 Quan Xu1†
Quan Xu1† Yu Yang
Yu Yang- 1Hunan Coal Science Research Institute Co., Ltd., Changsha, Hunan, China
- 2School of Minerals Processing and Bioengineering, Central South University, Changsha, Hunan, China
- 3Key Laboratory of Biohydrometallurgy of Ministry of Education, School of Minerals Processing and Bioengineering, Central South University, Changsha, Hunan, China
Abandoned coal mines cause severe soil degradation, water pollution, and biodiversity loss. This study characterizes microbial community dynamics across distinct microhabitats—slag-enriched soil (D-1), waterlogged sediment (W-3), and acidic wastewater (W-5)—in Hunan mining areas. High-throughput 16S rRNA sequencing revealed Proteobacteria (31%–60%) dominance across sites, with Firmicutes enriched in acidic W-5 (49%) and Bacteroidetes in organic-rich D-1 (21%). Community diversity significantly diverged along pH/metal gradients (db-RDA: pH explained 74.8% variation, p < 0.001), correlating with environmental stressors. Functional predictions (PICRUSt/FAPROTAX) suggest potential adaptations: sulfur oxidation increased in acidic zones (25.7%), while organic degradation peaked in contaminated soils (38.1%). These community-environment linkages support bioremediation design: augmenting acid-tolerant taxa in low-pH zones and pollutant-degrading microbes in metal-impacted soils to accelerate ecological restoration.
1 Introduction
Global industrialization has intensified coal mining, leaving behind vast abandoned sites that degrade land resources and trigger severe ecological damage. These areas face critical issues including soil degradation, water contamination by metals (e.g., Pb, Cd, Hg) and acidic sulfates, and vegetation loss, collectively threatening ecosystem stability (Bian et al., 2010; Tozsin et al., 2022). Abandoned coal mines cause severe soil degradation, water pollution, and biodiversity loss, with microbial communities significantly impacted by altered geochemical conditions (Aguinaga et al., 2018; Sajjad et al., 2024). Mining activities also drastically alter geochemical conditions—redox status, pH extremes, and metal accumulation—which cascade into profound disruptions of local biological communities (Quadros et al., 2016; Zhao et al., 2024).
Microbial remediation offers a sustainable solution for mine rehabilitation due to its efficiency, cost-effectiveness, and environmental compatibility (Ly et al., 2019; Pande et al., 2022). Microorganisms employ metabolic pathways to detoxify pollutants: certain bacteria immobilize metals via redox reactions or biosorption, while others degrade organic compounds, reducing COD/BOD in water bodies (Gaur et al., 2018). However, remediation efficacy hinges on microbial community structure and functional activity, which vary significantly across heterogeneous mining environments (Sui et al., 2021). Despite their critical role in biogeochemical cycles (C, N, S) and as bioindicators of environmental stress (Zhang et al., 2022), how microbial communities adapt to site-specific stressors (e.g., metal toxicity, acidity, hydrodynamic conditions) remains inadequately characterized, limiting targeted bioremediation strategies.
Pollutant distribution and environmental gradients (pH, redox, water mobility) impose strong selective pressures on microbial communities. Contaminated soils and acidic wastewater pits favor metal-resistant and acidophilic taxa, often reducing biodiversity but enriching specialized consortia (Wen et al., 2020). Microbial responses, such as shifts in diversity, functional gene expression, and stress-tolerant phenotypes, provide vital insights into ecosystem resilience and natural attenuation potential (Li et al., 2017; Graham and Knelman, 2023). Understanding these adaptations is essential for designing effective ecological restoration protocols in degraded mine landscapes.
Here, we investigate microbial community structure and adaptive mechanisms at three distinct sites within an abandoned Hunan Province coal mine: (D-1) metal-rich topsoil under oxidative erosion, (W-3) micro-oxidized, moderately mobile sediments with accumulated metals, and (W-5) an acidic (low pH), static wastewater pit enriched with sulfate. By integrating physicochemical analyses (pH, metal concentrations) with high-throughput 16S rRNA sequencing, we address: 1 How do microbial diversity and composition differ across these heterogeneous habitats? 2 Which environmental drivers (e.g., pH, metal ions, hydrology) govern community assembly? 3 What adaptive strategies enable microbial survival under extreme stresses? Our findings will elucidate microbial ecology in post-mining systems and inform tailored bioremediation approaches for ecological recovery.
2 Materials and methods
2.1 Sample collection and pre-processing
The sampling area is located in a mining area in Hunan Province, China, which is a typical acidic mining environment rich in pollutants such as metal ions and sulphates due to the long-term impact of mining activities. Three sampling sites were selected to be representative, including:
Tailings Impoundment (D-1): Yuxing inclined shaft (27°47′N 111°32′E), topsoil containing high concentrations of metal slag, affected by oxidation and precipitation erosion.
Mine Sediment (W-3): Yangguling flat lane (27°44′N 111°30′E), water-bearing sediments with some mobility and potentially high accumulation of heavy metals.
Acidic pit water (W-5): Yangguling inclined lane (27°44′N 111°30′E), a static wastewater pit at the bottom of the pit with low pH and high sulfate concentrations.
A 10 × 10 m2 core area is delineated at each sampling point, where three parallel samples are collected using the triangular point layout method (spacing ≥5 m) to avoid micro-topographic bias. Soil samples are taken vertically from the 0–15 cm profile using an ethanol flame-sterilized stainless steel ring knife (5 cm in diameter), with each parallel sample consisting of a mixture from five subsamples (after removing gravel by quartering and sieving through a 2 mm mesh). Water samples are collected at a 30 cm depth surface layer (avoiding sediment disturbance) using HDPE bottles (250 mL, Nalgene®) pre-rinsed with 0.22 μm filters. All samples are immediately stored in a cooler (4 °C) after collection and transported to the laboratory within 24 h for freezing (−80 °C).
2.2 Physicochemical analyses
Solid samples were air-dried (25 °C, 48 h), sieved (<2 mm), and homogenized prior to analysis. pH was measured following EPA 9045D (USEPA 2004) using a calibrated PHS-3C meter after suspension (1:2.5 w/v soil:water, 180 rpm, 30 min shaking). Suspended solids (SS) were quantified via EPA 160.2 gravimetry (USEPA 1983), and chemical oxygen demand (COD) by closed reflux colorimetry (ISO 15705:2002). Thirteen metals (Fe, Mn, Ca, Mg, Al, As, Sb, Pb, Cd, Cu, Cr, Tl, Zn) were analyzed for pseudototal content (Vidal et al., 1999). Samples were digested in a microwave system (MARS 6, CEM Corp.) with HNO3-H2O2 (10:2 mL; program: 25 °C–180 °C/20 min ramp, 180 °C hold/30 min), followed by ICP-MS (iCAP RQ, Thermo Scientific) detection. Recovery rates (92%–107%) were validated using certified materials (NIST 2711a soil, NIST 1643e water). Sulfate (SO42−) quantification employed ion chromatography (EPA 300.0; Dionex ICS-2100 with AS23 column, 4.5 mM Na2CO3/1.4 mM NaHCO3 eluent).
2.3 DNA extraction
Total community genomic DNA extraction was performed using a E.Z.N.A™ Mag Bind Soil DNA Kit (Omega, M5635-02, United States), following the manufacturer’s instructions. We measured the concentration of the DNA using a Qubit 4.0 (Thermo, USA) to ensure that adequate amounts of high-quality genomic DNA had been extracted.
