xinwen/2019-05/02/content_5388253.htm (accessed May 2, 2019).
25. Food Drug Administration. (FDA). Priority Area 9: Strengthening the Global Product Safety Net. (2013). Available online at: https://www.fda.gov/science-research/advancing-regulatory-science/priority-area-9-strengthening-global-product-safety-net (accessed August 20, 2022).
26. National Medical Products Administration. Notice of the NMPA on the Implementation of the Second Batch of Key Projects of China Drug Regulatory Science Action Plan. (2021). Available online at: https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20210628172854126.html (accessed June 28, 2022).
27. Tambuyzer E, Vandendriessche B, Austin CP, Brooks PJ, Larsson K, Miller Needleman KI, et al. Therapies for rare diseases: therapeutic modalities, progress and challenges ahead. Nat Rev Drug Discov. (2020) 19:93–111. doi: 10.1038/s41573-019-0049-9
28. Shimokawa M, Sato D, Wakao R, Arai H. PMDA's vision for horizon scanning of emerging technologies potentially relevant to the development of new medical products: the regulatory challenge. Clin Pharmacol Ther. (2021) 109:295–8. doi: 10.1002/cpt.1986
29. Bajaj G, Gupta M, Wang H-GH, Barrett JS, Tan M, Rupalla K, et al. Challenges and opportunities with oncology drug development in China. Clin Pharmacol Ther. (2019) 105:363–75. doi: 10.1002/cpt.1017
30. Food and Drug Administration. Critical path opportunities report. (2006). Available online at: http://www.slcmsr.net/download/flyer/Critical_Path_Initiative_Report_FDA_2006.pdf (accessed August 20, 2022).
31. Food and Drug Administration. Critical path opportunities list. (2006). Available online at: http://wayback.archive-it.org/7993/20180125035449/https://www.fda.gov/downloads/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/UCM077258.pdf (accessed August 20, 2022).
32. Food and Drug Administration. The critical path initiative: Report on Key Achievements in 2009. (2010). Available online at: https://fda.report/media/78814/Critical-Path-Report-on-Key-Achievements-in-2009.pdf (accessed August 20, 2022).
33. Food and Drug Administration. FDA Organization Charts. (2019). Available online at: https://www.fda.gov/about-fda/fda-organization/fda-organization-charts (accessed March 21, 2019).
34. Food and Drug Administration. Statement of Organization, Functions, and Delegations of Authority. (2022). Available online at: https://www.govinfo.gov/content/pkg/FR-2022-01-03/pdf/2021-28386.pdf (accessed March 1, 2022).
35. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA oncology center of excellence and precision medicine. Exp Biol Med. (2018) 243:308–12. doi: 10.1177/1535370217740861
36. Oner EBS Labra SR and Fehr S. FDA drug regulation: investigational new drug applications. in Academic Entrepreneurship for Medical and Health Sciences. (2021): 1-24. doi: 10.21428/b2e239dc.784553dd
37. Food Drug Administration. (FDA). Centers of Excellence in Regulatory Science and Innovation. (CERSIs)-Program Evaluation. (2016). Available online at: https://www.fda.gov/media/96457/download (accessed January 5, 2023).
38. Food Drug Administration. (FDA). Partnerships to Advance Innovation and Regulatory Science. (PAIRS). (2021). Available online at: https://www.fda.gov/about-fda/center-devices-and-radiological-health/partnerships-advance-innovation-and-regulatory-science-pairs (accessed July 21, 2021).
39. European Medicines Agency. Principles for publication of agendas and minutes of EMA scientific committees. (2013). Available online at: https://www.ema.europa.eu/en/documents/other/principles-publication-agendas-minutes-ema-scientific-committees_en.pdf (accessed December 17, 2022).
40. Pharmaceutical and Medical Devices Agency. Outcome Documents of the Science Board. (2022). Available online at: https://www.pmda.go.jp/english/rs-sb-std/sb/outcome-docs/0001.html (accessed December 20, 2022).
