 Haifeng Gao1,2†
Haifeng Gao1,2† Xun Zhu
Xun Zhu- 1Xinjiang Production and Construction Corps Key Laboratory of Special Fruits and Vegetables Cultivation Physiology and Germplasm Resources Utilization, Agriculture College of Shihezi University, Shihezi, China
- 2Key Laboratory of Integrated Pest Management on Crop in Northwestern Oasis, Institute of Plant Protection, Xinjiang Academy of Agricultural Sciences, Ministry of Agriculture and Rural Affairs, Urumqi, China
- 3Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 4State Key Laboratory of Integrated Management of Pest and Rodents, Institute of Zoology, Chinese Academy of Sciences, Beijing, China
Xinjiang (XJ) and Ningxia (NX) provinces are important agricultural regions in western China. Aphids are one kind of the most devastating pests in both the provinces. Aphids are typical phloem-feeding insects distributed worldwide and can severely damage crops. In this study, two representative Metopolophium dirhodum (Walker) (Hemiptera: Aphididae) populations were collected from the typical agricultural regions of XJ and NX, respectively for a high-throughput transcriptome sequencing analysis. A total of 5,265 differentially expressed genes (DEGs) were identified. The functional annotation of DEGs and the identification of enriched pathways indicated many of the DEGs are involved in processes related to energy metabolism, development, and insecticide resistance. Furthermore, an investigation of insecticide toxicity revealed the NX population is more resistant to insecticide treatments than the XJ population. Thus, the transcriptome data generated in present study can be used for functional gene characterization relevant to aphid development, metabolism, environmental adaptation, and insecticide resistance.
Introduction
Aphids, which are typical phloem-feeding insects found worldwide, can severely damage crops and other plants (Powell et al., 2006). The suitability of a plant host for an insect depends on plant surface features, including the wax layers (Walling, 2008) and leaf trichomes (Wang et al., 2001; Rodriguezlopez et al., 2011). Insects use their highly specialized mouthparts to probe vegetative tissue for phloem sap, which contains nutrient compounds including sugars and amino acids (Kaloshian and Walling, 2005). After establishing a feeding site, aphids can continue feeding for a prolonged period. Phloem feeding by aphids depletes crop photo assimilate contents and inhibits plant growth. Previous studies revealed that aphids feeding on phloem deposit honeydew on the leaf surface and encourage the growth of sooty mold, which can block the light required for photosynthesis and restrict crop growth (Filho and Paiva, 2006; Taylor et al., 2012). Aphids are also the vectors of numerous plant viruses (Chen et al., 2012, 2013). When insects penetrate plants' tissue with their mouthparts to enable phloem feeding, they continuously secrete saliva (Will et al., 2009), which is one of the efficient ways for viruses transmission (Weintraub and Beanland, 2006). To date, aphids are mainly controlled by the frequent application of insecticides, which is relatively inefficient and potentially damaging the environment (Devonshire and Field, 1991).
There are ~4,700 members of the superfamily Aphidoidea (https://www.aphidoidea.com), including Metopolophium dirhodum (Walker) (Hemiptera: Aphididae), which is a very important pest in the crop-growing regions of many countries (Winder et al., 2001; Carver, 2010). Like other aphid species, M. dirhodum annually reproduces multiple generations on different hosts. Rosaceae species (e.g., apple and peach) are the primary hosts of M. dirhodum in the fall, in which season male and female M. dirhodum morphs mate for sexual propagation. In the spring, after hatching from eggs, aphids fly away seeking a secondary host to produce colonies. Poaceae species, including wheat, are the secondary hosts for the summer generation comprising only female and parthenogenic aphids (Argandona et al., 1980; Garratt et al., 2010). During the summer, M. dirhodum (virginopara) can produce two or three generations on secondary host plants. The plant sap is the sole source of nutrients for M. dirhodum and other aphids. The aphid mouthpart is a highly specialized structure. After probing plants, the mouthpart can reach the phloem to feed on the sap (Kempema et al., 2006). Although sap feeding by aphids decreases the nutrient and water contents of plants, ultimately leading to wilting and yield losses, the collateral damages could be more severe.
An earlier investigation proved that M. dirhodum can serve as the vector for several viruses, including barley yellow dwarf virus, maize mosaic virus, and potato virus (Waterhouse and Helms, 1985). The puncturing of the host plant by aphids facilitates the infection of the inner plant parts. During sap feeding, aphids produce honeydew containing a substantial abundance of sugars, which promotes the growth of saprophytic fungi detrimental to plant health (Taylor et al., 2012).
Several kinds of insecticides are used to control M. dirhodum for instance beta-cypermethrin et al. However, these insecticides can also induce the expression of detoxification and metabolism-related genes in insects. Transcriptome sequencing based on new nucleotide sequencing platforms (e.g., Illumina and PacBio) has considerably enhanced molecular and mechanisms research in various life science fields. High-throughput and deep-sequencing technologies enable the rapid and inexpensive acquisition of massive amounts of data. The bioinformatics-based analysis of the sequencing data [e.g., annotation of gene functions, identification of differentially expressed genes (DEGs), and analyses of gene ontology] may further elucidate the metabolic and signal transduction pathways underlying specific phenotypes.
Xinjiang Uygur Autonomous Region (XJ) and Ningxia Hui Autonomous Region (NX) are very large and agriculturally important provinces in western China. In terms of area, XJ is the biggest province in China. Although parts of these provinces are covered by desert, large areas are used for agricultural production. The similarities in the climate conditions of XJ and NX enable the cultivation of the same crops in both provinces, including wheat, barely, rice, cotton, apple, pear, grape, and sweet melon. M. dirhodum has been detected in the agricultural regions of both provinces, resulting in severe damages to crops every year, especially wheat. Unfortunately, M. dirhodum genes, genomes, and transcriptomes remain relatively uncharacterized. In this study, we selected two M. dirhodum populations from the wheat fields of XJ and NX for a transcriptome analysis under laboratory conditions. The comparison of the resulting transcriptome data revealed the DEGs. Furthermore, differences in metabolic pathways and insecticide tolerance between the two aphid populations were examined and discussed further.
