 Markus Moehler1*†
Markus Moehler1*† Katrin Goepfert1†
Katrin Goepfert1† Bernd Heinrich1
Bernd Heinrich1 Caroline J. Breitbach2Maike Delic1Peter Robert Galle1
Caroline J. Breitbach2Maike Delic1Peter Robert Galle1 Jean Rommelaere3
Jean Rommelaere3
- 11st Department of Internal Medicine, University Medical Center of the Johannes Gutenberg, University of Mainz, Mainz, Germany
- 2Jennerex Biotherapeutics, Inc., San Francisco, CA, USA
- 3Division of Tumor Virology, German Cancer Research Center (DKFZ), Heidelberg, Germany
Human tumors develop multiple strategies to evade recognition and efficient suppression by the immune system. Therefore, a variety of immunotherapeutic strategies have been developed to reactivate and reorganize the human immune system. The recent development of new antibodies against immune check points may help to overcome the immune silencing induced by human tumors. Some of these antibodies have already been approved for treatment of various solid tumor entities. Interestingly, targeting antibodies may be combined with standard chemotherapy or radiation protocols. Furthermore, recent evidence indicates that intratumoral or intravenous injections of replicative oncolytic viruses such as herpes simplex-, pox-, parvo-, or adenoviruses may also reactivate the human immune system. By generating tumor cell lysates in situ, oncolytic viruses overcome cellular tumor resistance mechanisms and induce immunogenic tumor cell death resulting in the recognition of newly released tumor antigens. This is in particular the case of the oncolytic parvovirus H-1 (H-1PV), which is able to kill human tumor cells and stimulate an anti-tumor immune response through increased presentation of tumor-associated antigens, maturation of dendritic cells, and release of pro-inflammatory cytokines. Current research and clinical studies aim to assess the potential of oncolytic virotherapy and its combination with immunotherapeutic agents or conventional treatments to further induce effective antitumoral immune responses.
Introduction
Human tumors develop complex strategies to circumvent the human immune system and to become resistant to classical therapies like radiotherapy or chemotherapy (1). Besides the low immunogenicity of tumors, tumor-induced dysregulation of the immune response leads to loss of effective immune defense and uncontrolled tumor growth. Even though many classical chemotherapy or radiation strategies induce some extent of tumor surveillance (1), new approaches should be tested to overcome early tumor resistance and recurrence. Thus, the basic challenge of molecular immune targeting is to conquer local regulatory mechanisms in order to re-introduce tumor immune recognition and promote tumor cell apoptosis and immunogenic cell death (ICD) (2). Recently, loss of immune defense has been shown to be caused by expression of different immune suppressive receptors also called immune checkpoint pathways, such as cytotoxic T-lymphocyte antigen-4 (CTLA-4) (3). Its ligation is crucial to preventing immune overreaction by inhibiting T-cell activation (4). The inhibitory CTLA-4 antibody ipilimumab [Yervoy, Bristol Myers Squibb (BMS)], approved for the treatment of metastatic melanoma patients, blocks this negative immune stimulatory receptor, thereby preventing downregulation of T-cell activation (5).
Oncolytic virotherapy represents an emerging therapeutic modality that has achieved tumor regression in several pre-clinical models and in clinical trials (6). Preferential depletion of cancer cells by oncolytic viruses (OV) is based on the fact that more aggressive tumor cells show both impaired antiviral responses and higher permissiveness for virus replication. Therefore, these agents open up new horizons for the treatment of cancer types that commonly display poor prognosis (7, 8). Cancer virotherapy is an old concept that arose from observations of unexpected tumor regressions coinciding with virus infections. This can be exemplified by a report on Newcastle disease virus (NDV) in gastric cancer dating back to 1971 (9). It should be stated that viruses with natural or engineered effects on the immune system are highly potent candidates for cancer therapy (Table 1). Herein, oncolytic viruses can be engineered to deliver therapeutic transgenes to cancer cells, causing additional anti-tumor effects through cytokine secretion and induction of anti-tumor immune responses (10–14). For example, the oncolytic vaccinia virus pexastimogene devacirepvec (Jennerex, Inc., and Transgene SA; Pexa-Vec, JX-594) and herpes simplex virus (HSV) talimogene laherparepvec (T-VEC, Amgen) were “armed” with GM-CSF-expressing genes (15, 16) to initiate local and systemic immune responses. Recently a randomized, Phase III trial of talimogene laherparepvec or GM-CSF in patients (pts) with unresectable melanoma with regional or distant metastases (OPTiM) met its primary endpoint by improving durable response rates versus GM-CSF alone, and showed a tolerable safety profile (17). A Phase II study of Pexa-Vec in primarily first-line liver cancer (HCC) patients demonstrated survival improvement in patients receiving intratumoral (it) injections of high-dose Pexa-Vec (18). The following randomized Phase IIb study in second-line HCC patients did not meet its primary endpoint of survival improvement for Pexa-Vec compared to best supportive care (BSC) (19). However, this trial was comprised primarily of patients with end-stage disease and significant comorbidities such as liver cirrhosis, therefore likely not the optimal population for successful OV therapy. Therefore, further studies of Pexa-Vec in a less advanced HCC population as well as other indications are warranted. Besides above-mentioned agents, various other viruses were shown to have oncolytic and/or immunostimulating properties, and are presently used in clinical trials. These include Parvovirus, Adenovirus, Vesicular Stomatitis Virus, Reovirus, NDV, Measles Virus, Seneca Valley Virus, Poliovirus, and Coxsackie Virus (Table 1).