2.4 16S rRNA gene amplification by PCR
The target was the V3–V4 hypervariable region of the bacterial 16S rRNA gene. PCR was started immediately after the DNA was extracted.The 16S rRNA V3–V4 amplicon was amplified using 2×Hieff® Robust PCR Master Mix (Yeasen, 10105ES03,China). Two universal bacterial 16S rRNA gene amplicon PCR primers (PAGE purified) were used: the amplicon PCR forward primer 341F (5′-CCTACGGGNGGCWGCAG-3′) and amplicon PCR reverse primer 805R (5′-GACTACHVGGGTATCTAATCC-3′). The reaction was set up as follows: microbial DNA (10 ng/μL) 2 µL; amplicon PCR forward primer (10 µM) 1 µL; amplicon PCR reverse primer (10 µM) 1 µL; 2×Hieff® Robust PCR Master Mix (Yeasen, 10105ES03, China) (total 30 µL). The plate was sealed and PCR performed in a thermal instrument (Applied Biosystems 9,700, United States) using the following program: 1 cycle of denaturing at 95 °C for 3 min, first five cycles of denaturing at 95 °C for 30 s, annealing at 45 °C for 30 s, elongation at 72 °C for 30 s, then 20 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s, elongation at 72 °C for 30 s and a final extension at 72 °C for 5 min. The PCR products were checked using electrophoresis in 2% (w/v) agarose gels in TBE buffer (Tris, boric acid, EDTA) stained with ethidium bromide (EB) and visualized under UV light.
2.5 16S gene library construction, quantification, and sequencing
Using Hieff NGS™ DNA Selection Beads (Yeasen, 10105ES03, China) to purify the free primers and primer dimer species in the amplicon product.Samples were delivered to Sangon BioTech (Shanghai) for library construction using universal Illumina adaptor and index. Before sequencing, the DNA concentration of each PCR product was determined using a Qubit® 4.0 Green double-stranded DNA assay and it was quality controlled using a bioanalyzer (Agilent 2,100, United States). Depending on coverage needs, all libraries can be pooled for one run.The amplicons from each reaction mixture were pooled in equimolar ratios based on their concentration. Sequencing was performed using the Illumina MiSeq system (Illumina MiSeq, United States), according to the manufacturer’s instructions.
2.6 Sequence processing, OTU clustering, representative tags alignment and biological classification
After sequencing, The two short Illumina readings were assembled by PEAR software (version 0.9.8) (Zhang et al., 2014) according to the overlap and fastq files were processed to generate individual fasta and qual files, which could then be analyzed by standard methods. The effective tags were clustered into operational taxonomic units (OTUs) of ≥97% similarity using Usearch software (version 11.0.667) (Edgar, 2013; Edgar, 2016). Chimeric sequences and singleton OTUs (with only one read) were removed, after which the remaining sequences were sorted into each sample based on the OTUs. The tag sequence with the highest abundance was selected as a representative sequence within each cluster. Bacterial and fungal OTU representative sequences were classified taxonomically by blasting against the RDP Database and UNITE fungal ITS Database, respectively (Wang et al., 2007).
2.7 Statistical analysis
α-diversity (Chao1, Shannon indices) was calculated in Mothur (Schloss et al., 2009), using descriptive gradient comparisons. β-diversity analysis used Bray-Curtis dissimilarity matrices visualized via PCoA (vegan R package). Spearman’s rank correlations assessed metal-microbe relationships with FDR-adjusted p-values. Distance-based redundancy analysis (db-RDA) was performed to quantify the explanatory power of environmental factors on microbial community structure. The analysis followed a three-step protocol. 1 Pre-screening of variables: All measured physicochemical parameters were initially included (Table 1). Collinear variables (r >0.8) were identified via variance inflation factor (VIF) analysis and removed iteratively until all VIF <5. 2 Forward selection: Remaining variables were tested using the vegan::ordiR2step() function with 999 permutations. Factors significantly explaining community variation (p < 0.05 after Bonferroni correction) were retained. 3 Constrained ordination: The final model included pH, Ca2+, and Mg2+ as significant constraints. Visualization was implemented in vegan::capscale() with Bray-Curtis dissimilarity matrices.

Table 1. Physicochemical properties and metal content of samples.
2.8 Function prediction
Microbial metabolic functions were predicted using two complementary bioinformatic approaches:
1. PICRUSt v1.1.4 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) was employed to predict functional potential based on KEGG orthologs (KO), mapping 16S rRNA gene sequences to established metabolic pathways (Langille et al., 2013).
2. FAPROTAX v1.2.4 (Functional Annotation of Prokaryotic Taxa) was applied to decode biogeochemical cycles and other ecologically relevant functions, leveraging its curated database linking microbial taxa to 80+ metabolic traits (Louca et al., 2016). Functional profiles were generated by collapsing OTU tables into ecological functional groups (e.g., nitrification, metal respiration) using the collapse_table.py script with default parameters.
3 Results and discussion
3.1 Physicochemical properties and metal content of samples
As shown in Table 1, under the conditions where the samples exhibited acidity (pH 4.43-6.07) and extremely high sulfate (SO2-4) concentrations (1170–3280 mg/L), dissolved metal ions primarily existed in sulfate forms. The prevalence of sulfates in our samples reflects typical AMD geochemistry, driven by pyrite oxidation and mineral dissolution (She et al., 2023). Among them, iron (Fe(II/III), particularly the hydrolysis of ferric iron generated from the oxidation of ferrous iron, which exacerbated the acidic environment) and calcium (Ca(II)) were the dominant cations. Iron was prominent in typical acidic samples (D-1, W-5) with high concentrations (587–872 mg/L), while calcium consistently showed high levels across all samples (216–564 mg/L), especially becoming the predominant cation in the weakly acidic W-3 sample (pH 6.07) due to reduced iron content (89 mg/L, attributed to limited oxidation during migration). Aluminum (Al(III)), prone to precipitation at pH >4, was only significantly present in the lowest pH sample, W-5 (26.3 mg/L), while other metals were relatively low in concentration. This ion distribution pattern, dominated by iron and calcium sulfates, originates from sulfate/iron ions released by pyrite oxidation and the dissolution of carbonate/sulfate minerals in the surrounding rocks. pH profoundly influenced the dissolved concentrations of these metals by controlling the hydrolysis and precipitation of iron and aluminum.
3.2 16S rRNA sequencing results
The 16S rRNA sequencing results of the samples are shown in Supplementary Table S1. A total of 189,879 valid 16S rRNA sequences were obtained from the three samples, with an average sequence number of 63,293 and an average length of 423 bp. Cluster analysis with 97% similarity yielded a total of 1,138 OTUs with 98.1%–98.3% coverage for each sample, indicating a high probability of genes being detected in the samples. The direction of the rarefaction curve (Supplementary Figure S1) tends to be flat, indicating that the number of sequencing tends to be saturated and the data obtained from sequencing are sufficient.
3.3 Alpha diversity analysis
Table 2 presents a systematic comparison of three sample groups across alpha diversity metrics. W-5 consistently showed the lowest values in all indicators, significantly lower than D-1 and W-3. Although statistical significance was not reached (limited by sample size n = 3), the effect size (Cohen’s d >1.2) confirmed the inhibitory trend of acidity on microbial richness and evenness. Different calculation methods all demonstrated a gradient pattern of D-1 > W-3 > W-5, validating the progressive restructuring of community structure under environmental stress. Goods_coverage values all exceeded 0.999, confirming data reliability and particularly supporting that W-5’s low diversity was not due to technical bias.