41. Pharmaceuticals and Medical Devices Agency. Opening Center for Regulatory Science. (2018). Available online at: https://www.pmda.go.jp/rs-std-jp/rs-center/0001.pdf (accessed February 26, 2019).
42. Tatsuya Kondo. Op-ed: PMDA advances regulatory science-based “rational medicine”. (2017). Available online at: https://foreignpolicy.com/sponsored/op-ed-pmda-advances-regulatory-science-based-rational-medicine/ (accessed December 20, 2022).
43. People.cn. Establishment of China Society for Drug Regulation. (2013). Available online at: http://politics.people.com.cn/n/2013/0726/c70731-22332460.html (accessed December 20, 2022).
44. The State Council of PRC. Opinions of the State Council on Reforming the Review and Approval System for Drugs and Medical Devices. (2015). Available online at: http://www.gov.cn/zhengce/content/2015-08/18/content_10101.htm (accessed August 8, 2022).
45. Food and Drug Administration. FDA science and mission at risk: report of the Subcommittee on Science and Technology. (2007). Available online at: https://books.google.com/books/about/FDA_Science_and_Mission_at_Risk.html?id=mf8l69Qxw6YC (accessed November 20, 2022).
46. Food and Drug Administration. The critical path initiative: Report on Key Achievements in 2009. (2010). Available online at: https://fda.report/media/78814/Critical-Path-Report-on-Key-Achievements-in-2009.pdf (accessed December 20, 2022)
47. Food and Drug Administration. Advancing Regulatory Science for Public Health—A Framework for FDA'S Regulatory Science Initiative. (2010). Available online at: https://www.fda.gov/media/79184/download (accessed October 20, 2022).
48. Food and Drug Administration. Advancing regulatory science at FDA: a strategic plan. (2011). Available online at: https://www.fda.gov/media/81109/download (accessed August 20, 2022).
49. Food and Drug Administration. Strategy and implementation plan for advancing regulatory science for medical products. (2013). Available online at: https://www.fda.gov/media/86053/download (accessed July 8, 2022).
50. Food and Drug Administration. Advancing Regulatory Science at FDA: Focus Areas of Regulatory Science. (FARS). (2021). Available online at: https://www.fda.gov/media/145001/download (accessed January 11, 2022).
51. Food and Drug Administration. FDA In Brief: FDA Publishes Report on Focus Areas of Regulatory Science. (2021). Available online at: https://www.fda.gov/news-events/fda-brief/fda-brief-fda-publishes-report-focus-areas-regulatory-science (accessed January 11, 2022).
52. European Medicines Agency. EMA regulatory science to 2025- strategic reflection. (2018). Available online at: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf (accessed December 20, 2022).
53. Hamburg MA. Advancing regulatory science. Science. (2011) 331:987–987. doi: 10.1126/science.1204432
54. Food and Drug Administration. Driving Biomedical Innovation: Initiatives to Improve Products for Patient. (2011). Available online at: https://www.ipqpubs.com/wp-content/uploads/2012/02/FDA-Driving-Biomedical-Innovation.pdf (accessed December 20, 2022).
55. Food and Drug Administration. Mission Possible: How FDA Can Move at the Speed of Science. (2015). Available online at: https://www.fda.gov/files/about%20fda/published/Report–Mission-Possible–How-FDA-Can-Move-at-the-Speed-of-Science.pdf (accessed December 20, 2022).
56. Food and Drug Administration. FY 2015-2016: Regulatory Science Progress Report. (2017). Available online at: https://www.fdanews.com/ext/resources/files/2017/03/03-30-17-FDAReport.pdf?1495554120 (accessed December 20, 2022).
57. Food and Drug Administration. CDRH's Regulatory Science Priorities. (2019). Available online at: https://www.fda.gov/media/130213/download (accessed December 20, 2022).
58. Food and Drug Administration. Sentinel System: FIVE-YEAR STRATEGY 2019-2023. (2019). Available online at: https://www.fda.gov/media/120333/download (accessed January 1, 2022).