Materials and Methods
Insect Culture
Metopolophium dirhodum samples were collected from wheat fields in the XJ (N43°41′14″, E87°22′46″) and NX (N39°5′57″, E106°44′51″) provinces. The collected aphids were sub-cultured on fresh wheat seedlings in insect-rearing cages at 20 ± 1°C, with 60 ± 10% relative humidity and a 16-h light:8-h dark photoperiod, for more than 8 generations.
Sample Preparation and RNA Isolation
For the NX and XJ populations, total RNA was extracted from 10 wingless adults with the TRIzol reagent according to the manufacturer instructions (Invitrogen TRIZOL® Reagent) for the subsequent RNA sequencing (RNA-seq) analysis, which was completed with four replicates. The purity of all RNA samples was assessed based on the OD260/280 and OD260/230 absorbance ratios, which were determined with the NanoDrop 2000 spectrophotometer. Samples with OD260/280 and OD260/230 ratios exceeding 1.8 and 2.2, respectively, were considered sufficiently pure. The RNA integrity was evaluated by 1% agarose gel electrophoresis. The extracted RNA was treated with DNase I to eliminate any residual genomic DNA.
Library Preparation and Illumina Sequencing
The cDNA libraries were constructed and sequenced by OriGene Technologies, Beijing, China. Specifically, mRNA was isolated from two total RNA samples (Grabherr et al., 2011). To construct the paired-end RNA-seq libraries for the transcriptome analysis, 10 μg total poly(A) mRNA was isolated with Sera-mag Magnetic Oligo(dT) Beads (Thermo) and then fragmented in fragmentation buffer. The resulting fragments were used as templates for the first-strand cDNA synthesis with a random hexamer primer. The second cDNA strand was synthesized in 20 μl second strand buffer (Invitrogen) supplemented with 10 mM dNTP mix, 5 U/μL RNase H, and 10 U/μL DNA polymerase I. Short fragments were purified with the QIAquick PCR purification kit and resuspended in EB buffer for the end-repair step and addition of a poly(A) tail. Sequencing adapters were then added to the short fragments. After an agarose gel electrophoresis, suitable fragments were selected as templates for a PCR amplification. Finally, paired-end RNA-seq libraries were prepared as recommended by Illumina and then sequenced on the HiSeq™ X Ten platform (Illumina). The Illumina instrument software was used for data analysis and base calling.
RNA-seq Data Processing and Analysis
The RNA-seq data were assessed based on the Q30 standard, which confirmed that the sequencing data quality was acceptable. Raw reads with adapters or more than 5% unknown nucleotides as well as low-quality reads were eliminated. The remaining clean reads were used for the de novo assembly of the transcriptome with the Trinity short-read assembly program, which connects contigs and generates unigene sequences.
The unigenes were annotated by screening protein databases, including the non-redundant (NR), Swiss-Prot, KOG, KEGG, and GO databases, with the BLASTX algorithm (E-value < 1 × 10−5 and sequence identity >30%). The GO analysis (http://www.geneontology.org) grouped the unigenes into three major categories based on function (i.e., molecular function, cellular component, and biological process). The Blast2GO program was used for the GO annotation of unigenes, after which the WEGO software was used for the GO functional classification of all unigenes and for clarifying the distribution of gene functions. The KEGG pathway database was used to functionally characterize gene products regarding their effects on metabolic and cellular processes. The KEGG database was also applied to investigate the complex biological behaviors of the gene products. The KEGG pathways enriched among the unigenes were identified with the Blastall algorithm.
The DEGs were identified based on the fold-changes in expression levels (p < 0.05), which were determined with Cufflinks (version 2.0.2). The DEGs were clustered according to the KEGG analysis involving the Fisher and Chi-square tests.
Insecticide Toxicity
Insecticide toxicity was evaluated according to a published leaf dip method (Saska et al., 2016). The insecticide (Imidacloprid, Thiamethoxam, Omethoate, Avermectin) stock solutions (10,000 mg/L) were prepared with dimethylformamide. The stock solutions were diluted 5–7 times with 0.1% Tween 80. The 0.1% Tween 80 solution was used as a control. For each insecticide concentration, three replicates of at least 30 aphids were treated. Wheat bran and leaves carrying aphids were immersed in the insecticide solution for 3–5 s and then placed in Petri dishes containing moistened filter paper. The Petri dishes were placed in an incubator set at 20 ± 1°C, with 60 ± 10% relative humidity and a 16-h light:8-h dark photoperiod. After 24 h, the aphids were examined with a microscope. They were gently touched with an anatomical needle. Aphids that did not move and aphids with only one foot that moved were considered dead. The total number of aphids and the number of dead aphids were recorded for each Petri dish.
Statistical Analysis
Data were recorded as the average of two replicates ± standard deviation. The data underwent an analysis of variance with SPSS 20.0. In the figures presented herein, significantly different values (p < 0.05) are indicated by different lowercase letters (a–d).
Results
Illumina Sequencing and de novo Transcriptome Assembly
Each aphid population was analyzed with independently constructed libraries from four biological replicates. A total of 53.4, 44.1, 39.2, and 43.4 million raw reads were obtained for the XJ population, whereas 10.1, 10.1, 5.6, and 10.0 million raw reads were generated for the NX population (Table 1). The average raw read length varied from 148.2 to 149.3 bp. After the quality control step (Q30) and filtering of raw reads, the clean reads were used for the de novo assembly of 30,262 transcripts. After eliminating redundancies, 22,150 unigenes were retained for the XJ and NX populations, with N50 values of 2,491 and 2,479 bp (XJ population) and 1,356.3 and 1,443.0 bp (NX population). The unigene sequences are provided in Supplementary File 1.