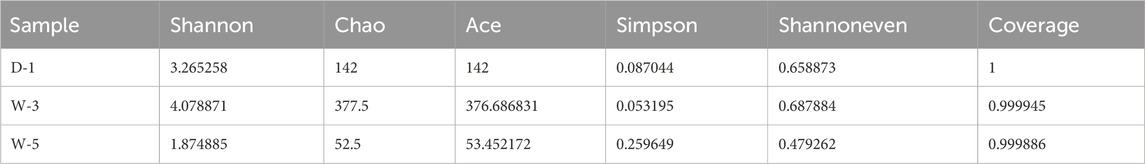
Table 2. The alpha diversity index of the community.
3.4 Community structure of samples at different taxonomic levels
As depicted in Figure 1, microbial community composition was analyzed at the phylum level across three sample groups. Proteobacteria exhibited the highest average relative abundance (48.65%), with distribution ranked as D-1 (60.45%) > W-3 (55.92%) > W-5 (31.21%). Firmicutes showed the second-highest abundance, dominating W-5 at 48.73%, while being minimally present in W-3 and undetected in D-1. Actinobacteria ranked third in average abundance (12.81%–17.01%), with relative abundances ordered as W-3 (17.01%) > D-1 (12.81%) > W-5 (8.10%). Bacteroidetes displayed the fourth-highest mean abundance (12.24%), distributed as D-1 (21.26%) > W-3 (15.45%) > W-5 (undetected). Nitrospirae appeared only in W-5 (7.30%), with minor contributions from unclassified taxa and other phyla. The circular phylogenetic tree in Figure 2 further revealed differentiation in community composition. Bacteroidetes accounted for 21% in D-1, correlating with organic degradation functions (Flavobacterium at 15%). W-3 showed the highest Actinobacteria abundance (17%), reflecting the ecological niche advantage of sediment-dwelling actinomycetes. In W-5, Firmicutes expanded to 49%, dominated by acid-tolerant spore-forming bacteria such as Bacillus (32% branch proportion). Nitrospirae appeared uniquely (7.3%), suggesting potential for acidic nitrogen cycling, while Proteobacteria were suppressed to 31% (a 48% decrease compared to D-1). At the class level (Figure 3), Proteobacteria subclasses showed distinct distributions. β-Proteobacteria (mean: 34.65%) peaked in D-1 (37.20%), followed by W-3 (36.93%) and W-5 (29.82%). α-Proteobacteria (mean: 7.90%) were highest in W-3 (13.79%), then D-1 (8.48%) and W-5 (1.40%). γ-Proteobacteria (mean: 6.10%) dominated in D-1 (14.76%), decreased in W-3 (3.54%), and were absent in W-5. δ-Proteobacteria (<2% average) emerged only in W-3 (1.66%). Proteobacteria’s dominance aligns with their prevalence in AMD environments, owing to their metabolic adaptability (Sajjad et al., 2024).
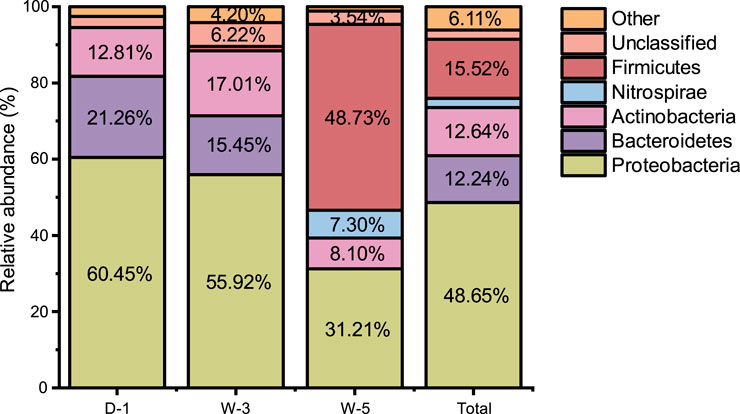
Figure 1. Relative abundance of major bacterial phyla across sampling sites. Stacked bar charts depict taxonomic composition at the phylum level for sites D-1, W-3, W-5, and a pooled “Total” dataset. The y-axis scale ranges from 0% to 80% relative abundance. Color codes: Proteobacteria (yellow-green), Bacteroidetes (purple), Actinobacteria (pink), Nitrospirae (light blue), Firmicutes (red), Unclassified (brown), Others (orange). Exact abundances for dominant phyla: • Proteobacteria: 60.45% (D-1), 55.92% (W-3), 31.21% (W-5), 48.65% (Total) • Firmicutes: 4.20% (D-1), 6.22% (W-3), 48.73% (W-5), 6.11% (Total) • Bacteroidetes: 21.26% (D-1), 17.01% (W-3), 15.52% (W-5), 15.45% (Total) • Nitrospirae: not detected in D-1/W-3, 7.30% (W-5), absent in Total.
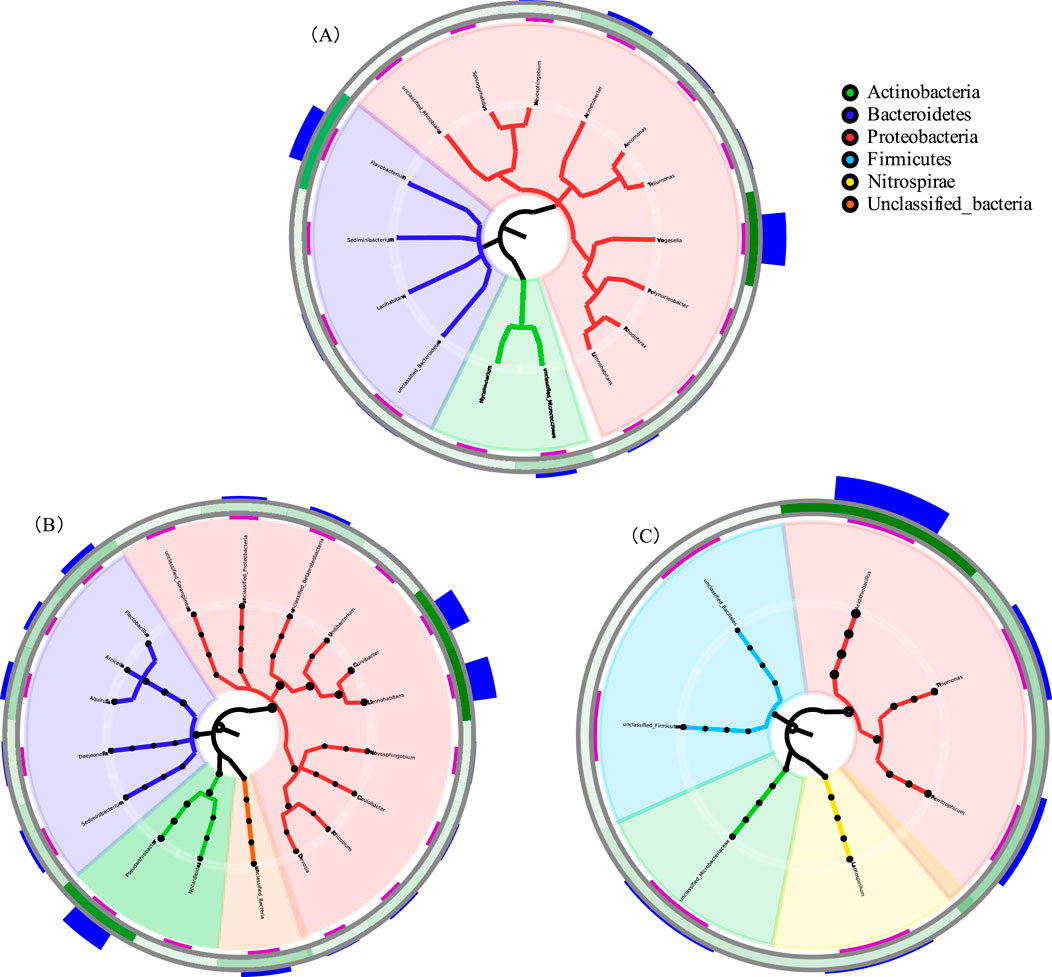
Figure 2. Phylogenetic tree analysis of microbial communities. Radial cladograms depict taxonomic hierarchies for (A) D-1, (B) W-3, and (C) W-5.