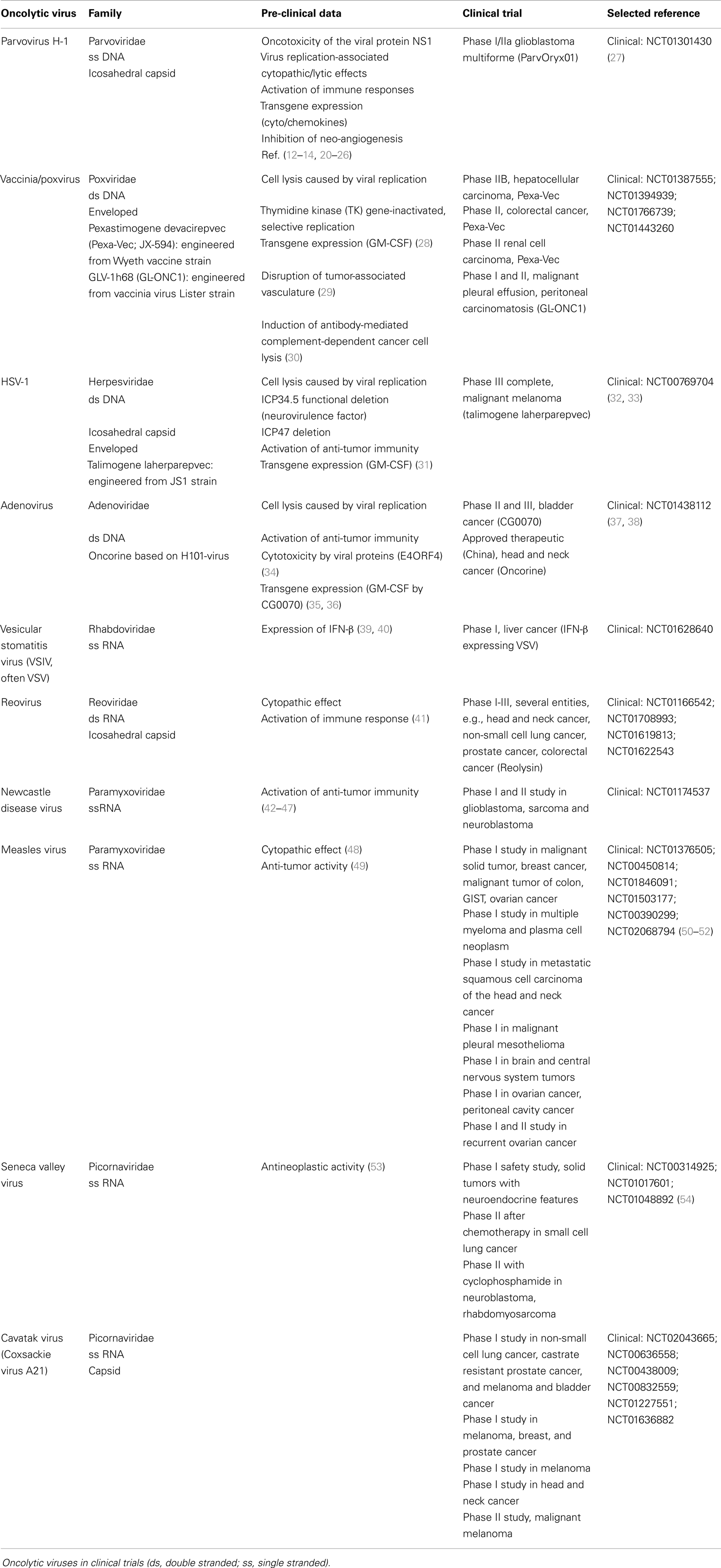
Table 1. Oncolytic viruses.
The aim of this article is to provide an overview of upcoming oncolytic viruses and their potential immunogenic therapeutic effects. A first insight into this issue is provided through our pioneer studies showing that infection with the autonomous parvovirus H-1 (H-1PV) generated immunogenic tumor cell lysates (TCLs) (14). H-1PV-infected TCLS proved able to induce maturation of dendritic cells (DCs), release of pro-inflammatory cytokines, tumor-associated antigens (TAA) cross-presentation, and T-cell stimulation in an ex vivo human melanoma model (see Figures 1 and 2) (7, 14, 55, 56). On the basis of these observations, we present the prospects of H-1PV and other OVs activating the human immune system either alone or in combination with immunomodulators, such as antibodies blocking immune suppressive receptors.
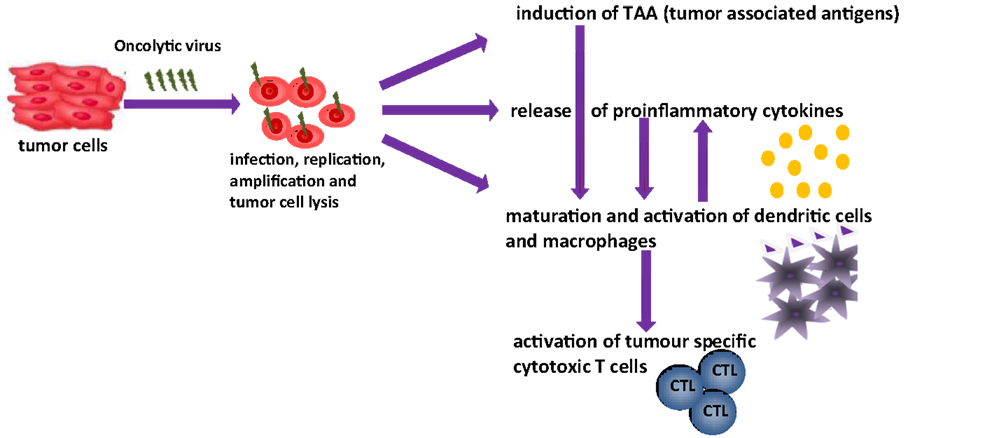
Figure 1. Oncolytic viruses and their possible function in tumor therapy [changed after Ref. (14)].
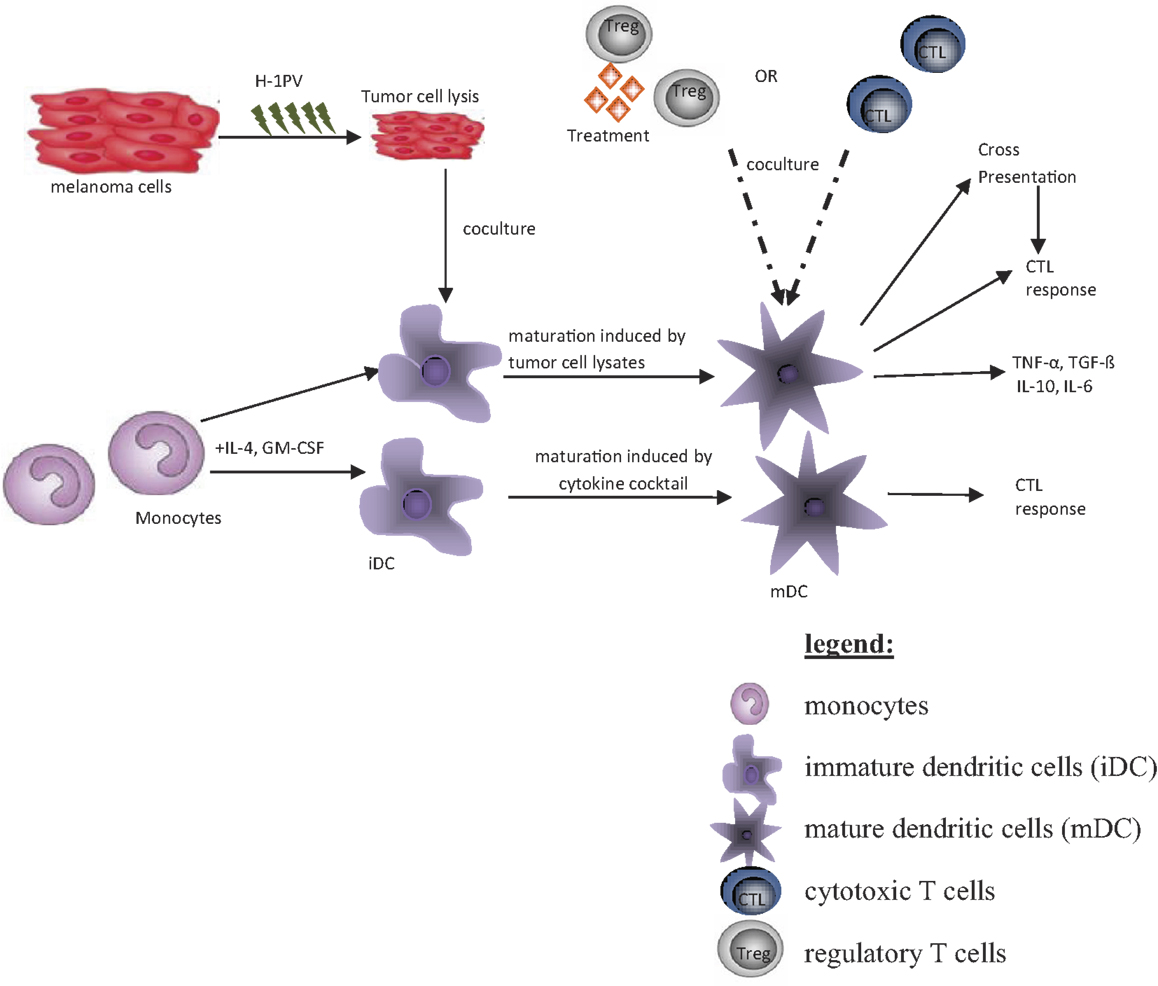
Figure 2. The ex vivo human melanoma model.