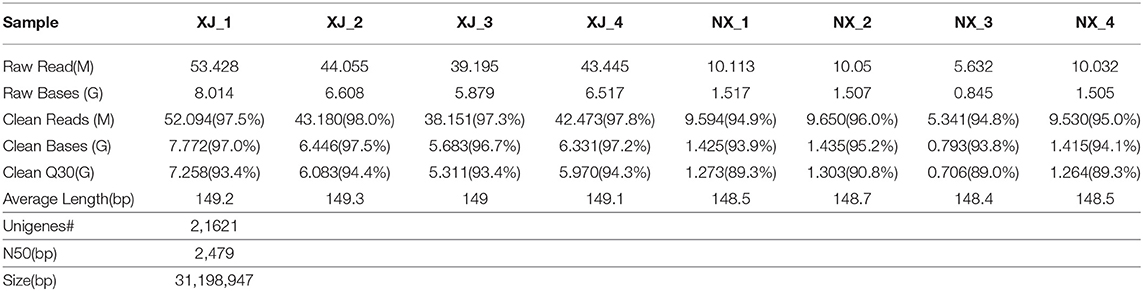
Table 1. Summary statistics of the Metopolophium dirhodum (Walker) transcriptomes.
Gene Identification and Annotation
The unigenes were used as queries to screen the NR (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/), Swiss-Prot (https://www.uniprot.org/), TrEMBL (https://www.uniprot.org/), Pfam (http://pfam.xfam.org/), and KOG databases with BLASTX. A total of 6,961 (32.2% of all distinct sequences), 8,646 (40.0%), 7,409 (34.3%), 6,836 (31.6%), and 4,968 (23.0%) unigenes were annotated based on the Swiss-Prot, NR, KOG, GO, and KEGG databases, respectively.
Regarding the KOG analysis, the orthologs were clustered based on their functions. The unigenes were clustered into the following four groups: information storage and processing, cellular processes and signaling, metabolism, and poorly characterized (Figure 1). The unigenes in these four groups were further divided into 24 subgroups. Similar KOG annotation trends were observed for both aphid populations. For example, the three largest subgroups for the two populations were “general function predicted only,” “signal transduction mechanism,” and “post translational modification, protein turnover, chaperones” (Supplementary File 2).
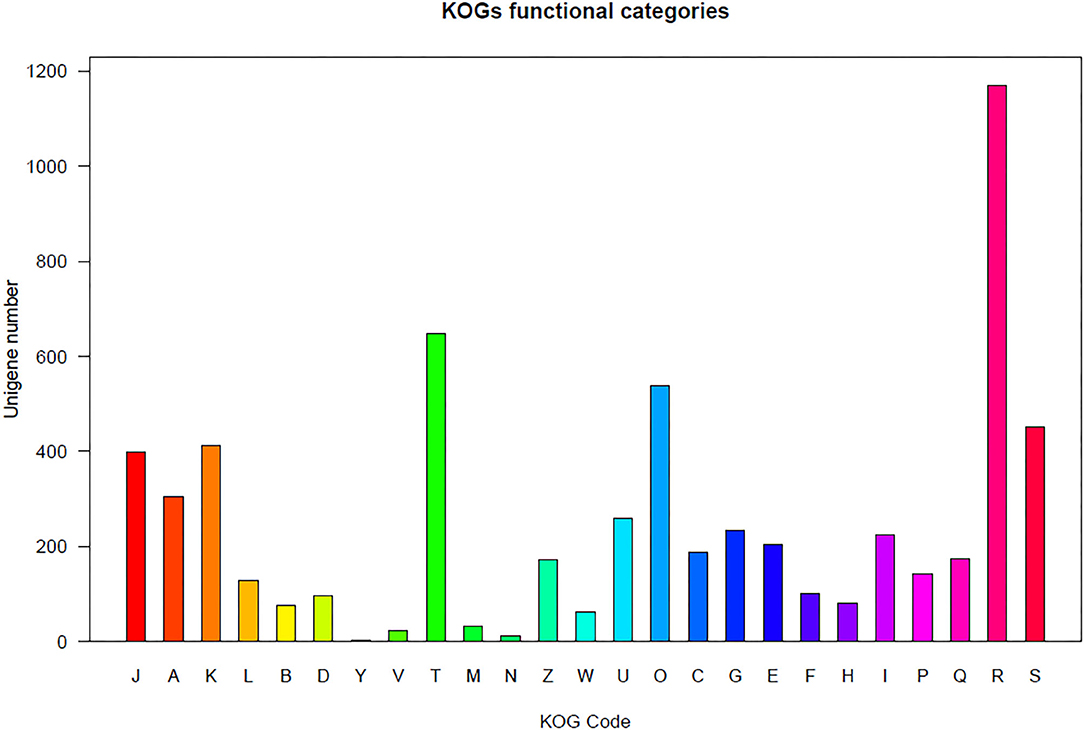
Figure 1. Unigene-function clusterification by KOG. INFORMATION STORAGE AND PROCESSING [J] Translation, ribosomal structure, and biogenesis; [A] RNA processing and modification; [K] Transcription; [L] Replication, recombination, and repair; [B] Chromatin structure and dynamics. CELLULAR PROCESSES AND SIGNALING: [D] Cell cycle control, cell division, chromosome partitioning; [Y] Nuclear structure; [V] Defense mechanisms; [T] Signal transduction mechanisms; [M] Cell wall/membrane/envelope biogenesis; [N] Cell motility; [Z] Cytoskeleton; [W] Extracellular structures; [U] Intracellular trafficking, secretion, and vesicular transport; [O] Posttranslational modification, protein turnover, chaperones. METABOLISM: [C] Energy production and conversion; [G] Carbohydrate transport and metabolism; [E] Amino acid transport and metabolism; [F] Nucleotide transport and metabolism; [H] Coenzyme transport and metabolism; [I] Lipid transport and metabolism; [P] Inorganic ion transport and metabolism; [Q] Secondary metabolites biosynthesis, transport and catabolism. POORLY CHARACTERIZED: [R] General function prediction only; [S] Function unknown.