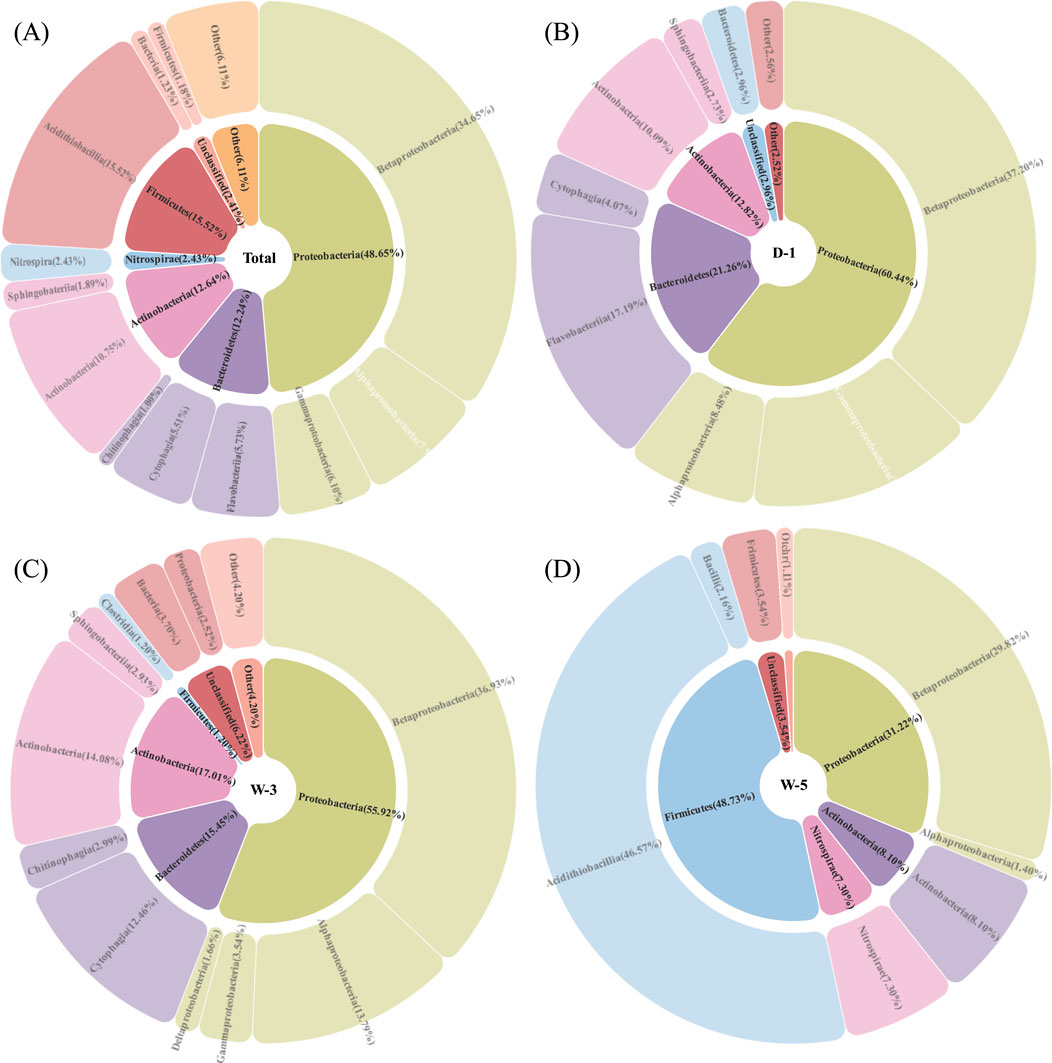
Figure 3. Microbial phylum and class classification level composition of samples. (A) Total sample composition; (B) D-1 sample dominated by β-Proteobacteria (37.2%), with γ-Proteobacteria exhibiting hydrocarbon-degrading traits (14.8%); (C) Hypoxic W-3 sample showing peak abundance of α-Proteobacteria (13.8%) and emergence of δ-Proteobacteria (1.7%); (D) W-5 sample lacking acid-sensitive γ-Proteobacteria. Sector size scales with relative abundance.
In sample D-1, the abundance of Bacteroidetes reached 21.26%. Studies have reported that Flavobacterium within this phylum encodes aromatic ring-hydroxylating dioxygenase (phnA1 gene), which can catalyze the degradation of coal tar pollutants (COD = 183 mg/L) (Kaur et al., 2023). Meanwhile, the dominance of Actinobacteria (17.01%) in W-3 reflects its adaptation to polymetallic environments, as its Streptomyces secretes siderophores that can chelate Fe/Cd and inhibit pathogens (Dimkpa et al., 2009). In W-5, Firmicutes was significantly enriched (48.73%). This phylum includes acid-resistant spore-forming bacteria (e.g., Bacillus), which regulate intracellular pH through proton pumps (Baker-Austin et al., 2007) and resist acidic environments (pH 4.43) via metallothionein chelation mechanisms. Proteobacteria sharply decreased to 31.21% (a 48% drop compared to D-1), with acid-sensitive γ-proteobacteria completely disappearing (0%). This group typically contains metal oxidases (e.g., copper oxidase), which are prone to inactivation at low pH (Zhang et al., 2019). The specific presence of Nitrospirae (7.30%) suggests potential for acidic nitrification, with its acid-resistant mechanisms including proton gradient reconstruction in cell membranes (Zhou et al., 2024), partially compensating for the missing nitrogen cycle functions in W-5. β-proteobacteria had the highest abundance in D-1 (37.20%), with its Cupriavidus containing the czcA cadmium resistance gene cluster (Dai et al., 2019), effectively binding Cd levels in D-1. γ-proteobacteria were significantly enriched in D-1 (14.76%), with its Pseudomonas expressing alkane monooxygenase (alkB gene) to degrade diesel pollutants (Rojas-Gätjens et al., 2022). α-proteobacteria were prominent in W-3 (13.79%), with its Bradyrhizobium potentially performing microaerobic nitrogen fixation (nifH gene), adapting to the low-oxygen sediment environment (Sulieman et al., 2019). δ-proteobacteria were only present in W-3 (1.66%), with its Desulfovibrio potentially performing sulfate reduction (dsrB gene), though limited by W-3’s low sulfate concentration (1170 mg/L).