Methods
The human ex vivo melanoma model (Figure 2) represents a system that mimics the in vivo situation (14). Thus, it was used to investigate effects of H-1PV-infected or tremelimumab-treated tumor cells on immune activation. The human melanoma cells MZ7-Mel, SK29-Mel-1, and SK29-Mel-1.22 used were a gift from T. Woelfel (Mainz, Germany) (57). The SK29-Mel-1.22 cell line (A2−) is an in vitro selected HLA-A2-loss variant of HLA-A2-positive SK29-Mel-1 (A2+) line (58, 59). The cytotoxic T-cell clones CTL2/9 and CTL IVSB recognize different antigens of SK29-Mel-1 cells in association with HLA-A2 (57, 58), lyse SK29-Mel cells, and release interferon γ (IFNγ) upon specific recognition of SK29-Mel-specific TAA (58).
Peripheral blood mononuclear cells (PBMCs) were derived from buffy coats of healthy blood donors. Monocytes were isolated via adherence, and differentiation into immature DCs (iDCs) was achieved by stimulation with GM-CSF and interleukin-4. Matured DCs (mDCs) were generated by stimulation with a cytokine cocktail for 2 days (60). For coculture experiments, melanoma cells were kept in FCS-free medium. For induction of maturation and phagocytosis, tumor cells were co-cultured with iDCs at a ratio of 1:3 for 2 days. CTL-Coculture with DC was performed at 1:10 ratio (60).
Results: Oncolytic Viruses are Able Not Only to Kill Human Tumor Cells but also to Stimulate Anti-Tumor Immune Responses: The Case of Parvovirus H-1PV
Over the last years, OV therapy has shown promising results in both pre-clinical and clinical studies against various solid tumors (61). It is worth noting that besides their own anti-tumor efficiency, OVs can resensitize resistant tumors to chemotherapeutics, thereby highlighting the potential of OVs in multimodal treatments (12, 13). We were particularly interested in the oncolytic parvovirus H-1PV [for reviews, see Ref. (20, 62)]. The mode of action of H-1PV involves both direct oncolytic and immune-mediated components, making this virus an attractive candidate for inclusion in the cancer immunotherapy armamentarium (60). H-1PV is a small nuclear-replicating DNA virus, which preferentially multiplies in oncogene-transformed and tumor-derived cells (7). This oncotropism results at least in part from the dependence of H-1PV on proliferation and differentiation factors that are dysregulated in neoplastic cells (20). In consequence, H-1PV exerts oncolytic effects, which were documented in human cells from various tumor entities including melanoma, pancreatic (PDAC), hepatocellular (HCC), colorectal or gastric carcinomas, sarcoma, glioma, and other neuroectodermal tumors (7, 20, 21, 62–64). Most interestingly, the death mechanisms activated by parvoviruses allow them to overcome resistance of tumor cells to conventional cytotoxic agents (22, 65). Another intriguing aspect of H-1PV-mediated OV lies in the possibility of combining H-1PV with conventional cytotoxic drugs to achieve synergistic tumor cell killing effects, as demonstrated for instance in the PDAC system (13, 21, 22, 66).
Though not or poorly infectious for humans under natural conditions, H-1PV can be administered experimentally to patients, resulting in viremia and seroconversion (67). Infections with H-1PV appear to be clinically silent (68). It should also be stated that recombinant parvoviruses can be constructed, for example to transduce immunostimulatory cytokines (62). This arming strategy was found to increase the anti-tumor effects of parvoviruses in certain models (69–71).
Bringing H-1PV from the Bench to the Bedside
Recent work using an immunocompetent rat glioma model showed that H-1PV was able to efficiently cure gliomas, while raising an anti-tumor memory immune response. This oncosuppressive effect appears to rely on both the direct oncolytic activity of H-1PV and its handover to the host immune system (23). These pre-clinical data led to the current clinical evaluation of H-1PV it and intravenous (iv) administration to patients with recurrent resectable GBM progressing in spite of conventional therapies (27).
H-1PV-Induced Tumor Cell Lysates Trigger Maturation of IDCs and Exert Immunostimulating Effects
H-1PV had little direct killing activity on human immune cells in vitro, in particular APCs and CTLs. Interestingly, the analysis of infected PBMCs revealed the induction of markers of both macrophage and Th1cell activation (Table 2). This Th1 bias is indicative of a possible direct immunostimulating capacity of the parvovirus. Nevertheless, a major impact of H-1PV on the immune system appears to be indirect, i.e., mediated by infected tumor cells, as discussed in the following sections. H-1PV caused the death of human melanoma cells in culture, including the above-mentioned SK29-Mel-1 and SK29-Mel-1.22 lines. The extent of cell killing varied between tested lines, was dependent on the multiplicity of infection (MOI) and correlated with expression of the replicative viral non-structural protein NS1. In this system, H-1PV induced an apoptotic cell death, which was accompanied with the release of immunogenic HSP72 (63).
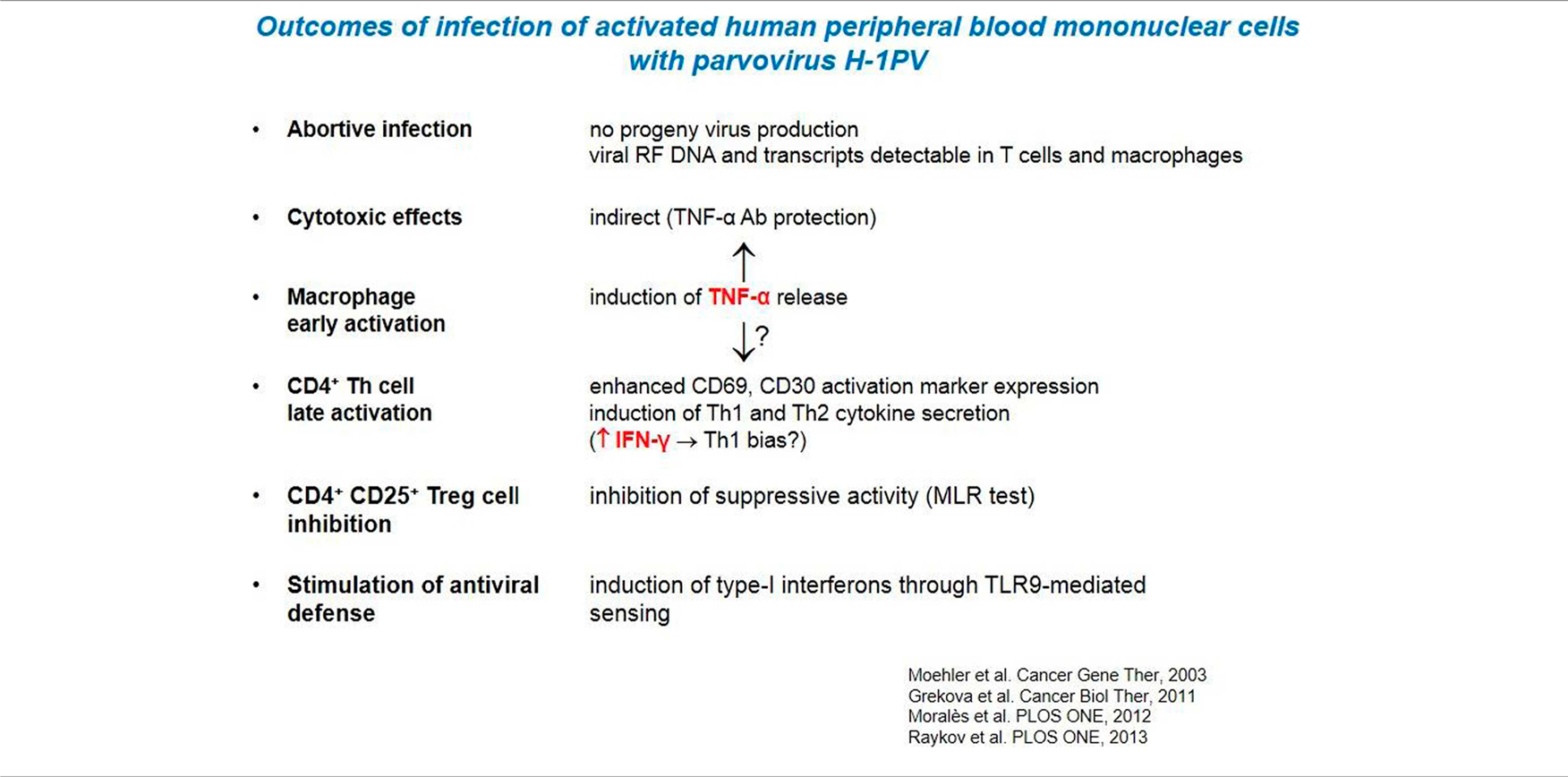
Table 2. Direct immunostimulating effects of parvovirus H-1PV.
In further experiments it was shown that H-1PV-infected melanoma TCLs were phagocytosed by iDCs and induced their maturation, in particular the secretion of pro-inflammatory cytokines such as TNFα and IL-6 (13, 63). Lysates of infected SK29-Mel-1.22 and MZ7-Mel cells were both competent for inducing DC maturation, although the former were more potent than the latter in this regard (13, 14). Primary immune cells were not permissive for H-1PV infection. Little direct killing effect, no apoptosis, and no progeny virus production could be detected in infected lymphocytes, monocytes, immature, and mature DCs (Table 2) (63).
We also demonstrated that human DCs coincubated with H-1PV-induced melanoma TCLs showed enhanced expression of TLR3, TLR9, and other maturation markers. This suggested that virus-induced TCLs contained molecular patterns triggering TLR signaling in DCs, as further evidenced by increased NF-κB levels and production of pro-inflammatory cytokines (12). Some of these immunostimulating patterns may consist of viral constituents, given the known ability of TLR3 and TLR9 for sensing viral determinants.
Combination of the oncolytic virus with cytostatic (cisplatin, vincristine) or targeted (sunitinib) drugs resulted in a further increase in melanoma cell apoptosis but failed to strengthen maturation of DCs. It was verified that the cytotoxic or targeted drug regimen used did not interfere with H-1PV infection (13). Interestingly, the interleukin profile of DCs was altered upon exposure to H-1PV plus sunitinib-cotreated TCLs. It therefore appears that H-1PV combination with this anti-angiogenic drug may reinforce its capacity not only for jeopardizing tumor cell survival but also for modulating the immune system.
H-1PV Induce Activation of Antigen-Specific Cytotoxic T-Cells and Other Anti-Tumor Immune Effectors
To further assess whether phagocytosis of H-1-infected TCLs by DCs induces cross-presentation of TAAs to antigen-specific CTLs in an HLA-class I-restricted manner, the above-mentioned human melanoma in vitro model was used (58, 72). Both melanoma-specific CTL clones tested were found to release increased levels of IFNγ after being co-cultured with DCs preincubated with H-1PV-infected SK29-Mel-1 or HLA-negative SK29-Mel-1.22 cells (14). Thus, H-1PV-induced TCLs stimulated cross-presentation of TAAs by DCs. This effect may contribute to reinforce the anti-tumor immune response by generating tumor-specific CTLs (14). In addition, several H-1PV-infected tumor cells were recently found to acquire an enhanced capacity for activating NK cells and getting killed by these cells (73, 74). The adjuvant effect of H-1PV was also evidenced in vivo by the virus-enhanced efficacy of an autologous tumor cell vaccine (24) and the adoptive transfer of anti-tumor immune cells from animals undergoing oncolytic H-1PV therapy (75).
Oncolytic H-1PV Virotherapy Can be Combined with Immunotherapeutic Agents to Enhance Treatment Efficacy
Recent evidence for the expression of the immunosuppressing molecule CTLA-4 on regulatory T-cells (Tregs) and tumors generated widespread interest in the role of CTLA-4 in tumor escape and peripheral tolerance (3, 58). In particular, the human colon adenocarcinoma line SW480 was found to express CTLA-4 on the cell surface. This prompted us to extend the analysis of H-1PV anti-tumor effects to the SW480 system in combination with the anti-CTLA-4 antibody tremelimumab. When applied alone, this antibody had no detectable effect on SW480 cell viability and DC maturation. On the other hand, H-1PV alone was able to kill SW480 cells in a MOI-dependent manner. H-1PV-induced SW480 TCLs triggered iDC maturation in coculture experiments, as revealed in particular by increased release of the pro-inflammatory cytokines IFNγ, TNFα, and IL-6 (64). The secretion of IFNγ was stimulated to a low extent by treatment of the coculture with tremelimumab, recommend the use of the H-1PV/tremelimumab combination treatment to enhance tumor immunogenicity through both DC activation and CTLA-4 masking. It should also be stated that other (immuno)modulators, namely IFNγ (75) and HDAC inhibitors (76), were recently reported to cooperate with H-1PV for tumor suppression in human carcinoma animal models.