Comparison of the Transcriptional Profiles of the XJ and NX Populations
A total of 5,265 DEGs were identified following a comparison of the XJ and NX aphid transcriptomes (XJ/NX), of which 2,447 were up-regulated and 2,791 were down-regulated (Figure 2, Supplementary File 3). The functional annotations revealed considerable differences between the two aphid populations, 47 KEGG pathways were enriched among the DEGs. The top 30 enriched pathways are presented in Figure 3. Among them, 22 pathways were relevant to metabolism, all of them were underlined and marked with hashtag. There were three (marked with an extra asterisk) metabolism pathways concerned with drug/xenobiotics degradation, and two of them were of P450 monooxygenase system. The top 10 most enriched pathways based on the –log2 p-value are listed in Table 2, including those related to amino acid biosynthesis, starch and sucrose metabolism, fatty acid biosynthesis and metabolism, and metabolism of xenobiotics by cytochrome P450.
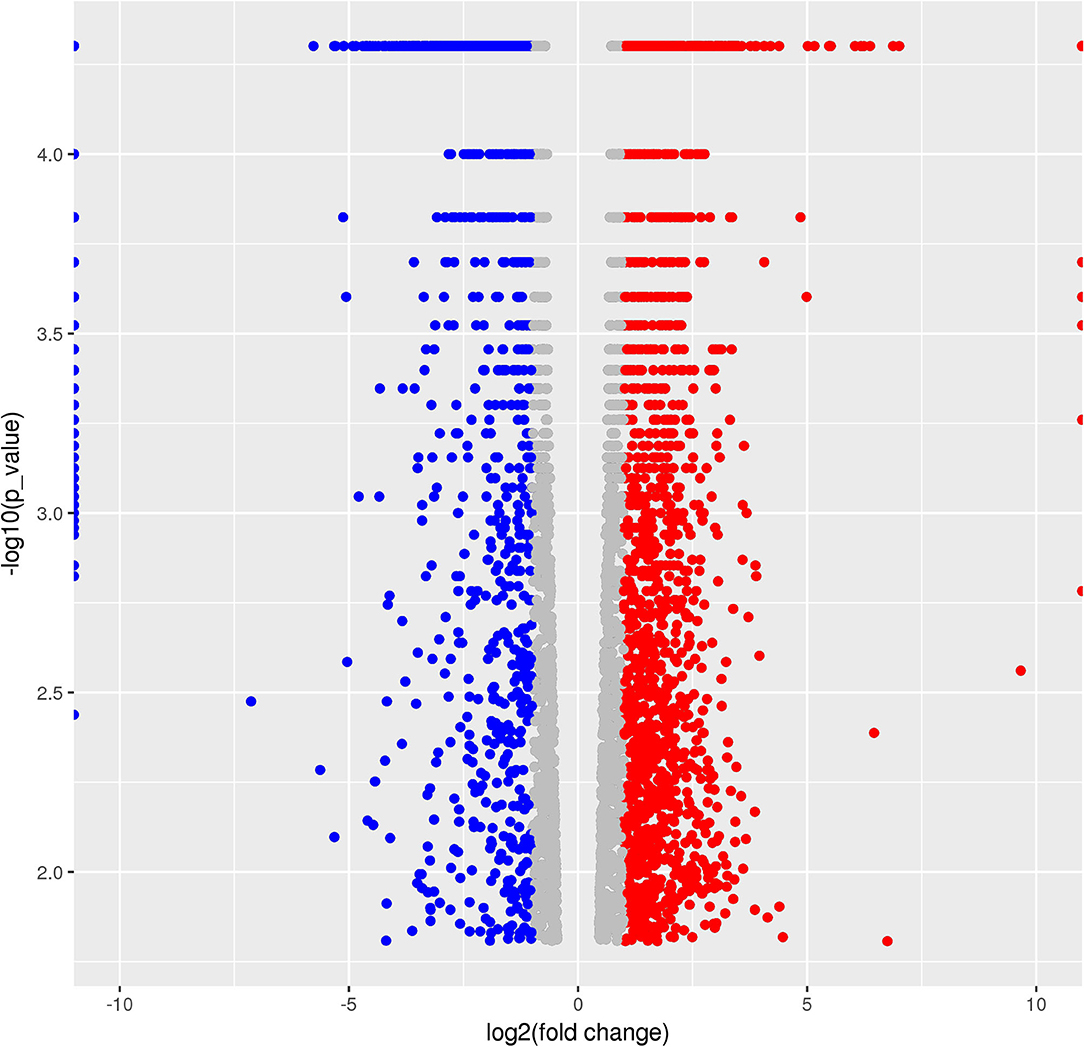
Figure 2. Volcano Graphic. In the figure each dot demonstrated a gene expression in two populations. The horizontal ordinate is the log2 value of folder change between populations (NX/JX). The y-coordinate is the –log10 of the p-value. Dots in red indicated the up-regulated, in blue were down-regulated, and in gray were those with no significant change.
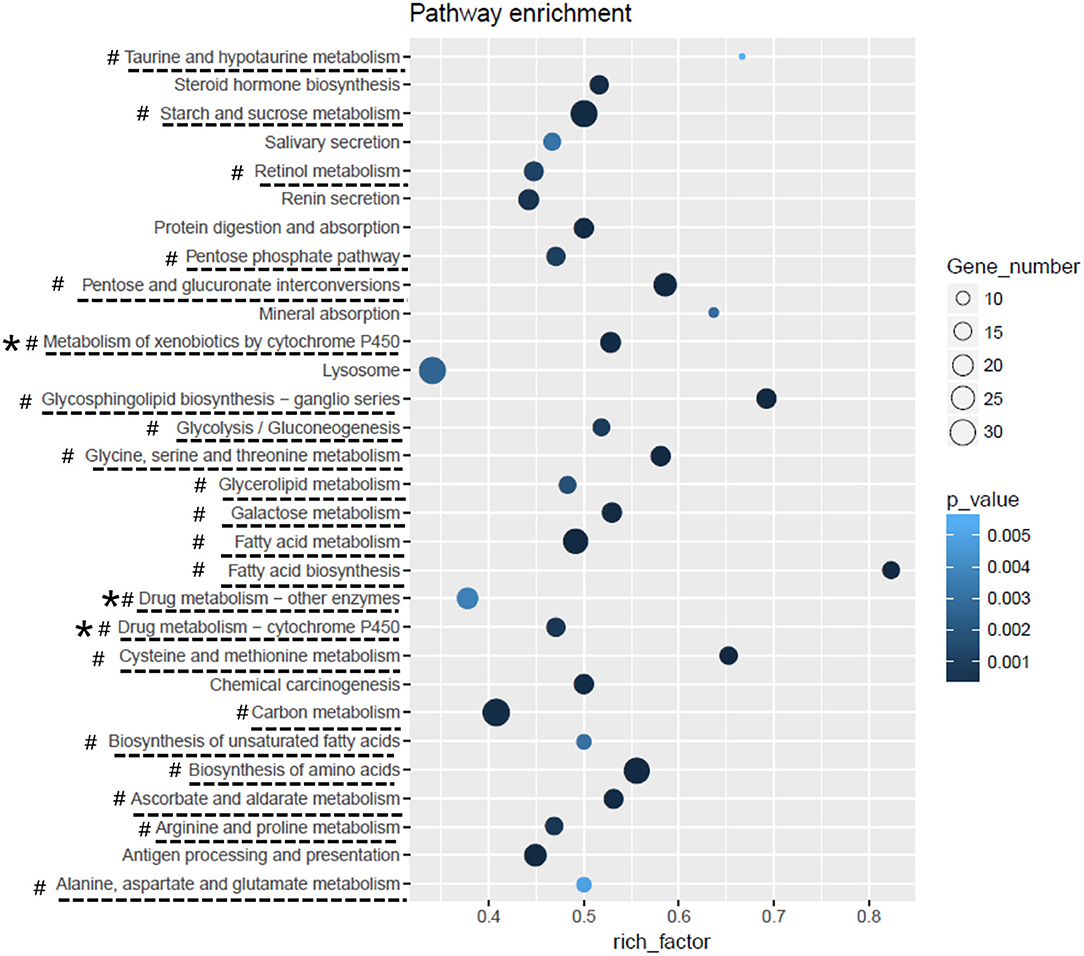
Figure 3. The top-30 pathways of DEGs KEGG enrichment analysis. The pathways list from the bottom to the top were in order of the first letter of the pathway name, from A to Z. Rich_facotor means (number of DEG enriched in the pathway/number of background genes).
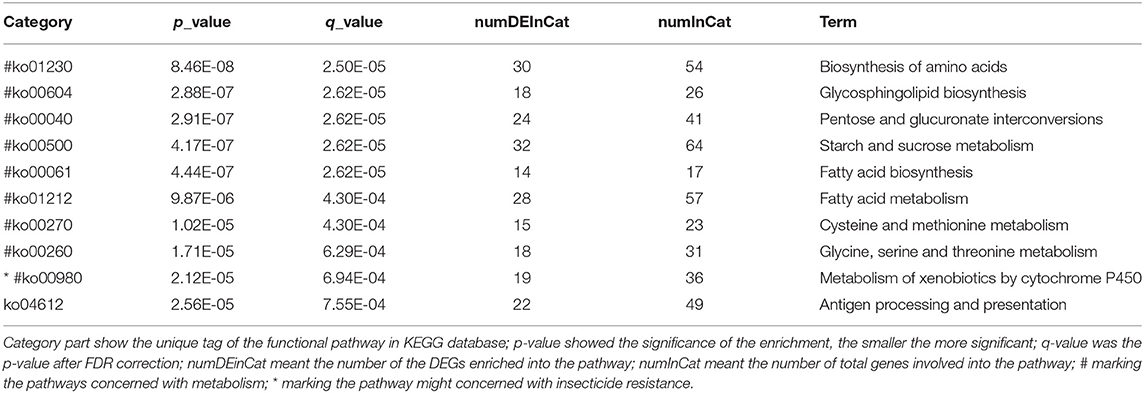
Table 2. Top-10 pathways DEGs involved in based on KEGG enrichment analysis.
Insecticide Toxicity
We evaluated the tolerance of the two aphid populations to the following beta-cypermethrin-containing insecticides: imidacloprid, thiamethoxam, omethoate, and avermectin. The median lethal dose (LC50) and toxicity regression equations indicated the NX population was substantially more tolerant to imidacloprid and thiamethoxam than the XJ population (Table 3). Additionally, the NX aphids were slightly more tolerant to avermectin than the XJ population, whereas there was no difference in the toxicity of omethoate. The LC50 of imidacloprid for the NX population was 56.63 mg/L, which was 5-fold higher than that for the XJ population. The LC50 of thiamethoxam for the NX aphids was as high as 573.29 mg/L, which was ~10-fold higher than that for the XJ aphids.
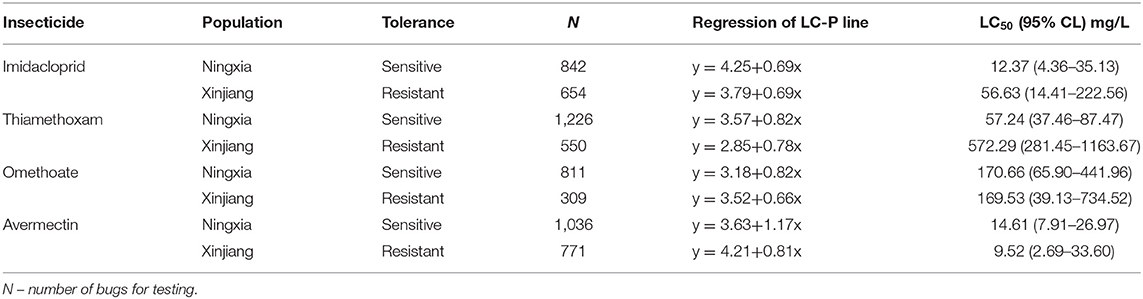
Table 3. Toxicology experiment of pesticides.
Discussion
In western China, both XJ and NX are large provinces of agriculture. Sustainable wheat production in both provinces is threatened by M. dirhodum. In the current study, we collected and analyzed two typical M. dirhodum populations reflecting the effects of the geographical and environmental conditions in the main agricultural regions of XJ and NX. The two aphid populations were compared based on a high-throughput transcriptome analysis. A total of 47 Gb data were generated. The subsequent data analysis identified 5,265 DEGs between the populations (XJ/NX).
The DEG functional annotation and pathway enrichment analysis indicated that a considerable proportion of the DEGs involved in the pathways related to energy metabolism, development, and insecticide resistance. There were several clear differences in the metabolic pathways between the two populations. For example, the amino acid biosynthesis (Chen, 1966; Wilson et al., 2010), starch and sucrose metabolism (Kunieda et al., 2006), and fatty acid biosynthesis and metabolism (Crippsc and De Renobales, 1988; Tan et al., 2011) varied between the XJ and NX aphids. This diversity might be due to the differences in the environmental conditions in the two provinces. The DEGs and their associated pathways may provide clues regarding how the aphids adapted to various environmental factors, including temperature, light, humidity, and nutrient availability (Gade, 2004; Contreras and Bradley, 2009). The significant enrichment of development-related pathways, such as the glycosphingolipid biosynthesis pathway, may help clarify the developmental differences between the aphid populations (Wandall et al., 2005), although these differences might also be due to the environment. Among these XJ/NX DEGs, there were only seven coding transcription factors (TFs). Their information was listed in Table 4. The one up-regulated DEG is TCF4_12 encoding a transcription factor (homolog of the Drosophila melanogaster daughterless gene) (Caudy et al., 1988). The expression levels of the other transcription factor-encoding DEGs were down-regulated. Of these DEGs, POU3F is reportedly important for regulating the neuron-specific expression of the gene encoding the diapause hormone and pheromone biosynthesis-activating neuropeptide in Bombyx mori (Zhang et al., 2004).
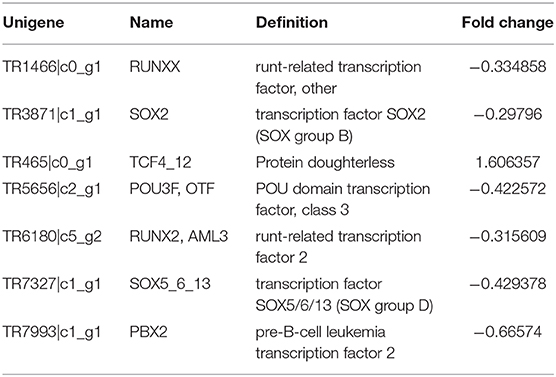
Table 4. Transcription factor coding DEGs.
The 30 most enriched KEGG pathways (Figure 3) included several associated with insecticide detoxification (e.g., metabolism of xenobiotics by cytochrome P450, drug metabolism – cytochrome P450, and drug metabolism – other enzymes) (Scott and Wen, 2001; Schwab, 2013). Previous studies revealed that the cytochrome P450 monooxygenase system (Berge et al., 1998; Wang et al., 2018; Zhang et al., 2018; Chiu et al., 2019) and carboxylesterases (Coppin et al., 2012; Zhang et al., 2014) help detoxify xenobiotics. In the current study, the insecticide treatments indicated that the NX aphids are substantially more tolerant to insecticides than the XJ aphids. To date, there is not yet transcriptomic research on M. dirhodum reported, neither any sequencing data available to the public. Accordingly, the transcriptome data generated in this study can enable researchers to further clarify M. dirhodum (aphid) gene functions and characterize the molecular mechanisms underlying M. dirhodum development, metabolism, adaptation, and insecticide resistance.
Conclusion
In the present study, two M. dirhodum populations collected from the main agricultural regions in western China underwent a transcriptome analysis. The resulting data suggest the genetic differences between the two aphid populations are primarily related to amino acid, sugar, and fatty acid metabolism as well as development. Moreover, the results also revealed differences in the cytochrome P450-related insecticide metabolism and detoxification. Toxicological experiment demostrated the insecticide resistance of the NX aphids was greater than that of the XJ aphids. These findings imply that the sequencing data can be relevant for future investigations regarding the classification, expression, and function of M. dirhodum detoxification enzyme genes that influence insecticide resistance. The data presented herein will be applicable for future functional genomics research and investigations on insect development, metabolism, environmental adaptation, and insecticide resistance. All the data generated in this study is free and available for any scientific research. The data has been uploaded to NCBI for releasing later (Accession numbers: SRR13447691, SRR13447690, SRR13447689, SRR13447688, SRR13447687, SRR13447686, SRR13447685).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject, accession no: PRJNA692554.
Author Contributions
HG, XZ, SZ, and FG conceived and designed the research. HG and XZ conducted the experiments. HG, XZ, and YS analyzed the data. HG and XZ wrote the manuscript. SZ, FG, GL, and EL revised the manuscript. All authors have read and approved the manuscript.
Funding
This study was sponsored by the National Key Research and Development program of China (2018YFD0200406), Tianshan Yonug Talents (2019Q051), SINOGRAIN II (CHN-17/0019): Technological Innovation to Support Environmentally-Friendly Food Production and Food Safety Under a Changing Climate-Opportunities and Challenges for Norway-China Cooperation, and the Technology Innovation and Key Cultivation Projects of Xinjiang Academy of Agricultural Sciences (xjkcpy-003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with one of the authors FG.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsufs.2021.639841/full#supplementary-material
References
Argandona, V. H., Luza, J. G., Niemeyer, H. M., and Corcuera, L. J. (1980). Role of hydroxamic acids in the resistance of cereals to aphids. Phytochemistry 19, 1665–1668. doi: 10.1016/S0031-9422(00)83790-5
Berge, J. B., Feyereisen, R., and Amichot, M. (1998). Cytochrome P450 monooxygenases and insecticide resistance in insects. Philosoph. Trans. Royal Soc. B 353, 1701–1705. doi: 10.1098/rstb.1998.0321
Carver, M. (2010). Metopolophium dirhodum (walker) (homoptera: aphididae) newly recorded from australia. Aust. J. Entomol. 23, 192–192. doi: 10.1111/j.1440-6055.1984.tb01944.x
Caudy, M., Vassin, H., Brand, M., Tuma, R., Jah, L. Y., and Jan, Y. N. (1988). daughterless, a Drosophila gene essential for both neurogenesis and sex determination, has sequence similarities to myc and the achaete-scute complex. Cell 55, 1061–1067. doi: 10.1016/0092-8674(88)90250-4
Chen, P. S. (1966). Amino acid and protein metabolism in insect development. Adv. Insect Phys. 3, 53–132. doi: 10.1016/S0065-2806(08)60186-1
Chen, X., Vosman, B., Visser, R. G. F., Der Vlugt, R. V., and Broekgaarden, C. (2012). High throughput phenotyping for aphid resistance in large plant collections. Plant Methods 8, 33–33. doi: 10.1186/1746-4811-8-33
Chen, X., Zhang, Z., Visser, R. G. F., Broekgaarden, C., and Vosman, B. (2013). Overexpression of IRM1 enhances resistance to aphids in Arabidopsis thaliana. PLoS ONE 8:e0070914. doi: 10.1371/journal.pone.0070914
Chiu, C. C., Keeling, C. I., and Bohlmann, J. (2019). Cytochromes P450 preferentially expressed in antennae of the mountain pine beetle. J. Chem. Ecol. 45, 178–186. doi: 10.1007/s10886-018-0999-0
Contreras, H. L., and Bradley, T. J. (2009). Metabolic rate controls respiratory pattern in insects. J. Exp. Biol. 212, 424–428. doi: 10.1242/jeb.024091
Coppin, C. W., Jackson, C. J., Sutherland, T. D., Hart, P. J., Devonshire, A. L., Russell, R. J., et al. (2012). Testing the evolvability of an insect carboxylesterase for the detoxification of synthetic pyrethroid insecticides. Insect Biochem. Mol. Biol. 42, 343–352. doi: 10.1016/j.ibmb.2012.01.004
Crippsc, C., and De Renobales, M. (1988). Developmental changes in fatty acid biosynthesis and composition in the house cricket, Acheta domesticus. Arch. Insect Biochem. Physiol. 9, 357–366. doi: 10.1002/arch.940090409
Devonshire, A. L., and Field, L. M. (1991). Gene amplification and insecticide resistance. Annu. Rev. Entomol. 36, 1–23. doi: 10.1146/annurev.en.36.010191.000245
Filho, J. P. D. L., and Paiva, E. A. S. (2006). The effects of sooty mold on photosynthesis and mesophyll structure of mahogany (Swietenia macrophylla King., Meliaceae). Bragantia 65, 11–17. doi: 10.1590/S0006-87052006000100003
Gade, G. (2004). Regulation of intermediary metabolism and water balance of insects by neuropeptides. Annu. Rev. Entomol. 49, 93–113. doi: 10.1146/annurev.ento.49.061802.123354
Garratt, M. P. D., Leather, S. R., and Wright, D. J. (2010). Tritrophic effects of organic and conventional fertilisers on a cereal-aphid-parasitoid system. Entomol. Exp. Appl. 134, 211–219. doi: 10.1111/j.1570-7458.2009.00957.x
Grabherr, M., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Kaloshian, I., and Walling, L. L. (2005). Hemipterans as plant pathogens. Annu. Rev. Phytopathol. 43, 491–521. doi: 10.1146/annurev.phyto.43.040204.135944
Kempema, L. A., Cui, X., Holzer, F. M., and Walling, L. L. (2006). Arabidopsis transcriptome changes in response to phloem-feeding silverleaf Whitefly Nymphs. Similarities and distinctions in responses to aphids. Plant Physiol. 143, 849–865. doi: 10.1104/pp.106.090662
Kunieda, T., Fujiyuki, T., Kucharski, R., Foret, S., Ament, S. A., Toth, A. L., et al. (2006). Carbohydrate metabolism genes and pathways in insects: insights from the honey bee genome. Insect Mol. Biol. 15, 563–576. doi: 10.1111/j.1365-2583.2006.00677.x
Powell, G., Tosh, C. R., and Hardie, J. (2006). Host plant selection by aphids: behavioral, evolutionary, and applied perspectives. Annu. Rev. Entomol. 51, 309–330. doi: 10.1146/annurev.ento.51.110104.151107
Rodriguezlopez, M. J., Garzo, E., Bonani, J. P., Fereres, A., Fernandezmunoz, R., and Moriones, E. (2011). Whitefly resistance traits derived from the wild tomato Solanum pimpinellifolium affect the preference and feeding behavior of Bemisia tabaci and reduce the spread of tomato yellow leaf curl virus. Phytochemistry 101, 1191–1201. doi: 10.1094/PHYTO-01-11-0028
Saska, P., Skuhrovec, J., Lukas, J., Chi, H., Tuan, S., and Honěk, A. (2016). Treatment by glyphosate-based herbicide alters life history parameters of the rose-grain aphid Metopolophium dirhodum. Sci. Rep. 6:27801. doi: 10.1038/srep27801
Schwab, M. (2013). Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Therap. 138, 103–141. doi: 10.1016/j.pharmthera.2012.12.007
Scott, J. G., and Wen, Z. (2001). Cytochromes P450 of insects: the tip of the iceberg. Pest Manag. Sci. 57, 958–967. doi: 10.1002/ps.354
Tan, L., Meesapyodsuk, D., and Qiu, X. (2011). Molecular analysis of Δ6 desaturase and Δ6 elongase from Conidiobolus obscurus in the biosynthesis of eicosatetraenoic acid, a ω3 fatty acid with nutraceutical potentials. Appl. Microbiol. Biotechnol. 90, 591–601. doi: 10.1007/s00253-010-3060-y
Taylor, S. H., Parker, W. E., and Douglas, A. E. (2012). Patterns in aphid honeydew production parallel diurnal shifts in phloem sap composition. Entomol. Exp. Appl. 142, 121–129. doi: 10.1111/j.1570-7458.2011.01206.x
Walling, L. L. (2008). Avoiding effective defenses: strategies employed by phloem-feeding insects. Plant Physiol. 146, 859–866. doi: 10.1104/pp.107.113142
Wandall, H. H., Pizette, S., Pedersen, J. W., Eichert, H., Levery, S. B., Mandel, U., et al. (2005). Egghead and brainiac are essential for glycosphingolipid biosynthesis in vivo. J. Biol. Chem. 280, 4858–4863. doi: 10.1074/jbc.C400571200
Wang, E., Wang, R., Deparasis, J., Loughrin, J. H., Gan, S., and Wagner, G. J. (2001). Suppression of a P450 hydroxylase gene in plant trichome glands enhances natural-product-based aphid resistance. Nat. Biotechnol. 19, 371–374. doi: 10.1038/86770
Wang, X., Chen, Y., Gong, C., Yao, X., Jiang, C., and Yang, Q. (2018). Molecular identification of four novel cytochrome P450 genes related to the development of resistance of Spodoptera exigua (Lepidoptera: Noctuidae) to chlorantraniliprole. Pest Manag. Sci. 74, 1938–1952. doi: 10.1002/ps.4898
Waterhouse, P. M., and Helms, K. (1985). Metopolophium dirhodum (Walker): a newly arrived vector of barley yellow dwarf virus in Australia. Aust. Plant Pathol. 14, 64–66. doi: 10.1071/APP9850064
Weintraub, P. G., and Beanland, L. (2006). Insect vectors of phytoplasmas. Annu. Rev. Entomol. 51, 91–111. doi: 10.1146/annurev.ento.51.110104.151039
Will, T., Kornemann, S. R., Furch, A. C. U., Tjallingii, W. F., and Van Bel, A. J. E. (2009). Aphid watery saliva counteracts sieve-tube occlusion: a universal phenomenon? J. Exp. Biol. 212, 3305–3312. doi: 10.1242/jeb.028514
Wilson, A., Ashton, P. D., Calevro, F., Charles, H., Colella, S., Febvay, G., et al. (2010). Genomic insight into the amino acid relations of the pea aphid, Acyrthosiphon pisum, with its symbiotic bacterium Buchnera aphidicola. Insect Mol. Biol. 19, 249–258. doi: 10.1111/j.1365-2583.2009.00942.x
Winder, L., Alexander, C. J., Holland, J. M., Woolley, C., and Perry, J. N. (2001). Modelling the dynamic spatio-temporal response of predators to transient prey patches in the field. Ecol. Lett. 4, 568–576. doi: 10.1046/j.1461-0248.2001.00269.x
Zhang, H., Zhao, M., Liu, Y., Zhou, Z., and Guo, J. (2018). Identification of cytochrome P450 monooxygenase genes and their expression in response to high temperature in the alligatorweed flea beetle Agasicles hygrophila (Coleoptera: Chrysomelidae). Sci. Rep. 8, 1–13. doi: 10.1038/s41598-018-35993-1
Zhang, J., Li, D., Ge, P., Guo, Y., Zhu, K. Y., Ma, E., et al. (2014). Molecular and functional characterization of cDNAs putatively encoding carboxylesterases from the migratory locust, Locusta migratoria. PLoS ONE 9:e94809. doi: 10.1371/journal.pone.0094809
Keywords: RNA-seq, Metopolophium dirhodum, insecticide, DEGs, resistance
Citation: Gao H, Zhu X, Li G, Liu E, Shen Y, Zhao S and Ge F (2021) RNA Sequencing Analysis of Metopolophium dirhodum (Walker) (Hemiptera: Aphididae) Reveals the Mechanism Underlying Insecticide Resistance. Front. Sustain. Food Syst. 5:639841. doi: 10.3389/fsufs.2021.639841
Received: 10 December 2020; Accepted: 08 February 2021;
Published: 11 March 2021.
Edited by:
Fang Ouyang, Chinese Academy of Sciences, ChinaReviewed by:
Wen Xie, Chinese Academy of Agricultural Sciences, ChinaLitao Guo, Chinese Academy of Agricultural Sciences, China
Copyright © 2021 Gao, Zhu, Li, Liu, Shen, Zhao and Ge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Ge, Z2V0QGlvei5hYy5jbg==; Sifeng Zhao, emhzZl9hZ3JAc2h6dS5lZHUuY24=
†These authors have contributed equally to this work