3.5 Beta diversity analysis and similarity analysis between samples
Distance-based Redundancy Analysis (db-RDA) is a constrained ordination method used to quantify the explanatory power of environmental factors on microbial community structure variation (Liu et al., 2024; Teng et al., 2017). As shown in the Figure 4, the microbial community structure is hierarchically controlled by pH and ionic gradients: db-RDA1 (74.8% variance) separates samples along a gradient from the strongly acidic zone (W-5, pH 4.43) to the near-neutral zone (D-1, pH 5.52), with the radial long arrow of pH (projection length >3 times other factors) confirming acidity as the core driver (R2 = 0.74, p < 0.001). The db-RDA analysis corroborates findings that pH shapes microbial diversity in acidic mining settings (Huang et al., 2021). db-RDA2 (25.2% variance) reflects the synergistic effect of Ca2+/Mg2+—their arrows align toward W-5 (Ca = 564 mg/L, Mg = 99.6 mg/L), significantly increasing acid-tolerant bacterial abundance (Firmicutes↑40%) through extracellular polymer cross-linking and ion channel regulation. Short arrows of Al3+/Fe2+ spatially coupled with pH indicate their dependence on acidity (e.g., Al3+ dissolves only at pH <4.5). This pattern reveals a tripartite adaptation strategy of mine microbial communities under acid stress: acid-base balance (pH-driven) → ion homeostasis (Ca2+/Mg2+ buffering) → metal transformation (Al/Fe secondary mineralization). The inter-sample population distance heatmap (Figure 5) revealed that W-3 and W-5 exhibited greater similarity to each other, which can be attributed to the presence of common environmental conditions, including comparable pH levels, similar metal concentrations, and analogous redox conditions. Conversely, sample D-1 exhibited significant disparities from the other samples, which may be attributable to its provenance from contaminated soil samples, resulting in a substantially divergent species composition from the other samples. These findings are consistent with our previously formulated hypotheses, suggesting that environmental factors, particularly acidic conditions and metal contamination, play a pivotal role in driving changes in microbial community composition. Further studies that integrate environmental monitoring data could offer more insight into the specific effects of these environmental stressors on microbial community structure. Due to spatial constraints, samples within each 10 × 10 m2 plot are technical replicates; conclusions are site-specific and not generalizable without broader spatial sampling.
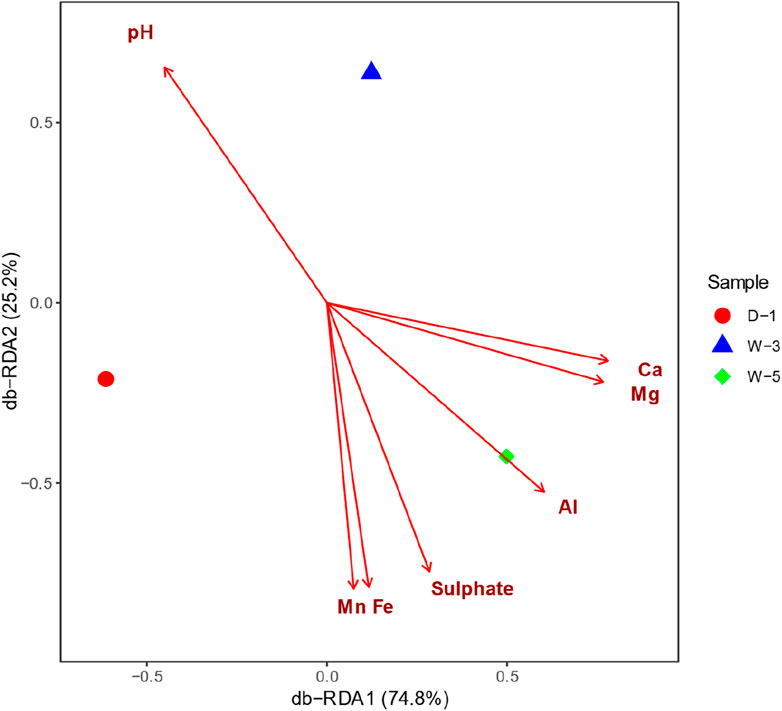
Figure 4. Distance-based redundancy analysis (db-RDA) of microbial communities constrained by environmental factors. Ordination plot reveals hierarchical environmental controls on microbial composition.
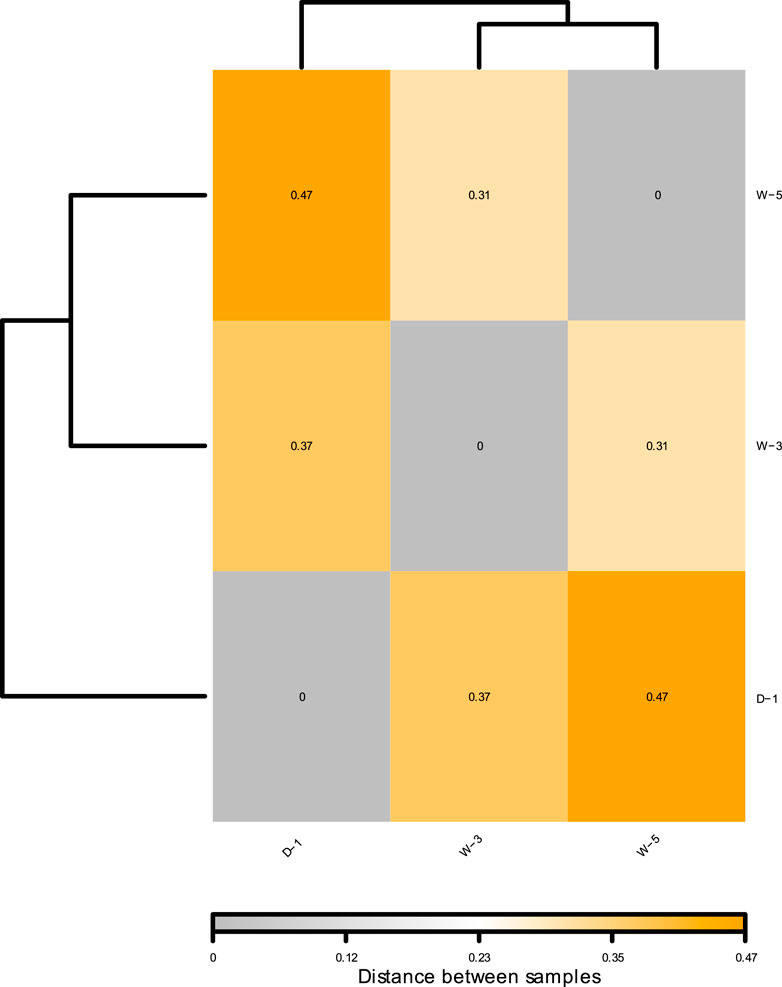
Figure 5. Heat map of microbial composition spacing for samples D-1, W-3, W-5. Distance values quantify compositional divergence: Dark orange (max distance: 0.47) for D-1 vs. W-5, light orange (0.37) for D-1 vs. W-3, light grey (0.31) for W-3 vs. W-5.
3.6 Functional predictive analytics
PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) is a metabolic function prediction tool for bacterial colonies developed by Curtis Huttenhower’s group at Harvard University, USA, which enables the prediction of bacterial and archaeal metabolic functions by comparing the existing 16S rRNA gene sequencing data with the reference genome databases of microorganisms whose metabolic functions are known (Langille et al., 2013). PICRUSt can predict 16S rRNA gene sequences in three functional profiling databases, namely, KEGG, COG and Rfam, and based on the prediction results of PICRUSt, we can obtain the annotation information of each sample corresponding to each functional profiling database, as well as the predicted abundance matrices of the functional taxa.The heat map is then plotted based on the abundance matrix. FAPROTAX, derived from Functional Annotation of Prokaryotic Taxa, is a prokaryotic functional annotation database based on the current manually compiled literature on culturable bacteria.The database contains more than 7,600 functional annotations collected from more than 4,600 prokaryotic microorganisms in more than 80 functional subgroups (Sansupa et al., 2021). The predicted functions predicted by FAPROTAX are focused on microbial functioning in oceans and lakes, particularly sulfur, carbon, hydrogen, and nitrogen cycling functions.