Clinical Evidence of OV-Mediated Activation of Immune Responses in Humans
Extensive analyses were performed to evaluate mechanisms-of-action of the oncolytic and immunotherapeutic vaccinia virus Pexa-Vec in patients. These include oncolysis (15, 77, 78), acute vascular disruption (29) as well as anti-tumor immune response induction. Pexa-Vec was engineered to express GM-CSF to stimulate white blood cell production and activate DCs. Detectable concentrations of GM-CSF in plasma were measured 4–15 days after treatment and associated with increased neutrophil, monocyte, and eosinophil production in patients receiving iv or it iPexa-Vec (77, 78). Inflammatory cell recruitment to tumors was confirmed on biopsy following Pexa-Vec administration in patients with melanoma (79, 80). Furthermore, functional anti-cancer immunity of Pexa-Vec treatment was demonstrated in patients by measuring induction of antibody-mediated complement-dependent cytotoxicity (CDC) utilizing a panel of tumor cell lines of different histologies (30). Low concentrations of serum ex vivo incubated with tumor cells resulted in a dramatic reduction in tumor cell viability; when normal cells did not exhibit decreased viability. This activity was shown to be dependent on both active complement as well as IgG antibody. Reproducible CDC activity was also observed in a Phase II study in HCC patient (18). Furthermore, T-cell responses to β-galactosidase peptides were detected in HCC patients treated with Pexa-Vec, as shown by ELISPOT analysis. In that way, the proof-of-concept provides that T-cell responses can be induced to transgenes encoded by oncolytic vaccinia viruses (18).
Talimogene laherparepvec is an oncolytic immunotherapy comprising a modified HSV type 1 engineered to selectively replicate in tumor cells and to express the immune-stimulating cytokine GM-CSF, while retaining sensitivity to antiherpetic agents (16). Local effects after intralesional injection include selective lysis of tumor cells and subsequent release of tumor antigen, as well as secretion of GM-CSF into the local environment, which results in the stimulation and maturation of DCs (32, 81). Antigen presentation by stimulated DCs to CD4+ and CD8+ cells may induce an adaptive systemic immune response (16, 82, 83). Recently a randomized, Phase III trial of talimogene laherparepvec in patients (pts) with unresected melanoma with regional or distant metastases (OPTiM) met its primary endpoint, demonstrating a significant improvement in durable response rate (defined as partial or complete responses that were maintained for ≥6 months starting within 12 months) versus GM-CSF alone (16 versus 2%, p < 0.0001) (17). Overall response rate was also higher in the talimogene laherparepvec arm (26.4 versus 5.7%, p < 0.0001). Subjects treated with talimogene laherparepvec showed a tolerable safety profile with the only grade 3/4 adverse event that occurred in >2% of patients being cellulitis (2.1%). A trend toward improved overall survival was seen based on a planned interim analysis (17). The primary overall survival results are pending. Evidence of durable responses together with the safety profile of talimogene laherparepvec supports evaluation of combinations with other immunotherapies, such as high-dose IL-2 or immune checkpoint blockade and with radiation therapy, chemotherapy, and/or targeted therapies that might amplify the anti-tumor response generated by talimogene laherparepvec (32).
Discussion: Potential of the Immunovirotherapy Concept
Despite recent improvements in surgical, locoregional, and systemic therapies, the prognosis of patients with gastrointestinal, hepatobiliary, and pancreatic cancers remains dismal, and treatment is limited to palliation in the majority of patients. These limitations indicate an urgent need for novel therapeutic strategies (13, 64, 66, 84). Combinations of oncolytic viruses with new targeted therapies draw much attention. It is however necessary to proceed with caution, as these therapies may interfere with pathways, which are needed for replication of genetically modified viruses. It was demonstrated that by interacting with the EGFR/RAS/RAF pathway, sorafenib inhibits replication of Pexa-Vec in liver cancer, when applied in combination. This is not surprising as Pexa-Vec replication is in part dependent on the EGFR/RAS/RAF pathway (85). Nevertheless, sequential therapy with Pexa-Vec followed by sorafenib resulted in decreased tumor perfusion and was associated with objective tumor responses for HCC (85). It is noteworthy that some oncolytic viruses such as parvovirus H-1PV also have potential to inhibit neo-angiogenesis. Therefore, OV-based combination treatments targeting both tumor cell proliferation and tumor angiogenesis represent a promising strategy for impeding the growth of various cancers (25).
Besides their low expression of TAA and low immunogenicity, tumors can induce an immune tolerance milieu by releasing anti-inflammatory cytokines such as IL-10 or TGF-β or recruiting Tregs to their microenvironment (86). T-cell activation relies on both, recognition of major histocompatibility complex (MHC) molecules by the T-cell receptor (TCR), and on costimulatory signals. Depending on the type of costimulatory receptor, T-cells can be activated or become anergic. For example, T-cell activation was prevented by engagement of CTLA-4 receptors with CD80 or CD86. In contrast, engagement of CD80 or CD86 with CD28 induced T-cell activation, often with a low affinity (87). Thus, a promising therapeutic option to achieve strong anti-tumor immune responses is the use of monoclonal antibodies against CTLA-4 and PD-1 alone or in combination. Herein, the constitutive expression of CTLA-4 and PD-1 on Tregs may play a crucial role in inhibiting anti-tumor T-cell responses. Tregs are often found in the peripheral blood of cancer patients and in the tumor microenvironment. These cells suppress an optimal anti-tumor immune response by preventing infiltrating CD8+ T-cells from proliferating and producing cytolytic granules (88). BMS developed an anti-CTLA-4 monoclonal antibody named ipilimumab and an anti-PD-1 monoclonal antibody named nivolumab. Both antibodies were already tested in Phase III trials and found to achieve clinically significant benefits in median overall survival (89, 90). First pre-clinical studies of the combination of these antibodies to achieve blockade of both CTLA-4 and PD-1 showed increased tumor infiltration by CD4+ and CD8+ T-cells, enhanced IFNγ and TNFα production, and reduced amounts of Tregs (91). A Phase I study of nivolumab and ipilimumab combination in advanced melanoma patients showed an outstanding activity in 65% of patients with an objective response rate of 40% (92). As part of their further development and mechanistic understanding, these antibodies against immune check points would certainly deserve to be combined with OV in order to optimize anti-tumor immune responses. Preliminary data from a Phase Ib trial combining talimogene laherparepvec with ipilimumab indicated that the combination was tolerable and devoid of unexpected toxicities (93). Exploiting these combinations represents a promising strategy to bring oncolytic viruses from bench to bedside and to establish oncolytic virotherapy as a new effective immunotherapeutic approach.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Christiane Dinsart from the DKFZ for her support. Eduard Gasal (Amgen Inc., Thousand Oaks, CA, USA) provided the text relating to talimogene laherparepvec clinical development and data.
