 Jie Wen1,2,3,4
Jie Wen1,2,3,4 Shanshan Liu1
Shanshan Liu1 Yuchun Wang1,2,3,4*
Yuchun Wang1,2,3,4* Mohd Yawar Ali Khan5Xuejian Zhou1,2
Mohd Yawar Ali Khan5Xuejian Zhou1,2 Shanze Li1,2,3,4
Shanze Li1,2,3,4 Yufei Bao1,2,3,4Xiaoyu Cui1,2Zhihua Huang1,2Meng Sun1,2Hanxiao He1,2
Yufei Bao1,2,3,4Xiaoyu Cui1,2Zhihua Huang1,2Meng Sun1,2Hanxiao He1,2- 1State Key Laboratory of Watershed Water Cycle Simulation and Regulation, China Institute of Water Resources and Hydropower Research, Beijing, China
- 2Department of Water Ecology and Environment, China Institute of Water Resources and Hydropower Research, Beijing, China
- 3Yangtze Three Gorges Reservoir Ecological Integrated Field Science Observation and Research Station, Ministry of Water Resources, Fengjie, China
- 4Yangtze River Ecological Environment Engineering Research Center, China Three Gorges Corporation, Wuhan, China
- 5Department of Hydrogeology, Faculty of Earth Sciences, King Abdulaziz University, Jeddah, Saudi Arabia
Introduction: Microorganisms are essential for cycling phosphorus and nitrogen and play a crucial role in maintaining the health and stability of river ecosystems. The reservoir operation changes the river's hydrological processes, forming reservoir inundation areas and influencing the diversity of microorganisms and their environmental functions.
Methods: To study the microbial composition and action mechanism in rivers affected by reservoir operation, Xiangxi River, the closest tributary to the Three Gorges Dam on the Yangtze River, was sampled to examine the spatiotemporal fluctuations of bacterial populations and functional genes in water and sediments. The physicochemical properties, microbial communities and functional genes were analyzed in August 2022 and June 2023.
Results: Spatially, except for conductivity, the chlorophyll (Chl), dissolved oxygen (DO), and pH values in the upper reaches of the basin were higher than those in the mouth (where it joins the Yangtze River). Specific physicochemical gradients created by the reservoir operation drove spatial and temporal shifts in bacterial community structure. In water samples (W), dominant microbial species included Exiguobacterium and Candidatus Fonsibacter, contributing to organic matter degradation and nutrient transformation. Nitrospira indicated their roles as nitrifiers or denitrifiers in sediment samples (S), essential for nitrogen cycling. In the mouth zone, Methyloceanibacter dominated in the transition zone, and they were involved in methane or organic metabolism.
Discussion: The dominance of Microcystis in the upstream region reflected its prevalence in nutrient-rich, algal-rich environments. Paralia in the middle of the river highlighted the favorable conditions of suitable light and moderate flow rates for diatom growth. Reservoir regulation also altered the functional gene composition, making it more similar to that found in lake ecosystems. The most abundant functional genes were those associated with Amino Acid Transport and Metabolism, while phosphorus-related genes predominantly involved energy production and conversion. The dominance of genes linked to electron transport underscored the pivotal role of microbial respiration and oxidative phosphorylation in energy metabolism, which was fundamental to ecosystem productivity.
1 Introduction
Microorganisms in water and sediment are closely involved in the phosphorus cycle, and their influence is complicated. In water bodies, microorganisms can decompose organic substances containing phosphorus, such as nucleic acids and phospholipids, and convert organic into inorganic phosphorus, further affecting the bioavailability of phosphorus (Caron, 1994; Van Mooy et al., 2009). Microorganisms can absorb dissolved phosphorus and participate in the phosphorus cycle through metabolic activities, thus affecting algae growth and algal blooms (Havens, 2008; Duhamel, 2025). Microorganisms in sediments participate in phosphorus transformation and cycling through various biogeochemical processes (Richardson et al., 2009). Phosphate-solubilizing microorganisms (PSMs) in sediments can dissolve insoluble phosphorus in sediments by secreting organic acids and other metabolites, converting them into forms that plants and other microorganisms can absorb and increasing the available phosphorus content in the water (Rawat et al., 2021; Sadiq et al., 2013).
Environmental factors affect the phosphorus cycle function genes in microorganisms: salinity, pH, and N/P ratio affect the characteristics of microbial communities and phosphorus metabolism processes in sediments (Jalali and Peikam, 2013). These affect the gene expression of phosphorus cycle function and the phosphorus biogeochemical cycle by influencing microbial communities’ structure and function (Zeglin, 2015). For example, the coupling mechanism of phosphorus components with bacterial and archaeal community succession in karst lake sediments in plateaus suggests that different active components of inorganic (Pi) and organic (Po) phosphorus have significant effects on the diversity and composition of microbial communities (Yuan et al., 2024). A study of the Danjiangkou Reservoir found that water temperature (Temp), redox potential, DO, and Chl-a were the main factors affecting the composition of the planktonic bacteria community. These environmental factors were significantly correlated with nitrogen and phosphorus cycle functional genes, resulting in the apparent vertical distribution of these genes in the planktonic bacteria (Chen et al., 2022).
Water conservancy project scheduling affects river habitats and the composition and function of river ecosystems. The construction of reservoirs has altered the natural flow characteristics of rivers, slowed the flow rate, and extended water retention times. These changes result in the enrichment of nutrients, such as nitrogen and phosphorus, which stimulate the rapid growth of algae, thus affecting water quality and the diversity of aquatic organisms (Zhu et al., 2022; Saunders, 2005). Reservoir regulation influences the concentration of nutrients in water bodies and modifies microbial community structure and ecological functions, particularly the release of endogenous phosphorus and its cycling (Dević, 2015). Microorganisms are involved in phosphorus mineralization, transformation, and absorption through phosphatase genes and phosphate transporter genes, while they also store phosphorus by synthesizing polyphosphates (Janati et al., 2023; Chen et al., 2022). These microorganisms play a critical role in the phosphorus cycle, and the expression of their functional genes is influenced by changes in the reservoir environment, thereby regulating the dynamic processes of phosphorus cycling (Chen et al., 2022; Wang et al., 2017). The response of river microbial communities to environmental changes may be more rapid because the hydrodynamic conditions of rivers are generally more dynamic (Wang et al., 2023). For example, there are significant differences in the composition of planktonic bacterial communities in river regions of the Yangtze River basin, in which Temp is the main influencing factor (Liu et al., 2018). Microbial communities may be more sensitive to long-term environmental changes in relatively static lakes because such environments are relatively stable (Liu et al., 2022). The archaea in lakes often depend on the type of lake, and the dominant microphyla vary significantly with different water bodies, sediments, and lake regions (Zhang et al., 2015). The backwater areas of rivers affected by reservoir operation tend to be lake-type environments (Xu et al., 2024). Studying the composition and functional characteristics of microbial communities in different regions is essential for understanding the characteristics of river-water ecological environments and taking adequate measures to protect ecological functions.
As a tributary significantly influenced by the operation of the Three Gorges Dam, the Xiangxi River faces a pronounced eutrophication issue with annual algal blooms. Analyzing microbial diversity and functional genes related to phosphorus cycling in the Xiangxi River Basin is crucial for elucidating the relationship between microorganisms and nutrient elements in eutrophic regions. This study addresses this issue by 1) investigating the distribution characteristics of physical and chemical parameters from the upper reaches to the mouth of the Xiangxi River Basin; 2) examining the differences and relationships of microbial diversity and phosphorus cycle-related functional genes across various media within the Xiangxi River Basin; 3) comparing the variations in microorganisms and functional genes in sediments from the upper reaches to the mouth of the Xiangxi River and analyzing the underlying causes of differences and the implications for river health.