Functional prediction was performed on the three sample groups based on the COG database. The results indicate that the microbial communities exhibit metabolic division of labor characteristics coupled with stress environments (Figure 6). The acid stress-dominated W-5 sample achieves dynamic responses to proton toxicity through high-abundance signal transduction functions, but at the cost of energy metabolism. The metal-contaminated D-1 sample, on the other hand, is enriched with resistance defense modules, supporting its adaptation to metal stress but simultaneously leading to suppressed organic matter degradation. Meanwhile, the W-3 sediment adapts to fluctuations in sediment carbon sources through the dominance of carbon hydrolases, but its functional deficiency in nitrogen cycling (undetected nitrogenase nifH) exposes its functional limitations in low-oxygen environments. This functional compensation pattern confirms the “energy reallocation theory,” where extreme environments force microorganisms to allocate resources toward survival and defense, resulting in deficits in fundamental metabolic functions. The FAPROTAX-based functional prediction (Figure 7) revealed that microbial communities undergo metabolic restructuring coupled with environmental stress. In the strongly acidic wastewater sample (W-5), the relative abundance of sulfur oxidation functions significantly increased to 25.7% (represented by the largest dark circle in bubble plot A), corroborating the earlier observation of enriched acid-tolerant sulfur-oxidizing bacteria (e.g., Acidithiobacillus). These bacteria adapt to the pH 4.43 environment and harvest energy through proton gradient-driven chemoautotrophy (H2S→SO2- 4). In contrast, the organically contaminated soil (D-1) was dominated by fermentative metabolism, where Bacteroidetes degraders synergistically break down coal tar pollutants, albeit with suppressed nitrogen cycling functions. Elevated sulfur oxidation in W-5 (25.7%) reflects acidophilic bacteria activity, while organic degradation in D-1 (38.1%) highlights Bacteroidetes’ role in pollutant breakdown (Tian et al., 2025; Gonçalves et al., 2024). The nitrate reduction function observed in the sediment sample (W-3) reflects the dominance of denitrification at the low-oxygen sediment interface. This metabolic trade-off-enhanced sulfur oxidation at the expense of weakened nitrogen cycling-highlights microbial energy allocation strategies under acid stress but exacerbates ecosystem nitrogen limitation risks.
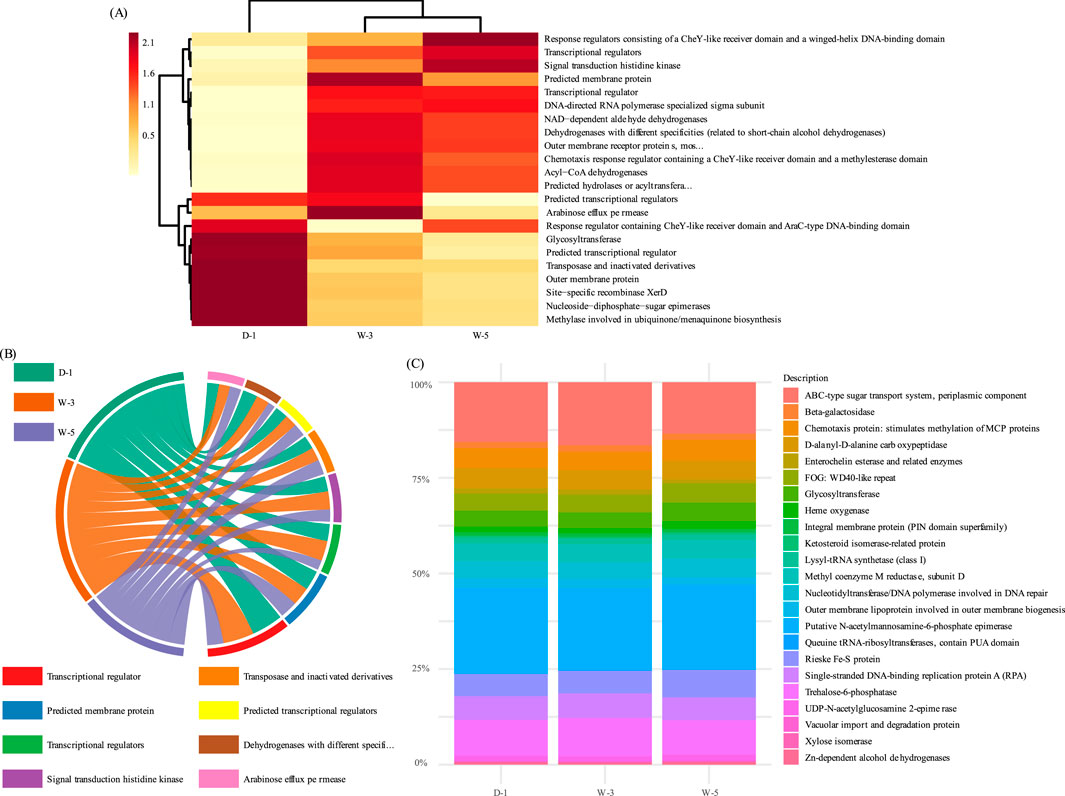
Figure 6. PICRUSt-based prediction of microbial functional profiles using the COG database. (A) Heatmap of COG abundance clustered by function and sample type. (B) Chord diagram displaying proportions of functional categories. (C) Stacked bar chart showing percentages of dominant gene families. Technical parameters: PICRUSt v1.1.4 software paired with COG database; color gradient normalized by Z-score.
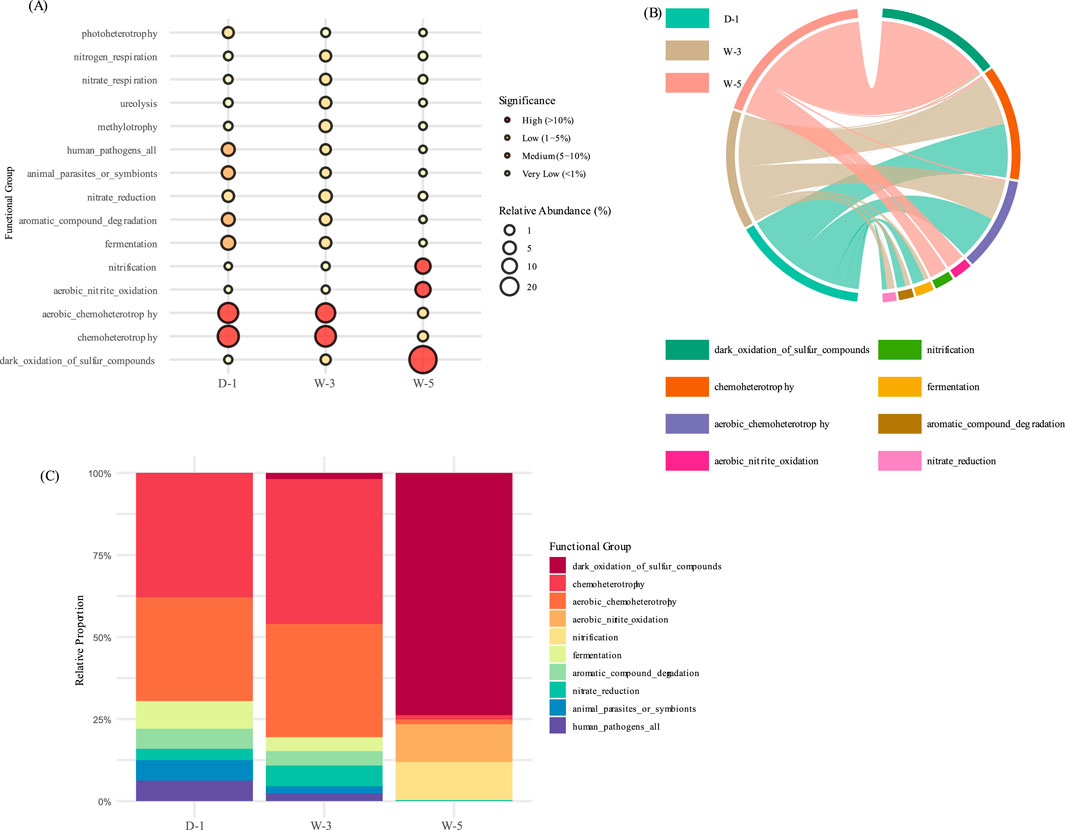
Figure 7. Predicted metabolic functions of contaminated sites by FAPROTAX. (A) Functional bubble chart (circle size indicates statistical significance). (B) Chord diagram of functional groups. (C) Percentage stacked bar chart of functional composition.