59. Food and Drug Administration. FDA's Predictive Toxicology Roadmap. (2019). Available online at: https://www.fda.gov/media/109634/download (accessed December 20, 2022).
60. Food and Drug Administration. 2022 Advancing Regulatory Science: Focus Areas of Regulatory Science Report. (2022). Available online at: https://www.fda.gov/media/161381/download (accessed December 20, 2022).
61. European Medicines Agency. Road map to 2015. (2010). Available online at: https://www.ema.europa.eu/en/documents/report/road-map-2015-european-medicines-agencys-contribution-science-medicines-health_en.pdf (accessed December 20, 2022).
62. European Medicines Agency. The European regulatory system for medicines: a consistent approach to medicines regulation across the EU. (2014). Available online at: https://www.ema.europa.eu/en/documents/leaflet/european-regulatory-system-medicines-european-medicines-agency-consistent-approach-medicines_en.pdf (accessed December 20, 2022).
63. European Medicines Agency. EU Medicines Agencies Network Strategy to 2020. (2015). Available online at: https://www.ema.europa.eu/en/documents/other/eu-medicines-agencies-network-strategy-2020-working-together-improve-health_en.pdf (accessed December 20, 2022).
64. European Medicines Agency. Regulatory science strategy. (2017). Available online at:https://www.ema.europa.eu/en/about-us/how-we-work/regulatory-science-strategy (accessed December 20, 2022).
65. European Medicines Agency. EU Medicines Agencies Network Strategy to 2025. (2020). Available online at: https://www.ema.europa.eu/en/documents/report/european-union-medicines-agencies-network-strategy-2025-protecting-public-health-time-rapid-change_en.pdf (accessed December 20, 2022).
66. Pharmaceutical and Medical Devices Agency. Strategy of SAKIGAKE. (2014). Available online at: https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/dl/140729-01-01.pdf (accessed December 20, 2022).
68. Pharmaceutical and Medical Devices Agency. PMDA Center for regulatory science. (2018). Available online at: https://www.pmda.go.jp/files/000235786.pdf (accessed December 20, 2022).
69. Peking University Clinical Research Institute. Peking University- Asia-Pacific Economic Cooperation. (APEC) Regulatory Science Center of Excellence. (2015). Available online at http://pucri.bjmu.edu.cn/cn/basic-106-106.html (accessed December 20, 2022).
70. The State Council of PRC. The Opinion on Deepening the Reform of the Review and Approval System and Encouraging the Innovation of Drugs and Medical Devices. (2017). Available online at: http://www.gov.cn/zhengce/2017-10/08/content_5230105.htm (accessed August 8, 2022).
71. The State Council of PRC. Implementation Opinions on Comprehensively Strengthening Drug Regulatory Capacity Building. (2021). Available online at: http://www.gov.cn/zhengce/content/2021-05/10/content_5605628.htm (accessed August 8, 2022).
72. Pharmaceutical and Medical Devices Agency. Strategy of SAKIGAKE. (2014). Available online at: https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/dl/140729-01-01.pdf (accessed December 20, 2022).
73. European Business Council in Japan. New Scheme Related To New Drug Review And Approval. (2022). Available online at: https://ebc-jp.com/digital-white-paper/issues/health-science/pharmaceuticals/new-scheme-related-to-new-drug-review-and-approval-2/ (accessed November 22, 2022).
74. Food and Drug Administration. Regulatory Science in FDA's CDRH: A Vital Framework for Protecting And Promoting Public Health. (2018). Available online at: https://www.fda.gov/media/120333/download (accessed September 20, 2022).
75. Mahdavi M, Khasraghi JS, Sajadi HS, Yazdizadeh B, Nikooee S, Ehsani-Chimeh E, et al. Developing framework and strategies for capacity building to apply evidence-informed health policy-making in Iran: mixed methods study of SAHSHA Project. Int J Health Policy Manag. (2022) 11:2236–47. doi: 10.34172/ijhpm.2021.142
76. Bowen S, Zwi AB. Pathways to “evidence-informed” policy and practice: a framework for action. PLoS Med. (2005) 2:e166. doi: 10.1371/journal.pmed.0020166
77. Haynes A, Rowbotham SJ, Redman S, Brennan S, Williamson A, Moore G. What can we learn from interventions that aim to increase policy- makers' capacity to use research? a realist scoping review. Health Res Policy Syst. (2018) 16:31. doi: 10.1186/s12961-018-0277-1“
78. Park J, Shin H, Kim J. Analysis of trends in regulatory science and regulatory science experts training projects: US, Japan, Singapore, and Korea. Korean J Clin Pharm. (2021) 31:257–67. doi: 10.24304/kjcp.2021.31.4.257
79. Maeda H, Ng DB. Regulatory approval with real-world data from regulatory science perspective in Japan. Front Med. (2022) 3:1082. doi: 10.3389/fmed.2022.864960
80. Romeu B, Rodríguez Y, Bendiner S. The role of regulatory sciences from the perspective of the Cuban medicines regulatory agency: the impact of COVID-19 in promoting innovation, cooperation and scientific thinking. Ther Innov Regul Sci. (2021) 55:1014–8. doi: 10.1007/s43441-021-00300-0
81. Azar FE, Solhi M, Nejhaddadgar N, Amani F. The effect of intervention using the PRECEDE-PROCEED model based on quality of life in diabetic patients. Electron Physician. (2017) 9:5024–30. doi: 10.19082/5024
82. Herrero-Martinez E, Hussain N, Roux NL, MacDonald J, Mayer M, Palacios R, et al. Dynamic regulatory assessment: evolving the european regulatory framework for the benefit of patients and public health-an EFPIA view. Clin Ther. (2022) 44:132–8. doi: 10.1016/j.clinthera.2021.11.001
83. Milko LV, Chen F. Chan K. FDA oversight of NSIGHT genomic research: the need for an integrated systems approach to regulation. NPJ Genom Med. (2019) 4:32. doi: 10.1038/s41525-019-0105-8
84. Peters DH. The application of systems thinking in health: why use systems thinking? Health Res Policy Syst. (2014) 12:51. doi: 10.1186/1478-4505-12-51
85. Naaldenberg J, Vaandrager L, Koelen M, Wagemakers AM, Saan H, de Hoog K. Elaborating on systems thinking in health promotion practice. Glob Health Promot. (2009) 16:39–47. doi: 10.1177/1757975908100749
86. Seaton TL. Dissemination and implementation sciences in pharmacy: a call to action for professional organizations. Res Soc Adm Pharm. (2017) 13:902–4. doi: 10.1016/j.sapharm.2017.05.021
87. World Health Organization. Global Benchmarking Tool (GBT) for Evaluation of National Regulatory Systems of Medical Products: Revision VI. WHO (2021).
88. Ministry of Food and Drug Safety. MFDS Achieves Highest Maturity Level in Regulatory System by WHO. (2022). Available online at: https://www.mfds.go.kr/eng/brd/m_61/view.do?seq=135&srchFr=&srchTo=&srchWord=&srchTp=&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&company_nm=&page=1 (accessed November 30, 2022).
Keywords: PRECEDE-PROCEED model (PPM), regulatory science, implementation science, drug regulatory authorities (DRAs), The United States (US), European Union, Japan, China
Citation: Shi J, Chen X, Hu H and Ung COL (2023) Application of implementation science framework to develop and adopt regulatory science in different national regulatory authorities. Front. Public Health 11:1172557. doi: 10.3389/fpubh.2023.1172557
Received: 23 February 2023; Accepted: 03 April 2023;
Published: 04 May 2023.
Edited by:
Shintaro Sengoku, Tokyo Institute of Technology, JapanCopyright © 2023 Shi, Chen, Hu and Ung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolina Oi Lam Ung, Y2Fyb2xpbmF1bmdAdW0uZWR1Lm1v
†These authors have contributed equally to this work and share first authorship