Key Concepts
• Key concept1: There is a consistent need for immunotherapies in the treatment of human cancer.
• Key concept2: Oncolytic viruses reduce tumor burden and show first clinical results in humans.
• Key concept3: Oncolytic viruses, such as parvovirus H-1PV, induce effective anti-tumor immune responses.
• Key concept4: Combinations of oncolytic viruses with immunotherapeutics are likely to achieve enhanced immune activation.
References
1. Duffy AG, Greten TF. Immunological off-target effects of standard treatments in gastrointestinal cancers. Ann Oncol (2014) 25(1):24–32. doi:10.1093/annonc/mdt349
2. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi:10.1038/nm1523
3. Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Fala F, Fabbi M, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer (2005) 117(4):538–50. doi:10.1002/ijc.21155
4. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res (2013) 73(12):3591–603. doi:10.1158/0008-5472.CAN-12-4100
5. Weber JS, Amin A, Minor D, Siegel J, Berman D, O’Day SJ. Safety and clinical activity of ipilimumab in melanoma patients with brain metastases: retrospective analysis of data from a phase 2 trial. Melanoma Res (2011) 21(6):530–4. doi:10.1097/CMR.0b013e32834d3d88
6. Carpenter SG, Carson J, Fong Y. Regional liver therapy using oncolytic virus to target hepatic colorectal metastases. Semin Oncol (2010) 37(2):160–9. doi:10.1053/j.seminoncol.2010.03.001
7. Moehler M, Blechacz B, Weiskopf N, Zeidler M, Stremmel W, Rommelaere J, et al. Effective infection, apoptotic cell killing and gene transfer of human hepatoma cells but not primary hepatocytes by parvovirus H1 and derived vectors. Cancer Gene Ther (2001) 8(3):158–67. doi:10.1038/sj.cgt.7700288
8. Donnelly O, Harrington K, Melcher A, Pandha H. Live viruses to treat cancer. J R Soc Med (2013) 106(8):310–4. doi:10.1177/0141076813494196
9. Csatary L. Viruses in the treatment of cancer. Lancet (1971) 298(7728):825. doi:10.1016/S0140-6736(71)92788-7
10. Bourke MG, Salwa S, Harrington KJ, Kucharczyk MJ, Forde PF, de Kruijf M, et al. The emerging role of viruses in the treatment of solid tumours. Cancer Treat Rev (2011) 37(8):618–32. doi:10.1016/j.ctrv.2010.12.003
11. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol (2012) 30(7):658–70. doi:10.1038/nbt.2287
12. Sieben M, Schafer P, Dinsart C, Galle PR, Moehler M. Activation of the human immune system via toll-like receptors by the oncolytic parvovirus H-1. Int J Cancer (2012) 132(11):2548–56. doi:10.1002/ijc.27938
13. Moehler M, Sieben M, Roth S, Springsguth F, Leuchs B, Zeidler M, et al. Activation of the human immune system by chemotherapeutic or targeted agents combined with the oncolytic parvovirus H-1. BMC Cancer (2011) 11:464. doi:10.1186/1471-2407-11-464
14. Moehler MH, Zeidler M, Wilsberg V, Cornelis JJ, Woelfel T, Rommelaere J, et al. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum Gene Ther (2005) 16(8):996–1005. doi:10.1089/hum.2005.16.996
15. Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther (2006) 14(3):361–70. doi:10.1016/j.ymthe.2006.05.008
16. Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther (2003) 10(4):292–303. doi:10.1038/sj.gt.3301885
17. Andtbacka RHI, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, et al. OPTiM: a randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol (2013) 31(suppl; abstr LBA9008).
18. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013) 19(3):329–36. doi:10.1038/nm.3089
19. Transgene Announces that its Phase 2 Study of Pexa-Vec in Second-line Advanced Liver Cancer did not Meet its Primary Endpoint (2013). Available from: http://www.transgene.fr/index.php?option=com_press_release&task=download&id=236&l=en
20. Nuesch JP, Lacroix J, Marchini A, Rommelaere J. Molecular pathways: rodent parvoviruses – mechanisms of oncolysis and prospects for clinical cancer treatment. Clin Cancer Res (2012) 18(13):3516–23. doi:10.1158/1078-0432.CCR-11-2325
21. Sieben M, Herzer K, Zeidler M, Heinrichs V, Leuchs B, Schuler M, et al. Killing of p53-deficient hepatoma cells by parvovirus H-1 and chemotherapeutics requires promyelocytic leukemia protein. World J Gastroenterol (2008) 14(24):3819–28. doi:10.3748/wjg.14.3819
22. Angelova AL, Aprahamian M, Grekova SP, Hajri A, Leuchs B, Giese NA, et al. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin Cancer Res (2009) 15(2):511–9. doi:10.1158/1078-0432.CCR-08-1088
23. Geletneky K, Kiprianova I, Ayache A, Koch R, Herrero YCM, Deleu L, et al. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol (2010) 12(8):804–14. doi:10.1093/neuonc/noq023
24. Raykov Z, Grekova S, Leuchs B, Aprahamian M, Rommelaere J. Arming parvoviruses with CpG motifs to improve their oncosuppressive capacity. Int J Cancer (2008) 122(12):2880–4. doi:10.1002/ijc.23472
25. Lavie M, Struyf S, Stroh-Dege A, Rommelaere J, Van Damme J, Dinsart C. Capacity of wild-type and chemokine-armed parvovirus H-1PV for inhibiting neo-angiogenesis. Virology (2013) 447(1–2):221–32. doi:10.1016/j.virol.2013.09.019
26. Enderlin M, Kleinmann EV, Struyf S, Buracchi C, Vecchi A, Kinscherf R, et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther (2009) 16(2):149–60. doi:10.1038/cgt.2008.62
27. Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, Dahm M, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer (2012) 12:99. doi:10.1186/1471-2407-12-99
28. Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther (2012) 20(4):749–58. doi:10.1038/mt.2011.276
29. Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res (2013) 73(4):1265–75. doi:10.1158/0008-5472.CAN-12-2687
30. Kim MK, Breitbach CJ, Moon A, Heo J, Lee YK, Cho M, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med (2013) 5(185):185ra63. doi:10.1126/scitranslmed.3005361
31. Carson J, Haddad D, Bressman M, Fong Y. Oncolytic herpes simplex virus 1 (HSV-1) vectors: increasing treatment efficacy and range through strategic virus design. Drugs Future (2010) 35(3):183–95. doi:10.1358/dof.2010.35.3.1470166
32. Kaufman HL, Bines SD. OPTIM trial: a phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol (2010) 6(6):941–9. doi:10.2217/fon.10.66
33. Kaufman H, Harrington K, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Secondary Endpoints from OPTiM: A Multicenter, Randomized Phase 3 Trial of Talimogene Laherparepvec vs GM-CSF for the Treatment of Unresected Stage IIIB/C and IV Melanoma. The European Cancer Congress (2013). Available from: http://eccamsterdam2013.ecco-org.eu/Scientific-Programme/Abstract-search.aspx?abstractid=7021
34. Li S, Szymborski A, Miron MJ, Marcellus R, Binda O, Lavoie JN, et al. The adenovirus E4orf4 protein induces growth arrest and mitotic catastrophe in H1299 human lung carcinoma cells. Oncogene (2009) 28(3):390–400. doi:10.1038/onc.2008.393
35. Fang L, Cheng Q, Bai J, Qi YD, Liu JJ, Li LT, et al. An oncolytic adenovirus expressing interleukin-24 enhances antitumor activities in combination with paclitaxel in breast cancer cells. Mol Med Rep (2013) 8(5):1416–24. doi:10.3892/mmr.2013.1680
36. Ramesh N, Ge Y, Ennist DL, Zhu M, Mina M, Ganesh S, et al. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor – armed oncolytic adenovirus for the treatment of bladder cancer. Clin Cancer Res (2006) 12(1):305–13. doi:10.1158/1078-0432.CCR-05-1059
37. Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst (2006) 98(5):298–300. doi:10.1093/jnci/djj111
38. Liang M. Clinical development of oncolytic viruses in China. Curr Pharm Biotechnol (2012) 13(9):1852–7. doi:10.2174/138920112800958760
39. D’Agostino PM, Amenta JJ, Reiss CS. IFN-beta-induced alteration of VSV protein phosphorylation in neuronal cells. Viral Immunol (2009) 22(6):353–69. doi:10.1089/vim.2009.0057
40. Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, et al. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther (2010) 21(1):51–64. doi:10.1089/hum.2009.088
41. Steele L, Errington F, Prestwich R, Ilett E, Harrington K, Pandha H, et al. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol Cancer (2011) 10:20. doi:10.1186/1476-4598-10-20
42. Ni J, Galani IE, Cerwenka A, Schirrmacher V, Fournier P. Antitumor vaccination by Newcastle disease virus hemagglutinin-neuraminidase plasmid DNA application: changes in tumor microenvironment and activation of innate anti-tumor immunity. Vaccine (2011) 29(6):1185–93. doi:10.1016/j.vaccine.2010.12.005
43. Jarahian M, Watzl C, Fournier P, Arnold A, Djandji D, Zahedi S, et al. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J Virol (2009) 83(16):8108–21. doi:10.1128/JVI.00211-09
44. Fournier P, Bian H, Szeberenyi J, Schirrmacher V. Analysis of three properties of Newcastle disease virus for fighting cancer: tumor-selective replication, antitumor cytotoxicity, and immunostimulation. Methods Mol Biol (2012) 797:177–204. doi:10.1007/978-1-61779-340-0_13
45. Fournier P, Schirrmacher V. Tumor antigen-dependent and tumor antigen-independent activation of antitumor activity in T cells by a bispecific antibody-modified tumor vaccine. Clin Dev Immunol (2010) 2010:84. doi:10.1155/2010/423781
46. Schirrmacher V, Fournier P. Newcastle disease virus: a promising vector for viral therapy, immune therapy, and gene therapy of cancer. Methods Mol Biol (2009) 542:565–605. doi:10.1007/978-1-59745-561-9_30
47. Fournier P, Arnold A, Wilden H, Schirrmacher V. Newcastle disease virus induces pro-inflammatory conditions and type I interferon for counter-acting Treg activity. Int J Oncol (2012) 40(3):840–50. doi:10.3892/ijo.2011.1265
48. Msaouel P, Iankov ID, Dispenzieri A, Galanis E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr Pharm Biotechnol (2012) 13(9):1732–41. doi:10.2174/138920112800958896
49. Russell SJ, Peng KW. Measles virus for cancer therapy. Curr Top Microbiol Immunol (2009) 330:213–41.
50. Heinzerling L, Kunzi V, Oberholzer PA, Kundig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood (2005) 106(7):2287–94. doi:10.1182/blood-2004-11-4558
51. Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res (2010) 70(3):875–82. doi:10.1158/0008-5472.CAN-09-2762
52. Mader EK, Butler G, Dowdy SC, Mariani A, Knutson KL, Federspiel MJ, et al. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J Transl Med (2013) 11:20. doi:10.1186/1479-5876-11-20
53. Reddy PS, Burroughs KD, Hales LM, Ganesh S, Jones BH, Idamakanti N, et al. Seneca valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J Natl Cancer Inst (2007) 99(21):1623–33. doi:10.1093/jnci/djm198
54. Rudin CM, Poirier JT, Senzer NN, Stephenson J Jr, Loesch D, Burroughs KD, et al. Phase I clinical study of seneca valley virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin Cancer Res (2011) 17(4):888–95. doi:10.1158/1078-0432.CCR-10-1706
55. Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat Med (2001) 7(7):781–7. doi:10.1038/89901
56. Cawood R, Hills T, Wong SL, Alamoudi AA, Beadle S, Fisher KD, et al. Recombinant viral vaccines for cancer. Trends Mol Med (2012) 18(9):564–74. doi:10.1016/j.molmed.2012.07.007
57. Wolfel T, Van Pel A, Brichard V, Schneider J, Seliger B, Meyer zum Buschenfelde KH, et al. Two tyrosinase nonapeptides recognized on HLA-A2 melanomas by autologous cytolytic T lymphocytes. Eur J Immunol (1994) 24(3):759–64. doi:10.1002/eji.1830240340
58. Wolfel T, Klehmann E, Muller C, Schutt KH, Meyer zum Buschenfelde KH, Knuth A. Lysis of human melanoma cells by autologous cytolytic T cell clones. Identification of human histocompatibility leukocyte antigen A2 as a restriction element for three different antigens. J Exp Med (1989) 170(3):797–810. doi:10.1084/jem.170.3.797
59. Wang Z, Seliger B, Mike N, Momburg F, Knuth A, Ferrone S. Molecular analysis of the HLA-A2 antigen loss by melanoma cells SK-MEL-29.1.22 and SK-MEL-29.1.29. Cancer Res (1998) 58(10):2149–57.
60. Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol (1997) 27(12):3135–42. doi:10.1002/eji.1830271209
61. Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol (2014) 12(1):23–34. doi:10.1038/nrmicro3140
62. Rommelaere J, Geletneky K, Angelova AL, Daeffler L, Dinsart C, Kiprianova I, et al. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev (2010) 21(2–3):185–95. doi:10.1016/j.cytogfr.2010.02.011
63. Moehler M, Zeidler M, Schede J, Rommelaere J, Galle PR, Cornelis JJ, et al. Oncolytic parvovirus H1 induces release of heat-shock protein HSP72 in susceptible human tumor cells but may not affect primary immune cells. Cancer Gene Ther (2003) 10(6):477–80. doi:10.1038/sj.cgt.7700591
64. Heinrich B, Goepfert K, Delic M, Galle PR, Moehler M. Influence of the oncolytic parvovirus H-1, CTLA-4 antibody tremelimumab and cytostatic drugs on the human immune system in a human in vitro model of colorectal cancer cells. Onco Targets Ther (2013) 6:1119–27. doi:10.2147/OTT.S49371
65. Di Piazza M, Mader C, Geletneky K, Herrero YCM, Weber E, Schlehofer J, et al. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J Virol (2007) 81(8):4186–98. doi:10.1128/JVI.02601-06
66. Wittwer C, Boeck S, Heinemann V, Haas M, Stieber P, Nagel D, et al. Circulating nucleosomes and immunogenic cell death markers HMGB1, sRAGE and DNAse in patients with advanced pancreatic cancer undergoing chemotherapy. Int J Cancer (2013) 133(11):2619–30. doi:10.1002/ijc.28294
67. Siegl G. Molecular biology and pathogenicity of human and animal parvoviruses. Behring Inst Mitt (1990) 85:6–13.
68. Guglielmino S, Tempera G, Pappalardo G, Castro A. H-1 and X14 parvovirus antibodies in women with abortions or still-births. Acta Virol (1978) 22(5):426–8.
69. Haag A, Menten P, Van Damme J, Dinsart C, Rommelaere J, Cornelis JJ. Highly efficient transduction and expression of cytokine genes in human tumor cells by means of autonomous parvovirus vectors; generation of antitumor responses in recipient mice. Hum Gene Ther (2000) 11(4):597–609. doi:10.1089/10430340050015789
70. Wetzel K, Menten P, Opdenakker G, Van Damme J, Grone HJ, Giese N, et al. Transduction of human MCP-3 by a parvoviral vector induces leukocyte infiltration and reduces growth of human cervical carcinoma cell xenografts. J Gene Med (2001) 3(4):326–37. doi:10.1002/jgm.191
71. Giese NA, Raykov Z, DeMartino L, Vecchi A, Sozzani S, Dinsart C, et al. Suppression of metastatic hemangiosarcoma by a parvovirus MVMp vector transducing the IP-10 chemokine into immunocompetent mice. Cancer Gene Ther (2002) 9(5):432–42. doi:10.1038/sj.cgt.7700457
72. Arienti F, Sule-Suso J, Belli F, Mascheroni L, Rivoltini L, Melani C, et al. Limited antitumor T cell response in melanoma patients vaccinated with interleukin-2 gene-transduced allogeneic melanoma cells. Hum Gene Ther (1996) 7(16):1955–63. doi:10.1089/hum.1996.7.16-1955
73. Bhat R, Rommelaere J. NK-cell-dependent killing of colon carcinoma cells is mediated by natural cytotoxicity receptors (NCRs) and stimulated by parvovirus infection of target cells. BMC Cancer (2013) 13:367. doi:10.1186/1471-2407-13-367
74. Bhat R, Dempe S, Dinsart C, Rommelaere J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int J Cancer (2011) 128(4):908–19. doi:10.1002/ijc.25415
75. Grekova S, Aprahamian M, Giese N, Schmitt S, Giese T, Falk CS, et al. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol Ther (2011) 10(12):1280–9. doi:10.4161/cbt.10.12.13455
76. Li J, Bonifati S, Hristov G, Marttila T, Valmary-Degano S, Stanzel S, et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol Med (2013) 5(10):1537–55. doi:10.1002/emmm.201302796
77. Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol (2008) 9(6):533–42. doi:10.1016/S1470-2045(08)70107-4
78. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature (2011) 477(7362):99–102. doi:10.1038/nature10358
79. Mastrangelo MJ, Maguire HC Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther (1999) 6(5):409–22. doi:10.1038/sj.cgt.7700066
80. Hwang TH, Moon A, Burke J, Ribas A, Stephenson J, Breitbach CJ, et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol Ther (2011) 19(10):1913–22. doi:10.1038/mt.2011.132
81. Eager R, Nemunaitis J. GM-CSF gene-transduced tumor vaccines. Mol Ther (2005) 12(1):18–27. doi:10.1016/j.ymthe.2005.02.012
82. Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res (2006) 12(22):6737–47. doi:10.1158/1078-0432.CCR-06-0759
83. Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol (2009) 27(34):5763–71. doi:10.1200/JCO.2009.24.3675
84. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet (2012) 379(9822):1245–55. doi:10.1016/S0140-6736(11)61347-0
85. Heo J, Breitbach CJ, Moon A, Kim CW, Patt R, Kim MK, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther (2011) 19(6):1170–9. doi:10.1038/mt.2011.39
86. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6(4):295–307. doi:10.1038/nri1806
87. Perez N, Karumuthil-Melethil S, Li R, Prabhakar BS, Holterman MJ, Vasu C. Preferential costimulation by CD80 results in IL-10-dependent TGF-beta1(+) -adaptive regulatory T cell generation. J Immunol (2008) 180(10):6566–76.
88. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi:10.1016/j.cell.2010.01.025
89. Weber JS, O’Day S, Urba W, Powderly J, Nichol G, Yellin M, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol (2008) 26(36):5950–6. doi:10.1200/JCO.2008.16.1927
90. Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol (2013) 31(34):4311–8. doi:10.1200/JCO.2013.51.4802
91. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A (2010) 107(9):4275–80. doi:10.1073/pnas.0915174107
92. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
93. Puzanov I, Milhem M, Andtbacka R, Minor D, Hamid O, Li A, et al. Phase 1 results of a phase 1b/2, multicenter, open-label trial to evaluate safety and efficacy of talimogene laherparepvec (T-VEC) and ipilimumab (ipi) vs ipi alone in previously untreated, unresected stage IIIB-IV melanoma. J Immunother Cancer (2013) 1(Suppl 1):84. doi:10.1186/2051-1426-1-S1-P84
Keywords: immunotherapy, autonomous parvovirus, H-1PV, talimogene laherparepvec, T-VEC, JX-594, dendritic cells, CTLA-4
Citation: Moehler M, Goepfert K, Heinrich B, Breitbach CJ, Delic M, Galle PR and Rommelaere J (2014) Oncolytic virotherapy as emerging immunotherapeutic modality: potential of parvovirus H-1. Front. Oncol. 4:92. doi: 10.3389/fonc.2014.00092
Received: 19 February 2014; Accepted: 14 April 2014;
Published online: 01 May 2014.
Edited by:
Philippe Fournier, DKFZ, GermanyReviewed by:
Valery Grdzelishvili, University of North Carolina at Charlotte, USAVolker Schirrmacher, DKFZ, Germany
Yong Lu, Cleveland Clinic Foundation, USA
William L. Redmond, Earle A. Chiles Research Institute, USA
Copyright: © 2014 Moehler, Goepfert, Heinrich, Breitbach, Delic, Galle and Rommelaere. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Moehler, 1st Department of Internal Medicine, University Medical Center of the Johannes Gutenberg, University of Mainz, Building 601, Langenbeckstrasse 1, Mainz 55131, Germany e-mail:bWFya3VzLm1vZWhsZXJAdW5pbWVkaXppbi1tYWluei5kZQ==
†Markus Moehler and Katrin Goepfert have contributed equally to this work.