4 Conclusion
Microorganisms are pivotal in coal mine drainage remediation, excelling in metal detoxification, organic pollutant degradation, and elemental cycling, with metal-resistant taxa like Proteobacteria and organic degraders like Bacteroidetes showing promise (Wang et al., 2025; Karnwal et al., 2024). Metal-resistant bacteria, such as Pseudomonas and sulfate-reducing bacteria (SRB), immobilise and transform toxic metals into less harmful forms through adsorption, precipitation and redox reactions. Organic degrading bacteria such as Pseudomonas and Acinetobacter efficiently break down aromatic hydrocarbons and flotation agents, reducing ecological damage. Spatial variation in microbial communities reflects their adaptive strategies to different mine microenvironments, ranging from aerobic zones near the entrance to anaerobic regions in the flooded goaf. In addition, microbial involvement in carbon, nitrogen, sulphur and iron cycles drives ecosystem recovery by regulating material balance and reducing pollution. These processes highlight the critical role of microorganisms as natural engineers in restoring and maintaining the ecological stability of coal mine drainage systems.
In this study, the physicochemical properties and metal content of the analyzed samples reveal distinct environmental conditions and compositional characteristics. Samples were uniformly acidic, with sulfate-bound Fe(II/III) and Ca(II) as predominant ions, although W-3 exhibited a higher pH and significantly lower Fe(II/III) levels compared to D-1 and W-5. The microbial communities, dominated by Proteobacteria, Firmicutes, and Bacteroidetes, showcased functional diversity and adaptability to environmental stressors, such as metal presence and organic pollutants. Notably, β-proteobacteria, α-proteobacteria, and γ-proteobacteria contributed to elemental cycling and pollutant mitigation. Functional predictions emphasized variations in chemoheterotrophy, sulfur oxidation, and other microbial processes across the samples. These findings underscore the influence of environmental factors on microbial community structure and functional diversity, offering a foundation for further exploration of microbial ecology in coal mine drainage systems.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: the NCBI BioProject database under accession number PRJNA1309875 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1309875).
Author contributions
QX: Conceptualization, Data curation, Investigation, Methodology, Software, Writing – original draft. DT: Data curation, Software, Validation, Visualization, Writing – original draft. JL: Methodology, Validation, Writing – review and editing. HQ: Validation, Visualization, Writing – review and editing. YY: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Hunan Provincial Department of Education Key Project (24A0009) and National Key Research and Development program Foundation of China (2022YFE0119600). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We sincerely acknowledge the Hunan Coal Science Research Institute Co., Ltd. for granting the sample collection permits critical to this study. Our gratitude also extends to Central South University for providing access to their advanced experimental facilities, which laid the foundation for the experiments. Special thanks are due to Yu Yang for his expert guidance in experimental design and methodology refinement, which significantly enhanced the reliability of our results.
Conflict of interest
Author QX was employed by Hunan Coal Science Research Institute Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2025.1590925/full#supplementary-material
References
Aguinaga, O. E., McMahon, A., White, K. N., Dean, A. P., and Pittman, J. K. (2018). Microbial community shifts in response to acid mine drainage pollution within a natural wetland ecosystem. Front. Microbiol. 9, 1445. doi:10.3389/fmicb.2018.01445
Baker-Austin, C., Dopson, M., Wexler, M., Sawers, R. G., Stemmler, A., Rosen, B. P., et al. (2007). Extreme arsenic resistance by the acidophilic archaeon ‘Ferroplasma acidarmanus’ Fer1. Extremophiles 11, 425–434. doi:10.1007/s00792-006-0052-z
Bian, Z., Inyang, H. I., Daniels, J. L., Otto, F., and Struthers, S. (2010). Environmental issues from coal mining and their solutions. Min. Sci. Technol. (China) 20, 215–223. doi:10.1016/S1674-5264(09)60187-3
Dai, M., Zhou, G., Ng, H. Y., Zhang, J., Wang, Y., Li, N., et al. (2019). Diversity evolution of functional bacteria and resistance genes (CzcA) in aerobic activated sludge under Cd(II) stress. J. Environ. Manag. 250, 109519. doi:10.1016/j.jenvman.2019.109519
Dimkpa, C. O., Merten, D., Svatos, A., Büchel, G., and Kothe, E. (2009). Siderophores mediate reduced and increased uptake of cadmium by Streptomyces tendae F4 and sunflower (Helianthus annuus), respectively. J. Appl. Microbiol. 107, 1687–1696. doi:10.1111/j.1365-2672.2009.04355.x
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi:10.1038/nmeth.2604
Edgar, R. (2016). SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv. doi:10.1101/074161
Gaur, N., Narasimhulu, K., and Y, P. (2018). Recent advances in the bio-remediation of persistent organic pollutants and its effect on environment. J. Clean. Prod. 198, 1602–1631. doi:10.1016/j.jclepro.2018.07.076
Gonçalves, O. S., Fernandes, A. S., Tupy, S. M., Ferreira, T. G., Almeida, L. N., Creevey, C. J., et al. (2024). Insights into plant interactions and the biogeochemical role of the globally widespread Acidobacteriota phylum. Soil Biol. Biochem. 192, 109369. doi:10.1016/j.soilbio.2024.109369
Graham, E. B., and Knelman, J. E. (2023). Implications of soil microbial community assembly for ecosystem restoration: patterns, process, and potential. Microb. Ecol. 85, 809–819. doi:10.1007/s00248-022-02155-w
Huang, Y., Li, X. T., Jiang, Z., Liang, Z. L., Wang, P., Liu, Z. H., et al. (2021). Key factors governing microbial community in extremely acidic mine drainage (pH <3). Front. Microbiol. 12, 761579. doi:10.3389/fmicb.2021.761579
Karnwal, A., Martolia, S., Dohroo, A., Al-Tawaha, A. R. M. S., and Malik, T. (2024). Exploring bioremediation strategies for heavy metals and POPs pollution: the role of microbes, plants, and nanotechnology. Front. Environ. Sci. 12, 1397850. doi:10.3389/fenvs.2024.1397850
Kaur, T., Lakhawat, S. S., Kumar, V., Sharma, V., and Neeraj, R. R. K. (2023). Polyaromatic hydrocarbon specific ring hydroxylating dioxygenases: diversity, structure, function, and protein engineering. Curr. Protein Pept. Sci. 24, 7–21. doi:10.2174/1389203724666221108114537