2 Materials and Methods
2.1 Study area
The Xiangxi River basin is located near Yichang City, Hubei Province, approximately 29 km from the Three Gorges Dam and the first tributary upstream of it. The river originates in the Shennongjia Forest area in the northwest of Hubei Province. Positioned at the head of the Three Gorges reservoir area, the Xiangxi River basin is affected by the upper support of the main stream of the Yangtze River. The hydraulic interaction reduces flow velocity, resulting in lentic (lake-like) water characteristics. When the reservoir area is at the lowest water level of 145 m, a permanent backwater area of 27.6 km is formed above the mouth of the Xiangxi River, within which two important tributaries, the Gaolan and the Qilixia Rivers, flow into the Xiangxi. Over 70% of the basin’s population engages in agriculture, the primary economic activity (Xiang et al., 2022). The upstream Xingshan phosphate rock reserves are approximately 460 million tons and are one of the three major phosphate rock bases in China. The pollution sources of the Xiangxi River primarily are both natural and anthropogenic. Natural pollution arises from the dissolution of local phosphorus-rich strata that release phosphorus into the water (Mainstone and Parr, 2002). Anthropogenic pollution comes from various human activities, including emissions from mining, chemical industries, and other enterprises, as well as agricultural non-point source pollution, which introduces excess nutrients such as nitrogen and phosphorus into the Xiangxi River (Chakraborty, 2021; Xu et al., 2022). These combined pollution sources significantly impact the river’s water quality and ecological balance.
2.2 Sample collection
Based on the topography, geomorphology, and extent of the backwater area within the Xiangxi River basin, water samples (W), overlying water samples (O), and sediment samples (S) were collected from key points (river mouth, major tributary confluences, and backwater zone) in August 2022 and June 2023. August and June were chosen because they are the periods when algal blooms are obvious and severe. The sample points are labeled XX00–XX08 accordingly; the specific sampling points are shown in Figure 1.

Figure 1. Map of the study area including sampling locations.
2.3 Determination of physicochemical properties
A portable multi-parameter water quality instrument (multi-parameter water quality monitoring system, Xylem (China) Company Limited) was used to measure water Temp, Chl, DO, pH, and conductivity values from surface to bottom at nine sites (Figure 1). W, S, and O were collected from points XX00, XX02, XX03, XX05, XX06, and XX08.
Triplicate W and S were collected at each point and composited to ensure representativeness. W is followed by sampling year and site number (e.g., W1_00 denotes a sample collected in 2022 at site XX00).
A cylindrical sampling device developed by the Institute of Water Ecology and Environment of the China Institute of Water Resources and Hydropower Research (national invention patent No.: ZL200810056757.7) was used to collect S. For each point, S is followed by sample time, site number, and depth (e.g., S1_00_1 denotes the 0–3 cm layer collected in 2022 at XX00; layers 2 and 3 were 3–6 cm and 6–9 cm, respectively). For the XX05 site, where algal blooms occurred at high frequency, samples were conducted every 2 cm from the surface to the bottom.
Disturbed sediment was avoided as far as possible during the sampling process. Overlying water samples were extracted by a negative pressure diversion pipe and stored in sterile glass bottles.
Due to sampling process loss and the quality evaluation of DNA extraction, not all samples obtained from the sampling points met the requirements of metagenomic analysis. Only the samples that passed quality inspection were considered in this study. All W and O samples were filtered through a 0.22-μm membrane on the same day of sampling; the membrane was stored in a liquid nitrogen environment before microbial diversity and metagenomic detection.
2.4 DNA extraction and metagenome sequencing
DNA extraction was performed using E. Z.N.A.® Soil DNA Kit (Omega Bio-Tek, United States). After genomic DNA extraction, TBS-380 detected DNA concentration, DNA purity was detected by NanoDrop2000, and DNA integrity was detected by 1% agarose gel electrophoresis. DNA was segmented by Covaris M220 (Genetics, China) screening for fragments interrupted by approximately 400 bp. PE libraries were constructed using the NEXTFLEX Rapid DNA-Seq (Bioo Scientific, United States) library building kit. After bridge PCR amplification, metagenomic sequencing was performed using the Illumina NovaSeq/HiSeq Xten (Illumina, United States) sequencing platform.
The raw data were controlled by Fastp software, and contaminated reads with high similarity were removed by BWA software. MEGAHIT, a splicing software based on the succinct de Bruijn graphs principle, assembled the optimized sequences. Contigs ≥300 bp were selected as the final assembly result. MetaGene was used to predict the ORF of the assembled contig, and then CD-HIT software was used to cluster the predicted gene sequences of all samples to construct the non-redundant gene set. Finally, SOAPaligner software was used to align the high-quality reads of each sample with the set of non-redundant genes (95% identity) so that the abundant information of genes in the corresponding sample could be measured.
2.4.1 Taxonomic notes on species
The amino acid sequence of the non-redundant gene set was compared with the NR database (the expected e-value of the BLASTP comparison parameter setting was 1e-5), and species annotation was obtained from the taxonomic information database corresponding to the NR database. The abundance of the species was then calculated using the sum of the corresponding gene abundance of the species to obtain species annotation information at the taxonomic levels of domain, kingdom, phylum, class, order, family, genus, and species.
2.4.2 Functional annotation of COG
The amino acid sequence of the non-redundant gene set was compared with the eggNOG database using BLASTP to obtain the functional information of the COG corresponding to the gene. Then, the COG abundance in the sample was calculated using the sum of the gene abundance corresponding to COG.
2.4.3 KEGG function annotation
The amino acid sequences of the non-redundant gene set were compared with the KEGG (Kyoto Encyclopedia of Genes and Genomes) gene database using BLASTP to obtain the corresponding KEGG function information. The sum of gene abundances corresponding to KO, pathway, EC and module was used to calculate the abundances of corresponding functional categories. Based on the hierarchical annotation of the KEGG function pathway, genes related to the phosphorus cycle were selected, and a set of functional genes related to the phosphorus cycle was established using the P_metabolism gene screening function of Megi Biology.
2.5 Species and functional composition analysis
Based on the annotation results of different databases, the co-occurrence relationship maps were applied to describe the abundance correspondence between the sample and species/functions, thus assisting in understanding of the proportion of species/functions in different groups and showing which microorganisms and functions are in the group and their relative abundance. The similarity of species and functions in various samples was determined using a Venn diagram, and the difference of species or functional components in each sample/group were analyzed using the Kruskal–Wallis H test.
3 Results
3.1 Physicochemical properties
Figure 2 shows the water Temp, Chl, DO, pH, and conductivity values. In August 2022, the water Temp was 7–8°C higher than in June 2023, and the DO, pH, and conductivity values were also elevated in August compared to June. Spatially, the Chl, DO, and pH values in the upper reaches of the basin were higher than those in the mouth, while conductivity values were higher in the mouth than in the upper reaches. Surface water Temp, Chl, DO, pH, and conductivity were generally high and decreased with increasing water depth.

Figure 2. Physicochemical properties, including Temp, Chl, DO, pH, and conductivity, in the Xiangxi River. Figures to the left are samples collected in 2022 and to the right in 2023.
Surface Temp was highest in August 2022, the highest Temp was 31.39°C, and the bottom Temp was the lowest. The minimum Temp was 25.22°C (Figure 2A). In June 2023, the highest surface Temp was 24.08°C, and the lowest bottom Temp was 17.63°C (Figure 2B). In the horizontal direction, from the mouth to the upstream, the overall change of water Temp was not noticeable. In the vertical direction, in August 2022 the vertical water Temp decreased significantly in the mouth and upstream sections. In June 2023, the vertical water Temp of the upstream section decreased significantly.
In 2022, the Chl value in the middle reaches of the study area was higher than that in the river’s mouth and upper reaches, ranging from 0.29 ug/L to 15.62 ug/L (Figure 2C). In 2023, the upstream value was higher than in other regions. The highest value was in the XX05 section, which reached 120.36ug/L (Figure 2D).
The DO value in the study area varied greatly. In 2022, the highest DO value was located at XX07 and XX09 upstream, with a value of 15.83 mg/L. The value in the water layer below 5 m was below 6.6 mg/L (Figure 2E) (Figure 2E). In 2023, the distribution of DO values at each point was relatively concentrated, the upstream DO value was relatively high, and the surface DO value of XX09 was the highest at 12.36 mg/L (Figure 2F).
The pH values in the study area varied greatly, ranging from 7.73 to 9.7. In 2022, the values of each point were significantly different, with the highest value at XX08 and the lowest at XX01 (Figure 2G). In 2023, the pH values were relatively concentrated, all above 8.5. The surface layer was 5 m. The pH value was large. The maximum value was 9.55, located in the upstream XX09 point, and the minimum value was 8.5 at XX03 (Figure 2H).
The conductivity values of the mouth zone in 2022 and 2023 had an extensive fluctuation range. In 2022, from the mouth zone to site XX04, the conductivity value was concentrated at 450 us/cm, and the upstream was relatively low, mainly concentrated at 380 us/cm (Figure 2I). In 2023, at position XX00–XX04 near the mouth zone, the conductivity value was mainly concentrated at 450 us/cm, and the upstream fluctuated greatly, ranging 266.3–455 us/cm (Figure 2J).
3.2 Microbial community
The microbial composition of the study area was quite rich. According to the results of species and abundance information of different taxonomic levels in the NR database, the most abundant species were Betaproteobacteria, Actinomycetia, and Alphaproteobacteria, followed by Gammaproteobacteria and Deltaproteobacteria. Lu et al. (2020) analyzed spatial variation in bacterial biomass in the Xiangxi River and found that Flavobacteriia, Betaproteobacteria, and Acidobacteria were abundant in the river.
Figure 3 compares the similarity of microbial populations classified by genes in samples of different groups. Compared with 2023, microbial populations in 2022 had 77.35% identical genes, 5.49% unique genes in 2022, and 17.16% unique genes in 2023. The results of microbial gene level richness in W, O, and S showed that all samples shared 49.94% of the genes, 1.42% of the samples were unique to W, 4.39% were unique to O, and 17.08% were unique to S. The differences in microbial genes in different sampling points from mouth to upstream were compared. Among them, 39.39% of microbial genes were standard to all sites. It was found that S_05 had the highest proportion, accounting for 9.59%, followed by S_02, accounting for 1.95%, and S_00, accounting for 0.79%. Comparing the proportion of microbial gene diversity from surface to bottom S at site XX05, the common genes accounted for 32.37%, the relatively high genes were in the surface layer and S_05_06, accounting for 4.37% and 6.45%, respectively, and the proportion of genes in other layers was similar.
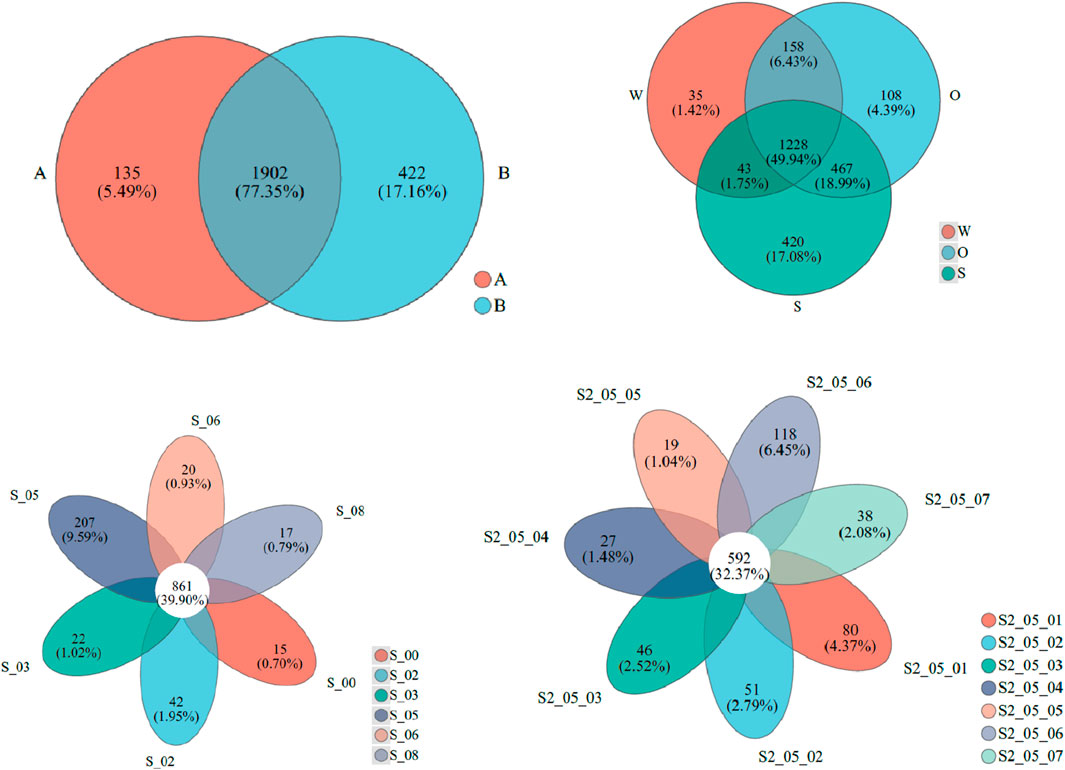
Figure 3. Similarity analysis results of microbial populations classified by gene in samples of different groups. A and B represent samples collected in 2022 and 2023, respectively, S_00 to S_08 were sediment samples (S) collected in XX00 to XX08, and S2_05_01 to S2_05_07 were S collected in XX05 from the surface to bottom layer.
3.3 Functional gene abundance
Compared to the Clusters of Orthologous Groups (COG) database, the highest proportions of functional genes in all samples collected from the basin were classified under categories E (amino acid transport and metabolism), C (energy production and conversion), and R (general function prediction) in the original data. After screening, the functional gene categories most related to the phosphorus cycle were C (energy production and conversion), G (carbohydrate transport and metabolism), and I (lipid transport and metabolism) (Figure 4). The results revealed that the highest microbial abundance in W and S was associated with amino acid transport, protein decomposition, and synthesis. At the same time, the primary function involved in phosphorus metabolism was energy production and conversion.
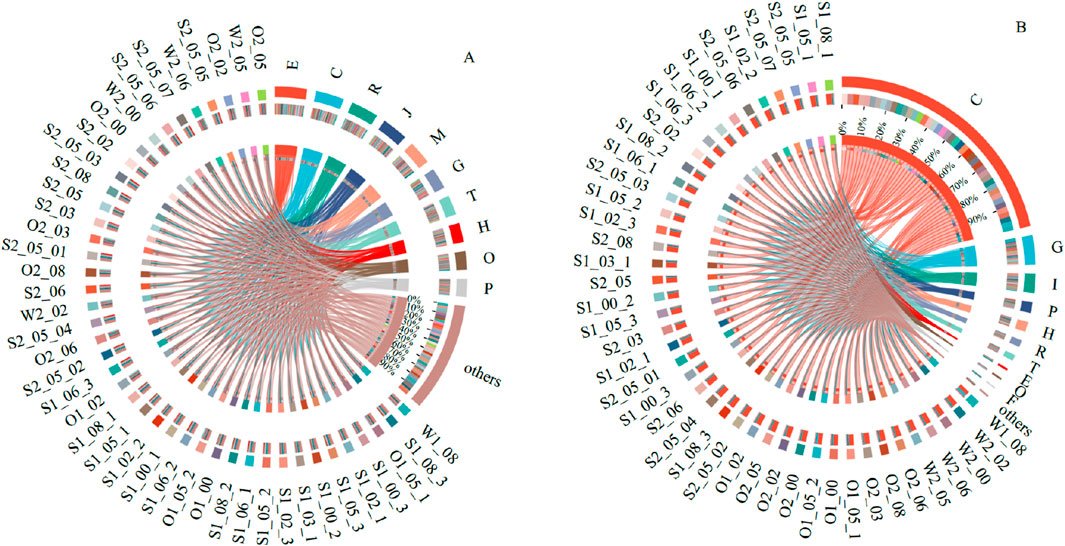
Figure 4. Top-ten function gene abundance in origin gene set (A) and phosphorus-cycle-related gene set (B).
To further analyze the distribution characteristics of phosphorus cycle functional genes, their proportion related to the phosphorus cycle in the study area was compared with the KEGG database. The results showed that the most abundant functional genes were K00341, K00342, and K02274 followed by K00335, K02111, and K02112. The most abundant gene names were nuoL, nuoM, and coxA, followed by ctaD, nuoF, and ATPF1A. K00341 (nuoL), K00342 (nuoM), and K02274 (coxA) are essential genes in the oxidative phosphorylation pathway. K00341 and K00342 encode subunits of nicotinamide adenine dinucleotide (reduced form, NADH): ubiquinone oxidoreductase (Complex I). K02274 encodes a subunit of cytochrome c oxidase (Complex IV). These proteins are critical for generating a proton gradient across the inner mitochondrial membrane through electron transport which drives adenosine triphosphate (ATP) synthesis. K02112 (ATP5A) directly catalyzes the phosphorylation of ADP (adenosine diphosphate) to ATP, forming high-energy phosphate bonds, and represents a central component of ATP synthase (Complex V) in the oxidative phosphorylation process (Chadwick et al., 2018).
Comparing functional gene similarity in 2023 and 2022, 88.45% of the functional genes were identical, 2.82% of the particular genes were in 2022, and 8.73% were in 2023. The results of functional gene richness in W, O, and S showed that all samples shared 85.63% of functional genes, 1.41% were unique to W, 0.85% were unique to O, and 2.25% were unique to S. The differences of functional genes in different sampling points from the mouth to upstream were compared. Of functional genes, 69.49% were common to all sites. It was found that the proportion of unique genes in S_05 was the highest, accounting for 4.23%, followed by S_02 and S_03, accounting for 1.81%, and S_00 being relatively low. The proportion of microbial functional gene diversity was compared across the surface to bottom S at site XX05, where the common genes accounted for 62.67%. In S_05_01 and S_05_02, the unique genes accounted for 4.45% and 1.71%, respectively, and the proportion was similar in other layers.
3.4 Spatial distribution of microbial community and functional genes
The distribution of microorganisms in different media environments was significantly different. The most abundant microorganisms included Exiguobacterium, Candidatus Fonsibacter, and Clavibacter, which were mainly distributed in water samples, followed by overlying water samples, and they were less distributed in sediment samples (Figure 5A). In addition to the three microorganisms mentioned above, Limnohabitans and Macrococcus occupied a high proportion of O. Nitrospira, Rubrivivax, and Nocardioides had many microorganisms in S.

Figure 5. Kruskal–Wallis H test bar plots. (A) Comparison of significant differences in microbial distribution among W, O, and S samples. (B) Comparison of microbial distribution in S from different locations of mouth zone. (C) Comparison of microbial function among W, O, and S samples. (D) Comparison of microbial function in S from different locations of mouth zone. (E) Comparison of functional genes in W, O, and S samples. (F) Comparison of functional gene differences in S from different locations of mouth zone.
The spatial distribution of microbial communities in S of the Xiangxi River, from the mouth zone to upstream regions, revealed distinct ecological patterns and reflected the underlying mechanisms driving these changes. The microbial species composition showed significant variations across different locations (Figure 5B), with Nitrospira, Methyloceanibacter, and Microcystis being the most abundant species in S. Nitrospira was the dominant species in the mouth zone S but decreased in abundance upstream. At the same time, Microcystis, Nocardioides, and Methyloceanibacter were more prevalent in the upstream region. The middle of the river was characterized by a higher diversity of species, including Paralia, Methanoregula, and Coscinodiscus. These shifts in microbial community structure from the mouth to upstream S were driven by environmental gradients and ecological functions that reflected the distinct conditions and processes in each area.
The main functional genes in W, O, and S were functions C, G, and 1: lipid transport and metabolism, followed by P: inorganic ion transport and metabolism and H: coenzyme transport and metabolism. In the three mediators, the difference in functional genes reached a very significant level. The dominant functions in W and S were functions G and C, respectively (Figure 5C).
The main functional genes collected from different spatial locations were functions G, 1: lipid transport and metabolism, P: inorganic ion transport and metabolism, followed by R: general function prediction, T: signal transduction mechanism, and E (Amino acid transport and metabolism). The differences in functional genes in different spatial locations were significant, and the differences in functional genes in the top four reached a significant level (Figure 5D).
The high richness of general function prediction genes in the middle of the river introduced an aspect of functional plasticity within microbial communities in these areas (Figure 5E). According to the KEGG database, the most abundant functional genes were nuoL (NADH-quinone oxidoreductase subunit L), nuoM (NADH-quinone oxidoreductase subunit M), and coxA ctaD (cytochrome c oxidase subunit I). The primary function was oxidative phosphorylation. Other abundant functional genes were ATPF1A atpA (F-type H+/Na+-transporting ATPase subunit alpha), ppk1 (polyphosphate kinase, RNA degradation; oxidative phosphorylation), and sdhA frdA (succinate dehydrogenase flavoprotein subunit, metabolic pathways). For nuoL, nuoM, coxA ctaD (cytochrome c oxidase subunit I), ATPF1A atpA, and sdhA frdA, abundance in S was richer than in O and W samples. For the functional gene ppk1, the abundance was higher in W and O than in S.
In the mouth zone environment, reservoir operations had a greater influence, and the higher availability of organic matter and oxygen supported more active aerobic respiration. This process was facilitated by the electron transport chain (ETS), with key genes such as nuoL, coxA, ctaD, ATPF1B, and atpD involved in ATP production and energy conversion (Figure 5F). These genes were part of the NADH: ubiquinone oxidoreductase complex (complex I), which was critical for energy production in aerobic conditions (Brandt, 2006). The high abundance of these functional genes in the mouth zone indicated that microorganisms in these areas relied heavily on aerobic processes to metabolize organic matter and convert organic phosphorus into inorganic forms—a key step in phosphorus cycling. Thus, mouth zone microbial communities played an active role in phosphorus mineralization, transforming organic phosphorus into the bioavailable inorganic phosphate essential to primary productivity (Lin et al., 2024).
Figure 6A shows the richness of functional genes in different microorganisms. Function C accounted for the highest proportion of functional genes among the top ten microorganisms. Unlike other microorganisms, the highest functional genes in Bacilli were function G. Among them, carbohydrate transport and metabolism function in W and O was higher than in S.
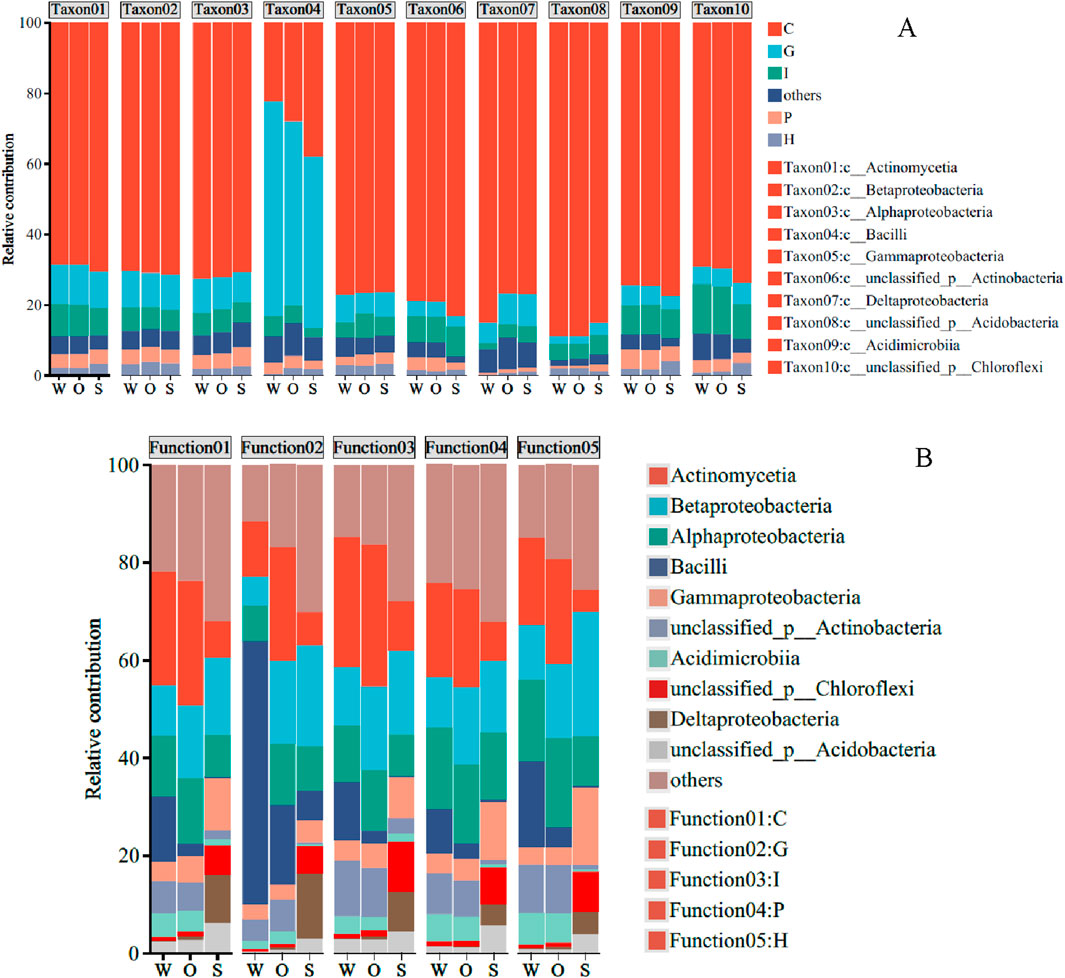
Figure 6. Functional distributions for specific microorganisms in W, O, and S samples (A), and comparison of differences in microbial abundance corresponding to specific functions in W, O, and S samples (B).
For the specific functional gene, the microbial composition in W, O, and S was significantly different (Figure 6B). For functional gene C, Gammaproteobacteria and Betaproteobacteria occupied the highest proportion in S, while Actinomycetia and Betaproteobacteria occupied the highest proportion in W and O. Bacilli had the highest proportion in W for gene function G, followed by Alphaproteobacteria. The microorganisms with the highest proportion in O were Actinomycetia, Betaproteobacteria, and Alphaproteobacteria. Gammaproteobacteria and Betaproteobacteria occupied the highest proportion in S. For functional genes 1, P, and H, Gammaproteobacteria and Betaproteobacteria occupied the highest proportion in S and Gammaproteobacteria and Actinomycetia occupied the highest proportion in W and O.
4 Discussion
4.1 Spatial differentiation of microbial communities and their responses to environmental gradients
The predominance of Exiguobacterium, Candidatus Fonsibacter, and Clavibacter in W underscores the importance of these microorganisms in environments rich in organic matter and nutrients. Exiguobacterium, known for thriving in nutrient-rich, low-oxygen conditions (García et al., 2025), shares functional significance with Candidatus Fonsibacter and Clavibacter. These latter genera exhibit metabolic versatility and degrade complex organic compounds, suggesting crucial roles in organic matter processing within these systems (Kumari and Das, 2023).
The high proportions of Limnohabitans, Macrococcus, and Methylobacterium in O reflect this zone’s dynamic nature. Limnohabitans, associated with freshwater organic matter degradation, likely responds to organic inputs from algal blooms or other sources (Ren et al., 2023). Macrococcus and Methylobacterium, key players in carbon and nitrogen cycling (Li et al., 2018), further highlight the importance of heterotrophic metabolism. The enrichment of these microorganisms suggests that O acts as a transitional zone for active organic matter processing before transport to S.
Nitrospira, Rubrivivax, and Nocardioides dominated sedimentary environments. Nitrospira, a key nitrifier, thrives in the low-oxygen conditions typical of sediments (Daims et al., 2011). Rubrivivax utilizes diverse organic and inorganic compounds, contributing to organic matter decomposition, while Nocardioides participates in denitrification and aromatic compound degradation (Yi et al., 2022). This microbial dominance reflects specialized metabolic strategies essential for survival in nutrient-rich, low-oxygen sediments.
Nitrospira was the most abundant genus within the mouth zone, emphasizing its critical role in the nitrogen cycle via nitrite oxidation (Daims et al., 2015). Its prevalence indicates that sediment samples in the mouth zone significantly influence nitrogen cycling, nutrient dynamics, and ecosystem function. Methyloceanibacter, abundant here and adapted to the nutrient-rich mouth zone, utilizes diverse carbon substrates (Walker, 2022; Pandey and Pande, 2023). The predominance of these species underscores the importance of nutrient availability and hydrodynamic conditions in creating a favorable environment for microbial growth and transformation.
In contrast, upstream sediment samples were dominated by Microcystis, Nocardioides, and Methyloceanibacter. The high abundance of the cyanobacterium Microcystis signals potential productivity, likely linked to increased nutrient inputs from land use or pollution that fuel algal blooms (Wurtsbaugh et al., 2019). The prevalence of Nocardioides (involved in organic matter degradation) and Methyloceanibacter reflects a shift in ecological functions as communities transition from the dynamic mouth to more stable upstream regions. This suggests that upstream sediment communities focus more on organic matter breakdown and are less influenced by tidal and salinity fluctuations (Morrissey et al., 2014).
Mid-river S exhibited notably higher microbial diversity, with Paralia, Methanoregula, and Coscinodiscus being abundant. The diatom Paralia, essential in siliceous phytoplankton communities, contributes to primary productivity and nutrient cycling; its abundance suggests favorable light and moderate flow conditions. Methanoregula, a methanogenic archaeon (Lyu et al., 2018), indicates active anaerobic processes like methane production. The presence of the diatom Coscinodiscus further emphasizes the role of diatoms in nutrient cycling and productivity at S, driven by specific hydrological and organic inputs.
Overall, the composition of these microbial communities reflects distinct ecological functions and environmental conditions at each spatial scale. The transition from the nutrient-rich mouth to the stable upstream regions involves microbial composition and functionality shifts: mouth zone communities focus on nitrogen cycling and carbon metabolism, upstream communities on organic matter degradation, and mid-river communities support diverse processes, including anaerobic metabolism and primary productivity. This diversity mirrors the hydrological and ecological variability across the river’s regions.
4.2 Spatial differentiation of functional genes and their responses to environmental gradients
Functional patterns aligned with expectations, as heterotrophic bacteria rapidly utilize dissolved organic matter for energy in aquatic environments (Wetzel, 2003). The higher proportion of genes dedicated to carbohydrate metabolism in W indicates enhanced metabolic potential and nutrient regeneration, supporting aquatic ecosystem productivity (Reddy et al., 2019).
Sediment samples exhibited the highest proportion of energy production and conversion genes, reflecting their role as microbial reservoirs where anaerobic processes (e.g., fermentation and sulfate reduction) predominate (Mallik et al., 2024). This elevated energy production potential likely stems from accumulated organic matter, which fosters diverse microbial activity that utilizes available substrates (Arndt et al., 2013).
Genes for lipid transport/metabolism (I), inorganic ion transport/metabolism (P), and coenzyme transport/metabolism (H) were high in proportion in both W and S. This dual prevalence underscores the importance of lipids as energy stores and membrane components—critical for growth under nutrient limitation (Rajpurohit and Eiteman, 2022)—and highlights the role of ion exchange in maintaining cellular homeostasis across habitats (Adhikary et al., 2024).
In upstream S, the high expression of carbohydrate transport/metabolism genes (G) suggests abundant organic carbon sources, facilitating robust microbial growth and diversity (Yu et al., 2023). This aligns with freshwater sediments being organic matter hotspots that promote carbohydrate degradation (Raza et al., 2023). Concurrently, the prevalence of genes P emphasizes nutrient cycling’s importance in these high-nutrient environments (Xu et al., 2018).
Mouth zone S showed a predominance of lipid transport/metabolism (I) and signal transduction (T) genes, indicating microbial adaptation to fluctuating salinity and nutrients. Lipids serve dual structural and energy-storage roles vital in dynamic environments (Beney and Gervais, 2001), while signal transduction mechanisms enable responsive regulation to environmental changes and resource competition (Crump and Bowen, 2024).
The dominance of oxidative phosphorylation genes (e.g., nuoL and coxA) reflects adaptation to anaerobic/microaerophilic sediment conditions, where electrons from NADH drive ATP synthesis (Yang and Chen, 2021). This is particularly advantageous given sediment oxygen fluctuations (Lasaga and Ohmoto, 2002). The higher abundance of these genes in S than W underscores the enhanced metabolic capacity of sedimentary microbes.
Elevated ATPF1A (encoding an ATP synthase subunit) indicates reliance on chemiosmotic gradients for ATP production, consistent with ATP synthase’s role in variable energy landscapes (Nirody et al., 2020). High sdhA (succinate dehydrogenase) expression further reveals complex metabolic networks that process carbon substrates via pathways like the citrate cycle (Pan et al., 2023).
The ppk1 gene (involved in polyphosphate metabolism and RNA degradation) was more abundant in W and O than S. This suggests roles in nutrient cycling and organic matter breakdown in dynamic environments, potentially facilitating polyphosphate accumulation and energy recycling (Tully and Ryals, 2017; Duhamel, 2025).
Upstream, decreased electron transport system (ETS) gene abundance implies a metabolic shift toward less oxygen-dependent pathways (e.g., fermentation and denitrification) suited to low-oxygen, organic-poor conditions (Hollingham, 2014). Such anaerobic adaptations in nutrient-scarce sediments can affect phosphorus cycling: denitrification may trigger internal phosphorus release from sediments, exacerbating eutrophication (Sun et al., 2022).
Functional gene distribution illustrates microbial adaptation to reservoir-induced environmental gradients. Mouth zone S with higher oxygen and nutrients supports communities optimized for aerobic respiration and efficient phosphorus cycling. Conversely, oxygen/nutrient-limited upstream S hosts anaerobically adapted communities that alter phosphorus dynamics through sediment release (Parsons et al., 2017).
This study highlights the central role of electron transport chain genes in aerobic microbial phosphorus cycling and demonstrates the metabolic flexibility of microbial communities in responding to reservoir-induced changes. Such adaptability is crucial for understanding phosphorus cycle regulation in impacted river ecosystems (Duhamel, 2025).
4.3 Interaction characteristics of microbial species and gene functional composition
The results highlight the fundamental role of energy metabolism across diverse microbial communities, which is essential for maintaining ecological balance and supporting biomass production in varying environments (Alvarenga et al., 2013). The prominence of energy metabolism genes across microorganisms suggests that these energy acquisition strategies are critical drivers of microbial diversity and community structure.
For bacilli, carbohydrate transport and metabolism genes were the most abundant functional category. This prevalence likely reflects their adaptation to organic-rich environments like sediments or water bodies (Trabucho Alexandre, 2015), suggesting a pivotal role in complex organic matter degradation and nutrient cycling.
G genes were more pronounced in W and O than in S. This aligns with the understanding that dissolved organic matter in aquatic environments is readily bioavailable (Dittmar et al., 2021). The rapid turnover of organic materials in these environments creates an enriched substrate pool, catalyzing higher metabolic activity. Consequently, this pattern indicates functional specialization within microbial communities for efficient resource utilization and dynamic ecosystem processes (Louca et al., 2018).
The disparity in functional gene abundance between S and W emphasizes the importance of habitat-specific adaptations. Sediments often serve as nutrient sinks with complex carbon sources requiring specialized degradation pathways (Leithold et al., 2016), while microorganisms in water access more readily bioavailable carbon, enhancing carbohydrate metabolism functions. This dynamic interaction underscores the importance of understanding spatial variations in microbial functional potential to predict ecosystem responses to environmental changes.
The distinct microbial compositions associated with specific functional genes across W, O, and S samples reflect nuanced community–environment interactions. This reveals significant differences in microbial diversity and functional gene representation, highlighting the importance of habitat-specific adaptations in shaping metabolic capabilities.
Regarding functional gene C, Gammaproteobacteria and Betaproteobacteria predominated in S. These groups are typically associated with anaerobic/microaerophilic sediment conditions, facilitating diverse energy utilization through processes like sulfate reduction and denitrification (Qian et al., 2019; Zhang et al., 2022). Their ability to occupy low-oxygen niches enhances their significance in sediment biogeochemistry.
In contrast, Actinomycetia and Betaproteobacteria were more represented in W and O, suggesting a functional shift influenced by physicochemical properties. Actinomycetia, known for degrading complex organic compounds, may be critical to nutrient cycling within the water column (Mallik et al., 2024). Their presence alongside Betaproteobacteria—involved in organic matter degradation and ammonia oxidation—emphasizes that microbial interactions regulate aquatic nutrient dynamics (Hui et al., 2022).
Bacilli showed the highest proportion of functional gene G in water samples, highlighting their role in processing readily available organic substrates. Alphaproteobacteria, the second most abundant group, further indicates specialization in carbohydrate metabolism within aquatic nutrient cycles. The higher representation of Actinomycetia, Betaproteobacteria, and Alphaproteobacteria in overlying water samples underscores the importance of functional diversity and their potentially facilitating efficient organic matter turnover (Bergauer et al., 2018).
For functional genes I, P, and H, the dominance of Gammaproteobacteria and Betaproteobacteria in sediment samples highlights their diverse metabolic potentials. These groups exhibit adaptability in sediment biogeochemistry processes like lipid metabolism and ion transport (Huettel et al., 2014). Conversely, a higher abundance of Gammaproteobacteria and Actinomycetia in W/O suggests a shift toward dynamic nutrient availability interactions.
Overall, the significant differences in microbial community composition across various habitats reveals a complex interplay of environmental factors driving the functional capacities of these microorganisms. Understanding these dynamics is critical for predicting ecosystem responses to changes in nutrient inputs and environmental conditions. Further research should focus on the specific ecological roles of these microbial communities and their functional genes in various aquatic systems, which might provide deeper insights into their contributions to biogeochemical cycling.
5 Conclusion
Dominant microbial species, including Exiguobacterium and Candidatus Fonsibacter, were found to contribute to organic matter degradation and nutrient transformation, while Nitrospira played a key role in nitrogen cycling as nitrifiers or denitrifiers. The mouth zone, enriched in nutrients, supported Methyloceanibacter, which was involved in methane or organic-matter metabolism. Microcystis dominated in the upstream region, indicative of nutrient-rich, algal-rich conditions. In the middle of the river, Paralia thrived due to favorable light and flow conditions.
Reservoir regulation altered functional gene composition, making it more like that of lake ecosystems. Genes related to amino acid transport and metabolism were most abundant. Phosphorus-related genes are primarily involved in energy production and conversion, highlighting the crucial role of microbial respiration and oxidative phosphorylation in supporting ecosystem productivity.
This study has demonstrated the effects of reservoir-induced changes on microbial diversity, suggesting impacts on microbial processes and nutrient cycling in river systems downstream from a large dam. In terms of limitations, temporal sampling during only 2 months may not fully capture the annual variability in microbial dynamics, and more frequent and extended sampling would provide a better understanding of seasonal changes. Secondly, the functional roles of microbial species, particularly in phosphorus cycling and their interactions within the community, were not fully explored. A deeper analysis of gene–environmental correlations would enhance the understanding of microbial functions and the implications for protecting river water quality and ecological conditions below dams.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Author contributions
JW: Data curation, funding acquisition, methodology, software, writing – original draft, and writing – review and editing. SsL: Data curation, formal analysis, investigation, methodology, supervision, and writing – original draft. YW: Conceptualization, funding acquisition, resources, and writing – original draft. MK: Supervision, validation, and writing – review and editing. XZ: Data curation, investigation, software, and writing – original draft. SzL: Software, supervision, validation, and writing – original draft. YB: Project administration, supervision, and writing – original draft. XC: Supervision, validation, and writing – original draft. ZH: Supervision, validation, and writing – original draft. MS: Data curation, investigation, and writing – original draft. HH: Data curation, investigation, and writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (42107283), the IWHR Research and Development Support Program (WE110145B0012024), and the Project of China Three Gorges Corporation (No. 201903144).
Conflict of interest
Authors JW, YW, SL, and YB were employed by China Three Gorges Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adhikary, S., Saha, J., Dutta, P., and Pal, A. (2024). Bacterial homeostasis and tolerance to potentially toxic metals and metalloids through diverse transporters: metal-specific insights. Geomicrobiol. J. 41 (5), 496–518. doi:10.1080/01490451.2024.2340517
Alvarenga, R. A. F., Dewulf, J., and Van Langenhove, H. (2013). A new natural resource balance indicator for terrestrial biomass production systems. Ecol. Indic. 32, 140–146. doi:10.1016/j.ecolind.2013.03.029
Arndt, S., Jørgensen, B. B., LaRowe, D. E., Middelburg, J., Pancost, R., and Regnier, P. (2013). Quantifying the degradation of organic matter in marine sediments: a review and synthesis. Earth-science Rev. 123, 53–86. doi:10.1016/j.earscirev.2013.02.008
Beney, L., and Gervais, P. (2001). Influence of the fluidity of the membrane on the response of microorganisms to environmental stresses. Appl. Microbiol. Biotechnol. 57, 34–42. doi:10.1007/s002530100754
Bergauer, K., Fernandez-Guerra, A., Garcia, J. A. L., Sprenger, R. R., Stepanauskas, R., Pachiadaki, M. G., et al. (2018). Organic matter processing by microbial communities throughout the Atlantic water column as revealed by metaproteomics. Proc. Natl. Acad. Sci. 115 (3), E400–E408. doi:10.1073/pnas.1708779115
Brandt, U. (2006). Energy converting NADH: quinone oxidoreductase (complex I). Annu. Rev. Biochem. 75 (1), 69–92. doi:10.1146/annurev.biochem.75.103004.142539
Caron, D. A. (1994). Inorganic nutrients, bacteria, and the microbial loop. Microb. Ecol. 28, 295–298. doi:10.1007/bf00166820
Chadwick, G. L., Hemp, J., Fischer, W. W., and Orphan, V. J. (2018). Convergent evolution of unusual complex I homologs with increased proton pumping capacity: energetic and ecological implications. ISME J. 12 (11), 2668–2680. doi:10.1038/s41396-018-0210-1
Chakraborty, S. K. (2021). River pollution and perturbation: perspectives and processes. Riverine ecology. Biodivers. Conservation, Conflicts Resolut. 2, 443–530. doi:10.1007/978-3-030-53941-2_5
Chen, Z. J., Liu, Y. Q., Li, Y. Y., Lin, L. A., Zheng, B. H., Ji, M. F., et al. (2022). The seasonal patterns, ecological function and assembly processes of bacterioplankton communities in the danjiangkou reservoir, China. Front. Microbiol. 13, 884765. doi:10.3389/fmicb.2022.884765
Crump, B. C., and Bowen, J. L. (2024). The microbial ecology of estuarine ecosystems. Annu. Rev. Mar. Sci. 16 (1), 335–360. doi:10.1146/annurev-marine-022123-101845
Daims, H., Lebedeva, E. V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., et al. (2015). Complete nitrification by nitrospira bacteria. Nature 528 (7583), 504–509. doi:10.1038/nature16461
Daims, H., Lücker, S., Paslier, D. L., and Wagner, M. (2011). Diversity, environmental genomics, and ecophysiology of nitrite-oxidizing bacteria. In: Nitrification. Hoboken, New Jersey: Wiley. p. 295–322.
Dević, G. (2015). Environmental impacts of reservoirs. Environ. Indic., 561–575. doi:10.1007/978-94-017-9499-2_33
Dittmar, T., Lennartz, S. T., Buck-Wiese, H., Hansell, D. A., Santinelli, C., Vanni, C., et al. (2021). Enigmatic persistence of dissolved organic matter in the ocean. Nat. Rev. Earth and Environ. 2 (8), 570–583. doi:10.1038/s43017-021-00183-7
Duhamel, S. (2025). The microbial phosphorus cycle in aquatic ecosystems. Nat. Rev. Microbiol. 23 (4), 239–255. doi:10.1038/s41579-024-01119-w
García, G. J. Y., Badotti, F., Ferreira-Silva, A., da Cruz Ferraz Dutra, J., Martins-Cunha, K., Gomes, R. F., et al. (2025). Microbial diversity of the remote trindade island, Brazil: a systematic review. PeerJ 13, e19305. doi:10.7717/peerj.19305
Havens, K. E. (2008). Cyanobacteria blooms: effects on aquatic ecosystems. Cyanobacterial harmful algal blooms state Sci. Res. needs 619, 733–747. doi:10.1007/978-0-387-75865-7_33
Hollingham, M. (2014). Effect of organic carbon substrates on denitrification rates in sediment. Waterloo, Ontario: University of Waterloo.
Huettel, M., Berg, P., and Kostka, J. E. (2014). Benthic exchange and biogeochemical cycling in permeable sediments. Annu. Rev. Mar. Sci. 6 (1), 23–51. doi:10.1146/annurev-marine-051413-012706
Hui, C., Li, Y., Zhang, W., Zhang, C., Niu, L., Wang, L., et al. (2022). Modelling structure and dynamics of microbial community in aquatic ecosystems: the importance of hydrodynamic processes. J. Hydrology 605, 127351. doi:10.1016/j.jhydrol.2021.127351
Jalali, M., and Peikam, E. N. (2013). Phosphorus sorption–desorption behaviour of river bed sediments in the abshineh river, hamedan, Iran, related to their composition. Environ. Monit. Assess. 185, 537–552. doi:10.1007/s10661-012-2573-5
Janati, W., Bouabid, R., Mikou, K., Ghadraoui, L. E., and Errachidi, F. (2023). Phosphate solubilizing bacteria from soils with varying environmental conditions: occurrence and function. Plos one 18 (12), e0289127. doi:10.1371/journal.pone.0289127
Kumari, S., and Das, S. (2023). Bacterial enzymatic degradation of recalcitrant organic pollutants: catabolic pathways and genetic regulations. Environ. Sci. Pollut. Res. 30 (33), 79676–79705. doi:10.1007/s11356-023-28130-7
Lasaga, A. C., and Ohmoto, H. (2002). The oxygen geochemical cycle: dynamics and stability. Geochimica Cosmochimica Acta 66 (3), 361–381. doi:10.1016/s0016-7037(01)00685-8
Leithold, E. L., Blair, N. E., and Wegmann, K. W. (2016). Source-to-sink sedimentary systems and global carbon burial: a river runs through it. Earth-Science Rev. 153, 30–42. doi:10.1016/j.earscirev.2015.10.011
Li, Y., Tang, K., Zhang, L., Zhao, Z., Xie, X., Chen, C. T. A., et al. (2018). Coupled carbon, sulfur, and nitrogen cycles mediated by microorganisms in the water column of a shallow-water hydrothermal ecosystem. Front. Microbiol. 9, 2718. doi:10.3389/fmicb.2018.02718
Lin, L., Xiong, J., Yue, T., Xu, W., Liu, L., Wang, F., et al. (2024). Phosphorus starvation response genes and function coupling: a mechanism to regulate phosphorus availability in a subtropical Estuary. Sci. Total Environ. 928, 172575. doi:10.1016/j.scitotenv.2024.172575
Liu, S., Yu, H., Yu, Y., Huang, J., Zhou, Z., Zeng, J., et al. (2022). Ecological stability of microbial communities in Lake donghu regulated by keystone taxa. Ecol. Indic. 136, 108695. doi:10.1016/j.ecolind.2022.108695
Liu, T., Zhang, A. N., Wang, J., Liu, S., Jiang, X., Dang, C., et al. (2018). Integrated biogeography of planktonic and sedimentary bacterial communities in the yangtze river. Microbiome 6, 16–14. doi:10.1186/s40168-017-0388-x
Louca, S., Polz, M. F., Mazel, F., Albright, M. B. N., Huber, J. A., O’Connor, M. I., et al. (2018). Function and functional redundancy in microbial systems. Nat. Ecol. and Evol. 2 (6), 936–943. doi:10.1038/s41559-018-0519-1
Lu, Q., Song, Y., Mao, G., Lin, B., Wang, Y., and Gao, G. (2020). Spatial variation in bacterial biomass, community composition and driving factors across a eutrophic river. Ecotoxicol. Environ. Saf. 205, 111113. doi:10.1016/j.ecoenv.2020.111113
Lyu, Z., Shao, N., Akinyemi, T., and Whitman, W. B. (2018). Methanogenesis. Curr. Biol. 28 (13), R727–R732. doi:10.1016/j.cub.2018.05.021
Mainstone, C. P., and Parr, W. (2002). Phosphorus in Rivers—Ecology and management. Sci. total Environ. 282, 25–47. doi:10.1016/s0048-9697(01)00937-8
Mallik, S. K., Pathak, R., and Shahi, N. (2024). Sediment microbiology in the aquatic environment[M].Handbook of aquatic microbiology. Boca Raton, FL: CRC Press. p. 56–76.
Morrissey, E. M., Gillespie, J. L., Morina, J. C., and Franklin, R. B. (2014). Salinity affects microbial activity and soil organic matter content in tidal wetlands. Glob. Change Biol. 20 (4), 1351–1362. doi:10.1111/gcb.12431
Nirody, J. A., Budin, I., and Rangamani, P. (2020). ATP synthase: Evolution, energetics, and membrane interactions. J. General Physiology 152 (11), e201912475. doi:10.1085/jgp.201912475
Pan, Y., Sun, R. Z., Wang, Y., Chen, G. L., Fu, Y. Y., and Yu, H. Q. (2023). Carbon source shaped microbial ecology, metabolism and performance in denitrification systems. Water Res. 243, 120330. doi:10.1016/j.watres.2023.120330
Parsons, C. T., Rezanezhad, F., O'Connell, D. W., and Van Cappellen, P. (2017). Sediment phosphorus speciation and mobility under dynamic redox conditions. Biogeosciences 14 (14), 3585–3602. doi:10.5194/bg-14-3585-2017
Qian, Z., Tianwei, H., Mackey, H. R., van Loosdrecht, M. C., and Guanghao, C. (2019). Recent advances in dissimilatory sulfate reduction: from metabolic study to application. Water Res. 150, 162–181. doi:10.1016/j.watres.2018.11.018
Rajpurohit, H., and Eiteman, M. A. (2022). Nutrient-limited operational strategies for the microbial production of biochemicals. Microorganisms 10 (11), 2226. doi:10.3390/microorganisms10112226
Rawat, P., Das, S., Shankhdhar, D., and Shankhdhar, S. C. (2021). Phosphate-solubilizing microorganisms: mechanism and their role in phosphate solubilization and uptake. J. Soil Sci. Plant Nutr. 21 (1), 49–68. doi:10.1007/s42729-020-00342-7
Raza, T., Qadir, M. F., Khan, K. S., Eash, N. S., Yousuf, M., Chatterjee, S., et al. (2023). Unraveling the potential of microbes in decomposition of organic matter and release of carbon in the ecosystem. J. Environ. Manag. 344, 118529. doi:10.1016/j.jenvman.2023.118529
Reddy, B., Pandey, J., and Dubey, S. K. (2019). Assessment of environmental gene tags linked with carbohydrate metabolism and chemolithotrophy associated microbial community in river ganga. Gene 704, 31–41. doi:10.1016/j.gene.2019.04.004
Ren, H., Wang, G., Ding, W., Li, H., Shen, X., Shen, D., et al. (2023). Response of dissolved organic matter (DOM) and microbial community to submerged macrophytes restoration in Lakes: a review. Environ. Res. 231, 116185. doi:10.1016/j.envres.2023.116185
Richardson, A. E., Barea, J. M., McNeill, A. M., and Prigent-Combaret, C. (2009). Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil, 1356–1365. doi:10.1007/s11104-009-9895-2
Sadiq, H. M., Jahangir, G. Z., Nasir, I. A., Iqtidar, M., and Iqbal, M. (2013). Isolation and characterization of phosphate-solubilizing bacteria from rhizosphere soil. Biotechnol. Biotechnol. Equip. 27 (6), 4248–4255. doi:10.5504/bbeq.2013.0091
Saunders, A. M. (2005). The physiology of microorganisms in enhanced biological phosphorous removal. Queensland, Australia: University of Queensland.
Sun, C., Wang, S., Wang, H., Hu, X., Yang, F., Tang, M., et al. (2022). Internal nitrogen and phosphorus loading in a seasonally stratified reservoir: implications for eutrophication management of deep-water ecosystems. J. Environ. Manag. 319, 115681. doi:10.1016/j.jenvman.2022.115681
Trabucho Alexandre, J. (2015). Organic matter-rich shale depositional environments. Fundam. Gas Shale Reservoirs, 21–45. doi:10.1002/9781119039228.ch2
Tully, K., and Ryals, R. (2017). Nutrient cycling in agroecosystems: balancing food and environmental objectives. Agroecol. Sustain. Food Syst. 41 (7), 761–798. doi:10.1080/21683565.2017.1336149
Van Mooy, B. A. S., Fredricks, H. F., Pedler, B. E., Dyhrman, S. T., Karl, D. M., Koblížek, M., et al. (2009). Phytoplankton in the ocean use non-phosphorus lipids in response to phosphorus scarcity. Nature 458 (7234), 69–72. doi:10.1038/nature07659
Walker, A. M. (2022). Exploring the north American arctic benthos: community structure and oil degradation potential of sediment bacteria and archaea. Fairbanks, AK: University of Alaska Fairbanks.
Wang, H., Zhang, W., Li, Y., Gao, Y., Niu, L., Zhang, H., et al. (2023). Hydrodynamics-driven community coalescence determines ecological assembly processes and shifts bacterial network stability in river bends. Sci. Total Environ. 858, 159772. doi:10.1016/j.scitotenv.2022.159772
Wang, Y., Zhang, R., He, Z., Van Nostrand, J. D., Zheng, Q., Zhou, J., et al. (2017). Functional gene diversity and metabolic potential of the microbial community in an estuary-shelf environment. Front. Microbiol. 8, 1153. doi:10.3389/fmicb.2017.01153
Wetzel, R. G. (2003). Dissolved organic carbon: detrital energetics, metabolic regulators, and drivers of ecosystem stability of aquatic ecosystems. Salt Lake City, UT: Academic Press. p. 455–477.
Wurtsbaugh, W. A., Paerl, H. W., and Dodds, W. K. (2019). Nutrients, eutrophication and harmful algal blooms along the freshwater to marine continuum. Wiley Interdiscip. Rev. Water 6 (5), e1373. doi:10.1002/wat2.1373
Xiang, H., Ma, Y., Zhang, R., Chen, H., and Yang, Q. (2022). Spatio-temporal evolution and future simulation of agricultural land use in xiangxi, central China. Land 11 (4), 587. doi:10.3390/land11040587
Xu, H., Tan, X., Liang, J., Cui, Y., and Gao, Q. (2022). Impact of agricultural non-point source pollution on river water quality: evidence from China. Front. Ecol. Evol. 10, 858822. doi:10.3389/fevo.2022.858822
Xu, R., Zhang, K., Liu, P., Khan, A., Xiong, J., Tian, F., et al. (2018). A critical review on the interaction of substrate nutrient balance and microbial community structure and function in anaerobic co-digestion. Bioresour. Technol. 247, 1119–1127. doi:10.1016/j.biortech.2017.09.095
Xu, Y., Yu, S., Liu, D., Ma, J., and Chuo, M. (2024). The impact of the three gorges reservoir operations on hydraulic characteristics in the backwater region: a comprehensive 2D modeling study. Water 16 (14), 2045. doi:10.3390/w16142045
Yang, X., and Chen, S. (2021). Microorganisms in sediment microbial fuel cells: ecological niche, microbial response, and environmental function. Sci. Total Environ. 756, 144145. doi:10.1016/j.scitotenv.2020.144145
Yi, M., Zhang, L., Qin, C., Lu, P., Bai, H., Han, X., et al. (2022). Temporal changes of microbial community structure and nitrogen cycling processes during the aerobic degradation of phenanthrene. Chemosphere 286, 131709. doi:10.1016/j.chemosphere.2021.131709
Yu, T., Wu, W., Liang, W., Wang, Y., Hou, J., Chen, Y., et al. (2023). Anaerobic degradation of organic carbon supports uncultured microbial populations in estuarine sediments. Microbiome 11 (1), 81. doi:10.1186/s40168-023-01531-z
Yuan, H., Zhang, R., Chen, J., Wu, J., Han, Q., Li, Q., et al. (2024). Phosphorus resource partitioning underpins diversity patterns and assembly processes of microbial communities in Plateau karst Lakes. Sci. Total Environ. 952, 175860. doi:10.1016/j.scitotenv.2024.175860
Zeglin, L. H. (2015). Stream microbial diversity in response to environmental changes: review and synthesis of existing research. Front. Microbiol. 6, 454. doi:10.3389/fmicb.2015.00454
Zhang, J., Yang, Y., Zhao, L., Li, Y., Xie, S., and Liu, Y. (2015). Distribution of sediment bacterial and archaeal communities in Plateau freshwater Lakes. Appl. Microbiol. Biotechnol. 99, 3291–3302. doi:10.1007/s00253-014-6262-x
Zhang, Z., Zhang, C., Yang, Y., Zhang, Z., Tang, Y., Su, P., et al. (2022). A review of sulfate-reducing bacteria: metabolism, influencing factors and application in wastewater treatment. J. Clean. Prod. 376, 134109. doi:10.1016/j.jclepro.2022.134109
Keywords: Xiangxi river, spatiotemporal variations, water and sediment, bacterial communities, functional gene
Citation: Wen J, Liu S, Wang Y, Khan MYA, Zhou X, Li S, Bao Y, Cui X, Huang Z, Sun M and He H (2025) Spatiotemporal variations of bacterial communities and functional genes in the water and sediments of a typical river influenced by reservoir operations. Front. Environ. Sci. 13:1568871. doi: 10.3389/fenvs.2025.1568871
Received: 31 January 2025; Accepted: 07 July 2025;
Published: 28 August 2025.
Edited by:
Xuwang Zhang, Dalian University of Technology, ChinaReviewed by:
Lunhui Lu, Chinese Academy of Sciences (CAS), ChinaSha Wu, Changsha University of Science and Technology, China
Copyright © 2025 Wen, Liu, Wang, Khan, Zhou, Li, Bao, Cui, Huang, Sun and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuchun Wang, d2FuZ3ljaXdockAxNjMuY29t