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi:10.1038/nbt.2676
Li, X., Meng, D., Li, J., Yin, H., Liu, H., Liu, X., et al. (2017). Response of soil microbial communities and microbial interactions to long-term heavy metal contamination. Environ. Pollut. 231, 908–917. doi:10.1016/j.envpol.2017.08.057
Liu, P., Yuan, H., Ning, Y., Chakraborty, B., Liu, N., and Peres, M. A. (2024). A modified and weighted Gower distance-based clustering analysis for mixed type data: a simulation and empirical analyses. BMC Med. Res. Methodol. 24, 305. doi:10.1186/s12874-024-02427-8
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi:10.1126/science.aaf4507
Ly, T., Wright, J. R., Weit, N., McLimans, C. J., Ulrich, N., Tokarev, V., et al. (2019). Microbial communities associated with passive acidic abandoned coal mine remediation. Front. Microbiol. 10, 1955. doi:10.3389/fmicb.2019.01955
Pande, V., Pandey, S. C., Sati, D., Bhatt, P., and Samant, M. (2022). Microbial Interventions in bioremediation of heavy metal contaminants in agroecosystem. Front. Microbiol. 13, 824084. doi:10.3389/fmicb.2022.824084
Quadros, P. D. D., Zhalnina, K., Davis-Richardson, A. G., Drew, J. C., Menezes, F. B., Camargo, F. A. O., et al. (2016). Coal mining practices reduce the microbial biomass, richness and diversity of soil. Appl. Soil Ecol. 98, 195–203. doi:10.1016/j.apsoil.2015.10.016
Rojas-Gätjens, D., Fuentes-Schweizer, P., Rojas-Jiménez, K., Pérez-Pantoja, D., Avendaño, R., Alpízar, R., et al. (2022). Methylotrophs and hydrocarbon-degrading bacteria are Key players in the microbial community of an abandoned century-old oil exploration well. Microb. Ecol. 83, 83–99. doi:10.1007/s00248-021-01748-1
Sajjad, W., Ilahi, N., Kang, S., Bahadur, A., Banerjee, A., Zada, S., et al. (2024). Microbial diversity and community structure dynamics in acid mine drainage: acidic fire with dissolved heavy metals. Sci. Total Environ. 909, 168635. doi:10.1016/j.scitotenv.2023.168635
Sansupa, C., Wahdan, S. F. M., Hossen, S., Disayathanoowat, T., Wubet, T., and Purahong, W. (2021). Can we use functional annotation of prokaryotic taxa (FAPROTAX) to assign the ecological functions of soil bacteria? Appl. Sci. 11, 688. doi:10.3390/app11020688
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi:10.1128/aem.01541-09
She, Z., Wang, J., He, C., Jiang, Z., Pan, X., Wang, M., et al. (2023). Molecular insights into the impacts of acid mine drainage on dissolved organic matter dynamics in pit lakes. Sci. Total Environ. 888, 164097. doi:10.1016/j.scitotenv.2023.164097
Sui, X., Wang, X., Li, Y., and Ji, H. (2021). Remediation of petroleum-contaminated soils with microbial and microbial combined methods: advances, mechanisms, and challenges. Sustainability 13, 9267. doi:10.3390/su13169267
Sulieman, S., Kusano, M., Ha, C. V., Watanabe, Y., Abdalla, M. A., Abdelrahman, M., et al. (2019). Divergent metabolic adjustments in nodules are indispensable for efficient N(2) fixation of soybean under phosphate stress. Plant Sci. 289, 110249. doi:10.1016/j.plantsci.2019.110249
Teng, W., Kuang, J., Luo, Z., and Shu, W. (2017). Microbial diversity and community assembly across environmental gradients in acid mine drainage. Minerals 7, 106. doi:10.3390/min7060106
Tian, S., Dong, Y., Pang, S., Yuan, G., Cai, S., Zhang, P., et al. (2025). Driving role of acid mine drainage on microbial community assembly and species coexistence in paddy soil profiles. J. Environ. Sci. (China) 156, 771–783. doi:10.1016/j.jes.2024.12.034
Tozsin, G., Ihsan, A. A., Sebnem, D., Hilal, S., and Torun, A. (2022). Effects of abandoned coal mine on the water quality. Int. J. Coal Prep. Util. 42, 3202–3212. doi:10.1080/19392699.2022.2044320
Vidal, M., López-Sánchez, J. F., Sastre, J., Jiménez, G., Dagnac, T., Rubio, R., et al. (1999). Prediction of the impact of the Aznalcóllar toxic spill on the trace element contamination of agricultural soils. Sci. Total Environ. 242 (1–3). doi:10.1016/S0048-9697(99)00380-0
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi:10.1128/aem.00062-07
Wang, C., Zhang, R., Xia, J., Liu, H. c., Sand, W., Etim, I. I. N., et al. (2025). Microbial community dynamics and elemental speciation in antimony-contaminated mining areas. J. Sustain. Metallurgy. doi:10.1007/s40831-025-01143-x
Wen, T., Yang, L., Dang, C., Miki, T., Bai, H., and Nagasaka, T. (2020). Effect of basic oxygen furnace slag on succession of the bacterial community and immobilization of various metal ions in acidic contaminated mine soil. J. Hazard Mater 388, 121784. doi:10.1016/j.jhazmat.2019.121784
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi:10.1093/bioinformatics/btt593
Zhang, Z., Yan, K., Zhang, L., Wang, Q., Guo, R., Yan, Z., et al. (2019). A novel cadmium-containing wastewater treatment method: bio-immobilization by microalgae cell and their mechanism. J. Hazard Mater 374, 420–427. doi:10.1016/j.jhazmat.2019.04.072
Zhang, L., Xu, Z., Sun, Y., Gao, Y., and Zhu, L. (2022). Coal mining activities driving the changes in microbial community and hydrochemical characteristics of underground mine water. Int. J. Environ. Res. Public Health 19, 13359. doi:10.3390/ijerph192013359
Zhao, J., Cao, C., Chen, X., Zhang, W., Ma, T., Irfan, M., et al. (2024). Source-specific ecological risk analysis and critical source identification of heavy metal(loid)s in the soil of typical abandoned coal mining area. Sci. Total Environ. 947, 174506. doi:10.1016/j.scitotenv.2024.174506
Zhou, M., Wang, J., Wang, H., Ran, X., Xue, H., Liu, C., et al. (2024). Revealing the comprehensive impact of organic compounds on the partial nitrification-anammox system during incineration leachate treatment: metabolic hierarchy and adaptation. Water Res. 255, 121534. doi:10.1016/j.watres.2024.121534
Keywords: microbial diversity, 16S rRNA sequencing, abandoned coal mines, metal pollution, microbial remediation, biogeochemical cycles
Citation: Xu Q, Tan D, Liu J, Qiu H and Yang Y (2025) Microbial community structure and potential functional diversity in a post-mining ecosystem of central China. Front. Environ. Sci. 13:1590925. doi: 10.3389/fenvs.2025.1590925
Received: 11 March 2025; Accepted: 04 August 2025;
Published: 02 September 2025.
Edited by:
Martin Graziano, CONICET and University of Buenos Aires, ArgentinaReviewed by:
Humberto Aponte, Universidad de O’Higgins, ChileXiaomeng Chen, Northeast Agricultural University, China
Copyright © 2025 Xu, Tan, Liu, Qiu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Yang, Y3N1eWFuZ3l1QGNzdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship